

Abstract
Polycystic kidney disease is typified by cysts in the kidney and extra-renal manifestations including hypertension and heart failure. The main genetic underpinning this disease are loss-of function mutations to the two polycystin proteins, polycystin 1 and polycystin 2. Molecularly, the disease is characterized by changes in multiple signaling pathways including down regulation of calcium signaling, which, in part, is contributed by the calcium permeant properties of polycystin 2. These signaling pathways enable the cystic cells to survive and avoid cell death. This review focuses on the studies that have emerged in the past 5 years describing how the structural insights gained from PC-1 and PC-2 inform the calcium dependent molecular pathways of autophagy and the unfolded protein response that are regulated by the polycystin proteins and how it leads to cell survival and/or cell death.
Keywords: Kidney cysts, autophagy, TRP channels, cell survival, ADPKD
Graphical Abstract
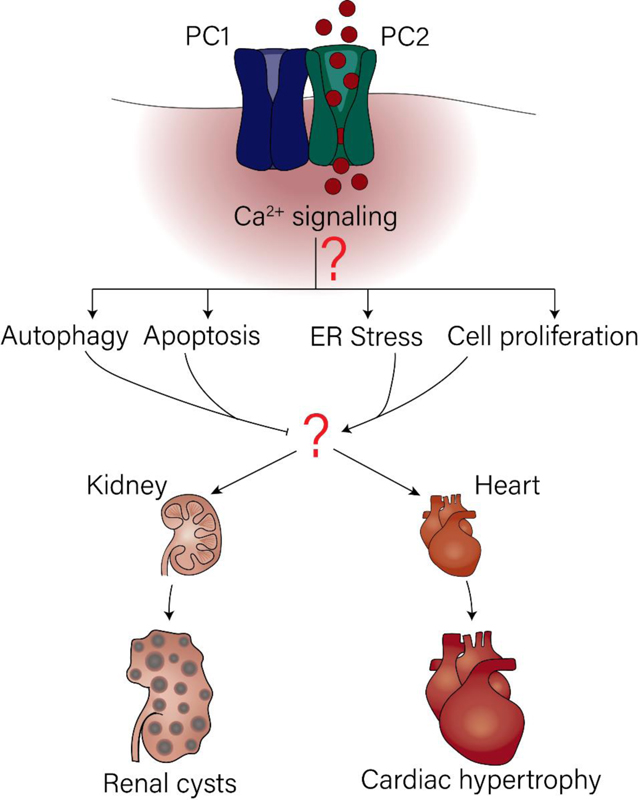
1. Introduction
Many inherited diseases arise due to mutations in the ion channels that mediate calcium flux, underscoring the importance of the intracellular calcium pathway in maintaining cellular physiology. One such disease, Autosomal Dominant Polycystic Kidney Disease (ADPKD) is the most common genetic causes of renal failure and has no cure [1]. Approximately 98% of ADPKD cases arise due to germline mutations in the PKD1 and PKD2 genes, which encode for the interacting proteins polycystin-1 and polycystin-2, respectively (PC-1 and PC-2). The remaining ADPKD cases are contributed by other genes that can regulate the polycystin genes (including HNF1β, PKHD1, GANAB and DNAJB11 amongst others) [2–4]. Currently, the “threshold model”, is the best explanation for renal cyst formation, which postulates that cysts form when the functional polycystin proteins get below a certain level [4–6]. Thus, cysts arise when the non-mutated allele acquires somatic mutations or when the proteins which interact with or regulate the polycystin proteins are affected. This review will focus on the polycystin proteins.
PC-1, with 11 transmembrane spans, has been described to function as a receptor with mechanosensory functions. In contrast, PC-2, known as TRRP1 (as defined by the International Union of Pharmacology [7] but also called TRPP2 in other literature) is a non-selective cation channel that belongs to the Transient Receptor Potential (TRP) channel family. PC-1 and PC-2 have been shown to localize to the plasma membrane, cilia, and ER where they allow for calcium influx or release as either a complex, or by PC-2 itself [8, 9]. There can also be interaction with other TRP channels and intracellular ion flux channels as discussed below [10–14]. The renal cysts result from the nephron-lining epithelial cells undergoing transformation to acquire proliferative, secretory and survival properties. Intriguingly, mutations in the PKD1/PKD2 gene also lead to extra-renal pathologies commonly found in the heart and liver. Indeed, in the current era of renal replacement therapy, the most common cause of mortality in ADPKD patients is cardiovascular complications [15]. Emerging studies have provided new insights that aid in our understanding into how the polycystins regulate important cellular signaling pathways under physiologic circumstances and how their disruption in ADPKD causes pathology. In this review we will discuss the commonalities and discrepancies in the signaling pathways that are disrupted when PC-1 or PC-2 are deleted or mutated. We will focus on the studies that have emerged in the past 5 years which have provided insights that aid in our understanding of how the polycystins regulate important calcium signaling pathways under physiologic circumstances and how their disruption in ADPKD causes renal and extra-renal pathology.
2. Renal pathologies in ADPKD
ADPKD is characterized by bilateral cyst formation in a small fraction of the kidney nephrons [5, 16]. Although only a small number of the one million nephrons per kidney are affected, the cysts can cause the kidney to increase from the size of a human fist to a basketball. As the name implies, ADPKD is inherited in an autosomal dominant fashion. Genotypic analysis of ADPKD patients shows there is a higher prevalence of PKD1 mutations than PKD2 mutations, with germline PKD1 mutations comprising ~80% of all ADPKD diagnoses, and 15–20% due to PKD2 mutations [6]. ADPKD is very common, with an incidence of 1 in 400–2,500 people and spans across all sexes and ethnic groups [17]. There is one FDA approved treatment, Tolvaptan, which targets the Vasopressin Receptor type 2 (V2R), and there is no cure [18, 19] although there are several ongoing clinical trials that target other promising pathways [20]. Before the molecular identity of PC-1 and PC-2 were discovered in 1994 and 1995 respectively [21–25], it was initially proposed that the molecular mechanism behind cyst development occurs due to abnormal secretion of chloride ions through the cystic fibrosis transmembrane conductance regulator (CFTR) into the cystic cavity. CFTR is a chloride-permeant channel phosphorylated in a cAMP-dependent manner, and its activity is increased in ADPKD [16, 26] [27]. Drugs targeting CFTR have been effective in slowing cyst growth in animal models [28–30] and clinical trials with CFTR inhibitors are ongoing [20]. The molecular identity of PKD1 and PKD2 as the genetic molecules which underlie ADPKD have since led to a better understanding of cyst development. It has emerged that multiple signaling pathways are affected by either being upregulated (cAMP signaling) or downregulated (Ca2+ signaling) in cystic cells following mutations to PC-1 and/or PC-2 [31]. One theory which originally gained traction but has since been questioned is that because of their localization to the plasma membrane in primary cilia, PC-1 and PC-2 can sense fluid flow inducing calcium flux through PC-2, which leads to subsequent downstream calcium signaling [32]. The caveat with the ciliary hypothesis is that it discounts a role for polycystin dependent calcium signaling originating in the ER or at the plasma membrane. In addition, it has been recently shown that the cilia are largely impermeant to calcium, opposing the ciliary hypothesis [33]. Irrespective of the initiating source of calcium, it has been consistently found that loss or dysfunction of the polycystins leads to impaired fluid secretion and disrupted calcium signaling [31]. Yet, explaining the exact contribution of disrupted calcium in leading to renal cysts has still been a matter of debate.
As addressed above, cAMP, which is elevated in ADPKD, enhances CFTR activity and fluid secretion. Recent studies have added a new potential secretory pathway mediated by TMEM16A, a calcium-dependent chloride channel. In kidney specific Pkd2−/− mice and ADPKD patient cells, TMEM16A expression increased through the hypoxia induced factor 1 (HIF1α) and additionally, lipid peroxidation was shown to directly increase the activity of TMEM16A [34, 35]. This suggests that the cystic environment causes transcriptional changes which allow for the polycystin deficient cells to adapt to the hypoxic environment in a non-calcium dependent manner by regulating both TMEM16A expression and activity. Importantly, subsequent studies showed that Pkd1/Pkd2 deficient organoids generated from murine collecting duct (CD) cells had increased Cl− secretion and proliferation promoting the cystic phenotype, which was dependent on TMEM16A activity [36]. Contrary to most ADPKD literature, it has been shown that in Pkd1/Pkd2 deficient cells and Pkd1 deficient cells there was an increase in ATP-mediated calcium signaling, but a calcium signaling decrease in Pkd2 knock out cells [37]. In all cases, knockdown of TMEM16A restored the basal calcium phenotype, suggesting a feedback mechanism between TMEM16A and ATP-mediated signaling. As FDA approved inhibitors to TMEM16A were efficacious in mitigating cyst growth in vitro and in vivo animal models, it is possible that TMEM16A inhibition may be a viable therapeutic target for ADPKD [38]. Interestingly, the dual knock out of Pkd1 and Cftr did not decrease cyst growth, challenging the traditional understanding that CFTR is the major contributor of chloride secretion that promotes cyst expansion [39], although it must be noted that these studies were done in animal models and they do not negate the ongoing clinical trials using CFTR inhibitors.
