

Abstract
Stereoselective construction of conjugated dienes and polyenes has remained an enduring synthetic problem, due to the central roles they play in natural product synthesis, methodology, and medicine. This review focuses on the recent developments in dienylation as an emerging strategy for the direct installation of unsaturated four carbon atom units of conjugated π-systems, outlining the regio- and stereoselectivity, as well as the synthetic scope of reactions with various dienylating reagents and the mechanistic implications of the catalytic cross-coupling processes that are used to enable dienylation.
1. Introduction
Conjugated dienes and polyenes are broadly represented among natural products that have been used extensively in drug discovery and medicine (Figure 1).1 Conjugated π-systems also play increasingly important roles in materials science as semiconductors and precursors to functional polymers.2 Notably, the unsaturated polyene motif is crucial for the biological activity of the natural products containing conjugated π-systems, and the configuration of each C=C bond is important for the molecule to attain the three-dimensional shape required for the optimal interaction with a target protein.3 Conjugated dienes are also key synthetic precursors to a variety of functionalized intermediates, because the reactive C(sp2)4 dienyl group can be transformed to a C4 unit with up to four stereocenters and four functional groups in a stepwise fashion by a variety of cycloaddition and addition reactions (Figure 2).4 The regio- and stereochemistry of the substituents in conjugated dienes and polyenes is also important for synthetic applications, since only one regioisomer with specific configurations and substitution patterns of the C=C bonds is typically required to access a target molecule. Given the importance of conjugated dienes and polyenes, a number of strategies have emerged for their stereo- and regioselective synthesis (Figure 3). Olefination of carbonyl compounds (Cx–C1 + C3) has historically been one of the major synthetic approaches to conjugated dienes and polyenes.5 This strategy is suitable when the corresponding carbonyl precursor is readily accessible but does require additional synthetic steps for the preinstallation of the appropriately configured and substituted C=C bond in the olefination reagent. Transition metal-catalyzed cross-coupling reactions have emerged as another major direction in the stereoselective synthesis of conjugated dienes and polyenes by a Cx + C2 + C2 strategy. The typically high stereoselectivity of the C–C bond-forming cross-coupling step ensures the retention of the configurations of the reacting alkenyl moieties, permitting a predictable and reliable stepwise construction of conjugated π-systems.6 Combined with the development of the MIDA protection for boronates, this strategy has recently been successfully expanded into a broad synthetic, automatable platform by the Burke group,7 facilitating access to a wide array of polyenes. The Cx + C2 + C2 strategy calls for a stepwise installation of each C=C unit that is introduced with a predefined configuration and substitution pattern. In addition, other methods have also been developed, for example metathesis,8 elimination reactions in the allylic systems,4b and carbometallation9 that can facilitate access to conjugated dienes and polyenes from structurally unique synthetic precursors. In contrast to these well-established approaches, a dienylation strategy (Cx + C4) has until recently remained virtually unexplored in the context of the stereoselective construction of conjugated π-systems. Dienylation enables the direct installation of the four-carbon unit with the stereo- and regiochemistry established during the cross-coupling step. This approach can reduce the number of synthetic manipulations and streamline access to conjugated synthetic targets. However, dienylation presents a number of challenges that need to be overcome for it to become a widely used synthetic tool. The four-carbon synthons that deliver the dienyl payload should be readily accessible from simple precursors. The regio- and stereoselectivity of the dienylation reaction should be sufficiently high for synthetic applications, as separation of review will discuss the recent advances in dienylation showcased by select examples with a focus on addressing the structurally close regio- and diastereomeric dienes and polyenes can present a significant problem. On the other hand, because the configurations of the two C=C bonds and the position of the incoming coupling counterpart are established during the dienylation step, the strategy can present an opportunity for a development of stereo- and regiodivergent construction of dienes and polyenes, if they can be altered in a predictable manner by changing the reaction conditions. This review will discuss the recent advances in dienylation with a focus on addressing the aforementioned challenges and opportunities. Given the key role of the four-carbon atom synthons in enabling the installation of dienyl units, the review is organized by classes of dienylation reagents.
Figure 1.

Representative natural products featuring conjugated diene and polyene units
Figure 2.

Synthetic roles of conjugated dienes
Figure 3.

