

A frequent narrative is that no medical therapy modifies the natural history of hypertrophic cardiomyopathy (HCM), but evidence shows valsartan and mavacamten modify disease progression in HCM. Yet, no medical therapy has been shown to modify fibrosis in HCM. Given the role of fibrosis in the risk of adverse outcomes, there is a need to identify pathways that may influence HCM development and progression.
Neprilysin (NEP) is the primary enzyme that degrades natriuretic peptides. Myocardial expression and activity of NEP is elevated in dilated cardiomyopathy (DCM),1 and NEP inhibition with the angiotensin receptor blocker/neprilysin inhibitor (ARNi) sacubitril/valsartan reduces the risk of heart failure (HF) hospitalization and death in patients with HF with reduced ejection fraction (HFrEF). However, in patients with HF with preserved ejection fraction (HFpEF) there was no reduction in mortality. Patients with HCM were excluded from trials evaluating ARNi in HF pointing to an unmet clinical need to determine the efficacy of ARNi in HCM. Furthermore, myocardial expression of NEP in patients with HCM has not been previously described. Herein, we examine myocardial expression of NEP in HCM.
We extracted publicly available gene expression data from three independent cohorts of patients with HCM. In all three cohorts, the expression of MME, the gene encoding NEP, was significantly higher in patients with HCM versus controls (unused healthy donor hearts).2–4 MME expression was higher in obstructive HCM (oHCM) versus controls using both microarray2 (Fig A) and RNAseq3 (Fig B). In patients with end-stage non-obstructive HCM (nHCM) and DCM at the time of transplant, snRNAseq revealed higher MME expression only in fibroblasts, and the degree of MME elevation was not significantly different in nHCM versus DCM (Fig C). A prior report demonstrated higher myocardial MME expression and NEP activity in the setting of severe aortic senosis.1 To further determine if MME expression is altered in myocardial hypertrophy not due to HCM, we examined patients with HFpEF and mild-moderate hypertrophy without HCM. MME expression was also slightly higher versus controls, but significantly lower compared with patients with HFrEF at transplant. (Fig D).5 Of note, the latter study analyzed RV septal samples, but gene expression profiles strongly correlated between RV and LV tissue.5 Collectively, these data suggest convergence of MME transcription is a feature of cardiomyopathy independent of etiology.
Figure.
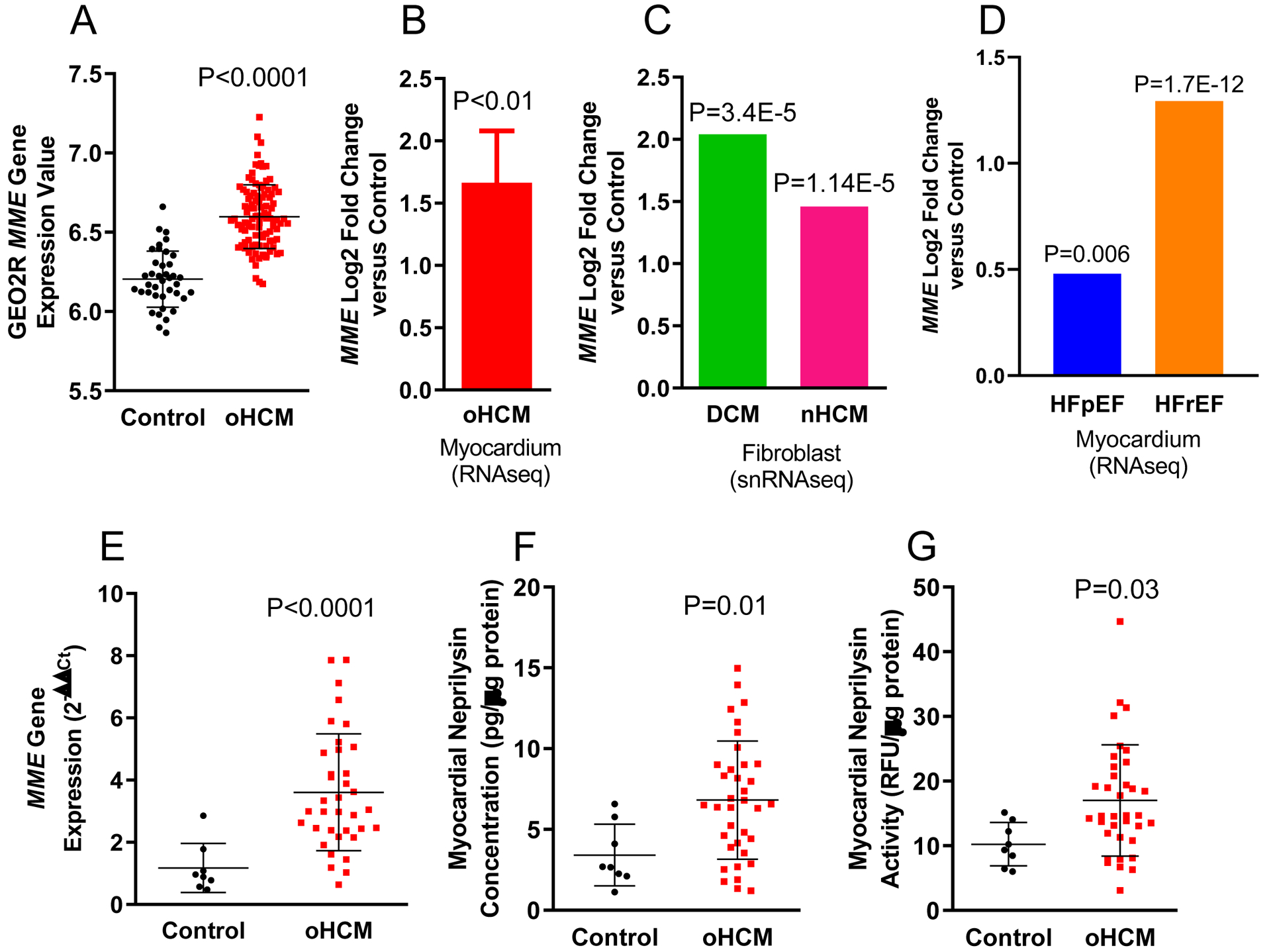