Despite the advances in understanding how renal epithelial cells acquire a cystic phenotype, how inactivation of the polycystin proteins ultimately leads to cyst development is still contested. It is known that the regulation of metabolic pathways and cell differentiation through the remodeling of the extracellular matrix by the polycystin proteins can facilitate cyst development [40]. This was highlighted when re-expression of either Pkd1 or Pkd2 in a polycystin deficient mouse model was able to revert cyst growth and restore kidney function [41]. Evidentially, even though extensive research has provided a basis for understanding how cysts develop in ADPKD, we still do not have a full grasp how loss of polycystin function regulate and activate these pathways that lead to cyst growth and development. Nonetheless, research in the last few years have given us an insight into the different calcium dependent pathways that could be contributing to the pathophysiology of ADPKD. The remainder of this review will focus on how the calcium signaling pathways regulated and mediated by PC-1 and PC-2 allow cystic cells to avoid apoptosis and engage cell proliferation.
3. Calcium conduction of the polycystin-1 and polycystin-2 complex
3.1. Polycystin 1
PC-1 is an 11 transmembrane domain protein that consists of a long N-terminus and a short C-terminal. The last 6 transmembrane spans of PC-1 have homology to the 6 transmembrane spans of PC-2. The N-terminus of PC-1 contains multiple domains like the leucine-rich repeats, low-density lipoprotein, receptor for egg jelly, and C-type lectin (CTL) domain which undergoes auto proteolytic cleavage and non-covalently reattaches. Several studies have demonstrated how auto-cleavage and re-attachment by non-covalent bonding of the N-termini regulates a variety of cellular functions including cell-cell interactions and mitochondrial function [42, 43]. Research on PC-1 has focused on the last few years on understanding the contribution of PC-1 in the ionic flux within the PC-1/PC-2 complex and dissecting the role of the different domains within the N and C-termini.
Based on the structural insight gained from the structure of the PC-1/PC-2 complex [44], recent work dissected the potential of PC-1 to function as an ion channel when complexed with PC-2. Co-expression of PC-1 in oocytes with a gain-of-function (GOF) PC-2 mutant (mutations on the S4-S5 linker, which does not arise from ADPKD pathogenesis, as all disease-causing mutations disrupt function) produced robust calcium currents [45]. The resulting calcium currents from the PC-1/PC-2-GOF complex were more robust than the PC-2-GOF mutant alone, providing evidence that PC-1 can contribute to the flux of ions [45]. Additionally, expression of PC-1 after cleavage of the N-terminus or the last 6 transmembrane domains of PC-1, evoked calcium currents when complexed with PC-2-GOF [45]. The strongest piece of evidence to demonstrate that PC-1 forms part of the pore, was shown when a key amino acid in the pore of PC-1, R4090W, was mutated and the currents produced by the complex were abolished [45].
The N-terminus of PC-1, which contains the CTL domain (a stretch of 108 amino acids), is cleaved under normal physiological conditions and remains non-covalently attached to the PC-1 membrane fraction [46]. Recent work proposed that the role of the cleaved PC-1 N-terminus is regulating the conductivity and open probability of PC-2 [47]. Application of the CTL domain to excised ciliary patches prolonged the opening of PC-2 and showed a five-fold increase in the open probability of PC-2 channels [47]. It is proposed that the cleaved PC-1 CTL domain binds to a glycan localized to N375 on the TOP domain of PC-2 where it regulates ion permeability [47], and that this could represent a new regulatory mode of PC-1 in PC-2 gating or ion permeation. A summary of the different regions of PC-1 which regulate PC-2 channel function are shown in Fig. 1.
Figure 1. Topological structure of PC-1 and PC-2.

PC-1 contains 11 transmembrane spans with a long N terminus. Domains within PC-1 N termini have been described to regulate channel activity of PC-2, a member of the Transient Receptor Potential Channel family. Red arrow indicates regions within PC-1 that have been described to regulate PC-2 channel activity. See text under Section 3.1 for details.
On the other hand, the C-terminus tail of PC-1 contains a G-protein binding domain (GBD) and a coiled-coil domain [48]. These two domains (the GBD and coiled-coil) enable PC-1 to bind to other channels and receptors like PC-2 and G-coupled receptors [49]. A recent study demonstrated the importance of the GBD site not only in the regulation of PC-2 activity but in disease progression [50]. Knock-in heterozygous mice with a single amino acid deletion in L4132, identified from a human ADPKD sample and found within the GBD domain, resulted in severe cyst growth [50]. When the Pkd1 ΔL4132 version was co-expressed with PC-2, there was a significant decrease in the currents mediated by PC-2 in comparison to the WT PC-1 [50]. It is important to highlight that the GBD domain also activates downstream G-protein signaling pathways [50] and thus it cannot be ruled out that additional regulatory mechanisms downstream of channel activation also contributes to the resulting cystic phenotype.
While the previous studies have elucidated a potential role of PC-1 to membrane calcium flux under in vitro conditions, the question remains as to the contribution of PC-1 under normal physiologic conditions. A key limitation of the studies discussed above is that functional analysis of PC-1 was achieved only when expressed with the GOF PC-2 mutant or when pore mutations to PC-1 were introduced [45] [47]. Evidence of PC-1 ionic conductance was only achieved by mutating residues within the potential pore of PC-1 suggesting that PC-1 in its native state is unlikely to flux ions [45]. Future studies will be essential to uncover the link between structure and function of PC-1 that has not yet been revealed and connected in the studies discussed above.
3.2. Polycystin 2
Unlike PC-1, PC-2 is part of the Transient Receptor Potential (TRP) family and contains 6 transmembrane domains. PC-2 is a well described non-selective cation channel that when localized with its interacting partner PC-1 in the cilia allows for calcium entry [51–53]. Nonetheless, PC-2 contains an ER retention signal in the C-termini which localizes the protein in the ER, its predominant localization [54], as well as an EF-Hand domain followed by a coiled-coil domain [55, 56]. There have been contradictory findings as to the conductive nature of PC-2. The earliest studies decoding PC-2 ionic conductivity were conducted on lipid bilayer preparations [57], however, subsequent electrophysiological studies have primarily focused on ciliary PC-2 [11, 33, 52, 58].
ADPKD is classified as a ciliopathic disease as mutations to the PC-1 and PC-2 proteins leads to defect in the primary cilia [59]. The primary cilia are a single, non-motile organelle projecting from the apical side of a range of cell types including renal epithelial cells [60]. A variety of diseases that range from liver, kidney to retinal disease have been described due to abnormalities within the cilia [61]. The cilia are known to be a signaling hubs, and calcium signaling is one the primary signaling pathway related to ADPKD ciliopathy.
As previously mentioned, most of the work done to decode PC-2 cation conductivity has focused on using direct ciliary electrophysiological approaches. There are two main advantages of this approach: bypassing the expression of PC-2 in artificial systems, and with genetic manipulation, there can be dissection of the specific ionic contribution by PC-2 through gene deletion. PC-2 has been described to produce a large conductance current which can be activated by an increase of calcium concentration [51, 52]. It is worth highlighting that under normal physiologic experimental conditions PC-2 is primarily closed in the cilia and requires large depolarization events to activate cation flux, a condition that would be likely improbable in a renal context [10]. Additionally, the primary cilia have a very small volume (0.5 fL) and calculated to have a resting concentration of ~600 nM [Ca2+], which would be equivalent to ~100 free calcium ions [62]. The low number of calcium ions within the cilia by volume does not seem sufficient to activate cytosolic calcium release to amplify and sustain the signal [63].