Synthetic approaches to conjugated dienes and polyenes
2. Sulfolenes
Sulfolenes can be readily prepared by a cheletropic reaction between 1,3-dienes and excess sulfur dioxide gas. Given the toxicity of sulfur dioxide and the impracticality of monitoring the exact amount of gas required for the reaction, several methods were developed that use bench-stable sulfur dioxide surrogates, e.g., DABCO-bis(sulfur dioxide) (DABSO)10 at an elevated temperature, or using an adduct of sulfur dioxide with 4-dimethylaminopyridine (DMAP) in the presence of boron trifluoride–diethyl ether complex (Scheme 1a).11 Alternatively, the synthesis of 3-sulfolenes can be accomplished from 1,3-dienes and allylic alcohols using sodium metabisulfite as a sulfur dioxide equivalent in hexafluoroisopropanol (HFIP) or in aqueous methanol in the presence of potassium hydrogen sulfate,12 affording a variety of substituted sulfolenes (Scheme 1b).
Scheme 1.
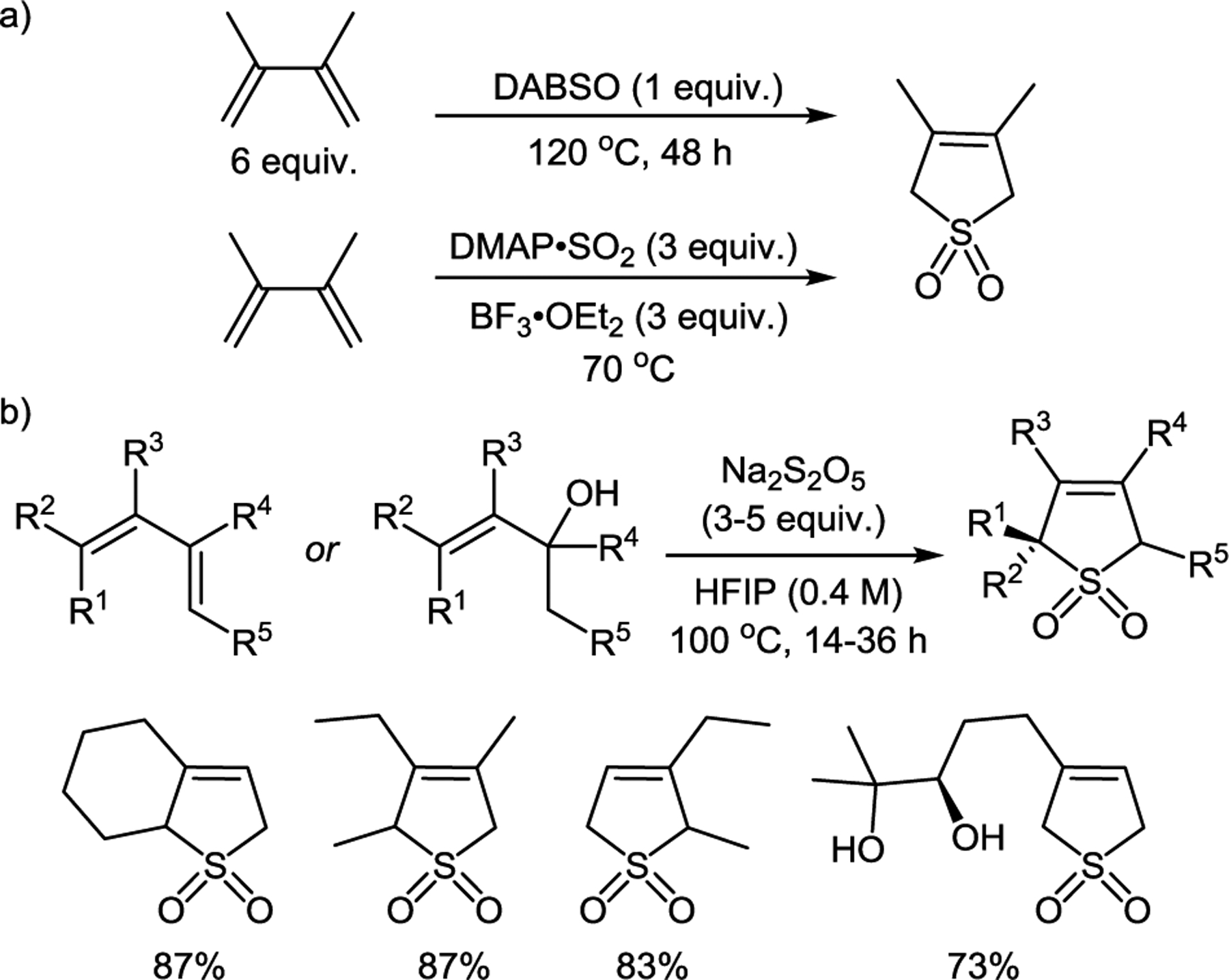
Synthesis of sulfolenes
In 2018, Larionov and co-workers introduced sulfolenes as dienylation reagents for the palladium catalyzed regio- and stereoselective construction of conjugated dienes and polyenes13 to overcome the problems associated with the use of dienylboronic acids that are unstable and difficult to prepare.14 Air- and moisture-stable, easily synthesizable sulfolenes are known to produce sulfinates in the presence of a base via regio- and stereoselective ring opening of sulfolenes (Scheme 2a). The in situ-generated sulfinates undergo a desulfitative Pd-catalyzed coupling reaction with aryl halides to afford regio- and stereoselective dienes and polyenes. A combination of Pd(OAc)2 and dppbz (1,2-bis(phenylphosphino)benzene) catalyzed a highly E- and Z-selective dienylation of aryl, heteroaryl, and vinyl chlorides and bromides (Scheme 2b) with the position of the newly-formed C–C bond and the configurations of the double bonds in the diene unit determined by the substitution pattern in the sulfolene and in line with the direction of the base-mediated sulfolene ring opening (Scheme 2a). The dienylation demonstrated a broad scope with respect to aryl, heteroaryl, and vinyl bromides and chlorides as well as a variety of diversely substituted sulfolenes. Notably, the dienylation with the unsubstituted sulfolene produced the 1E-diene product, pointing to an efficient isomerization during the reaction. The scope of the reaction was further extended to (hetero)aryl sulfonates by the replacement of potassium methoxide with potassium tert-butoxide and potassium carbonate with potassium bicarbonate.15 Aryl iodides were found to be recalcitrant under the optimized reaction conditions but underwent the dienylation with silver oxide as a basic additive in place of potassium bicarbonate. Interestingly, the stereoselectivity of the dienylation with the unsubstituted sulfolene can be inverted to >30:1 in favor of the 1Z-diene when Xantphos is used as a ligand instead of dppbz (Scheme 3).16 The Z-selective dienylation afforded an array of 1Z-dienes from the corresponding (hetero)aryl halides and sulfonates in good to excellent yields, demonstrating a broad tolerance of a variety of functional groups. Combined with the Pd/dppbz-catalyzed E-dienylation, the Pd/Xantphos protocol represents the first example of a stereodivergent dienylation with the configuration the dienyl unit determined by the ligand. Kinetic studies revealed that both the Z-selective Pd/Xantphos and E-selective Pd/dpbbz catalytic systems operated through an intrinsically Z-selective mechanism featuring a turnover- limiting transmetalation of Z-dienylsulfinate Z-1 (Scheme 4). While the Pd/Xantphos system was consistently Z-selective throughout the course of reaction, the initially Z-selective Pd/dppbz system exhibited eventual reversal of selectivity in favor of the E-isomer, attributed to an efficient Z → E isomerization of the Z-diene product catalyzed by colloidal palladium present exclusively in the Pd/dppbz system. DFT calculations confirmed that Pd(0)/dppbz exhibited a lower Gibbs free energy of ligand dissociation than the corresponding Pd(0)/Xantphos complex, suggesting an environment more amenable to Pd aggregation. Additional calculations in the PdL2 series indicated that Pd/Xantphos featured a more thermodynamically favorable first ligand dissociation than the Pd/dppbz system, in agreement with the experimentally observed increased rate of reaction of the Pd/Xantphos system relative to the Pd/dppbz system.
Scheme 2.

