

Abstract
Targeted protein degradation induced by heterobifunctional compounds and molecular glues presents an exciting avenue for chemical probe and drug discovery. To date, small-molecule ligands have been discovered for only a limited number of E3 ligases, which is an important limiting factor for realizing the full potential of targeted protein degradation. We report herein the discovery by chemical proteomics of azetidine acrylamides that stereoselectively and site-specifically react with a cysteine (C1113) in the E3 ligase substrate receptor DCAF1. We demonstrate that the azetidine acrylamide ligands for DCAF1 can be developed into electrophilic PROTACs (proteolysis-targeting chimeras) that mediated targeted protein degradation in human cells. We show that this process is stereoselective and does not occur in cells expressing a C1113A mutant of DCAF1. Mechanistic studies indicate that only low fractional engagement of DCAF1 is required to support protein degradation by electrophilic PROTACs. These findings, taken together, demonstrate how the chemical proteomic analysis of stereochemically defined electrophilic compound sets can uncover ligandable sites on E3 ligases that support targeted protein degradation.
Keywords: activity-based protein profiling, chemical proteomics, cysteine, DCAF1, electrophile, PROTAC, targeted protein degradation
Graphical Abstract
Introduction
Most small molecules affect the functions of proteins through binding-mediated agonism or antagonism. Targeted protein degradation has emerged as a distinct type of pharmacology wherein small molecules induce the formation of complexes between a substrate protein and a component of the ubiquitin-proteasome system (typically, an E3 ligase) to promote the physical turnover of the substrate protein1–3. Targeted protein degradation is often enacted by compounds termed PROTACs (proteolysis-targeting chimeras), which are heterobifunctional molecules that contain two independent recognition units – one that binds the substrate protein and the other that binds an E3 ligase – connected by a linker to form a ternary complex that brings the substrate protein into proximity of the E3 ligase, resulting in substrate ubiquitination and proteasome-mediated degradation2, 4. PROTACs have the potential to address gaps in chemical probe and drug development by, for instance, providing a means to i) eliminate multidomain or multifunctional proteins for which small-molecule antagonism proves insufficient to block the full scope of protein activities, and ii) convert silent ligand-protein interactions into functional (degradation) outcomes2. The successful design of PROTACs and, for that matter, molecular glues (an attractive alternative class of degrader compounds that form direct contacts with both the substrate and E3 ligase without requiring linkers5–7) depends on identifying ligands for E3 ligases. Even though human cells express hundreds of E3 ligases, small-molecule ligands have been discovered so far for only a small number of these proteins2, 8, and most PROTACs utilize one of two E3 ligases – cereblon (CRBN) or VHL2, 9–10. Recent work indicates that CRBN and VHL display distinct and restricted substrate specificities for executing targeted protein degradation11–14. Discovering ligands for additional E3 ligases therefore represents an important objective for realizing the full potential of targeted protein degradation.
So far, efforts to expand the scope of E3 ligases addressable with small-molecule ligands have largely relied on phenotypic screening15–17 or focused studies of purified E3 ligases18–19. This work has uncovered the potential to target a range of E3 ligases with covalent chemistry20, including DCAF16, DCAF11, RNF4, FEM1B, and RNF11415–19, which has enabled the design of electrophilic PROTACs that can promote targeted protein degradation at remarkably low stoichiometric engagement of the E3 ligase15–16 (reflecting the high catalytic potential of electrophilic PROTACs9). Nonetheless, the E3 ligase-targeting components of most of these PROTACs represent simple fragments bearing highly reactive cysteine-directed electrophiles such as α-chloroacetamides, and it therefore remains unclear whether such small molecule-E3 ligase interactions can be progressed to more advanced, selective chemical probes. Indeed, in multiple cases (e.g., DCAF11 and DCAF16), initial data suggest that the electrophilic PROTACs may have the capacity to engage more than one cysteine on the E3 ligase itself, underscoring the persistent challenges facing the discovery of selective electrophilic ligands for E3 ligases.
We recently introduced an activity-based protein profiling (ABPP) strategy for the chemical proteomic discovery of small molecule-protein interactions in human cells that leverages sets of stereoisomeric electrophilic compounds (or ‘stereoprobes’), wherein the interactions are prioritized based on a combination of cellular potency, stereochemical selectivity, and site-specificity21. Using these criteria, we discovered a striking number of cysteines on structurally and functionally diverse proteins that were stereoselectively engaged in primary human T cells by a focused set of tryptoline acrylamide stereoprobes. Here, we screened a distinct set of stereoisomeric azetidine acrylamides in human T cells using cysteine-directed mass spectrometry (MS)-ABPP21 These experiments identified a stereoselectively engaged cysteine (C1113) in the substrate binding domain of the Cullin4-RING E3 ligase (CRL4) substrate receptor DCAF1 (or VPRBP). We show that azetidine acrylamide-derived PROTACs promote the degradation of multiple target proteins (FBKP12, BRD4) in a stereoselective manner that depends on DCAF1 and is blocked by mutation of C1113. We further demonstrate that the azetidine acrylamide PROTACs form a ternary complex with FKBP12 and DCAF1 and promote selective FBKP12 degradation at low fractional engagement (~20%) of DCAF1_C1113. These findings, taken together, designate CRL4DCAF1 as an E3 ligase capable of supporting targeted protein degradation mediated by electrophilic PROTACS that act in a stereo- and site-selective manner.
Results and Discussion
Discovery of a stereo- and site-selective electrophilic compound-cysteine interaction in DCAF1.
Guided by the principles of diversity-oriented synthesis22, specifically the design of compounds with densely functionalized and entropically constrained sp3-rich cores bearing one or more stereocenters, we generated a set of azetidine acrylamide stereoprobes (Figure 1A) and screened these compounds by cysteine-directed MS-ABPP23–26 in primary human T cells. Across more than 10000 quantified cysteines, these experiments uncovered several azetidine acrylamide-cysteine interactions (Figure 1B and Dataset S1), including the stereo- (Figure 1B, C) and site- (Figure 1D) selective engagement of C1113 on the E3 ligase substrate adaptor DCAF1. Among the azetidine acrylamides, MY-1B (2) showed the strongest reactivity with DCAF1_C1113, while the enantiomer MY-1A (1) was inactive (Figure 1B, C). A weaker enantioselective interaction was also observed for DCAF1_C1113 with MY-3B (4) compared to MY-3A (3) (Figure 1B, C). Other quantified cysteines in DCAF1 were unaffected by MY-1B (Figure 1D).
Figure 1. Chemical proteomic discovery of a stereo- and site-selective covalent ligand for DCAF1.
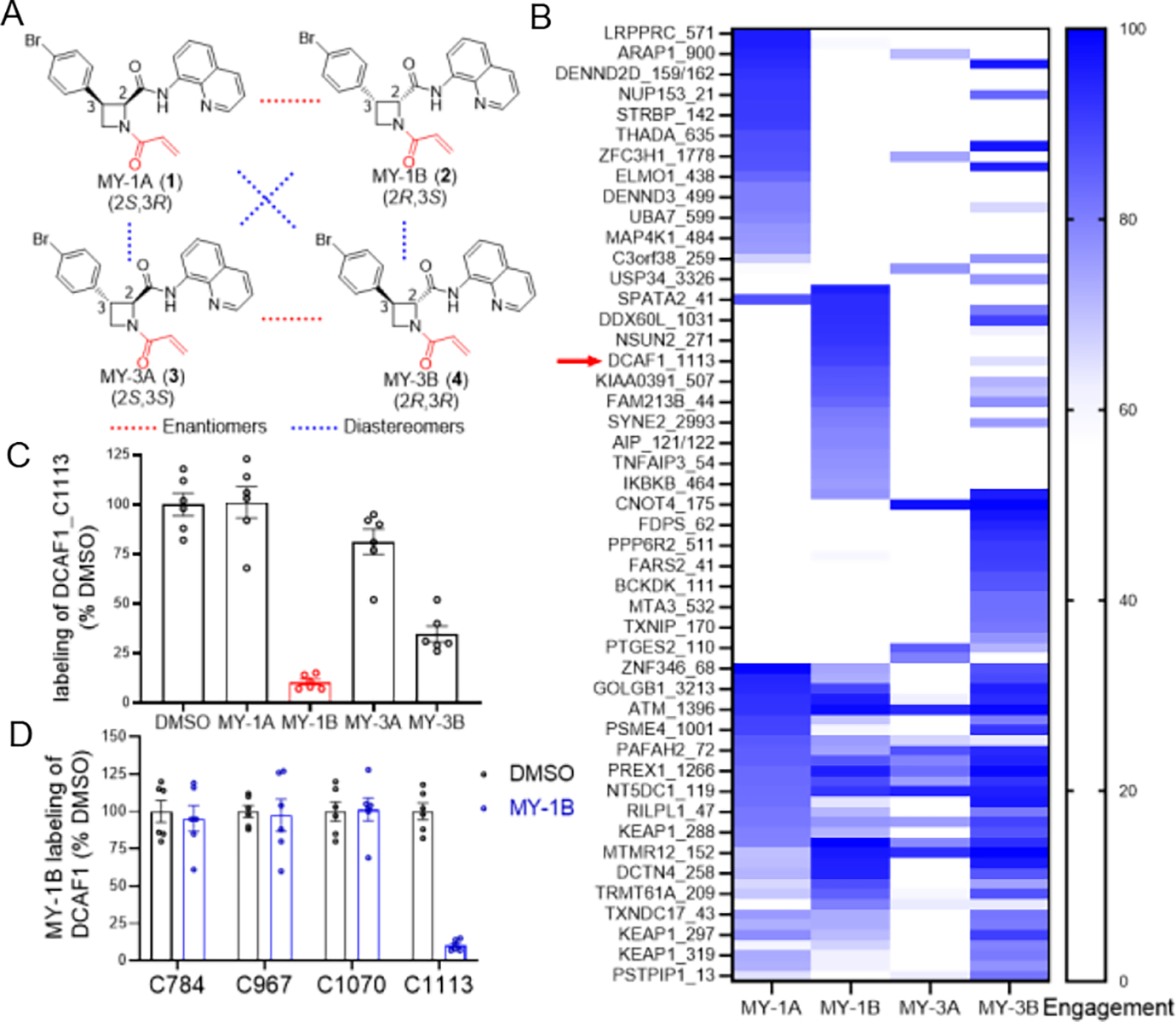
(A) Structures of a set of stereochemically defined azetidine acrylamides MY-1A (1), MY-1B (2), MY-3A (3), and MY-3B (4). (B) Heat map showing cysteines that were substantially engaged by azetidine acrylamides (> 75% by at least one compound) in human T cells (20 μM compound, 3 h) as determined by MS-ABPP using an iodoacetamide desthiobiotin (IA-DTB) probe following previously described methods21. Red arrow marks DCAF1_C1113. (C) MS-ABPP quantification of IA-DTB labeling of DCAF1_C1113 from T cells treated with the indicated azetidine acrylamides (20 μM, 3 h) relative to control T cells treated with DMSO. (D) MS-ABPP quantification of IA-DTB labeling of the indicated cysteines in DCAF1 from T cells treated with MY-1B (20 μM, 3 h) relative to control T cells treated with DMSO. For (B-D), data represent average values (for C and D, average values ± SEM) from three independent experiments each performed with two technical replicates, where cysteines were required to have been quantified in at least two experiments for interpretation.