If the environmental conditions of the primary cilia do not favor calcium flux via PC-2, which cation (Na+, K+ or Ca2+) is being fluxed by PC-2? To resolve which ion is the most likely to be fluxed by PC-2 in primary cilia, recent work in ciliary patches has been performed in experimental conditions that maximized calcium conductivity of the channel [64]. Based on these experimental conditions, a calcium permeability of 0.09 was calculated with an approximate conductance of 20 pS [64]. To reach this conduction value would require hundreds of ciliary PC-2 channels to simultaneously open, and this high number of channels has not been demonstrated [64]. Moreover, the number of ions (millions) that would be calculated to flow through the hundreds of open PC-2 channels would exceed physical capacity of the cilia [64]. These results further accentuate the outstanding question of what are then the conditions that allow for calcium influx through PC-2 in the cilia. The authors of the previously mentioned study also emphasize key points that should be taken into consideration when comparing existing literature to determine PC-2 ionic conductivity [64]. Experimental approaches have significantly differed amongst the studies that have characterized PC-2 channel conduction [10, 51–54, 64, 65]. Characterization of ionic permeability greatly depends on the ionic composition, depolarizing conditions and excising of ciliary patches, all of which represent unique conditions that can alter the behavior of the channel. Thus, the imposed experimental conditions throughout the different studies can confound the physiological conditions that lead to PC-2 activation.
To summarize this discussion on PC-2 channel conductance of calcium, future studies are required to determine ER PC-2 function vs ciliary PC-2 function, and the contribution of these PC-2 mediated calcium currents in signaling pathways that govern cell adaptability and are impaired in ADPKD. So far, no studies have clearly delineated the relative contribution of PC-2 channel activity in the ER vs in the cilia. This becomes especially relevant as one considers that PC-2 predominantly localizes to the ER. The next section will focus on the interactions and regulation of PC-1 and PC-2 signaling in the ER and plasma membrane.
4. Interaction of polycystin 1 and polycystin 2 with other calcium channels
In addition to the role of calcium influx the PC-1/PC-2 complex on the cilia, these proteins can also regulate calcium activity in the plasma membrane and ER by interaction with other known calcium channels, like L-type voltage calcium channels, TRP channels, Ryanodine Receptor (RyR) and the Inositol triphosphate Receptor (InsP3R). In cardiomyocytes it has been shown that PC-1 stabilizes and regulates the degradation of L-type calcium channel (LTCC), where deletion of PC-1 in cardiomyocytes, resulted in decreased levels of α1C LTCC, which leads to cardiac hypertrophy and fibrosis [66]. In contrast, in kidney epithelial cells PC-2 directly binds the InsP3R, although the actual stoichiometry of this interaction is unknown [12]. The loss of PC-2 in kidney epithelial cells diminished calcium dependent calcium release of the InsP3R whereas the overexpression of PC-2 restored the calcium signal [12, 13]. PC-2 can also alter the distribution of the InsP3R subtypes within ER-mitochondrial contact sites, known as the mitochondrial associated membranes (MAMs) [67].
PC-1 was similarly found to enhance InsP3R, however, as less has been studied on PC-1 in the ER, the direct mechanism of this interaction remains unsolved [68]. Additionally, it has been shown that PC-2 can associate in heterotetramers complexes with other channels like TRPV4 and TRPM3 [10, 11, 14]. These ciliary plasma membrane channels can regulate the frequency and time of opening of PC-2, as calcium influx through these TRP channels can represent a positive feedback loop to activate PC-2 calcium influx [10]. Ultimately, the loss of calcium signaling both directly (by the polycystins themselves) and indirectly (by regulation or associating with other calcium channels) is expected to culminate in downstream signaling pathways that drive cyst growth by regulating cellular processes that lead to cell death or survival. In the following section we will discuss the recent data that has emerged of how calcium signaling mediated by the polycystins can regulate proteins and signaling pathways that activate cell death and survival through the unfolded protein response (UPR) pathway, autophagy, extracellular stress, and cell proliferation (Fig. 2A–C).
Figure 2. The role of polycystin 1 and polycystin 2 in regulating signaling pathways that lead to cell death and or survival. A.

PC-1 and PC-2 have been described to interact and/or regulate molecules within the Unfolded Protein Response (UPR) which is activated by ER stress. Red arrows indicates when upon activation of ER stress there is upregulation of PC-1 or PC-2. B. Diagram of the autophagic pathways implicated in PC-1/PC-2 pathways that are activated through cell stress, calcium-dependent pathways, or hypoxia. Red spheres indicate upregulation of the indicated molecules while blue-red gradient spheres indicate contradicting data upon deletion or mutation of PC-1 or PC-2. C. Model of the regulation of the polycystin proteins in autophagy. Dash arrows indicates uncharacterized mechanism. See text under Sections 4 and 5 for details. Molecules within each panel are labeled with the corresponding color.
5. Polycystins role in cell death and cell survival
5.1. Polycystins and ER stress
The studies described in the previous sections have characterized the ionic conductance of PC-2 (Fig. 1). However, an aspect that remains unclear is the contribution of ciliary vs cytosolic calcium fluxing through the PC-1/PC-2 complex in regulating these calcium-dependent signaling pathways. An example of this intracellular calcium regulation is by the EF-hand motif that is present on the PC-2 C-terminus. Previous work has demonstrated that the EF-hand motif can regulate PC-2 channel activity, linking previous studies where a rise in intracellular calcium activated PC-2 channel conductance [51, 52, 55, 56]. Recent work questions this hypothesis as mutations to the Ca2+ coordinating sites of the EF hand did not alter PC-2 channel activity in primary cilia [65]. Additionally, mice with the EF hand mutation did not develop cystic kidneys, although it should be noted that mutations causing ADPKD have not been found within the EF domain [65]. Thus, the intracellular calcium regulation of PC-2 by the EF-hand may not contribute to cystogenesis, but it remains an open question whether calcium can regulate other functional roles of PC-2, and the downstream consequences of decreased PC-2 dependent calcium signaling as discussed below.
Calcium is an integral second messenger that is implicated to regulate a variety of cellular processes including cell death and survival [69]. Calcium is primarily stored in the ER where one of its roles is to maintain the calcium-dependent folding of chaperones and regulate protein quality control. Thus, calcium imbalance can contribute to the accumulation of unfolded protein promoting ER stress and activating the UPR pathway which is known to regulate cell death and survival [70]. This pathway is activated to halt translation and restore homeostasis, however prolonged activation of the UPR response can activate pro-apoptotic pathways. The UPR pathway consists of three branches: PERK, IRE1α, and ATF6. In response to ER stress, PERK dimerizes and phosphorylates eIF2α which contributes to reduce protein translation to aid with the overload of unfolded and misfolded proteins [71]. IRE1α is also activated, inducing splicing of the transcription factor XBP1s which then will activate the transcription of target UPR genes to restore homeostasis. Lastly, activated ATF6 translocate to the golgi where it undergoes proteolytic cleavage which allows for its interaction with ER stress elements [71].
In recent years, ER stress has been implicated to contribute to the pathogenesis of different diseases including renal and cardiovascular related pathologies [71]. Thus far, there are very few studies that investigate directly whether ER stress contributes to both renal and extra-renal related pathologies in ADPKD. In 2008, PC-2 was first implicated to regulate the UPR by activating the PERK signaling axis in HEK293 and iMCD3 cells [72]. It was shown that PC-2 forms a complex with PERK and eIF2α which promotes their activity in regulating ER stress, ultimately repressing cell growth [72]. Consistent with the above finding, expression of pathogenic mutants of PC-2 in immortalized murine collecting duct (imCD3) cells, including mutants that lack the C and/or N terminus led to decreased phosphorylation of eIF2α [73]. Moreover, ER stress increased PC-2 degradation by the association of PC-2 to the endoplasmic reticulum associated degradation (ERAD) machinery and its components [73]. Considering that the EF-hand domain is found on the C-terminus of PC-2, it brings to question whether calcium regulation of PC-2 through this domain can also regulate the UPR pathway.