Dienylation with sulfolenes
Scheme 3.

Pd/Xantphos-catalyzed Z-selective dienylation of (hetero)aryl halides and sulfonates
Scheme 4.

Mechanism of the stereodivergent dienylation
In 2020, Hu and co-workers extended the sulfolene-mediated dienylation by developing a palladium-catalyzed cyclization/dienylation cascade process that provides access to 2-indolinone-substitued aliphatic 1,3-dienes with high Z-selectivity (Scheme 5).17 The reaction could also be extended to 3- and 3,4-substitued sulfolenes. The proposed mechanism (Scheme 6) entails a sequence of the oxidative addition and cyclization, producing alkylpalladium intermediate 2 that subsequently undergoes transmetalation, extrusion of sulfur dioxide, and reductive elimination in line with the catalytic pathway established for the dienylation of haloarenes.
Scheme 5.

Cascade cyclization/dienylation with sulfolenes
Scheme 6.

Mechanism of the cascade cyclization/dienylation with sulfolenes
The development of the stereodivergent dienylation and the cyclization/dienylation cascade point to opportunities for the extension of the sulfolene-mediated dienylation to other classes of coupling partners and improved control over the stereoselectivity of construction of conjugated dienes and polyenes.
3. 1,3-Dienes
1,3-Dienes can potentially serve as atom-economical dienylation reagents. However, dienylation reactions with 1,3-dienes have suffered from reduced functional group compatibility owing to a propensity to undergo facile isomerization, polymerization, and other side reactions, as well as non-regioselective reactions under typical conditions of transition metal-catalyzed transformations.18 Despite these challenges, a number of recent methods have taken advantage of the reactivity of 1,3-dienes,19 as showcased by the two representative examples discussed in this section.
In 2019, Zhu and co-workers reported a stereoconvergent dienylation of alkyl bromides with 1,3-dienes and 1,3,5-trienes (Scheme 7).20 Advantageously, mixtures of E- and Z-alkenes were shown to undergo a radical-mediated reaction with bromoacetate and bromomalonate esters in the presence of a copper-based catalyst, producing alkylated E,E-dienes or E,E,E-trienes with excellent stereoselectivity. Aryl dienes bearing a variety of functionalities, ranging from electron-donating (e.g., alkyl, methoxy) to electron-withdrawing groups (e.g., ester, trifluoromethyl) were tolerated. Heterocyclic dienes including thiophene, thiazole, pyridine, quinoline were also suitable substrates. The ester group in alkyl bromides was indispensable since no reaction was observed in the absence of ester group.
Scheme 7.

