

Abstract
Herein is reported a robust and general method for the preparation of N-acylsulfenamides, important functionalities that have recently been utilized as central inputs for the asymmetric synthesis of high oxidation state sulfur compounds. This straightforward transformation proceeds by reaction of primary amides, carbamates, sulfonamides, sulfinamides, and ureas with stable N-thiosuccinimides or N-thiophthalimides, which in turn are prepared in a single step from commercial thiols. The use of stable N-thiosuccinimide and N-thiophthalimide reactants is desirable because it obviates the use of highly reactive sulfenyl chlorides.
Keywords: sulfenamide, amide, thiosuccinimide, thiophthalimide, thiol, asymmetric catalysis
Graphical Abstract

Sulfenamides, which incorporate a divalent sulfur bonded to nitrogen and carbon substituents, have enjoyed numerous uses ranging from industrial application for the vulcanization of rubber to prodrug synthesis.1 Sulfenamides are most often utilized as sulfenylating reagents due to the lability of the S–N bond. Recently, our lab has also demonstrated that N-acylsulfenamides are key reactants for asymmetric S-alkylation en route to enantioenriched sulfoximines (Scheme 1a).2 Moreover, the Ma lab has reported on their asymmetric oxidation for the preparation of chiral N-acylsulfinamides (Scheme 1b).3 Despite their increased relevance to asymmetric catalysis, few general methods have been developed for the synthesis of N-acylsulfenamides. The most common approaches towards N-acylsulfenamides typically involve the reaction of amides with sulfenyl chlorides,4 which are usually prepared by reaction of thiols with chlorine gas or sulfuryl chloride, both corrosive and toxic reagents (Scheme 1c). These methods are also often low yielding due to the high reactivity of the sulfenyl chloride intermediates. In recent years, new approaches towards the synthesis of N-acylsulfenamides have involved the reaction of primary amides with disulfides,5 disulfanyl derivatives of phosphorodithioic acid,6 and sulfonic acid thioesters.3 Several other specialized methods for the generation of N-acylsulfenamides have been reported as well.7 After submission of this manuscript, Wang, Chen, and coworkers disclosed an unrelated copper-catalyzed approach for the generation of N-acylsulfenamides.8
Scheme 1.

Background on the synthesis and application of N-acylsulfenamides
We hypothesized that thiosuccinimides9 and thiophthalimides10 could serve as optimal electrophilic coupling partners for displacement by amide nucleophiles because of their ease of generation, stability under ambient conditions, and the excellent leaving group character of the succinimide or phthalimide, respectively (Scheme 1d). Moreover, thiosuccinimides are readily prepared from commercially abundant thiol feedstocks and N-chlorosuccinimide.9 Two examples of the use of thiophthalimide as a leaving group for the synthesis of N-acylsulfenamides had been previously provided by Guarino and co-workers, albeit employing butyllithium as a base.11
We began our investigation by exploring the reaction between various amides and analogous functionalities with N-(phenylthio)phthalimide (1a) as the standard coupling partner (Scheme 2). We chose to employ the phthalimide rather than the succinimide leaving group due to the commercial availability of 1a. Based upon our previously developed conditions to prepare N-acylsulfinamides,12 we determined that sodium hydride was the optimal base for this transformation, leading to product formation in 88% yield. NaHMDS could also be employed but led to a depressed yield of 62% under the reaction conditions. The use of weaker organic bases such as triethylamine led to no conversion. When the reaction was performed at the higher concentration of 0.2 M, a comparable yield of 3a of 90% was obtained. We first examined the scope of the reaction using alkyl amides and found that pivalamide and acetamide provided sulfenamide products 3a and 3b, respectively, in excellent yields. We also employed a branched N-Boc-protected piperidine amide input, which provided 3c in excellent yield. Benzamides with varying electronics were compatible, including benzamide (3d), 4-methoxybenzamide (3e), 4-trifluoromethylbenzamide (3f), and 2-chlorobenzamide (3g). After demonstrating scope for amides, we were curious whether different, amide-related functional groups could also act as nucleophiles in the transformation. We were pleased to observe high conversion to product using carbamates as nucleophiles to provide Boc-protected (3h) and Cbz-protected (3i) sulfenamides in excellent yields. The tosyl-protected sulfenamide 3j could also be prepared in good yield from the corresponding p-toluenesulfonamide input. Moreover, the enantiomerically pure (S)-tert-butylsulfinamide was an effective coupling partner, giving sulfenamide 3k in good yield and in a 96–98% enantiomeric excess. Consequently, sulfenamide 3k could potentially find use as a chiral reagent in asymmetric synthesis. Finally, we found that urea functionality was effective in this transformation, with 3l obtained in good yield. Due to its decreased acidity, the urea required a higher temperature to achieve acceptable reaction conversion.
Scheme 2.