In addition to PC-2 positively regulating the UPR pathway, the induction of ER stress itself increased PC-2 expression in kidney epithelial cell lines [74]. Intriguingly, there is a positive correlation between the expression of the UPR protein component CHOP and PC-2 in the left ventricle from heart failure patients [74]. This increase in PC-2 protein levels is suggested to confer protection against stress induced cell death [74]. Moreover, inductions of ER stress in a variety of cell lines increased phospho-eIF2α leading to upregulation of PC-2 due to ribosomes suppressing inhibitory μORF, allowing for PKD2 transcription [75]. The role of PC-2 participating in ER stress was further demonstrated when deletion of Pkd2 in renal cortical tubule epithelial cells suppressed amino acid biosynthesis by disruption of the interaction of PC-2 with the adaptor protein TBL2 and impaired the ER stress axis [76]. Association of PC-2 with TBL2 was found to be necessary for the recruitment of eIF2α to TBL2 and the subsequent phosphorylation of eIF2α [76]. These interesting studies shed light on yet another role of PC-2 in ER stress that is largely understudied.
PC-1 can also negatively regulate the UPR pathway through the regulation of PC-2 expression [77]. Deletion of PC-1 in imCD3 cells increased expression of PERK and ATF6 [78]. It was shown that activation of these factors increased GRP94 expression, which then allowed for more efficient folding of PC-2, increasing PC-2 expression [78]. Exciting new studies have also shown that activation of the IRE1α activated the transcription factor, XBP1, in a hypomorphic PC-1 ADPKD murine model which delayed cyst formation [79]. Separately, another study demonstrated a reciprocal interaction of XBP1 to PC-1 as activation of XBP1 improved cleavage and trafficking of PC-1 [80]. While these studies demonstrate that both PC-1 and PC-2 are involved in promoting the UPR response, there are still many things that remain to be uncovered. First, there are no in-vivo studies that directly investigate the role of PC-1 and PC-2 in ER stress. Second, it remains largely unknown whether ER stress contributes to cyst development or to any of the known extra-renal complications. Finally, it is of great interest to consider whether the PC-2 mediated calcium release is responsible in regulating the UPR pathway. As ER stress contributes to the pathologies of many different diseases, we need to further understand how loss or mutations to the polycystin channels activate ER stress and the role in the pathology of ADPKD.
5.2. Polycystins and autophagy
To maintain cellular homeostasis, the cell constantly recycles and degrades macromolecules and organelles, which occurs through the process of autophagy [81]. Under physiological conditions, autophagy occurs at a steady rate, which increases under cellular stress to maintain cell survival [81, 82]. Macroautophagy can be induced by different cellular stresses such as hypoxia and ER stress as described in the previous section (Fig. 2B). Here, we will describe how the polycystins proteins regulate the autophagic flux, to maintain cellular homeostasis. In spite of the different initiators of autophagy all culminate in the activation of autophagy related genes (ATGs), which aid in the delivery of the cargo to the lysosome [83], and then bring about the engulphment of the autophagosome by the lysosome. While is not disputed that autophagic flux is changed in ADPKD models, understanding how autophagy contributes to cystogenesis is controversial, as contradictory findings (some suggesting increased autophagy, others suggesting decreased autophagy) have been reported, which will be further described below.
A Pkd1-hypomorphic murine model, mimicking mutations derived from patients with PKD1 mutations, showed decrease in autophagic genes such as beclin1, Atg3, Atg12 and p62 leading to decreased autophagic flux [84]. Other investigators have examined the mTOR pathway, a major regulator of autophagy which suppresses autophagy under basal conditions. Under cellular stress, however, the expression of mTOR is decreased, allowing for autophagic rate to increase [81]. In a zebrafish model, mutations to the pkd1 orthologue had increased mTOR levels which lead to decreased autophagy [85]. To further demonstrate that decreased autophagy is associated with increased cyst burden, the knockdown of an integral component of autophagy, ATG5, which lies downstream of mTOR, in the pkd1 mutant zebrafish, led to increased cyst burden [85]. Consistent with this pathway, treatment with a beclin1-peptide (which activates autophagy) led to increased autophagy, decreased cysts, and improved kidney function in the pkd1 zebrafish model [85]. Previous studies have demonstrated that rapamycin, a well-established inhibitor of mTOR, which would be expected to increase autophagy, improved cyst burden in ADPKD patients and animal models [86–89]. Collectively, these studies suggest that cysts have decreased levels of autophagy due to upregulation of the mTOR pathway by the polycystin proteins, and activation of autophagy diminishes cyst burden [90–92].
A possible mechanism by which polycystin proteins regulate autophagy is through calcium-dependent regulation of the mTOR pathway. In support of this, in stem cell derived cardiomyocytes PC-2 was shown to promote autophagy under glucose starvation, and knockdown of PC-2 diminished autophagy [93]. These authors suggested that PC-2 was required to provide the calcium-dependent repression of mTOR and activation of AMPK, an inducer of autophagy [93]. Similarly, a cardiac specific PC-2 KO study showed decreased autophagic flux under nutrient deprivation conditions. When the cardiomyocytes were treated with a Ca2+ chelator, the stress-induced autophagy was repressed [94]. The important role of calcium from PC-2 in this pathway was shown as over-expression of wild-type PC-2 but not a pathogenic variant which impedes cation and thus calcium movement by PC-2 was able to improve autophagic flux [94].
PC-2 has been shown by multiple investigators to regulate the autophagy pathway by direct interaction with beclin-1, a key regulator of the formation of the autophagosome. PC-2 induced autophagy by physical and functional interaction with beclin-1 in a PC-2 channel activity channel manner [95]. Thus, mutant versions of PC-2 without the C-terminal tail which contains the coiled-coil domain did not bind to beclin-1, and a version which did not flux calcium had decreased binding to beclin [95]. Our recent study supported this theory as deletion of PC-2 in a C2C12 myoblast cell line decreased autophagic flux and beclin-1 protein levels [96]. We suggest that PC-2 mediated calcium release may be necessary for beclin-1 activated autophagy [96]. Taken together, these results suggest that PC-2 mediated calcium signaling is required for autophagy, but if these same pathways are function in PC-1 mutations is unclear [97].
In contrast to the findings discussed above, other studies have found that cystic environments induce autophagy. In both human and rat PKD models it was found that beclin-1 and LC3, markers of autophagy, had increased expression [98]. Additionally, HIF1α, a transcription factor (TF) that promotes autophagy, was upregulated. However, inhibition of HIF1α, did not show a therapeutic effect [98]. In murine kidney epithelial iMCD3 cell lines, with PC-2 or PC-1 KO and in proximal tubule epithelial cells derived from patients with PKD1 mutations, there was increased autophagy which promoted enhanced cell survival in the cystic kidney upon starvation [99]. Indeed, activation of autophagy increased cyst development in a PKD1 deficient mouse model [100]. These data suggest that the role of the polycystins in autophagy may be cell dependent.
Given the above contradictory findings regarding autophagy in the setting of ADPKD, it is therefore not surprising that there have been mixed results in targeting autophagy as a global therapy for PKD patients [100]. Moreover, expression of autophagic genes can vary among ADPKD patients [100]. Such disparate clinical findings within the ADPKD patient population may explain why mTOR inhibitors have not been successful in clinical trials as treatment for ADPKD patients [101].
What can be concluded about autophagy and ADPKD? It appears that polycystin proteins play a role in the activation of autophagy, however, the mechanistic insight of how this is achieved is not fully understood. It may be that the disparate results of how autophagy is regulated is due to the type of stimulus (i.e.: cellular stress, calcium dependent pathway) and cell specificity. Perhaps most importantly, the stage of the cyst development will alter the autophagy status, thus, it is more likely to find a hypoxic environment when cysts are well established compared to the earliest stage of cytogenesis. Additionally, the different germline mutations in the polycystins may confer different levels of impairment to the downstream autophagy pathways. Thus, although clinical trials targeting the mTOR pathway in ADPKD patients have not been promising [20, 101], a precision medicine approach which includes measurement of the autophagy genes expression levels may be more beneficial.
5.3. Polycystin-2 dependent protective cellular mechanisms
In the absence of an ADPKD setting, PC-2 seems to have a protective role as it has been shown to be upregulated at both the mRNA and protein level under various disease states not related to ADPKD [74, 102]. Thus, it has been shown that PC-2 protein is increased in both ischemic and non-ischemic cardiomyopathy patients which are also under ER and oxidative stress [74]. In the brain it has been suggested that PC-2 could play a protective role when PC-2 expression was increased after inducing acute seizures in mice by injection of kainic acid known to induce epileptic seizure in the hippocampus [74]. This is of importance as PC-1 and PC-2 can form a plasmalemmal ion channel signaling complex in neurons like what has been observed in the cilia [103]. While these data are very interesting and point to the fact that PC-2 is not only important in the kidney, the mechanisms of how PC-2 is upregulated and confers a protective effect in other organs remain elusive.