Myocardial MME gene expression is higher in patients with oHCM at septal myectomy versus controls using A) microarray (N=106 oHCM and N=39 controls) and B) RNAseq (N=18 oHCM and N=5 controls). C) Higher myocardial MME gene expression is specific to fibroblasts in patients with end-stage nHCM (N=15) and DCM (N=11) at the time of transplant compared to controls (N=16). D) Higher myocardial MME expression in patients with both HFpEF (N=41) and HFrEF (N=30) compared to controls N=30), with MME expression being significantly higher in HFrEF compared to HFpEF (P=4.88E-7). Note, the data in B-D are presented as Log2 Fold Change Compared to Control, hence, there are no bars for control groups. In the VUMC cohort of patients with oHCM (N=36), MME expression is higher using qPCR (E) compared to controls (N=8). The increase in MME expression parallels higher NEP concentration (using AlphaLISA, PerkinElmer) and activity (using solid-phase activity assay) compared to controls (E-G). Collectively, these data indicate convergence of MME transcription is a feature of cardiomyopathy independent of etiology, and NEP may be a viable therapeutic target in patients with HCM. For panels A and E-G, t-test or Mann-Whitney U test were used when appropriate. Details regarding statistical analyses for panels B-D can be found in references 3–5. Data are presented as mean ± standard deviation where error bars are present.
Next, we tested whether higher MME transcription was associated with higher myocardial NEP expression and activity in patients with oHCM. Myocardial tissue was obtained from the Vanderbilt University Medical Center (VUMC) Cardiovascular Core Laboratory, consisting of 36 patients with oHCM at myectomy and 8 non-transplanted healthy donor hearts. This study was approved by the VUMC IRB; all participants gave informed consent. Among patients with oHCM, mean age was 51±15 years, 23 (64%) were female, mean resting LVOT gradient was 54±49 mmHg, mean provocable peak gradient was 106±70 mmHg, mean septal thickness was 2.2±0.5cm, mean LVEF was 65±10% (range 53–88%). Among 15 patients who underwent genetic screening, 7 (46%) had a pathogenic sarcomere mutation. A defibrilaltor was present in 10 (27%) at the time of surgery. Cardiac MRI was performed in 25 (69%) prior to surgery, of whom 20 (80%) had late gadolinium enhancement; none were on mavacamten. Consistent with aforesaid data, MME expression was significantly higher in patients with oHCM versus controls using qPCR (Fig D). Myocardial NEP protein expression and activity were also significantly higher in patients with oHCM versus controls (Fig E–F).