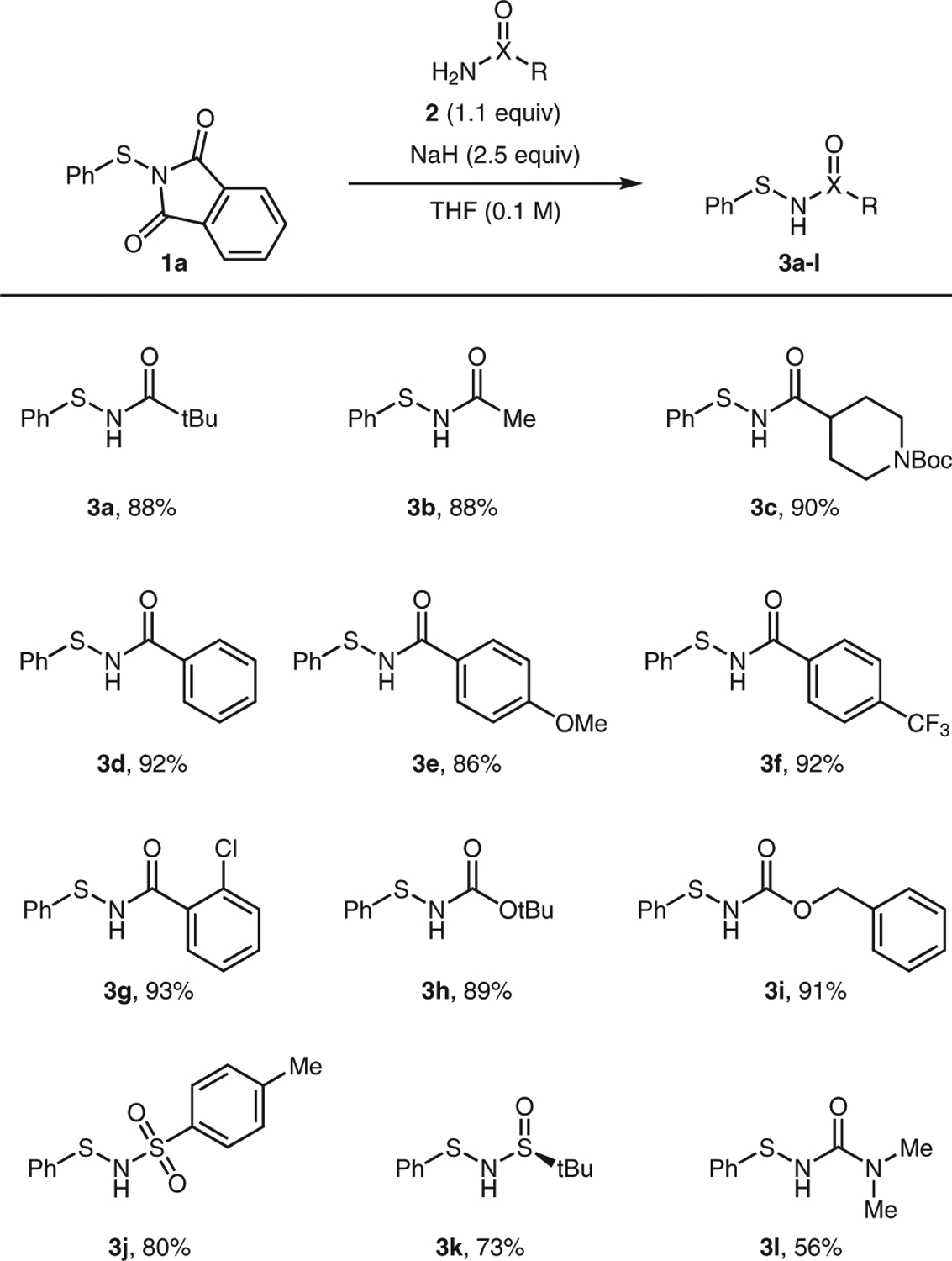
Scope of amide input
After exploring the amide scope, we turned our attention to the thiol scope (Scheme 3). Preparation of various succinimide analogues was easily accomplished using a literature procedure9 to explore a broad range of substituents on the sulfur side of the sulfenamide. We first explored linear alkyl N-thiosuccinimides and found that butylsulfenamide 3m could be prepared in excellent yield (Scheme 3). This chemistry was compatible with ester functionality as demonstrated by the preparation of alkyl ester 3n. For this substrate, it was necessary that the reaction be performed at 0 °C to minimize undesired elimination of the N-mercaptopivalamide. Branched alkyl N-thiosuccinimides were effective coupling partners, with both cyclopentyl (3o) and N-Boc-azetidine (3p) sulfenamides isolated in good yields. Moreover, the sterically congested tert-butylthiosuccinimide was efficiently converted to sulfenamide 3q. Benzylsulfenamide 3r was also readily accessed. We next explored reactivity with different aromatic and heteroaromatic N-thiosuccinimides. A range of electronics were compatible with this reaction, with both the strongly donating 4-methoxy (3s) and the highly electron-deficient 3,5-ditrifluoromethylphenyl (3t) N-thiosuccinimides providing the corresponding products in good yields. Moreover, the highly sterically hindered 2,6-dichlorobenzene N-thiosuccinimide could be employed effectively, giving sulfenamide 3u. We also demonstrated that both electron-poor and electron-rich heterocyclic systems could be employed, as demonstrated for the 2-pyridyl- (3v) and 2-thiophenyl (3w) sulfenamides, respectively.