5.4. Polycystins and cell proliferation
Renal cysts are also marked by the transformed ability to maintain proliferation. This alteration in proliferation has been linked to the primary cilia, as loss of cilia had initially been noted in cystic cells [104]. As already discussed, the ability of primary cilia to mediate calcium signaling is under debate. However, there is substantial evidence that the primary cilia does function to enable other non-calcium signaling pathways linked to cell proliferation by activation of Wnt, Notch and Hedgehog signaling pathways amongst others (the reader is directed to the following reviews and papers [105–108]).
As previously mentioned, one of the major pathways driving cyst growth is the cAMP signaling pathway, which activates the PKA dependent phosphorylation of CFTR [5, 26]. In renal cells, one way cAMP synthesis can occur is by activation of the vasopressin 2 receptor (V2R), located on the basolateral site of the collecting duct cells. The V2R is the primary target of tolvaptan which slows cyst growth by decreasing cAMP levels [18, 20, 109, 110]. It has been also shown that there is decreased calcium signaling in primary epithelial cells derived from human patients which activates the RAS/RAF-1/MEK/ERK pathway and leads to cell proliferation [111]. Restoring cytosolic calcium increased AKT expression, inhibiting the cAMP activation of RAF and ERK [111]. To further determine the role of polycystins in regulating AKT, it has been shown that in a cardiac setting PC-1 activates AKT during ischemia/reperfusion events which further activates CTFG expression, a marker for fibrosis and cardiac remodeling [112]. A recent study also demonstrated that selective activation of the calcium sensing receptor (CaSR) in a PKD1 mutant model restored cytosolic calcium levels and therefore decreased cAMP levels [113]. In addition, it was shown that loss of PC-2 and its partner protein PC-1 induces proliferation of neural progenitor cells, indicating that PC-2 also negatively regulates proliferation in the brain [114]. These studies collectively demonstrate that PC-2 acts as a negative regulator of proliferation not only in the kidney but also in the heart and brain. While we know that loss of polycystins, and increased proliferation contributes to cyst growth in the renal system, future studies are needed to characterize how loss of proliferation contributes to pathology.
5.5. Polycystins role in cell-matrix signaling pathways
ADPKD cystic cells are characterized by impaired migration, extracellular matrix (ECM), and increased fibrosis which has been seen in cells with PKD1 and PKD2 deficiency [115, 116]. Cell migration occurs through the activation of focal adhesion points that allow the cells to anchor and move utilizing the ECM [117]. Previous work has demonstrated that PC-1 localizes and interacts with proteins within the focal adhesions like integrins and cadherins in human renal epithelial cells [118]. Additionally, it was shown that PC-1 regulates focal adhesion dynamics in madin-darby canine kidney (MCDK) cells through the regulation of the actin cytoskeleton through the focal adhesion kinase (FAK) [119]. This was further validated in imCD3 cells where PC-1 was found to regulate the activation of FAK, which is integral for focal adhesions turnover and in turn, in cell migration [120]. This is important as a recent study demonstrated that inhibition of FAK in 3-D models of MCDK cells decreased cyst development [121]. However, how mutations to PC-1 and its role in regulation focal adhesion complexes lead to cyst development remains unclear. Interestingly, much less is known about the role of PC-2 in focal adhesions. Future studies are necessary to investigate if PC-2, like PC-1, plays a role in focal adhesions dynamics. This is especially relevant since both focal adhesions assembly and disassembly are calcium-dependent.
5.6. Polycystins and apoptosis
A final calcium dependent pathway affected in ADPKD models is apoptosis, which has been implicated to contribute to cyst pathogenesis [122, 123]. More than 20 years ago, it was suggested that apoptosis occurs in polycystic kidneys, and contributes to the pathogenesis in ADPKD [124, 125]. A later study suggested that PC-2 is an anti-apoptotic player by mediating calcium release from the ER, thereby regulating the signals sent to the mitochondria, and its sensitivity to apoptosis [124]. In addition, a separate study showed that PC-2 knockdown in LLC-PK1 and imCD3 also led to increased levels of caspase-3 when the cells are serum starved for 24 hours [74]. Curiously, a different study showed that cells subjected to 48 hours serum starvation in PC-2 KO imCD3 cells led to decreased apoptosis [99]. As the section on autophagy discussed, there are contradictory results regarding the function of polycystins in these pathways. These conflicting results may be a result of different experimental approaches which could lead to the activation of different signaling pathways.
6. Are polycystins pro-apoptotic or pro-survival?
What is the exact role of PC-1 and PC-2 in cell death and survival? The literature on polycystin protein being anti-proliferative seems undisputed. Thus, loss of polycystin proteins results in the upregulation of proliferation across the renal and extra-renal context. However, what remains ambiguous is the exact contribution of the polycystin proteins in activating and contributing to autophagic and cell-death pathways. As discussed in the sections above, these results are dependent on the tissue/organ type under investigation, and, in the context of renal cysts, the stage of the disease progression. The discrepancy noted among the studies described in this review discussing the role of the polycystins in cell death and/or survival underscore the different experimental approaches used to answer the question at hand. In some studies, the genetic approach consists of gene deletion, knock-down or mutations. The different genetic backgrounds can then confound the role of the polycystins in activating or regulating molecules within these signaling pathways. Additionally, different signaling pathways are activated in the context of the kidney, where cysts form, compared with the heart, where cysts do not form. Future studies investigating whether this phenomenon is cell and/or organ dependent and what are the conditions that lead to the activation of one pathway versus the other.
Finally, it is essential to understand that ADPKD is a progressive disorder even when considering a single organ, such as the kidney. This contrasts with other diseases such as cancer where both proliferation and apoptosis are regulated by calcium signaling (as discussed in other papers within this collection and [126]). Thus, the early cystogenic phase is not the same as the late cystic stage. Indeed, an elegant study that decreased the levels of PC-1 in only 8% of the tubules demonstrated that the kidneys were able to maintain a near-normal renal status for over 6 months after knockout [127]. However, the cysts that did form were in clusters, and these clustered cysts then preceded the rapid cystic progression, suggesting that the initial cysts caused the snowballing of more cysts [127]. What causes this change from mildly cystic to severely cystic? It should be remembered that the cystic areas become increasingly hypoxic and have decreased availability of metabolic substrate. Moreover, the late cystic phase is typified by increased cellular transformation (that enables cells to survive apoptotic conditions), increased extracellular matrix deposition, increased macrophage infiltration and increased fibrosis, all of which could contribute to the snowball cystic effect [127].
7. Conclusions and future directions
We have discussed the new emerging data about the function and role of the polycystins proteins in cell death and survival. Recent studies have open new avenues to understanding the signaling pathways PC-1 and PC-2 regulate like autophagy, the UPR pathway, and cellular stress (Fig. 2). Moreover, we discuss which of these processes are found to be dependent on the channel activity of the PC-1/PC-2 complex. Nevertheless, there are still gaps in the literature that need to be addressed. While previously it was thought that cyst development was primarily driven by the activation of CFTR, recent data demonstrates that other potential molecules such as TMEM16A may be involved. Furthermore, the novel role of PC-2 in the UPR response may be important in the ADPKD phenotype, as ER stress can contribute to both renal and extra-renal pathologies. In addition, both apoptosis and cell growth are well described to occur in ADPKD cell and mouse and patient derived samples. Polycystins play a critical role in regulating all these signaling pathways. However, there are key question that remain unanswered that limit our understanding of the biological impact of these proteins. What are the physiological conditions that activate the polycystin proteins to mediate ionic flux, and are the activating conditions specific to the intracellular location of PC-1/PC-2 and/or organ specific? PC-2 is known to localize primarily on the ER and yet, its role on this highly dynamic organelle is largely understudied. With the advancement of methodologies like high-resolution imaging, the exact contribution of ciliary PC-2 vs ER PC-2 might be discerned. Future studies focusing on generating organ specific conditional KOs along with in-vitro studies may identify the timing points as to when the polycystins are pro-survival and when they contribute to cell death. In addition, with new molecular techniques, pathological variants can be used to determine what occurs in a more physiological context and to distinguish the calcium dependency of these pathways. In conclusion, the polycystins play a critical role in these cellular pathways that regulates cell death and cell survival. However, there is a missing link between how loss of the PC-1 and PC-2 through all these calcium dependent processes contribute to renal and extra-renal pathologies.