Stereoconvergent dienylation of alkyl bromides
It was proposed that alkyl bromide 3 undergoes a single electron transfer (SET) reduction by Cu(I) catalyst, generating alkyl radical 4 that adds to a diene en route to allylic radical 5 (Scheme 8). Further conversion to the product diene can proceed along three pathways, i.e., oxidation of 5 by Cu(II) catalyst to cation 6 with subsequent deprotonation (path a), a capture of cation 6 with the bromide followed by a base-mediated elimination of hydrogen bromide (path b), and a radical chain process via a halogen atom transfer mediated by radical 5 (path c).
Scheme 8.
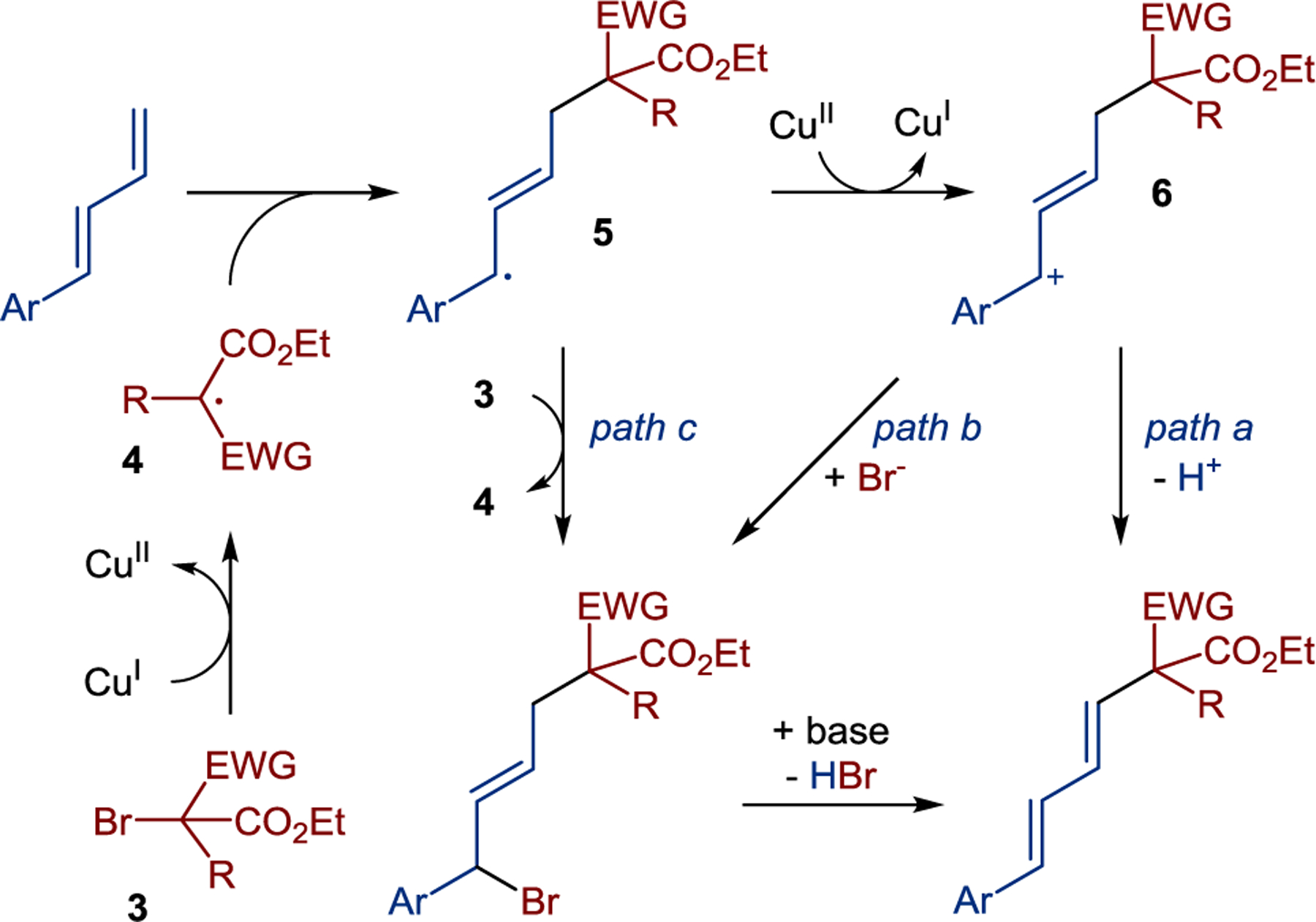
Proposed mechanism for stereoconvergent Heck-type alkylation of 1,3-dienes
In 2020, He, Feng, and Lin reported an efficient stereoselective synthesis of trisubstituted 1,3,5-trienes from 1,3-dienes via an aryl to alkenyl 1,4-palladium migration/Heck sequence, proceeding via indirect route through alkenylpalladium intermediates, while obviating the need to prepare alkenyl halides of specific geometry (Scheme 9a).21
Scheme 9.

Stereoselective dienylation via a 1,4-palladium migration/Heck cross coupling cascade
A range of trisubstituted 1,3,5-trienes were prepared in moderate to excellent yields from aryl halides and dienes bearing a range of electron-donating and electron-withdrawing substituents. Of note, the method afforded access to substrates with two distinct aryl groups at the terminal positions of the conjugated trienes. The cascade process follows a previously established22 sequence of an oxidative addition and 1,4-palladium migration en route to vinylpalladium intermediate 7 that can subsequently be engaged in a stereoselective reaction with 1,3-dienes (Scheme 9b).
As demonstrated by these two examples, the use of 1,3-dienes as dienylation reagents can present significant opportunities when implemented within recently developed catalytic pathways that are based both on one-electron and two-electron processes.
4. Small ring dienylation reagents
The high strain energy of three- and four-membered ring systems has provided a significant impetus to the development of new synthetic methodologies23 and remains largely untapped in the context of dienylation. Nevertheless, several recent reports have underscored the synthetic potential of strained dienylation reagents.
In 2018, Carrow and co-workers developed a palladium catalyzed regioselective dienylation of aryl- and alkenylboronic acids with cyclobutene (8), producing substituted 1,3-dienes or 1,3,5-trienes under aerobic conditions (Scheme 10).24 The limitations associated with previously developed synthesis of 1-aryl-1,3-butadienes in Heck type reactions with 1,3-butadiene (e.g., overarylation and a sluggish β-hydride elimination from the π-allylpalladium intermediate)25 were resolved using cyclobutene as a surrogate for 1,3-butadiene. An array of functionalized carbocyclic and heterocyclic dienes and trienes were prepared in good yields and with broad functional group tolerance that encompassed cyano, hydroxy, methoxy, thiomethoxy, halogen, amide, and trifluoromethyl groups.
Scheme 10.

Aerobic Heck-type dienylation of boronic acids with cyclobutene
A mechanistic proposal supported by computational studies suggests that Pd(II) species 9 generated by the aerobic oxidation of Pd(0) undergoes transmetalation with the boronic acid to form intermediate 10 (Scheme 11). Stereospecific syn-migratory insertion of intermediate 10 to cyclobutene affords cyclobutylpalladium intermediate 11 that can undergo β-alkyl (path a) or β-hydride elimination (path b). DFT calculations suggest a highly unfavorable barrier for the β-alkyl elimination (>30.0 kcal/mol) to alkylpalladium intermediate 12. Therefore, syn-β-hydride elimination is proposed to be the operative mechanism for the formation of 3-substituted cyclobutene 13 that undergoes a pericyclic ring opening, proceeding over a 28.7 kcal/mol barrier and furnishing the linear conjugated diene as a sole product.
Scheme 11.