DCAF1 is a multi-domain substrate receptor for CRL4 ligases (Figure 2A) that has diverse physiological and disease functions, including being co-opted by the HIV-2 virus to promote degradation of the antiviral host protein SAMHD127–28. This outcome is achieved by interactions between DCAF1 and the viral protein Vpx, and, interestingly, C1113, which is located in the WD40 domain of DCAF1, resides in close proximity to the Vpx interface29–30 (Figure 2B). We verified the stereo- and site-selective engagement of DCAF1_C1113 by azetidine acrylamides using alkynylated analogues of MY-1A and MY-1B (MY-11A (5) and MY-11B (6), respectively; Figure 2C). For these experiments, we doped into HEK293T cell lysates a recombinantly expressed and purified form of DCAF1 containing the WD40 domain (amino acids (aa) 1046–1396), in complex with the CRL4 adaptor protein DDB131, and exposed these samples to varying concentrations of MY-11A or MY-11B. After 1 h, we assessed MY-11A and MY-11B engagement of DCAF1 by copper-catalyzed azide-alkyne cycloaddition (CuAAC)32 with an azide-rhodamine reporter tag followed by SDS-PAGE and in-gel fluorescence scanning33. This gel-ABPP experiment revealed much greater concentration-dependent labeling of recombinant DCAF1 by MY-11B versus MY-11A, and this labeling was blocked by mutation of C1113 to alanine (DCAF1-C1113A mutant) (Figure 2D) or by pre-treatment with MY-1B, but not MY-1A (6.25 – 100 μM, 2 h pre-treatment; Figure 2E, right). We used gel-ABPP to quantify an in vitro target engagement value (TE50) of approximately 25 μM (95% C.I. = 13 – 38 μM) for MY-1B labeling of DCAF1 (Figure 2E, left). Modeling studies also supported a preferred interaction of DCAF1_C1113 with MY-1B over the other stereoisomers MY-1A, MY-3A, and MY-3B (Figure S1). These data, combined with our original MS-ABPP studies in T cells, indicate that MY-1B serves as a stereo- and site-selective covalent ligand for C1113 of DCAF1.
Figure 2. Azetidine acrylamides stereo- and site-selectively engage recombinant DCAF1_C1113.
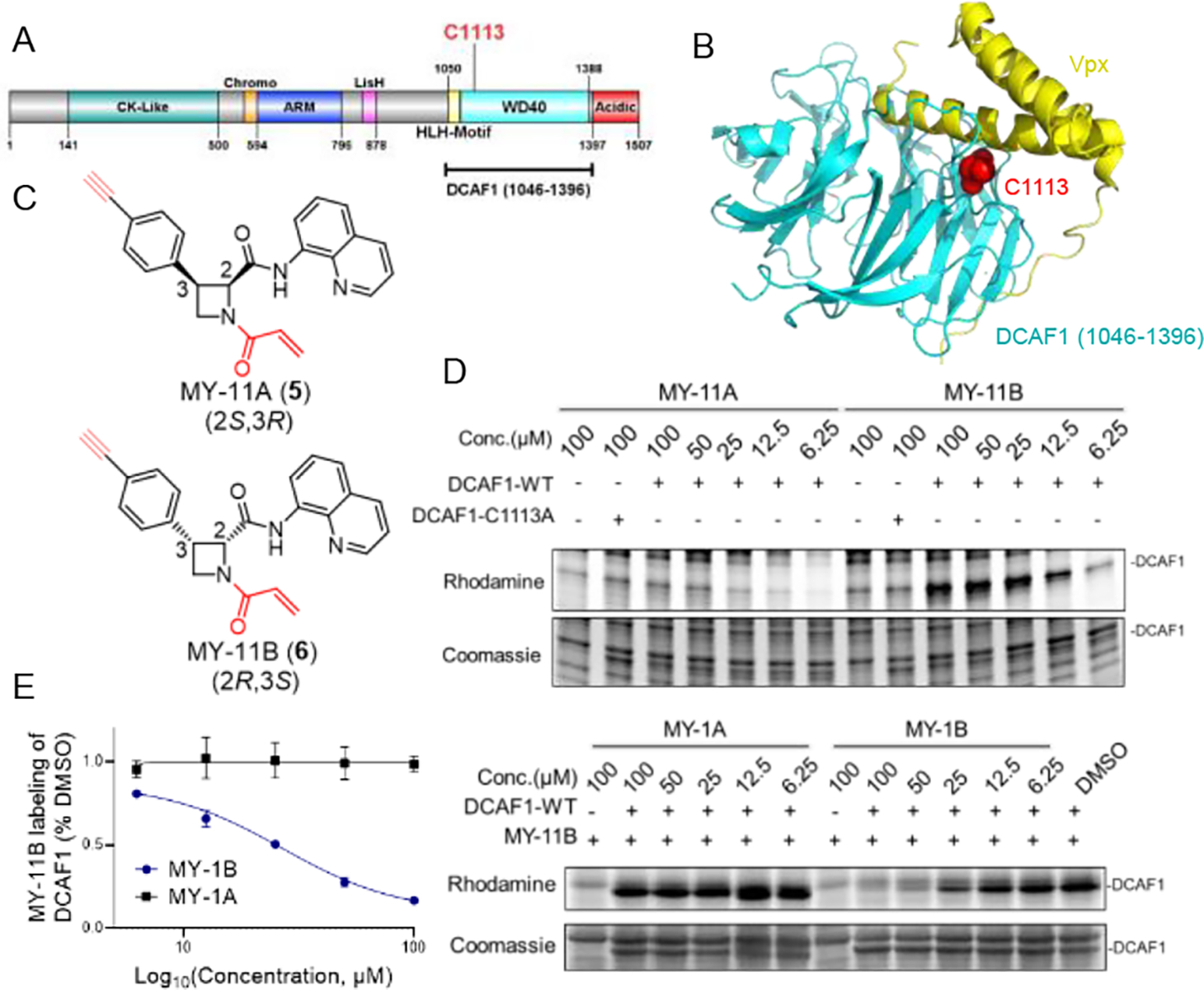
(A) Domain map of DCAF1 with the region used for recombinant protein studies marked in a bracket (aa 1046–1396) and C1113 highlighted in red. (B) Crystal structure of a DCAF1-Vpx complex (pdb: 5AJA). DCAF1 (aa 1046–1396) is shown in cyan, Vpx is shown in yellow, and C1113, which is located at the DCAF1-Vpx interface is shown in red. (C) Structures of alkyne-modified azetidine acrylamides MY-11A (5) and MY-11B (6). (D) Gel-ABPP showing the concentration-dependent reactivity of MY-11A and MY-11B (1 h) reactivity with recombinant, purified DCAF1-WT or the DCAF1-C1113A mutant (0.06 μg/μL of DCAF1 protein per sample) doped into HEK293T cell proteome (1 μg/μL). MY-11A/B labeling of proteins was visualized by CuAAC conjugation to an azide-rhodamine tag followed by SDS-PAGE and in-gel fluorescence scanning. Results are from a single experiment representative of two independent experiments. (E) Gel-ABPP showing the concentration-dependent effects of MY-1A and MY-1B on MY-11B reactivity with DCAF1-WT. Samples were treated with MY-1A/B for 2 h, followed by MY-11B (25 μM, 1 h) and analyzed as described in (D). Left, quantification of data presented as mean values ± SEM from two independent experiments. Right, representative gel-ABPP results. (D, E) Lower images are Coomassie blue-stained gels corresponding to ABPP gels shown in upper images.