Highlights:
Polycystin 2 functions as a calcium permeant ion channel in ciliary plasma membrane and endoplasmic reticulum but its conductivity for cations may be location specific.
The unfolded protein release pathway and endoplasmic reticulum stress pathways are emerging ways in which polycystins can contribute to cell survival.
Polycystin proteins are novel interactors and regulators of organelle specific and macro-autophagic pathways.
Acknowledgements
Research reported in this publication was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Numbers U2CDK129917 and TL1DK132769. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations:
- ADPKD
autosomal dominant polycystic kidney disease
- CFTR
cystic fibrosis transmembrane conductance regulator
- GBD
G-protein binding domain
- GOF
gain-of-function
- HIF1α
hypoxia induced factor 1
- MAM
mitochondrial associated membranes
- polycystin 1
PC-1
- polycystin 2
PC-2
- TRP
Transient Receptor Potential
Footnotes
Credit Author statement:
Karla M. Marquez-Nogueras: Conceptualization; Funding acquisition; Roles/Writing – original draft; Writing – review & editing.
Virdjinija Vuchkovska: Conceptualization; Roles/Writing – original draft; Writing – review & editing.
Ivana Y. Kuo: Conceptualization; Supervision; Roles/Writing – original draft; Writing – review & editing.
Declarations of interest: None
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bergmann C, et al. , Polycystic kidney disease. Nat Rev Dis Primers, 2018. 4(1): p. 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Audrezet MP, et al. , Comprehensive PKD1 and PKD2 Mutation Analysis in Prenatal Autosomal Dominant Polycystic Kidney Disease. J Am Soc Nephrol, 2016. 27(3): p. 722–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergmann C, et al. , Mutations in multiple PKD genes may explain early and severe polycystic kidney disease. J Am Soc Nephrol, 2011. 22(11): p. 2047–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lanktree MB, et al. , Insights into Autosomal Dominant Polycystic Kidney Disease from Genetic Studies. Clin J Am Soc Nephrol, 2021. 16(5): p. 790–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Torres VE and Harris PC, Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J Am Soc Nephrol, 2014. 25(1): p. 18–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossetti S, et al. , Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol, 2007. 18(7): p. 2143–60. [DOI] [PubMed] [Google Scholar]
- 7.Clapham DE, et al. , International Union of Pharmacology. XLIX. Nomenclature and structure-function relationships of transient receptor potential channels. Pharmacol Rev, 2005. 57(4): p. 427–50. [DOI] [PubMed] [Google Scholar]
- 8.Cantiello HF, A tale of two tails: ciliary mechanotransduction in ADPKD. Trends Mol Med, 2003. 9(6): p. 234–6. [DOI] [PubMed] [Google Scholar]
- 9.Tian PF, et al. , TRPP2 ion channels: The roles in various subcellular locations. Biochimie, 2022. 201: p. 116–127. [DOI] [PubMed] [Google Scholar]
- 10.Kleene SJ, Regenerative Calcium Currents in Renal Primary Cilia. Front Physiol, 2022. 13: p. 894518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kleene SJ, et al. , The TRPP2-dependent channel of renal primary cilia also requires TRPM3. PLoS One, 2019. 14(3): p. e0214053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sammels E, et al. , Polycystin-2 activation by inositol 1,4,5-trisphosphate-induced Ca2+ release requires its direct association with the inositol 1,4,5-trisphosphate receptor in a signaling microdomain. J Biol Chem, 2010. 285(24): p. 18794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuo IY, et al. , Cyst formation following disruption of intracellular calcium signaling. Proc Natl Acad Sci U S A, 2014. 111(39): p. 14283–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saigusa T, et al. , Loss of primary cilia increases polycystin-2 and TRPV4 and the appearance of a nonselective cation channel in the mouse cortical collecting duct. Am J Physiol Renal Physiol, 2019. 317(3): p. F632–F637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fick GM, et al. , Causes of death in autosomal dominant polycystic kidney disease. J Am Soc Nephrol, 1995. 5(12): p. 2048–56. [DOI] [PubMed] [Google Scholar]
- 16.Grantham JJ, Geiser JL, and Evan AP, Cyst formation and growth in autosomal dominant polycystic kidney disease. Kidney Int, 1987. 31(5): p. 1145–52. [DOI] [PubMed] [Google Scholar]
- 17.Harris PC and Torres VE, Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest, 2014. 124(6): p. 2315–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gattone VH 2nd, et al. , Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med, 2003. 9(10): p. 1323–6. [DOI] [PubMed] [Google Scholar]
- 19.Torres VE, et al. , Rationale and design of the TEMPO (Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and its Outcomes) 3–4 Study. Am J Kidney Dis, 2011. 57(5): p. 692–9. [DOI] [PubMed] [Google Scholar]
- 20.Bais T, Gansevoort RT, and Meijer E, Drugs in Clinical Development to Treat Autosomal Dominant Polycystic Kidney Disease. Drugs, 2022. 82(10): p. 1095–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mochizuki T, et al. , PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science, 1996. 272(5266): p. 1339–42. [DOI] [PubMed] [Google Scholar]
- 22.Geng L, et al. , Identification and localization of polycystin, the PKD1 gene product. J Clin Invest, 1996. 98(12): p. 2674–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hughes J, et al. , The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet, 1995. 10(2): p. 151–60. [DOI] [PubMed] [Google Scholar]
- 24.Palsson R, et al. , Characterization and cell distribution of polycystin, the product of autosomal dominant polycystic kidney disease gene 1. Mol Med, 1996. 2(6): p. 702–11. [PMC free article] [PubMed] [Google Scholar]
- 25.The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. The European Polycystic Kidney Disease Consortium. Cell, 1994. 77(6): p. 881–94. [DOI] [PubMed] [Google Scholar]
- 26.Sullivan LP, Wallace DP, and Grantham JJ, Chloride and fluid secretion in polycystic kidney disease. J Am Soc Nephrol, 1998. 9(5): p. 903–16. [DOI] [PubMed] [Google Scholar]
- 27.Jouret F. and Devuyst O, Targeting chloride transport in autosomal dominant polycystic kidney disease. Cell Signal, 2020. 73: p. 109703. [DOI] [PubMed] [Google Scholar]
- 28.O’Sullivan DA, et al. , Cystic fibrosis and the phenotypic expression of autosomal dominant polycystic kidney disease. Am J Kidney Dis, 1998. 32(6): p. 976–83. [DOI] [PubMed] [Google Scholar]
- 29.Magenheimer BS, et al. , Early embryonic renal tubules of wild-type and polycystic kidney disease kidneys respond to cAMP stimulation with cystic fibrosis transmembrane conductance regulator/Na(+),K(+),2Cl(−) Co-transporter-dependent cystic dilation. J Am Soc Nephrol, 2006. 17(12): p. 3424–37. [DOI] [PubMed] [Google Scholar]
- 30.Yang B, et al. , Small-molecule CFTR inhibitors slow cyst growth in polycystic kidney disease. J Am Soc Nephrol, 2008. 19(7): p. 1300–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Torres VE and Harris PC, Progress in the understanding of polycystic kidney disease. Nat Rev Nephrol, 2019. 15(2): p. 70–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nauli SM, et al. , Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet, 2003. 33(2): p. 129–37. [DOI] [PubMed] [Google Scholar]
- 33.Delling M, et al. , Primary cilia are not calcium-responsive mechanosensors. Nature, 2016. 531(7596): p. 656–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kraus A, et al. , HIF-1alpha promotes cyst progression in a mouse model of autosomal dominant polycystic kidney disease. Kidney Int, 2018. 94(5): p. 887–899. [DOI] [PubMed] [Google Scholar]
- 35.Schreiber R, et al. , Lipid Peroxidation Drives Renal Cyst Growth In Vitro through Activation of TMEM16A. J Am Soc Nephrol, 2019. 30(2): p. 228–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cabrita I, et al. , TMEM16A drives renal cyst growth by augmenting Ca(2+) signaling in M1 cells. J Mol Med (Berl), 2020. 98(5): p. 659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cabrita I, et al. , Loss of PKD1 and PKD2 share common effects on intracellular Ca(2+) signaling. Cell Calcium, 2021. 97: p. 102413. [DOI] [PubMed] [Google Scholar]