The proposed catalytic cycle for the dienylation with cyclobutene.
In 2019, Zhu and co-workers developed a rhodium-catalyzed 2-pyridyl group-directed C–H activation to enable a dienylation of 2-isoquinolinones and 2-pyridones using methylenecyclopropane as a dienylation reagent with Cu(OAc)2 as an oxidant (Scheme 12).26 Distinctively, the reaction enables introduction of a substituent to an internal position in the dienyl moiety, in contrast to other dienylation methods that introduce the substituent in a terminal position. While some products were formed as single diastereomers, albeit without an assignment of the double bond configuration, most products were produced as 1:1 mixtures of E- and Z-isomers. The reaction scope encompasses a range of structurally diverse 1-arylmethylenecyclopropanes and N-pyridyl-substituted 2-isoquinolinones and 2-pyridones. Based on deuterium exchange and parallel kinetic isotope effect (KIE) studies, it was proposed that the reaction proceeds via a reversible rate-limiting C–H metalation, producing rhodacycle 14 (Scheme 13). Subsequent migratory insertion to the methylenecyclopropane followed by a β-carbon elimination and a ring scission give rise to alkylrhodium intermediate 15 that releases the diene product upon β-hydride elimination. A similar Rh-catalyzed, directed dienylation of N-phenoxyacetamides with methylenecyclopropanes was recently described by Volla and co-workers.27
Scheme 12.

Rhodium catalyzed dienylation of 2-isoquinones and 2-pyridones with methylenecyclopropane
Scheme 13.

Mechanism of rhodium catalyzed dienylation of 2-isoquinones and 2-pyridones with methylenecyclopropane
In 2021, Jiang and co-workers introduced a rhodium-catalyzed dienylation mediated by cyclopropene-based dienylation reagents, affording dicarbonyl functionalized 1,3-dienes from enaminones by an unconventional skeletal editing process (Scheme 14).28 A range of functionalized aryl- and alkyl-substituted enaminones participated in the reaction to deliver the desired products in good to moderate yields under mild reaction conditions and with a low catalyst loading. A noteworthy feature of the reaction is the cleavage of the α,β-C=C bond in enaminones accompanied by an insertion of the cyclopropane-derived C4 unit, resulting in a geminal disubstitution at one of the diene termini.
Scheme 14.

Cyclopropene-mediated dienylation of enaminones
Based on preliminary mechanistic studies, it was proposed that the cyclopropene reagent undergoes ring opening to form rhodium carbenoid species 16 that further engages the enaminone in a cyclopropanation reaction, affording aminocyclopropane intermediate 17 (Scheme 15). Subsequent ring opening of the push-pull cyclopropane with a water molecule capture produced intermediate 18 that released the conjugated diene product upon acetic acid elimination.
Scheme 15.
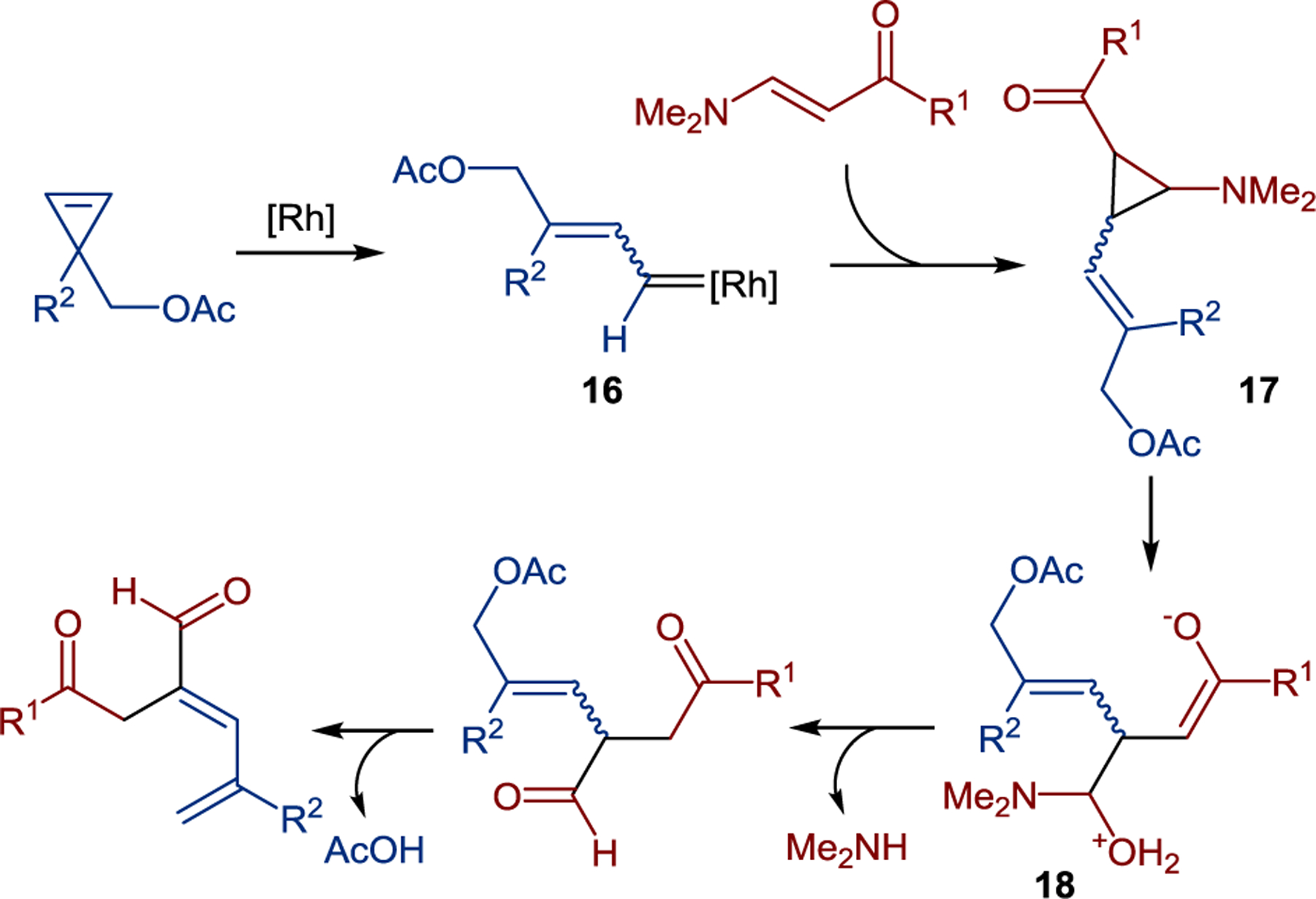
Mechanism of the cyclopropene-mediated dienylation of enaminones
The examples presented in this section clearly demonstrate the unique advantages offered by small ring dienylation reagents that include facile access to carbenoid intermediates that can engage new classes of substrates, high reactivity due to the release of the angular strain, and access to both linear and branched conjugated dienes. Further work in this area will likely lead to new dienylation reactions with broader scope and improved stereoselectivity, extending the synthetic reach of the synthetic methodologies based on the reactivity of small ring systems.
5. Pyrones, alkynes, and homoallenylboronates
A number of other structurally diverse dienylation reagents have recently emerged as promising synthetic tools for the construction of conjugated dienes through both metal-catalyzed and organocatalytic manifolds, exemplified by pyrones, alkynes, and homoallenylboronates.
In 2015, Tunge and co-workers developed a palladium catalyzed dienylation of allylic carbonates and carbamates with pyrone 19 serving as a C4 diene synthon (Scheme 16).29 A broad range of substituted allylic carbonates participated in the reaction, furnishing conjugated dienes in good to moderate yields and with an E/Z selectivity ranging from 3.8 : 1 to 9.1 : 1. Notably, the alkoxy, pyrrole and imidazole residues of the carbonates and carbamates were appended to the opposite terminus of the dienyl unit during the double decarboxylative cascade process.
Scheme 16.