Design and characterization of DCAF1-directed electrophilic PROTACs.
Considering the location of C1113 at the interface of DCAF1 binding to the viral Vpx protein, which in turn recruits the host antiviral protein SAMHD1 for ubiquitination and degradation30, we hypothesized that covalent ligands targeting this cysteine might serve as the basis for PROTAC-mediated targeted protein degradation. The structural model for the MY-1B-DCAF1_C1113 complex suggested multiple potential exit vectors for PROTAC construction, including the para position of the phenyl ring and the acrylamide electrophile (Figure S2). For our initial studies, we elected to focus on constructing PROTACs that extended from the phenyl ring so that the acrylamide electrophile was left intact, as exchanging acrylamide for a butynamide, which could have provided an additional exit vector (i.e., off of the methyl group of the butynamide), created a compound that was unable to engage DCAF1 (Figure S3). We synthesized two heterobifunctional compounds – YT41R (7) and YT47R (9) – that connected MY-1B to the small molecule SLF (Figure 3A), which is a high-affinity ligand for FKBP12 that is frequently used for assessing PROTAC performance15–16, 34–37. YT41R and YT47R differed in the length of the PEG (polyethylene glycol) linker connecting the MY-1B and SLF units, and we also prepared two analogous control probes based on the inactive enantiomer MY-1A (YT41S (8) and YT47S (10); Figure 3A).
Figure 3. Azetidine acrylamide-based heterobifunctional compounds stereoselectively engage DCAF1.
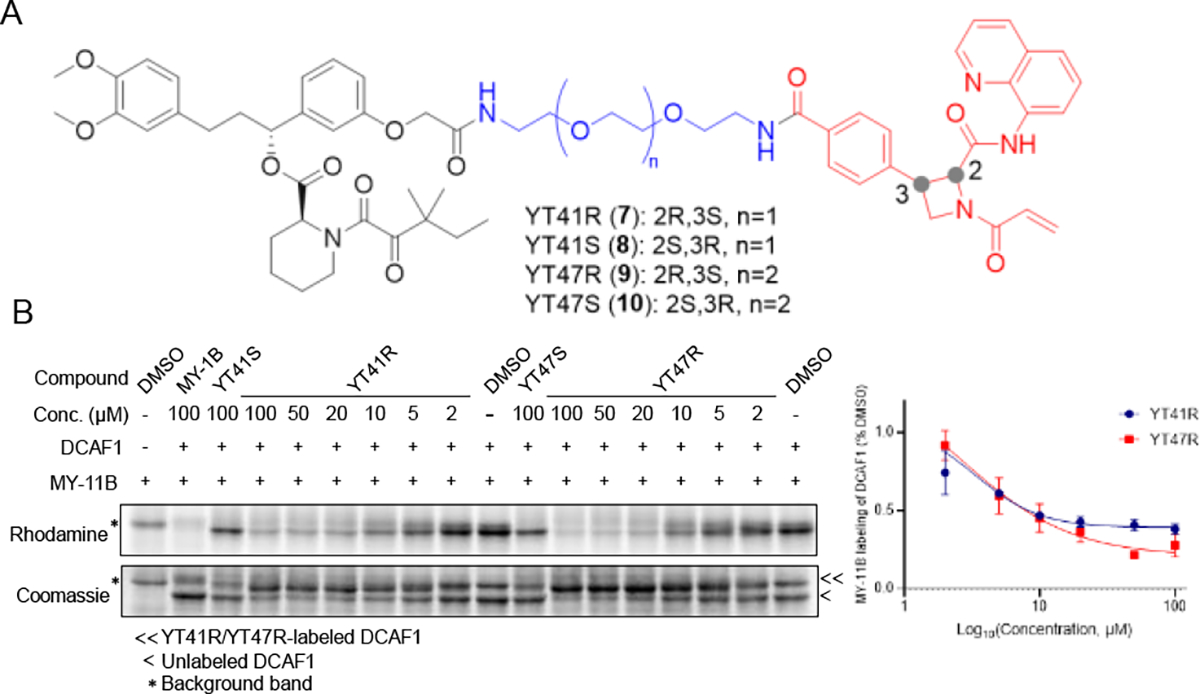
(A) Structures of candidate electrophilic PROTACs YT41R (7), YT41S (8), YT47R (9), and YT47S (10). (B) Gel-ABPP showing the concentration-dependent effects of YT41R and YT47R on MY-11B reactivity with DCAF1-WT protein (0.06 μg/μL of DCAF1 protein per sample) doped into HEK293T cell proteome (1 μg/μL). Also shown are the effects of YT41S and YT47S tested at a single concentration (100 μM). Samples were treated with heterobifunctional compounds for 2 h, followed by MY-11B (25 μM, 1 h) and analyzed as described in Figure 2D. Left, representative gel-ABPP results. Lower image is Coomassie blue-stained gel corresponding to ABPP gel shown in upper image. Right, quantification of data presented as mean values ± SEM from two independent experiments.
Gel-ABPP experiments revealed that both YT41R and YT47R produced a concentration-dependent inhibition of MY-11B probe reactivity with recombinant DCAF1 (Figure 3B). The blockade of MY-11B labeling plateaued at ~75% at 20 μM test concentrations of YT41R and YT47R, which could reflect the limited solubility of these candidate PROTACs at higher concentrations. In contrast, YT41S and YT47S did not impair the MY-11B-DCAF1 interaction (Figure 3B). Interestingly, we also observed an apparent gel-shift for recombinant DCAF1 by Coomassie blue staining in the presence of YT41R or YT47R (but not YT41S or YT47S), likely reflecting the substantial molecular weight change in the protein caused by covalent reaction with these compounds, which have MWs > 1000 Da (Figure 3B, lower image).
We next treated HEK293T cells co-transfected with FLAG epitope-tagged full length DCAF1 (aa 1 – 1507) and HA (hemagglutinin) epitope-tagged FKBP12 with a concentration range of YT41R or YT47R and, after 24 h, measured HA-FKBP12 abundance by Western blotting. Robust concentration-dependent degradation of HA-FKBP12 was observed for both YT41R and YT47R in co-transfected cells, but not HEK293T cells transfected only with HA-FKBP12 (Figure 4A). Evidence of YT41R/YT47R-induced degradation of FKBP12 was observed at concentrations of YT41R and YT47R as low at 0.25 μM (Figure S4) with a possible modest hook effect emerging at 5 μM (Figure 4A). Time-course studies revealed limited evidence of HA-FKBP12 degradation until ~24 h after treatment of FLAG-DCAF1-transfected cells with YT41R and YT47R (Figure S5). We also found that YT47R promoted the degradation of endogenous FBKP12 in HEK293T cells expressing WT-, but not C1113A-DCAF1 (Figure S6).
Figure 4. DCAF1-directed electrophilic PROTACs promote FKBP12 degradation in a stereo- and site-selective manner.