- 38.Cabrita I, et al. , Cyst growth in ADPKD is prevented by pharmacological and genetic inhibition of TMEM16A in vivo. Nat Commun, 2020. 11(1): p. 4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Talbi K, et al. , The chloride channel CFTR is not required for cyst growth in an ADPKD mouse model. FASEB J, 2021. 35(10): p. e21897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calvet JP, Polycystic kidney disease: primary extracellular matrix abnormality or defective cellular differentiation? Kidney Int, 1993. 43(1): p. 101–8. [DOI] [PubMed] [Google Scholar]
- 41.Dong K, et al. , Renal plasticity revealed through reversal of polycystic kidney disease in mice. Nat Genet, 2021. 53(12): p. 1649–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arac D, et al. , A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. EMBO J, 2012. 31(6): p. 1364–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weinmann HJ, Press WR, and Gries H, Tolerance of extracellular contrast agents for magnetic resonance imaging. Invest Radiol, 1990. 25 Suppl 1: p. S49–50. [DOI] [PubMed] [Google Scholar]
- 44.Su Q, et al. , Structure of the human PKD1-PKD2 complex. Science, 2018. 361(6406). [DOI] [PubMed] [Google Scholar]
- 45.Wang Z, et al. , The ion channel function of polycystin-1 in the polycystin-1/polycystin-2 complex. EMBO Rep, 2019. 20(11): p. e48336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qian F, et al. , Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1-associated mutations. Proc Natl Acad Sci U S A, 2002. 99(26): p. 16981–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ha K, et al. , The heteromeric PC-1/PC-2 polycystin complex is activated by the PC-1 N-terminus. Elife, 2020. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferreira FM, Watanabe EH, and Onuchic LF, Polycystins and Molecular Basis of Autosomal Dominant Polycystic Kidney Disease, in Polycystic Kidney Disease, Li X, Editor. 2015: Brisbane (AU). [PubMed] [Google Scholar]
- 49.Maser RL, Calvet JP, and Parnell SC, The GPCR properties of polycystin-1- A new paradigm. Front Mol Biosci, 2022. 9: p. 1035507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parnell SC, et al. , A mutation affecting polycystin-1 mediated heterotrimeric G-protein signaling causes PKD. Hum Mol Genet, 2018. 27(19): p. 3313–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu X, et al. , Polycystin-2 is an essential ion channel subunit in the primary cilium of the renal collecting duct epithelium. Elife, 2018. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kleene SJ and Kleene NK, The native TRPP2-dependent channel of murine renal primary cilia. Am J Physiol Renal Physiol, 2017. 312(1): p. F96–F108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vien TN, et al. , Molecular dysregulation of ciliary polycystin-2 channels caused by variants in the TOP domain. Proc Natl Acad Sci U S A, 2020. 117(19): p. 10329–10338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cai Y, et al. , Identification and characterization of polycystin-2, the PKD2 gene product. J Biol Chem, 1999. 274(40): p. 28557–65. [DOI] [PubMed] [Google Scholar]
- 55.Kuo IY, et al. , The number and location of EF hand motifs dictates the calcium dependence of polycystin-2 function. FASEB J, 2014. 28(5): p. 2332–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Petri ET, et al. , Structure of the EF-hand domain of polycystin-2 suggests a mechanism for Ca2+-dependent regulation of polycystin-2 channel activity. Proc Natl Acad Sci U S A, 2010. 107(20): p. 9176–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koulen P, et al. , Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol, 2002. 4(3): p. 191–7. [DOI] [PubMed] [Google Scholar]
- 58.Delling M, et al. , Primary cilia are specialized calcium signalling organelles. Nature, 2013. 504(7479): p. 311–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hildebrandt F, Benzing T, and Katsanis N, Ciliopathies. N Engl J Med, 2011. 364(16): p. 1533–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wheway G, Nazlamova L, and Hancock JT, Signaling through the Primary Cilium. Front Cell Dev Biol, 2018. 6: p. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Waters AM and Beales PL, Ciliopathies: an expanding disease spectrum. Pediatr Nephrol, 2011. 26(7): p. 1039–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.DeCaen PG, et al. , Direct recording and molecular identification of the calcium channel of primary cilia. Nature, 2013. 504(7479): p. 315–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yuan S, et al. , Intraciliary calcium oscillations initiate vertebrate left-right asymmetry. Curr Biol, 2015. 25(5): p. 556–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kleene SJ and Kleene NK, Inward Ca(2+) current through the polycystin-2-dependent channels of renal primary cilia. Am J Physiol Renal Physiol, 2021. 320(6): p. F1165–F1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vien TN, et al. , Disrupting polycystin-2 EF hand Ca(2+) affinity does not alter channel function or contribute to polycystic kidney disease. J Cell Sci, 2020. 133(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pedrozo Z, et al. , Polycystin-1 Is a Cardiomyocyte Mechanosensor That Governs L-Type Ca2+ Channel Protein Stability. Circulation, 2015. 131(24): p. 2131–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kuo IY, et al. , Polycystin 2 regulates mitochondrial Ca(2+) signaling, bioenergetics, and dynamics through mitofusin 2. Sci Signal, 2019. 12(580). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mekahli D, et al. , Polycystin-1 and polycystin-2 are both required to amplify inositoltrisphosphate-induced Ca2+ release. Cell Calcium, 2012. 51(6): p. 452–8. [DOI] [PubMed] [Google Scholar]
- 69.Clapham DE, Calcium signaling. Cell, 2007. 131(6): p. 1047–58. [DOI] [PubMed] [Google Scholar]
- 70.Wiseman RL, Mesgarzadeh JS, and Hendershot LM, Reshaping endoplasmic reticulum quality control through the unfolded protein response. Mol Cell, 2022. 82(8): p. 1477–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ren J, et al. , Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat Rev Cardiol, 2021. 18(7): p. 499–521. [DOI] [PubMed] [Google Scholar]
- 72.Liang G, et al. , Polycystin-2 down-regulates cell proliferation via promoting PERK-dependent phosphorylation of eIF2alpha. Hum Mol Genet, 2008. 17(20): p. 3254–62. [DOI] [PubMed] [Google Scholar]
- 73.Liang G, et al. , Polycystin-2 is regulated by endoplasmic reticulum-associated degradation. Hum Mol Genet, 2008. 17(8): p. 1109–19. [DOI] [PubMed] [Google Scholar]
- 74.Brill AL, et al. , Polycystin 2 is increased in disease to protect against stress-induced cell death. Sci Rep, 2020. 10(1): p. 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang J, et al. , Translational up-regulation of polycystic kidney disease protein PKD2 by endoplasmic reticulum stress. FASEB J, 2013. 27(12): p. 4998–5009. [DOI] [PubMed] [Google Scholar]
- 76.Zhou X, et al. , PKD2 deficiency suppresses amino acid biosynthesis in ADPKD by impairing the PERK-TBL2-eIF2a-ATF4 pathway. Biochem Biophys Res Commun, 2021. 561: p. 73–79. [DOI] [PubMed] [Google Scholar]
- 77.Cebotaru V, et al. , Polycystin-1 negatively regulates Polycystin-2 expression via the aggresome/autophagosome pathway. J Biol Chem, 2014. 289(10): p. 6404–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yao Q, et al. , Polycystin-1 dependent regulation of polycystin-2 via GRP94, a member of HSP90 family that resides in the endoplasmic reticulum. FASEB J, 2021. 35(10): p. e21865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Krappitz M, et al. , XBP1 Activation Reduces Severity of Polycystic Kidney Disease due to a Nontruncating Polycystin-1 Mutation in Mice. J Am Soc Nephrol, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fedeles SV, et al. , Sec63 and Xbp1 regulate IRE1alpha activity and polycystic disease severity. J Clin Invest, 2015. 125(5): p. 1955–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Parzych KR and Klionsky DJ, An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal, 2014. 20(3): p. 460–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yorimitsu T. and Klionsky DJ, Autophagy: molecular machinery for self-eating. Cell Death Differ, 2005. 12 Suppl 2(Suppl 2): p. 1542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Levine B. and Kroemer G, Biological Functions of Autophagy Genes: A Disease Perspective. Cell, 2019. 176(1–2): p. 11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Atwood DJ, et al. , The effect of trehalose on autophagy-related proteins and cyst growth in a hypomorphic Pkd1 mouse model of autosomal dominant polycystic kidney disease. Cell Signal, 2020. 75: p. 109760. [DOI] [PubMed] [Google Scholar]