Pd-catalyzed double decarboxylative addition to pyrone
It is proposed that the allylic carbonate first undergoes oxidative addition to Pd(0) to generate π-allylpalladium complex 20 along with a nucleophile emerging from the carbonate residue upon decarboxylation (Scheme 17). π-Allylpalladium complex 20 then participates in the reaction with pyrone to form intermediate 21 that captures the nucleophile to generate intermediate 22. The ensuing oxidative addition of allylic carbonate 22 to Pd(0) affords intermediate 23 that gives rise to the conjugated diene product upon decarboxylation.
Scheme 17.

Mechanism of the Pd-catalyzed dienylation with pyrone
In 2021, Wang and co-workers developed a phosphine/Brønsted acid-cocatalyzed dienylation method combining p-quinone methides and δ-substituted alkynoates via a 1,6-conjugate addition and isomerization cascade sequence (Scheme 18).30 A range of substituted and functionalized p-quinone methides as well as alkynoates were examined, albeit with the limitation of the double o-alkyl substitution in the quinone moiety, affording 1,3-dienes with >20:1 stereoselectivity and in good to moderate yields. Notably, protected 2-indolinones can be incorporated in the diene product. Based on deuterium labelling experiments, a mechanism was proposed that entails an initial phosphine addition to the alkynoate to give intermediate 24 (Scheme 19). Following a 1,3-proton shift and an elimination of the acid catalyst, a 1,4 proton shift affords zwitterionic intermediate 25.
Scheme 18.

Phosphine/Brønsted acid-cocatalyzed dienylation of p-quinone methides with alkynoates
Scheme 19.

Mechanism of the phosphine/Brønsted acid-cocatalyzed dienylation of p-quinone methides with alkynoates
Subsequent 1,6-conjugate addition to the p-quinone methide produces phenolate 26 that undergoes a proton shift and an elimination of the phosphine catalyst, furnishing the dienylation product.
In 2021, Fu, Xu and co-workers reported the construction of multi-substituted 1,3-dienes by dienylation of (hetero)aryl iodides, allyl halides, and alkynyl bromides with homoallenylboronates (Scheme 20).31 Notably, the method provided access to branched 1,3-dienes via the introduction of an incoming substituent at an internal position in the dienyl unit. The reactions were Z-selective with respect to the trisubstituted C=C bond. While exclusive Z-stereoselectivity was observed for (heteroaryl) iodides and alkynyl bromides, somewhat lower but still synthetically useful diastereomeric ratios (9–20 : 1) were observed for allylic halides. The use of alkyl-substituted homoallenylboronates also resulted in reduced stereoselectivity.
Scheme 20.

Dienylation with homoallenylboronates
It was proposed that the reaction commences with an oxidative addition of the electrophilic coupling partner to the Pd(0) catalyst, leading to vinylpalladium intermediate 27 via an SE2’ process that proceeds through a cyclic transition state 28 (path a, Scheme 21). Subsequent reductive elimination releases the major Z-diastereomeric product. However, a minor pathway (path b, Scheme 21) that proceeds via a homoallenylpalladium species 29 undergoing a sequence of a σ-π-σ rearrangements to isomeric vinylpalladium intermediate 30 may account for the formation of the minor diastereomer of the product.
Scheme 21.