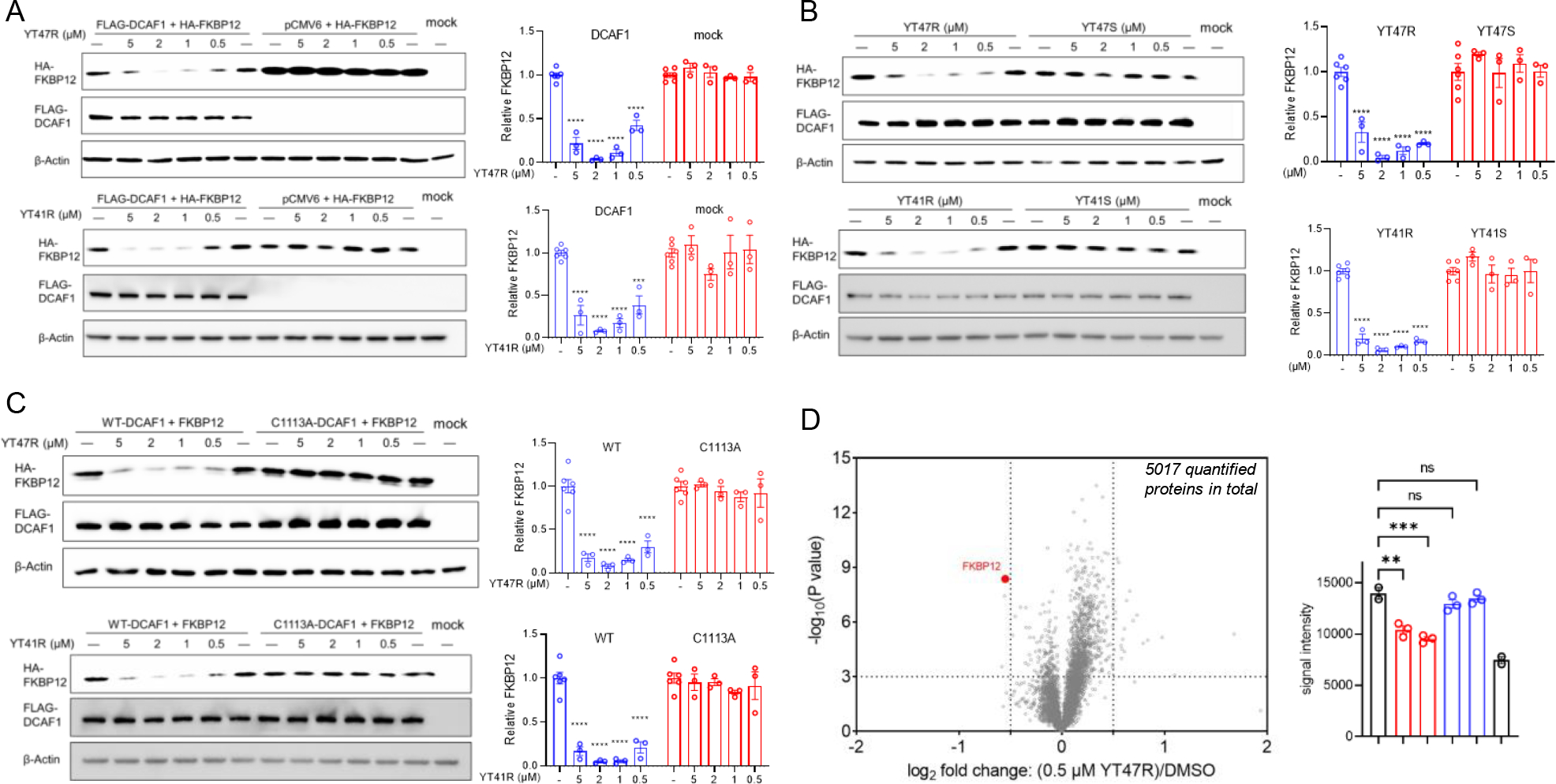
(A) Left, western blots showing concentration-dependent effects of YT47R (top) and YT41R (bottom) on FKBP12 abundance in HEK293T cells expressing HA-FBKP12 with or without co-expression of FLAG-DCAF1. Right, quantification of HA-FKBP12 abundance in the indicated experimental groups. (B) Left, western blots showing effects of YT47R versus YT47S (top) and YT41R versus YT41S (bottom) on FKBP12 abundance in HEK293T cells co-expressing HA-FBKP12 and FLAG-DCAF1. Right, quantification of HA-FKBP12 abundance in the indicated experimental groups. (C) Western blots showing effects of YT47R (top) and YT41R (bottom) on FKBP12 abundance in HEK293T cells co-expressing HA-FBKP12 and either FLAG-DCAF1-WT or FLAG-DCAF1-C1113A mutant. Quantification of HA-FKBP12 abundance in the indicated experimental groups is shown in the adjacent bar graphs. For (A-C), FKBP12 abundance was determined at 24 h post-treatment with compounds, and data are mean values ± SEM for three independent experiments. (D) Left, volcano plot showing quantitative MS-based proteomics results comparing the protein abundance profiles of HEK293T cells co-expressing HA-FBKP12 and FLAG-DCAF1 treated with YT47R (0.5 μM) or DMSO for 24 h. FKBP12 is highlighted in red. The x-axis and y-axis correspond to the log2 fold change of average relative abundance (0.5 μM YT47R/DMSO) and -log10(P value), respectively, from two (DMSO treatment) or three (compound treatment) independent experiments. Right, FKBP12 abundance in the indicated experimental groups quantified by MS-based proteomics. Statistical significance was calculated with unpaired two-tailed Student’s t-tests comparing YT41R/YT47R- and DMSO-treated cells. **P < 0.01, ***P < 0.001, ****P < 0.0001.
YT41R and YT47R acted in a stereoselective manner, as the control enantiomeric probes YT41S and YT47S did not support HA-FKBP12 degradation (Figure 4B), and neither compound promoted the degradation of FKBP12 in cells expressing a DCAF1-C1113A mutant (Figure 4C). We found that YT47R showed minimal additional effects on the HEK293T proteome, as, across >5000 proteins quantified by MS-based proteomics, only FKBP12 showed a substantial and significant stereoselective change in abundance in YT47R-treated HEK293T cells expressing WT-DCAF1 (Figure 4D, Table S1, Dataset S3). We should note that a handful of other proteins were also altered by YT47R, but these proteins were similarly changed by the inactive enantiomer YT47S (Table S1, Dataset S3), and we therefore conclude these protein changes were independent of DCAF1. While the YT47R-induced FKBP12 degradation measured by MS-proteomics appeared lower in magnitude than that observed in previous Western blotting experiments (compared bar graphs in Figure 4D and Figure 4A–C), we also observed a similarly attenuated degree of degradation when analyzing the samples prepared for MS-based proteomics by Western blotting (Figure S6). We interpret this difference to indicate that the adjusted conditions (cell density, media volume) required to prepare sufficient material for MS-based proteomics may have altered the potency and efficacy of YT47R. Finally, we found that YT47R also promoted the degradation endogenous FKBP12 in cells expressing DCAF1-WT protein, but not in cells expressing DCAF1-C1113A mutant protein (Figure S7).
Taken together, these data indicate YT41R and YT47R act as electrophilic PROTACs that promote the degradation of FKBP12 through the stereoselective engagement of C1113 of DCAF1.
Mechanistic studies of DCAF1-directed electrophilic PROTACs.
YT41R/YT47R-mediated degradation of HA-FKBP12 was blocked by pre-treatment with the proteasome inhibitor MG132 (5 μM, 1 h pre-treatment followed by 24 h co-treatment with YT41R/YT47R) (Figure 5A); however, interpretation of these experiments was complicated by the increased quantities of both HA-FKBP12 and FLAG-DCAF1 observed in MG132-treated cells (Figure 5A). On the other hand, treatment with the NAE inhibitor MLN4924, which blocks the Neddylation and activity of cullin-RING E3 ligases38, also impaired YT41R/YT47R-mediated degradation of HA-FKBP12 without causing alterations in the quantity of HA-FKBP12 or FLAG-DCAF1 in control cells (Figure 5A). These data support that the targeted protein degradation activity of YT41R and YT47R is proteasome-and cullin-RING E3 ligase-dependent.
Figure 5. FKBP12 degradation induced by DCAF1-directed electrophilic PROTACs is dependent on the ubiquitin-proteasome system.
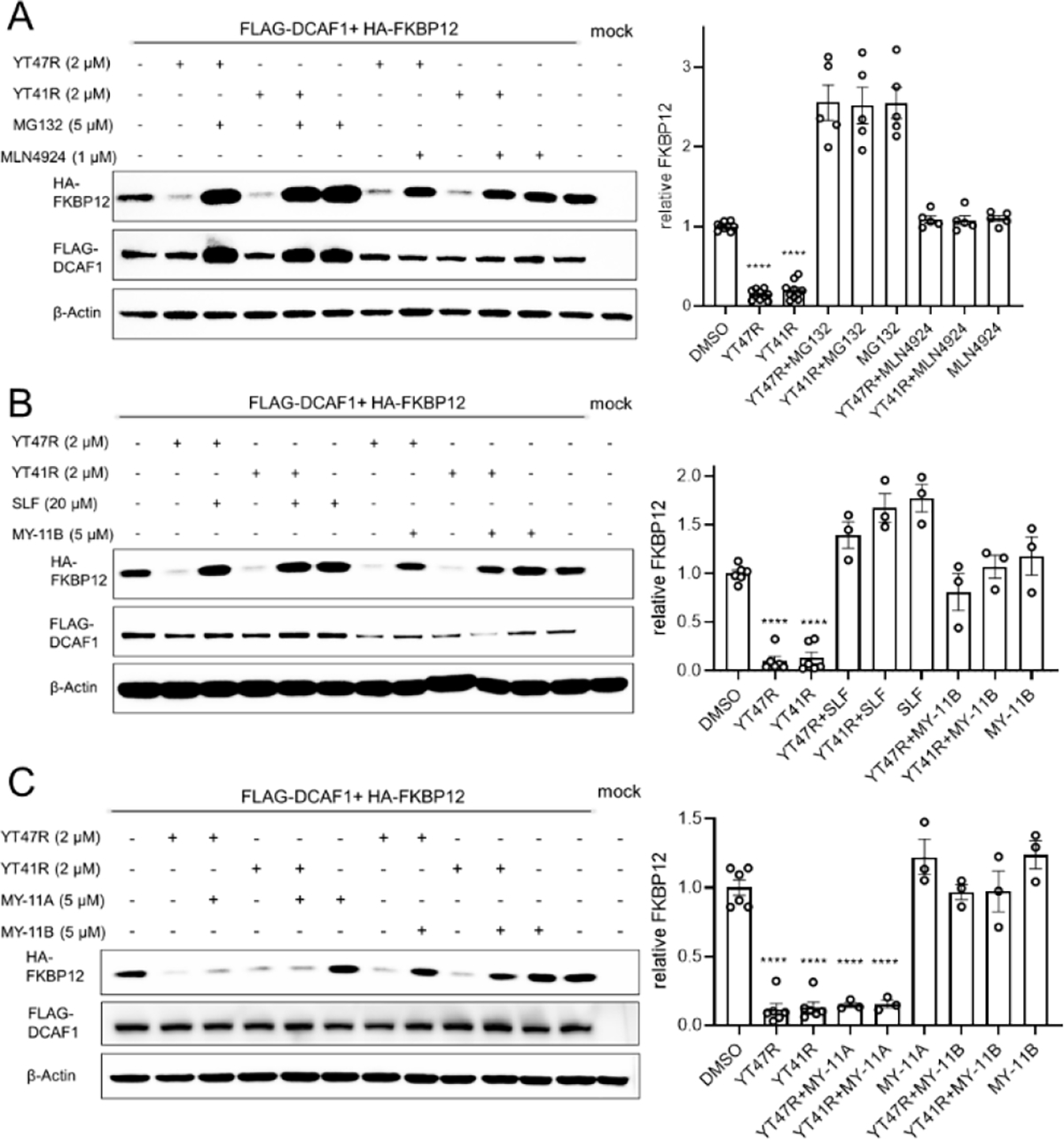
(A) Left, western blots showing effects of proteasome (MG132) and Neddylation (MLN4924) inhibitors (1 h pre-treatment) on YT47R- and YT41R-dependent degradation of FKBP12 in HEK293T cells co-expressing HA-FBKP12 and FLAG-DCAF1. FKBP12 abundance was determined at 24 h post-treatment with YT47R or YT41R. Right, quantification of HA-FKBP12 abundance in the indicated experimental groups. (B) Left, western blots showing effects of the FKBP12 ligand SLF (20 μM) or DCAF1 ligand MY-11B (1 h pre-treatment with 20 μM SLF or 5 μM MY-11B) on YT47R- and YT41R-dependent degradation of FKBP12 in HEK293T cells co-expressing HA-FBKP12 and FLAG-DCAF1. FKBP12 abundance was determined at 24 h post-treatment with YT47R or YT41R. Right, quantification of HA-FKBP12 abundance in the indicated experimental groups. (C) Left, western blots showing effects of MY-11A or MY-11B (5 μM, 1 h pre-treatment) on YT47R- and YT41R-dependent degradation of FKBP12 in HEK293T cells co-expressing HA-FBKP12 and FLAG-DCAF1. FKBP12 abundance was determined at 24 h post-treatment with YT47R or YT41R. Right, quantification of HA-FKBP12 abundance in the indicated experimental groups. For (A-C), data are mean values ± SEM for three-five independent experiments. Statistical significance was calculated with unpaired two-tailed Student’s t-tests comparing YT41R/YT47R- and DMSO-treated cells. ****P < 0.0001.