- 85.Zhu P, et al. , Autophagy activators suppress cystogenesis in an autosomal dominant polycystic kidney disease model. Hum Mol Genet, 2017. 26(1): p. 158–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tao Y, et al. , Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J Am Soc Nephrol, 2005. 16(1): p. 46–51. [DOI] [PubMed] [Google Scholar]
- 87.Perico N, et al. , Sirolimus therapy to halt the progression of ADPKD. J Am Soc Nephrol, 2010. 21(6): p. 1031–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shillingford JM, et al. , Folate-conjugated rapamycin slows progression of polycystic kidney disease. J Am Soc Nephrol, 2012. 23(10): p. 1674–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wahl PR, et al. , Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol Dial Transplant, 2006. 21(3): p. 598–604. [DOI] [PubMed] [Google Scholar]
- 90.Boletta A, Emerging evidence of a link between the polycystins and the mTOR pathways. Pathogenetics, 2009. 2(1): p. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dere R, et al. , Carboxy terminal tail of polycystin-1 regulates localization of TSC2 to repress mTOR. PLoS One, 2010. 5(2): p. e9239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Distefano G, et al. , Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Mol Cell Biol, 2009. 29(9): p. 2359–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lu J, et al. , Polycystin-2 Plays an Essential Role in Glucose Starvation-Induced Autophagy in Human Embryonic Stem Cell-Derived Cardiomyocytes. Stem Cells, 2018. 36(4): p. 501–513. [DOI] [PubMed] [Google Scholar]
- 94.Criollo A, et al. , Polycystin-2-dependent control of cardiomyocyte autophagy. J Mol Cell Cardiol, 2018. 118: p. 110–121. [DOI] [PubMed] [Google Scholar]
- 95.Pena-Oyarzun D, et al. , PKD2/polycystin-2 induces autophagy by forming a complex with BECN1. Autophagy, 2021. 17(7): p. 1714–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Marquez-Nogueras KM, et al. , Polycystin-2 (PC2) is a key determinant of in vitro myogenesis. Am J Physiol Cell Physiol, 2022. 323(2): p. C333–C346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ramirez-Sagredo A, et al. , Polycystin-1 regulates cardiomyocyte mitophagy. FASEB J, 2021. 35(8): p. e21796. [DOI] [PubMed] [Google Scholar]
- 98.Belibi F, et al. , Hypoxia-inducible factor-1alpha (HIF-1alpha) and autophagy in polycystic kidney disease (PKD). Am J Physiol Renal Physiol, 2011. 300(5): p. F1235–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Decuypere JP, et al. , Interdependent Regulation of Polycystin Expression Influences Starvation-Induced Autophagy and Cell Death. Int J Mol Sci, 2021. 22(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lee EJ, et al. , Autophagy induction promotes renal cyst growth in polycystic kidney disease. EBioMedicine, 2020. 60: p. 102986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kou P, Wei S, and Xiong F, Recent Advances of mTOR Inhibitors Use in Autosomal Dominant Polycystic Kidney Disease: Is the Road Still Open? Curr Med Chem, 2019. 26(16): p. 2962–2973. [DOI] [PubMed] [Google Scholar]
- 102.Kuo IY and Chapman AB, Polycystins, ADPKD, and Cardiovascular Disease. Kidney Int Rep, 2020. 5(4): p. 396–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Delmas P, et al. , Gating of the polycystin ion channel signaling complex in neurons and kidney cells. FASEB J, 2004. 18(6): p. 740–2. [DOI] [PubMed] [Google Scholar]
- 104.Patel V, et al. , Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum Mol Genet, 2008. 17(11): p. 1578–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Avasthi P, Maser RL, and Tran PV, Primary Cilia in Cystic Kidney Disease. Results Probl Cell Differ, 2017. 60: p. 281–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Simons M. and Walz G, Polycystic kidney disease: cell division without a c(l)ue? Kidney Int, 2006. 70(5): p. 854–64. [DOI] [PubMed] [Google Scholar]
- 107.Pan J, Seeger-Nukpezah T, and Golemis EA, The role of the cilium in normal and abnormal cell cycles: emphasis on renal cystic pathologies. Cell Mol Life Sci, 2013. 70(11): p. 1849–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pazour GJ, et al. , Cilia in cystic kidney and other diseases. Cell Signal, 2020. 69: p. 109519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang X, et al. , Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol, 2005. 16(4): p. 846–51. [DOI] [PubMed] [Google Scholar]
- 110.Chebib FT, et al. , A Practical Guide for Treatment of Rapidly Progressive ADPKD with Tolvaptan. J Am Soc Nephrol, 2018. 29(10): p. 2458–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yamaguchi T, et al. , Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J Am Soc Nephrol, 2006. 17(1): p. 178–87. [DOI] [PubMed] [Google Scholar]
- 112.Aranguiz P, et al. , Polycystin-1 mitigates damage and regulates CTGF expression through AKT activation during cardiac ischemia/reperfusion. Biochim Biophys Acta Mol Basis Dis, 2021. 1867(1): p. 165986. [DOI] [PubMed] [Google Scholar]
- 113.Di Mise A, et al. , Activation of Calcium-Sensing Receptor increases intracellular calcium and decreases cAMP and mTOR in PKD1 deficient cells. Sci Rep, 2018. 8(1): p. 5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Winokurow N. and Schumacher S, A role for polycystin-1 and polycystin-2 in neural progenitor cell differentiation. Cell Mol Life Sci, 2019. 76(14): p. 2851–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang Y, Reif G, and Wallace DP, Extracellular matrix, integrins, and focal adhesion signaling in polycystic kidney disease. Cell Signal, 2020. 72: p. 109646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nigro EA and Boletta A, Role of the polycystins as mechanosensors of extracellular stiffness. Am J Physiol Renal Physiol, 2021. 320(5): p. F693–F705. [DOI] [PubMed] [Google Scholar]
- 117.Paluch EK, Aspalter IM, and Sixt M, Focal Adhesion-Independent Cell Migration. Annu Rev Cell Dev Biol, 2016. 32: p. 469–490. [DOI] [PubMed] [Google Scholar]
- 118.Wilson PD, et al. , The PKD1 gene product, “polycystin-1,” is a tyrosine-phosphorylated protein that colocalizes with alpha2beta1-integrin in focal clusters in adherent renal epithelia. Lab Invest, 1999. 79(10): p. 1311–23. [PubMed] [Google Scholar]
- 119.Castelli M, et al. , Regulation of the microtubular cytoskeleton by Polycystin-1 favors focal adhesions turnover to modulate cell adhesion and migration. BMC Cell Biol, 2015. 16: p. 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Joly D, et al. , The polycystin 1-C-terminal fragment stimulates ERK-dependent spreading of renal epithelial cells. J Biol Chem, 2006. 281(36): p. 26329–39. [DOI] [PubMed] [Google Scholar]
- 121.He J, et al. , Inhibiting Focal Adhesion Kinase Ameliorates Cyst Development in Polycystin-1-Deficient Polycystic Kidney Disease in Animal Model. J Am Soc Nephrol, 2021. 32(9): p. 2159–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ibraghimov-Beskrovnaya O. and Bukanov N, Polycystic kidney diseases: from molecular discoveries to targeted therapeutic strategies. Cell Mol Life Sci, 2008. 65(4): p. 605–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Malekshahabi T, et al. , Autosomal dominant polycystic kidney disease: Disrupted pathways and potential therapeutic interventions. J Cell Physiol, 2019. 234(8): p. 12451–12470. [DOI] [PubMed] [Google Scholar]
- 124.Wegierski T, et al. , TRPP2 channels regulate apoptosis through the Ca2+ concentration in the endoplasmic reticulum. EMBO J, 2009. 28(5): p. 490–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Woo D, Apoptosis and loss of renal tissue in polycystic kidney diseases. N Engl J Med, 1995. 333(1): p. 18–25. [DOI] [PubMed] [Google Scholar]
- 126.Seeger-Nukpezah T, et al. , The hallmarks of cancer: relevance to the pathogenesis of polycystic kidney disease. Nat Rev Nephrol, 2015. 11(9): p. 515–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Leonhard WN, et al. , Scattered Deletion of PKD1 in Kidneys Causes a Cystic Snowball Effect and Recapitulates Polycystic Kidney Disease. J Am Soc Nephrol, 2015. 26(6): p. 1322–33. [DOI] [PMC free article] [PubMed] [Google Scholar]