Mechanism of the dienylation with homoallenylboronates
The examples discussed in this section highlight the structural and mechanistic diversity that can be exploited to develop stereoselective dienylation reactions and expand the scope of suitable coupling partners. The distinctive reactivities of pyrones, alkynes, and homoallenylboronates present opportunities for further expansion of the dienylation methodology, especially for the construction of branched conjugated dienes, for Z-selective dienylation, and to facilitate stereoselective access to reactive dienyl groups (e.g., dienyl ethers and dienamines).
6. Conclusion and outlook
The past decade has witnessed the emergence of catalytic dienylation for the regio- and stereoselective construction of conjugated π-systems. The examples presented in this Short Review demonstrate the advantages of the one-step installation of the dienyl unit using a number of structurally diverse dienylation reagents, with each class showing promising reactivity and stereoselectivity profiles. While sulfolenes provided an example of regiodivergent dienylation and regioselective C–C bond formation, the reactions of 1,3-dienes also indicated that useful levels of stereoselectivity can be achieved using both radical and traditional cross-coupling conditions. Similarly, small ring systems offered enhanced reactivity and unusual regioselectivity. On the other hand, alkyne-, allene-, and pyrone-based reagents engaged a broad scope of coupling partners by way of diverse mechanistic pathways. The discussed reactions, however, also reveal some limitations of the current dienylation methods. Most of the methods demonstrate synthetically useful levels of E-selectivity for the newly formed C=C bonds, while limited examples of the Z-selective dienylation (e.g., the Z-selective dienylation with sulfolene) are currently available. Further, the dienylations typically occur at a terminal carbon of the diene moiety. By contrast, methods that can introduce substituents in the internal positions of the conjugated π-systems remain underexplored. These limitations point to opportunities for the development of dienylation methods that have broader synthetic impact. Further development of stereodivergent and with highly stereoselective methods that can produce diastereomeric dienes with the same dienylation reagent can simplify access to a variety of conjugated dienes and polyenes. Similarly, the development of new dienylation reagents and further fine-tuning and synthetic scope exploration with the already developed reagents can enable reactions with other coupling partners, for example in the aliphatic series. The reactions discussed in this review clearly indicate that such advances are well within reach.
Funding Information
This work was supported by the NIGMS (GM134371) and the Welch Foundation (AX-0047).
Biography