Future studies are required to define the mechanism of NEP elevation in HCM. Higher myocardial NEP may degrade natriuretic peptides, which exert anti-hypertrophic and anti-fibrotic effects that would be favorable in HCM. Future studies are required to determine whether increased NEP contributes to natriuretic peptide deficiency and whether ARNi modifies myocyte and fibroblast biology in HCM. Alternatively, ARNi may prevent degradation of other substrates with favorable biologic actions such as adrenomedullin and glucagon-like peptide-1. Conversely, ARNi may increase endothelin, which would be deleterious in HCM. These data also suggest that innovative natriuretic peptide-based therapies for HCM may require analogs with resistance to enzymatic degradation by NEP. The efficacy of ARNi in HCM has been demonstrated in two case reports in patients with HCM who progressed to develop systolic dysfunction. The Phase 2 SILICOFCM trial (NCT03832660) will evaluate the effect of ANRi in HCM with change in peak VO2 as the primary outcome.
Limitations of our study include exclusion of patients with nHCM in evaluating NEP protein expression and activity, exclusion of patients with less severe disease/symptomatic burden, and lack of samples available to examine systemic NEP concentration and activity, which may be altered in HCM.
Overall, we report HCM is associated with elevation of myocardial NEP. These data provide insight into the potential role of NEP in the pathophysiology of HCM.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgements:
Thanks to Julie A. Bastarache MD, Division of Allergy, Pulmonary, and Critical Care Medicine, VUMC for the use of the BioTek plate reader; to Ashish Chougule PhD, PerkinElmer, Tim Prickett PhD, Christchurch Heart Institute, University of Otago, Oi Wah Liew PhD, Department of Medicine, National University of Singapore for helpful discussions regarding the AlphaLISA assay; to J. Scott Miners PhD, Institute of Clinical Neurosciences, University of Bristol, for helpful discussion regarding the solid-phase NEP activity assay.
Sources of Funding:
This work was supported by a grant from the Vanderbilt Institute for Clinical and Translational Research (#VR55836). Dr. Armstrong is supported by NIH F32HL165917. Dr. Merryman is supported by NIH R35HL135790.
Disclosures:
Dr. Gupta has received research support from Imara, Inc. The remaining authors have no relevant disclosures.
Nonstandard Abbreviations and Acronyms
- ANP
atrial natriuretic peptide
- ARNi
angiotensin receptor blocker/neprilysin inhibitor
- HCM
hypertrophic cardiomyopathy
- HF
heart failure
- HFpEF
heart failure with preserved ejection fraction
- HFrEF
heart failure with reduced ejection fraction
- LV
left ventricle
- LVEF
left ventricular ejection fraction
- LVOT
left ventricular outflow tract
- MME
the gene encoding neprilysin
- NEP
neprilysin
- nHCM
non-obstructive hypertrophic cardiomyopathy
- oHCM
obstructive hypertrophic cardiomyopathy
- RV
right ventricle
References
- 1.Fielitz J, Dendorfer A, Pregla R, et al. Neutral endopeptidase is activated in cardiomyocytes in human aortic valve stenosis and heart failure. Circulation. 2002;105(3):286–289. [DOI] [PubMed] [Google Scholar]
- 2.Bos JM, Hebl VB, Oberg AL, et al. Marked up-regulation of ACE2 in hearts of patients with obstructive hypertrophic cardiomyopathy: implications for SARS-CoV-2–mediated COVID-19. In: Mayo Clin Proc. Vol 95. Elsevier; 2020:1354–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maron BA, Wang R-S, Shevtsov S, et al. Individualized interactomes for network-based precision medicine in hypertrophic cardiomyopathy with implications for other clinical pathophenotypes. Nat Commun. 2021;12(1):873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaffin M, Papangeli I, Simonson B, et al. Single-nucleus profiling of human dilated and hypertrophic cardiomyopathy. Nature. 2022;608(7921):174–180. [DOI] [PubMed] [Google Scholar]
- 5.Hahn VS, Knutsdottir H, Luo X, et al. Myocardial Gene Expression Signatures in Human Heart Failure With Preserved Ejection Fraction. Circulation. 2021;143(2):120–134. [DOI] [PMC free article] [PubMed] [Google Scholar]