PROTAC-mediated protein degradation requires ternary complex formation between the target protein, the heterobifunctional compound, and the E3 ligase, and this process can often be competitively disrupted by monovalent ligands targeting individual components of the complex. Consistent with this model for YT41R/YT47R action, we found that pre-treatment of HA-FKBP12 and FLAG-DCAF1 co-transfected HEK293T cells with either the FKBP12 ligand SLF (20 μM) or the DCAF1 ligand MY-11B (5 μM) blocked YT41R and YT47R-mediated degradation of HA-FKBP12 (Figure 5B). The control enantiomer MY-11A, which does not engage DCAF1 (Figure 2D), did not block YT41R/YT47R-mediated HA-FKBP12 degradation (Figure 5C).
Finally, we investigated whether electrophilic PROTACs induce a stable ternary complex of FKBP12 with DCAF1 and promote FKBP12 ubiquitination. These co-immunoprecipitation experiments were performed in HEK293T cells co-transfected with HA-FKBP12 and FLAG-DCAF1 and treated with YT47R (or the control enantiomer YT47S) in the presence of MG132 (to block FKBP12 degradation) and revealed clear evidence of a ternary complex of FKBP12 and DCAF1 with YT47R, but not YT47S (Figure 6A). A ternary complex was not observed in cells expressing the DCAF1-C1113A mutant (Figure 6A), supporting that YT47R induces this complex through site-specifically reacting with C1113. We also detected robust polyubiquitination of the immunoprecipitated HA-FKBP12 in YT47R-treated, DCAF1-WT-expressing cells compared to YT47S- or DMSO-treated, DCAF1-WT-expressing cells or YT47R-treated, DCAF1-C1113A-expressing cells (Figure 6A). Interestingly, we also observed increased FKBP12 ubiquitination in YT47R- versus YT47S-treated HEK293T cells that did not express exogenous DCAF1 (Figure 6A). We wondered whether this outcome might be mediated by endogenous DCAF1, and, consistent with this possibility, we found that endogenous DCAF1 formed a more robust ternary complex with HA-FKBP12 in HEK293T cells treated with YT47R compared to YT47S or DMSO (Figure 6B). In considering possible reasons why this ternary complex formation with endogenous DCAF1 was insufficient to support the degradation of recombinantly expressed FKBP12 (Figure 4A), we speculate that our current PROTACs (YT41R, YT47R) may require further improvements in cellular properties to enable robust targeted protein degradation mediated by physiological concentrations of DCAF1. Possibly supporting this hypothesis, MS-ABPP experiments revealed that YT47R (5 μM) only engaged a maximum of ~20% of C1113 of recombinantly expressed DCAF1 evaluated at 3 or 24 h post-treatment (Figure 6C and Dataset S2). This minor engagement, which is consistent with previous studies of other electrophilic PROTACs that also act sub-stoichiometrically (and presumably catalytically) to degrade proteins15–16, was both stereo- and site-selective (Figure 6C) and suggests more optimized PROTACs that engage a greater fraction of DCAF1_C1113 in cells may enable targeted protein degradation by endogenous DCAF1. Alternatively, however, we cannot exclude at this time that the YT47R-induced ubiquitination of FKBP12 in HEK293T cells that lack ectopic DCAF1 is mediated by an alternative, DCAF1-independent mechanism.
Figure 6. Evidence for ternary complex formation and ubiquitination of HA-FKBP12 by DCAF1-directed electrophilic PROTACs.
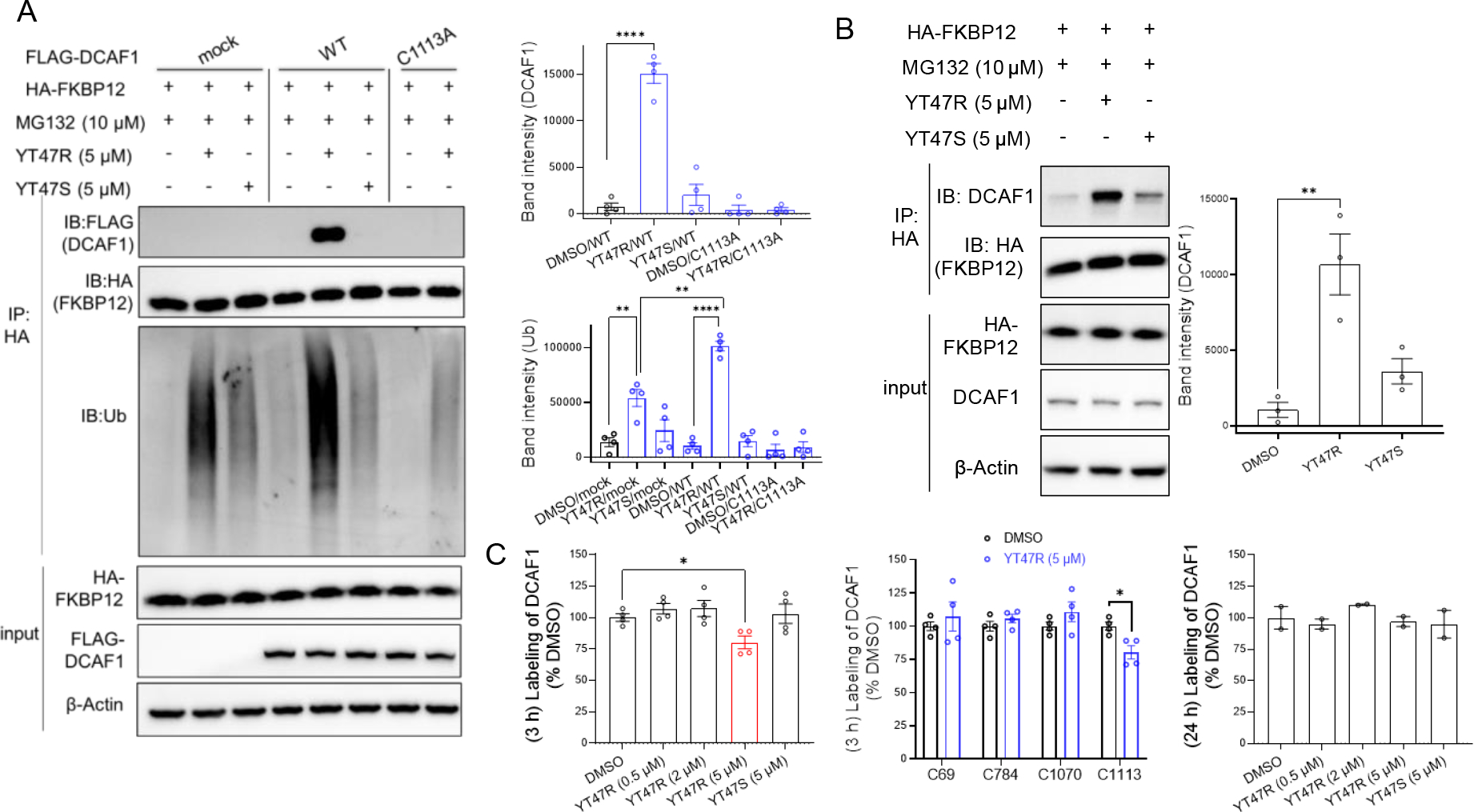
(A) Left, western blots showing effects of YT47R and YT47S on the immunoprecipitation (IP) of FLAG-DCAF1-WT or FLAG-DCAF1-C1113A with HA-FKBP12 in HEK293T cells co-expressing these proteins. Cells were treated with YT47R or YT47S (5 μM) for 2 h in the presence of MG132 (10 μM) prior to lysis and IP with an anti-HA antibody and western blotting for the indicated proteins (DCAF1, FKBP12, and ubiquitinated protein). Right, bar graphs quantifying DCAF1 (top) and ubiquitination (Ub) (bottom) in the indicated IP groups. (B) Left, western blots showing effects of YT47R and YT47S on the IP of endogenous DCAF1 with HA-FKBP12 in HEK293T cells expressing HA-FKBP12. Cells were treated with YT47R or YT47S (5 μM) for 2 h in the presence of MG132 (10 μM) prior to lysis and IP with an anti-HA antibody and western blotting for the indicated proteins (DCAF1, FKBP12). Right, bar graph quantifying DCAF1 in the indicated IP groups. For (A, B), data are mean values ± SEM for three independent experiments. Statistical significance was calculated with unpaired two-tailed Student’s t-tests comparing YT41R/YT47R- and DMSO-treated cells. **P < 0.01, ****P < 0.0001. (C) MS-ABPP quantification of IA-DTB labeling of the indicated cysteines in DCAF1 from FLAG-DCAF1-WT-expressing HEK293T cells treated with the indicated concentrations of YT47R for 3 h (left, middle) or 24 h (right) relative to cells treated with DMSO. Data represent average values ± SEM from four independent experiments. Statistical significance was calculated with unpaired two-tailed Student’s t-tests comparing compound to DMSO treated cells. *P < 0.1.