Oleg Larionov obtained his PhD degree from the University of Göttingen under the tutelage of Prof. Armin de Meijere. In 2010, after postdoctoral studies with Prof. Alois Fürstner at the Max-Planck-Institut für Kohlenforschung and Prof. E. J. Corey at Harvard University, he joined the faculty at the University of Texas at San Antonio, where he is now Professor and Robert A. Welch Distinguished University Chair in Chemistry. His research interests focus on the development of synthetic methods for the construction of carbon–carbon and carbon–heteroatom bonds, transition metal catalysis, organoboron chemistry, and organic photochemistry.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- (1).(a) Inano H; Suzuki K; Ishii-Ohba H; Yamanouchi H; Takahashi M; Wakabayashi K Carcinogenesis 1993, 14, 2157. [DOI] [PubMed] [Google Scholar]; (b) Rychnovsky SD Chem. Rev 1995, 95, 2021. [Google Scholar]; (c) Thirsk C; Whiting AJ Chem. Soc., Perkin Trans 1 2002, 999–1023. [Google Scholar]; (d) Paik IH; Xie SJ; Shapiro TA; Labonte T; Sarjeant AAN; Baege AC; Posner GH J. Med. Chem 2006, 49, 2731. [DOI] [PubMed] [Google Scholar]; (e) Davis SA; Vincent BM; Endo MM; Whitesell L; Marchillo K; Andes DR; Lindquist S; Burke MD Nat. Chem. Biol 2015, 11, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Endo MM; Cioffi AG; Burke MD Synlett 2016, 27, 337. [Google Scholar]
- (2).Hoeben FJM; Jonkheijm P; Meijer EW; Schenning APHJ Chem. Rev 2005, 105, 1491. [DOI] [PubMed] [Google Scholar]
- (3).Zotchev SB Curr. Med. Chem 2003, 10, 211. [DOI] [PubMed] [Google Scholar]
- (4).(a) Corey EJ Angew. Chem., Int. Ed 2002, 41, 1650. [DOI] [PubMed] [Google Scholar]; (b) Fringuelli F; Taticchi A The Diels-Alder Reaction: Selected Practical Methods; John Wiley & Sons, 2002. [Google Scholar]; (c) Bar GLJ; Lloyd-Jones GC; Booker-Milburn KI J. Am. Chem. Soc 2005, 127, 7308. [DOI] [PubMed] [Google Scholar]; (d) Du H; Zhao B; Shi YJ Am. Chem. Soc 2007, 129, 762. [DOI] [PubMed] [Google Scholar]; (e) Liao L; Jana R; Urkalan KB; Sigman MS J. Am. Chem. Soc 2011, 133, 5784. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Leung JC; Geary LM; Chen T-Y; Zbieg JR; Krische MJ J. Am. Chem. Soc 2012, 134, 15700. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) McNeill E; Ritter T Acc. Chem. Res 2015, 48, 2330. [DOI] [PubMed] [Google Scholar]; (h) Timsina YN; Sharma RK; RajanBabu TV Chem. Sci 2015, 6, 3994. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Yang X-H; Dong VM J. Am. Chem. Soc 2017, 139, 1774. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Sardini SR; Brown MK J. Am. Chem. Soc 2017, 139, 9823. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Tortajada A; Ninokata R; Martin RJ Am. Chem. Soc 2018, 140, 2050. [DOI] [PubMed] [Google Scholar]
- (5).Modern Carbonyl Olefination; Takeda T, Ed.; Wiley-VCH, Weinheim, 2004. [Google Scholar]
- (6).(a) Mehta G; Prakash Rao HS In Patai’s Chemistry of Functional Groups; Rappoport Z, Ed.; John Wiley & Sons, Ltd: Chichester, 1997. [Google Scholar]; (b) Delcamp JH; Gormisky PE; White MC J. Am. Chem. Soc 2013, 135, 8460. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) De Paolis M; Chataigner I; Maddaluno J Top. Curr. Chem 2012, 327, 87. [DOI] [PubMed] [Google Scholar]; (e) Hu X-H; Zhang J; Yang X-F; Xu YH; Loh T-PJ Am. Chem. Soc 2015, 137, 3169. [DOI] [PubMed] [Google Scholar]
- (7).(a) Lee SJ; Gray KC; Paek JS; Burke MD J. Am. Chem. Soc 2008, 130, 466. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lee SJ; Anderson TM; Burke MD Angew. Chem., Int. Ed 2010, 49, 8860. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Woerly EM; Roy J; Burke MD Nat. Chem 2014, 6, 484. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Li J; Ballmer SG; Gillis EP; Fuji S; Schmidt MJ; Palazzolo AME; Lehmann JW; Morehouse GF; Burke MD Science 2015, 347, 1221. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Li J; Grillo AS; Burke MD Acc. Chem. Res 2015, 48, 2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Maishal TK; Sinha-Mahapatra DK; Paranjape K; Sarkar A Tetrahedron Lett 2002, 43, 2263. [Google Scholar]; (b) Diver ST; Giessert AJ Chem. Rev 2004, 104, 1317. [DOI] [PubMed] [Google Scholar]; (c) Luo SX, Cannon JS, Taylor BHL, Engle KM, Houk KN, Grubbs RH J. Am. Chem. Soc 2016, 138, 14039. [DOI] [PubMed] [Google Scholar]
- (9).Liu Y; Wang L; Deng LJ Am. Chem. Soc 2016, 138, 112. [DOI] [PubMed] [Google Scholar]
- (10).Woolven H; Gonzalez-Rodriguez C; Marco I; Thompson AL; Willis MC Org. Lett 2011, 13, 4876. [DOI] [PubMed] [Google Scholar]
- (11).Martial L; Bischoff L Synlett 2015, 26, 1225. [Google Scholar]
- (12).Dang HT; Nguyen VT; Nguyen VD; Arman HD; Larionov OV Org. Biomol. Chem 2018, 16, 3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Nguyen VT; Dang HT; Pham HH; Nguyen VD; Flores-Hansen C; Arman HD; Larionov OV J. Am. Chem. Soc 2018, 140, 8434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Miyaura N; Suzuki A Chem. Rev 1995, 95, 2457. [Google Scholar]
- (15).Dang HT; Nguyen VD; Hoang HP; Arman HD; Larionov OV Tetrahedron 2019, 75, 3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dang HT; Nguyen VD; Haug GC; Vuong NTH; Arman HD; Larionov OV ACS Catal 2021, 11, 1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wu X-X; Ye H; Dai H; Yang B; Wang Y; Chen S; Hu. L Org. Chem. Front 2020, 7, 2731. [Google Scholar]
- (18).Deagostino A; Prandi C; Tobasso S; Venturello P; Molecules 2010, 15, 2667–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).(a) See also: Kiyota S; In S; Saito R; Komine N; Hirano M Organometallics 2016, 35, 4033. [Google Scholar]; (b) Xiao B-X; Jiang B; Yan R-J; Zhu J-X; Xie K; Gao X-Y; Ouyang Q; Du W; Chen Y-CJ Am. Chem. Soc 2021, 143, 4809. [DOI] [PubMed] [Google Scholar]
- (20).Zhang H; Wu X; Wei Y; Zhu C Org. Lett 2019, 21, 7568. [DOI] [PubMed] [Google Scholar]
- (21).Xue Z-J; Li M-Y; Zhu B-B; He Z-T; Feng C-G; Lin G-Q; Chem. Commun 2020, 56, 14420. [DOI] [PubMed] [Google Scholar]
- (22).(a) Hu T-J; Li M-Y; Zhao Q; Feng C-G; Lin G-Q Angew. Chem., Int. Ed 2018, 57, 5871. [DOI] [PubMed] [Google Scholar]; (b) Hu T-J; Zhang G; Chen Y-H; Feng C-G; Lin G-QJ Am. Chem. Soc 2016, 138, 2897. [DOI] [PubMed] [Google Scholar]
- (23).(a) Pohlmann T; de Meijere A Org. Lett 2000, 2, 3877. [DOI] [PubMed] [Google Scholar]; (b) Namyslo JC; Kaufmann DE Chem. Rev 2003, 103, 1485; [DOI] [PubMed] [Google Scholar]; (c) Rubin M; Rubina M; Gevorgyan V Chem. Rev 2007, 107, 3117. [DOI] [PubMed] [Google Scholar]; (d) Korotkov VS; Larionov OV; Hofmeister A; Magull J; de Meijere AJ Org. Chem 2007, 72, 7504. [DOI] [PubMed] [Google Scholar]; (e) Ogawa S; Urabe D; Yokokura Y; Arai H; Arita M; Inoue M Org. Lett 2009, 11, 3602. [DOI] [PubMed] [Google Scholar]; (f) Simlandy AK; Lyu M-Y; Brown MK ACS Catal 2021, 11, 12815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).McAlpine NJ; Wang L; Carrow BP J. Am. Chem. Soc 2018, 140, 13634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).(a) Mitsudo T; Fischetti W; Heck RF J. Org. Chem 1984, 49, 1640; [Google Scholar]; (b) Jeffery T. Tetrahedron Lett 1992, 33, 1989. [Google Scholar]
- (26).Zhu YQ; Niu YX; Hui LW; He JL; Zhu K Adv. Synth. Catal 2019, 361, 2897. [Google Scholar]
- (27).Singh A; Dey D; Volla CMR J. Org. Chem 2021, 86, 10474. [DOI] [PubMed] [Google Scholar]
- (28).Jiang C; Wu J; Han J; Chen K; Qian Y; Zhang Z; Jiang Y Chem. Commun 2021, 57, 5710. [DOI] [PubMed] [Google Scholar]
- (29).Maji T; Tunge JA Org. Lett 2015, 17, 4766. [DOI] [PubMed] [Google Scholar]
- (30).Song Z; Wang W; Liu Z; Lu Y; De Wang. J.Org. Chem 2021, 86, 8590. [DOI] [PubMed] [Google Scholar]
- (31).Yang C; Dai D; Lu H; Zhang F; Fu Y; Xu Y Org. Chem. Front 2021, 8, 2480. [Google Scholar]