DCAF1-directed electrophilic PROTACs degrades BRD4.
Encouraged by the stereoselective and site-specific degradation of FKBP12 by DCAF1-directed electrophilic PROTACs, we wonder if this strategy could be applied to other endogenous proteins. The BET domain protein BRD4 has been frequently used as a prototype target for PROTAC design due to the availability of high-quality ligands and the important biological roles played by this protein39–47. We synthesized a pair of heterobifunctional probes connecting MY-1B or MY-1A to the BRD4 ligand JQ-148 through a PEG linker (YT117R and YT117S, respectively) (Figure 7A). Gel-ABPP experiments confirmed the concentration-dependent and stereoselective blockade of MY-11B labeling of recombinant DCAF1 by YT117R (Figure 7B), albeit with lower potency than YT47R (Figure S8). We next found that YT117R (0.05–1 μM, 12 h) promoted the concentration-dependent degradation of BRD4 (Figure 7C). The induced degradation of BRD4 was submaximal compared to positive control compound dBET639, but was nonetheless stereoselective (Figure 7D, E), and only observed in DCAF1-WT-expressing HEK293T cells (Figure 7C–E), but not mock (Figure S9) or DCAF1-C1113A mutant-expressing (Figure 7D) HEK293T cells. Furthermore, YT117R-induced BRD4 degradation was blocked by the Neddylation inhibitor MLN4924 and the proteasome inhibitor MG132, consistent with a CRL4-mediated process (Figure 7E). These data, combined with the degradation of FBKP12 mediated by YT41R and YT47R, indicate that DCAF1-directed PROTACs have the potential to promote degradation of multiple endogenous proteins in human cells.
Figure 7. DCAF1-directed electrophilic PROTAC promotes degradation of endogenous BRD4.
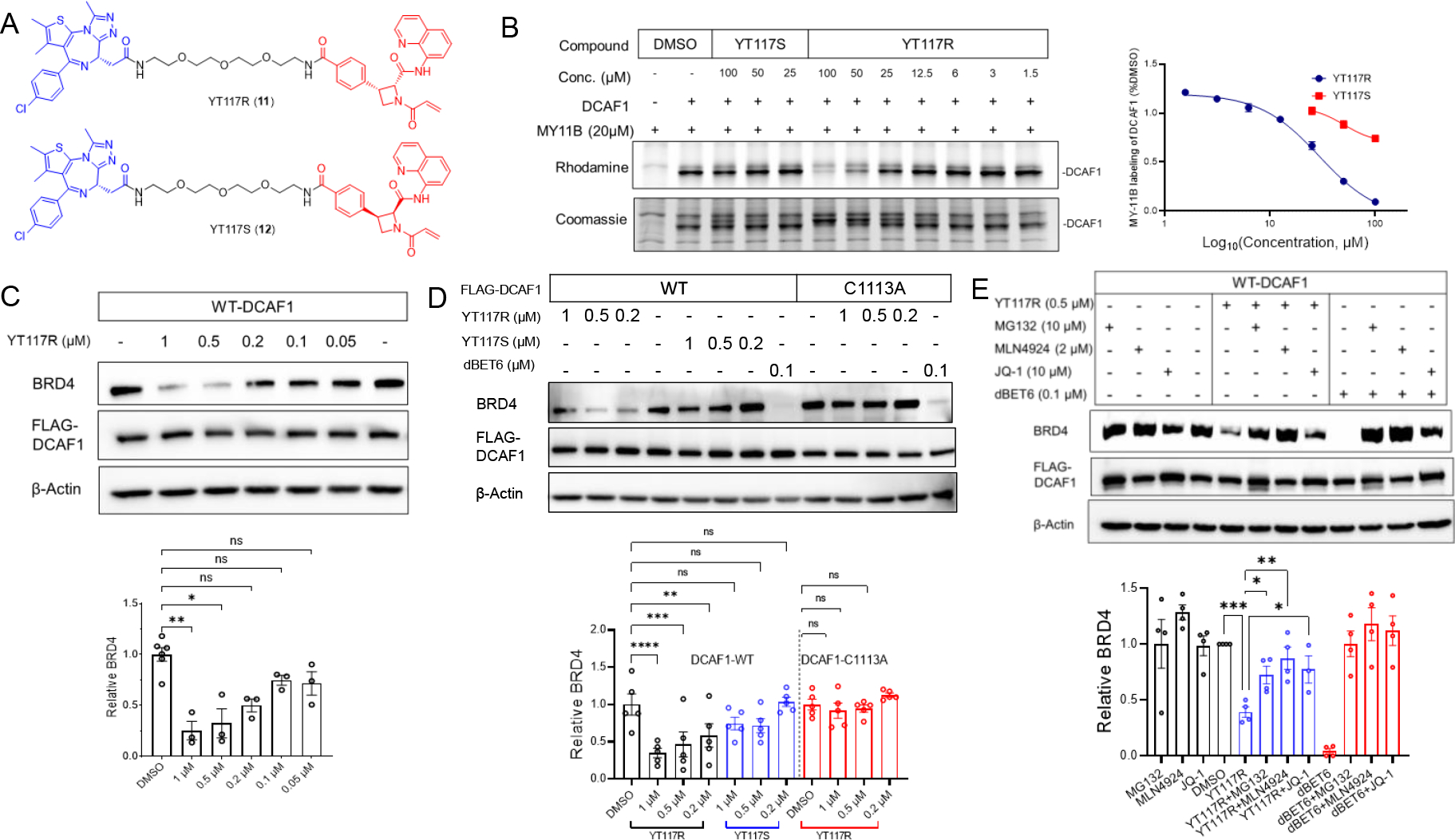
(A) Left, structures of BRD4-targeting PROTAC YT117R (11) and its stereoisomeric control probe YT117S (12). (B) Gel-ABPP showing the concentration-dependent effects of YT117R and YT117S on MY-11B reactivity with DCAF1-WT. Samples were treated with YT117R/S for 2 h, followed by MY-11B (20 μM, 1 h) and analyzed as described in Figure 2D. Left, representative gel-ABPP results. Lower images are Coomassie blue-stained gels corresponding to ABPP gels shown in upper images. Right, quantification of data presented as mean values ± SEM from two independent experiments. (C) Top, western blots showing concentration-dependent effects of YT117R on BRD4 abundance in HEK293T cells expressing FLAG-DCAF1. Bottom, quantification of BRD4 abundance in the indicated experimental groups. (D) Top, western blots showing effects of YT117R on BRD4 abundance in FLAG-DCAF1-WT- or mock-transfected HEK293T cells. Bottom, quantification of BRD4 abundance in the indicated experimental groups. (E) Top, western blots showing effects of YT117R (0.5 μM) or dBET6 (0.1 μM) alone or following co-treatment with the indicated concentrations of MG132, MLN4924, or JQ-1 on BRD4 abundance in HEK293T cells expressing FLAG-DCAF1-WT. Bottom, quantification of BRD4 abundance in the indicated experimental groups (lower). Data are mean values ± SEM for three independent experiments (C and D) and four independent experiments for (E). Statistical significance was calculated with unpaired two-tailed Student’s t-tests comparing YT117R/S- and DMSO-treated cells. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Conclusion
Here, we have described the chemical proteomic discovery of a ligandable cysteine (C1113) in the E3 ligase substrate receptor protein DCAF1 and the conversion of azetidine acrylamides targeting this cysteine into electrophilic PROTACs. These findings are significant because, to our knowledge, they describe the first electrophilic PROTACs that have been shown to act in a stereo- and site-selective manner. The properties of stereoselectivity and site-specificity provide convenient ways to establish controls to verify on-target activity for electrophilic PROTACs, as we have shown herein for DCAF1 and others have demonstrated for reversibly binding PROTACs that engage VHL in a stereoselective manner49. We further interpret these features to indicate the presence of a high-quality druggable pocket in proximity to DCAF1_C1113. Also supportive of this conclusion is the recent report of a noncovalent DCAF1 ligand that binds a pocket near C1113 (PDB code: 7SSE, Figure S10). While we have taken advantage of the druggability of DCAF1_C1113 to create electrophilic PROTACs, we also imagine that more advanced covalent ligands might serve as molecular glues5–7 or antagonists of the various physiological and pathological functions of DCAF150–54. Indeed, small-molecule antagonists of DCAF1 could counteract the pathogenesis of viruses like HIV-1 and HIV-2 that co-opt this E3 ligase substrate receptor to suppress immune cell responses30, 54–57. More generally, the rich dataset of stereoselective interactions reported herein between azetidine acrylamides and cysteines in the human T-cell proteome (Figure 1B and Dataset S1) should offer attractive starting points for chemical probe discovery for additional proteins from structurally and functionally diverse classes.
As has been described previously15–19, electrophilic PROTACs have the potential to maximally leverage the catalytic potential of targeted protein degradation by creating “neo”-E3 ligases that are permanently modified (until physical turnover) with a substrate-binding compound. However, key variables can impact the success of such endeavors, including the half-life and substrate compatibility of the E3 ligase11–14, as well as the quality of the chemical probes that target it. On the latter point, we view the azetidine acrylamide reactive group found herein to stereoselectively engage DCAF1_C1113 as an encouraging starting point for probe optimization, especially in comparison to electrophilic PROTACs reported to target other E3 ligases, which mostly use high-reactivity groups such as α-chloroacetamides15–16, 18–19. Nonetheless, we have, so far, only observed targeted protein degradation for electrophilic PROTACs in cells expressing recombinant DCAF1, and it will be important in future studies to determine if this activity can be extended to endogenous DCAF1. That we observed evidence of electrophilic PROTACs promoting a ternary complex with endogenous DCAF1 and FBKP12, as well as inducing FKBP12 ubiquitination, is encouraging, even though these effects were not apparently robust enough to lead to substantial FKBP12 degradation in cells expressing endogenous DCAF1 (possibly due to counteracting cellular deubiquitinases9). Further improvements in electrophilic PROTAC performance may require greater levels of cellular engagement of DCAF1, as our first-generation compounds only appear to modify ≤ 20% of recombinant DCAF1 at functional concentrations in cells. A recent study has also described an autoinhibitory oligomerization mechanism for DCAF1 that may not be shared by other CRL4 substrate receptors58. If a substantial proportion of endogenous DCAF1 is in an autoinhibited tetrameric state, then a greater quantity of total DCAF1 may need to be modified by electrophilic PROTACs to support targeted protein degradation. On the other hand, we wonder whether this autoregulatory feature of DCAF1 might also be exploited to create electrophilic PROTACs and/or molecular glues that carry out context-dependent protein degradation only in those cell types where DCAF1 is in an activated state.
Supplementary Material
ACKNOWLEDGMENT
We thank the Cravatt lab and Vividion scientists for helpful discussions.
Funding Sources
This work was supported by the NIH (AI142784, CA231991).
X.Z. was supported by NIH-NCI (R00CA248715) and the Damon-Runyon Cancer Research Foundation (DFS-53–22).
Footnotes
The authors declare the following competing financial interest(s): Dr. Cravatt is a founder and scientific advisor to Vividion Therapeutics, and Drs. Cravatt, Melillo, and Schreiber are founders and/or scientific advisors to Magnet Therapeutics. Vividion and Magnet are biotechnology companies interested in developing small-molecule medicines.
ASSOCIATED CONTENT
Supporting Information.
The supporting information is available free of charge via the Internet at http://pubs.acs.org.”
Supplementary Figures, Materials and Methods, Synthetic Chemistry (PDF)
Supplementary Dataset 1–3 (XLS)
REFERENCES
- 1.Bondeson DP; Crews CM, Targeted Protein Degradation by Small Molecules. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 107–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schapira M; Calabrese MF; Bullock AN; Crews CM, Targeted protein degradation: expanding the toolbox. Nat Rev Drug Discov 2019, 18 (12), 949–963. [DOI] [PubMed] [Google Scholar]
- 3.Kostic M; Jones LH, Critical Assessment of Targeted Protein Degradation as a Research Tool and Pharmacological Modality. Trends Pharmacol. Sci. 2020, 41 (5), 305–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun X; Gao H; Yang Y; He M; Wu Y; Song Y; Tong Y; Rao Y, PROTACs: great opportunities for academia and industry. Signal Transduct Target Ther 2019, 4 (1), 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schreiber SL, The Rise of Molecular Glues. Cell 2021, 184 (1), 3–9. [DOI] [PubMed] [Google Scholar]
- 6.Kozicka Z; Thoma NH, Haven’t got a glue: Protein surface variation for the design of molecular glue degraders. Cell Chem Biol 2021, 28 (7), 1032–1047. [DOI] [PubMed] [Google Scholar]
- 7.Dong G; Ding Y; He S; Sheng C, Molecular Glues for Targeted Protein Degradation: From Serendipity to Rational Discovery. J. Med. Chem. 2021, 64 (15), 10606–10620. [DOI] [PubMed] [Google Scholar]
- 8.Bekes M; Langley DR; Crews CM, PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discovery 2022, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bondeson DP; Mares A; Smith IE; Ko E; Campos S; Miah AH; Mulholland KE; Routly N; Buckley DL; Gustafson JL; Zinn N; Grandi P; Shimamura S; Bergamini G; Faelth-Savitski M; Bantscheff M; Cox C; Gordon DA; Willard RR; Flanagan JJ; Casillas LN; Votta BJ; den Besten W; Famm K; Kruidenier L; Carter PS; Harling JD; Churcher I; Crews CM, Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11 (8), 611–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE, DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348 (6241), 1376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lai AC; Toure M; Hellerschmied D; Salami J; Jaime-Figueroa S; Ko E; Hines J; Crews CM, Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew. Chem., Int. Ed. 2016, 55 (2), 807–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bondeson DP; Smith BE; Burslem GM; Buhimschi AD; Hines J; Jaime-Figueroa S; Wang J; Hamman BD; Ishchenko A; Crews CM, Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem Biol 2018, 25 (1), 78–87 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang H-T; Dobrovolsky D; Paulk J; Yang G; Weisberg EL; Doctor ZM; Buckley DL; Cho J-H; Ko E; Jang J, A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader. Cell chemical biology 2018, 25 (1), 88–99. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donovan KA; Ferguson FM; Bushman JW; Eleuteri NA; Bhunia D; Ryu S; Tan L; Shi K; Yue H; Liu X, Mapping the degradable kinome provides a resource for expedited degrader development. Cell 2020, 183 (6), 1714–1731. e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X; Luukkonen LM; Eissler CL; Crowley VM; Yamashita Y; Schafroth MA; Kikuchi S; Weinstein DS; Symons KT; Nordin BE; Rodriguez JL; Wucherpfennig TG; Bauer LG; Dix MM; Stamos D; Kinsella TM; Simon GM; Baltgalvis KA; Cravatt BF, DCAF11 Supports Targeted Protein Degradation by Electrophilic Proteolysis-Targeting Chimeras. J. Am. Chem. Soc. 2021, 143 (13), 5141–5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X; Crowley VM; Wucherpfennig TG; Dix MM; Cravatt BF, Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat. Chem. Biol. 2019, 15 (7), 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spradlin JN; Hu X; Ward CC; Brittain SM; Jones MD; Ou L; To M; Proudfoot A; Ornelas E; Woldegiorgis M; Olzmann JA; Bussiere DE; Thomas JR; Tallarico JA; McKenna JM; Schirle M; Maimone TJ; Nomura DK, Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat. Chem. Biol. 2019, 15 (7), 747–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ward CC; Kleinman JI; Brittain SM; Lee PS; Chung CYS; Kim K; Petri Y; Thomas JR; Tallarico JA; McKenna JM; Schirle M; Nomura DK, Covalent Ligand Screening Uncovers a RNF4 E3 Ligase Recruiter for Targeted Protein Degradation Applications. ACS Chem. Biol. 2019, 14 (11), 2430–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henning NJ; Manford AG; Spradlin JN; Brittain SM; Zhang E; McKenna JM; Tallarico JA; Schirle M; Rape M; Nomura DK, Discovery of a Covalent FEM1B Recruiter for Targeted Protein Degradation Applications. J. Am. Chem. Soc. 2022, 144 (2), 701–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gabizon R; London N, The rise of covalent proteolysis targeting chimeras. Curr. Opin. Chem. Biol. 2021, 62, 24–33. [DOI] [PubMed] [Google Scholar]
- 21.Vinogradova EV; Zhang X; Remillard D; Lazar DC; Suciu RM; Wang Y; Bianco G; Yamashita Y; Crowley VM; Schafroth MA; Yokoyama M; Konrad DB; Lum KM; Simon GM; Kemper EK; Lazear MR; Yin S; Blewett MM; Dix MM; Nguyen N; Shokhirev MN; Chin EN; Lairson LL; Melillo B; Schreiber SL; Forli S; Teijaro JR; Cravatt BF, An Activity-Guided Map of Electrophile-Cysteine Interactions in Primary Human T Cells. Cell 2020, 182 (4), 1009–1026 e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerry CJ; Schreiber SL, Recent achievements and current trajectories of diversity-oriented synthesis. Curr. Opin. Chem. Biol. 2020, 56, 1–9. [DOI] [PubMed] [Google Scholar]
- 23.Parker CG; Galmozzi A; Wang Y; Correia BE; Sasaki K; Joslyn CM; Kim AS; Cavallaro CL; Lawrence RM; Johnson SR; Narvaiza I; Saez E; Cravatt BF, Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell 2017, 168 (3), 527–541 e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matthews ML; He L; Horning BD; Olson EJ; Correia BE; Yates JR 3rd; Dawson PE; Cravatt BF, Chemoproteomic profiling and discovery of protein electrophiles in human cells. Nat Chem 2017, 9 (3), 234–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bar-Peled L; Kemper EK; Suciu RM; Vinogradova EV; Backus KM; Horning BD; Paul TA; Ichu TA; Svensson RU; Olucha J; Chang MW; Kok BP; Zhu Z; Ihle NT; Dix MM; Jiang P; Hayward MM; Saez E; Shaw RJ; Cravatt BF, Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell 2017, 171 (3), 696–709 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y; Dix MM; Bianco G; Remsberg JR; Lee HY; Kalocsay M; Gygi SP; Forli S; Vite G; Lawrence RM; Parker CG; Cravatt BF, Expedited mapping of the ligandable proteome using fully functionalized enantiomeric probe pairs. Nat Chem 2019, 11 (12), 1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schabla NM; Mondal K; Swanson PC, DCAF1 (VprBP): emerging physiological roles for a unique dual-service E3 ubiquitin ligase substrate receptor. J Mol Cell Biol 2019, 11 (9), 725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z; Zhang L; Ying S, SAMHD1: a novel antiviral factor in intrinsic immunity. Future Microbiol 2012, 7 (9), 1117–26. [DOI] [PubMed] [Google Scholar]
- 29.Schwefel D; Groom HC; Boucherit VC; Christodoulou E; Walker PA; Stoye JP; Bishop KN; Taylor IA, Structural basis of lentiviral subversion of a cellular protein degradation pathway. Nature 2014, 505 (7482), 234–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laguette N; Sobhian B; Casartelli N; Ringeard M; Chable-Bessia C; Segeral E; Yatim A; Emiliani S; Schwartz O; Benkirane M, SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474 (7353), 654–U132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Banchenko S; Krupp F; Gotthold C; Bürger J; Graziadei A; O’Reilly FJ; Sinn L; Ruda O; Rappsilber J; Spahn CM, Structural insights into Cullin4-RING ubiquitin ligase remodelling by Vpr from simian immunodeficiency viruses. PLoS Pathog. 2021, 17 (8), e1009775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rostovtsev VV; Green LG; Fokin VV; Sharpless KB, A stepwise huisgen cycloaddition process: copper (I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angewandte Chemie 2002, 114 (14), 2708–2711. [DOI] [PubMed] [Google Scholar]
- 33.Speers AE; Adam GC; Cravatt BF, Activity-based protein profiling in vivo using a copper (i)-catalyzed azide-alkyne [3+ 2] cycloaddition. J. Am. Chem. Soc. 2003, 125 (16), 4686–4687. [DOI] [PubMed] [Google Scholar]
- 34.Nalawansha DA; Li K; Hines J; Crews CM, Hijacking Methyl Reader Proteins for Nuclear-Specific Protein Degradation. BioRxiv 10.1101/2022.01.24.477424 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reynders M; Matsuura BS; Bérouti M; Simoneschi D; Marzio A; Pagano M; Trauner D, PHOTACs enable optical control of protein degradation. Science advances 2020, 6 (8), eaay5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nabet B; Roberts JM; Buckley DL; Paulk J; Dastjerdi S; Yang A; Leggett AL; Erb MA; Lawlor MA; Souza A, The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018, 14 (5), 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE, Selective target protein degradation via phthalimide conjugation. Science (New York, NY) 2015, 348 (6241), 1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Soucy TA; Smith PG; Milhollen MA; Berger AJ; Gavin JM; Adhikari S; Brownell JE; Burke KE; Cardin DP; Critchley S; Cullis CA; Doucette A; Garnsey JJ; Gaulin JL; Gershman RE; Lublinsky AR; McDonald A; Mizutani H; Narayanan U; Olhava EJ; Peluso S; Rezaei M; Sintchak MD; Talreja T; Thomas MP; Traore T; Vyskocil S; Weatherhead GS; Yu J; Zhang J; Dick LR; Claiborne CF; Rolfe M; Bolen JB; Langston SP, An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458 (7239), 732–U67. [DOI] [PubMed] [Google Scholar]
- 39.Winter GE; Mayer A; Buckley DL; Erb MA; Roderick JE; Vittori S; Reyes JM; di Iulio J; Souza A; Ott CJ, BET bromodomain proteins function as master transcription elongation factors independent of CDK9 recruitment. Mol. Cell 2017, 67 (1), 5–18. e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakamaki J.-i.; Wilkinson S; Hahn M; Tasdemir N; O’Prey J; Clark W; Hedley A; Nixon C; Long JS; New M, Bromodomain protein BRD4 is a transcriptional repressor of autophagy and lysosomal function. Mol. Cell 2017, 66 (4), 517–532. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Filippakopoulos P; Knapp S, Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discovery 2014, 13 (5), 337–356. [DOI] [PubMed] [Google Scholar]
- 42.Phelps MA; Lin TS; Johnson AJ; Hurh E; Rozewski DM; Farley KL; Wu D; Blum KA; Fischer B; Mitchell SM, Clinical response and pharmacokinetics from a phase 1 study of an active dosing schedule of flavopiridol in relapsed chronic lymphocytic leukemia. Blood, The Journal of the American Society of Hematology 2009, 113 (12), 2637–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dey A; Nishiyama A; Karpova T; McNally J; Ozato K, Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Mol. Biol. Cell 2009, 20 (23), 4899–4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang Z; He N; Zhou Q, Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol. Cell. Biol. 2008, 28 (3), 967–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.French CA; Miyoshi I; Kubonishi I; Grier HE; Perez-Atayde AR; Fletcher JA, BRD4-NUT fusion oncogene: a novel mechanism in aggressive carcinoma. Cancer Res. 2003, 63 (2), 304–307. [PubMed] [Google Scholar]
- 46.Zeng L; Zhou M-M, Bromodomain: an acetyl-lysine binding domain. FEBS Lett. 2002, 513 (1), 124–128. [DOI] [PubMed] [Google Scholar]
- 47.French CA; Miyoshi I; Aster JC; Kubonishi I; Kroll TG; Dal Cin P; Vargas SO; Perez-Atayde AR; Fletcher JA, BRD4 bromodomain gene rearrangement in aggressive carcinoma with translocation t (15; 19). The American journal of pathology 2001, 159 (6), 1987–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Filippakopoulos P; Qi J; Picaud S; Shen Y; Smith WB; Fedorov O; Morse EM; Keates T; Hickman TT; Felletar I, Selective inhibition of BET bromodomains. Nature 2010, 468 (7327), 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bond MJ; Chu L; Nalawansha DA; Li K; Crews CM, Targeted Degradation of Oncogenic KRAS(G12C) by VHL-Recruiting PROTACs. ACS Cent Sci 2020, 6 (8), 1367–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schabla NM; Mondal K; Swanson PC, DCAF1 (VprBP): emerging physiological roles for a unique dual-service E3 ubiquitin ligase substrate receptor. Journal of molecular cell biology 2019, 11 (9), 725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nakagawa T; Mondal K; Swanson PC, VprBP (DCAF1): a promiscuous substrate recognition subunit that incorporates into both RING-family CRL4 and HECT-family EDD/UBR5 E3 ubiquitin ligases. Bmc Molecular Biology 2013, 14 (1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li W; You L; Cooper J; Schiavon G; Pepe-Caprio A; Zhou L; Ishii R; Giovannini M; Hanemann CO; Long SB, Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4DCAF1 in the nucleus. Cell 2010, 140 (4), 477–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCall CM; de Marval PLM; Chastain PD; Jackson SC; He YJ; Kotake Y; Cook JG; Xiong Y, Human immunodeficiency virus type 1 Vpr-binding protein VprBP, a WD40 protein associated with the DDB1-CUL4 E3 ubiquitin ligase, is essential for DNA replication and embryonic development. Mol. Cell. Biol. 2008, 28 (18), 5621–5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hrecka K; Gierszewska M; Srivastava S; Kozaczkiewicz L; Swanson SK; Florens L; Washburn MP; Skowronski J, Lentiviral Vpr usurps Cul4–DDB1 [VprBP] E3 ubiquitin ligase to modulate cell cycle. Proceedings of the National Academy of Sciences 2007, 104 (28), 11778–11783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Planelles V; Barker E, Roles of Vpr and Vpx in modulating the virus-host cell relationship. Mol Aspects Med 2010, 31 (5), 398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Greenwood EJ; Williamson JC; Sienkiewicz A; Naamati A; Matheson NJ; Lehner PJ, Promiscuous targeting of cellular proteins by Vpr drives systems-level proteomic remodeling in HIV-1 infection. Cell reports 2019, 27 (5), 1579–1596. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu Y; Zhou X; Barnes CO; DeLucia M; Cohen AE; Gronenborn AM; Ahn J; Calero G, The DDB1–DCAF1–Vpr–UNG2 crystal structure reveals how HIV-1 Vpr steers human UNG2 toward destruction. Nat. Struct. Mol. Biol. 2016, 23 (10), 933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mohamed WI; Schenk AD; Kempf G; Cavadini S; Basters A; Potenza A; Abdul Rahman W; Rabl J; Reichermeier K; Thomä NH, The CRL4DCAF1 cullin-RING ubiquitin ligase is activated following a switch in oligomerization state. The EMBO journal 2021, 40 (22), e108008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.