Scheme 3.

Scope of thiosuccinimide input
In conclusion, we have developed a practical and general synthesis of N-acylsulfenamides from amides and N-thiosuccinimide derivatives. We established a very broad scope of amide coupling partners, including alkyl and aryl primary amides, carbamates, sulfonamides, sulfinamides, and ureas. We further established an impressive scope of alkyl and aryl N-thiosuccinimides, all derived from commercially abundant thiol inputs. This method enables the general synthesis of different N-acylsulfenamides under mild, basic conditions without requiring the intermediacy of unstable and difficult to prepare sulfenyl chlorides. We anticipate the use of N-acylsulfenamides will increase given the recent disclosures of their application towards the asymmetric synthesis of high oxidation state sulfur compounds. The robust method reported here for the preparation of N-acylsulfenamides should facilitate these endeavors.
All glassware were oven-dried (130 °C) and evacuated while hot prior to use. Unless otherwise indicated, all reactions for substrate preparation were carried out on the benchtop under a N2 atmosphere. Solvents were sparged with argon and purified by elution through a column of activated alumina under argon before use. Unless otherwise noted, all reagents were purchased from commercial sources and used without further purification. Product purification was performed by either flash column chromatography with SiliaFlash P60 (230–400 mesh) silica gel, or reverse phase chromatography with a Teledyne Isco automated chromatography system using C-18 gold columns. 1H, 13C, and 19F- NMR spectra were recorded on either a 400, 500, or 600 MHz instrument. Data are reported in the following format: chemical shift in ppm, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br s = broad singlet, m = multiplet, dd = doublet of doublets, etc.), coupling constant J in Hz, and integration. NMR solvents were used as received. NMR chemical shifts are reported in ppm relative to CDCl3 (7.26 ppm for 1H and 77.16 ppm for 13C), CD3OD (3.31 ppm for 1H and 49.00 ppm for 13C), acetone-d6 (2.05 ppm for 1H and 29.84 and 206.26 ppm for 13C), or C6D6 (7.16 ppm for 1H and 128.06 ppm for 13C). Partial IR spectra are reported. High-resolution mass spectra (HRMS) were obtained using electrospray ionization (ESI) on a time of flight (TOF) mass spectrometer or were sent to UIUC for analysis by the electron impact (EI) or chemical ionization (CI) methods.
N-Thiosuccinimides 1
N-Thiosuccinimides were synthesized according to a published literature procedure.9 Spectral data for known compounds 1m,13 1n,13 1r,14 1s,9 and 1w15 matched those reported in the literature. N-Thiophthalimide 1a was purchased commercially.
1-(Cyclopentylthio)pyrrolidine-2,5-dione (1o)
The compound was synthesized according to the literature procedure,9 employing cyclopentanethiol (0.54 mL, 5.0 mmol, 1.0 equiv). Purification by silica gel chromatography (40% EtOAc in hexanes) afforded the product 1o as a white solid; yield: 848 mg (85%); mp 74–76 °C; Rf = 0.31 (silica gel, 40% EtOAc/hexanes, UV).
IR (neat): 2961, 1709, 1302, 1134, 812, 658 cm−1.
1H NMR (400 MHz, CDCl3): δ = 3.74−3.62 (m, 1 H), 2.81 (s, 4 H), 1.91−1.73 (m, 4 H), 1.62−1.40 (m, 4 H).
13C NMR (101 MHz, CDCl3): δ = 177.4, 48.7, 31.2, 28.6, 24.7.
HRMS (ESI): m/z [M + H]+ calcd for C9H14NO2S+: 200.0740; found: 200.0745.
tert-Butyl 3-((2,5-Dioxopyrrolidin-1-yl)thio)azetidine-1-carboxylate (1p)
The compound was synthesized according to the literature procedure,9 employing tert-butyl 3-mercaptoazetidine-1-carboxylate (1.69 mL, 10.0 mmol, 1.0 equiv). After purification by silica gel chromatography (30% EtOAc in hexanes, then 60% EtOAc in hexanes) and concentration of the appropriate fractions, the product was diluted with CH2Cl2 followed by extraction with sat. aq NaHCO3 to remove a co-polar succinimide by-product. Concentration of the organic layer then provided the product as a highly viscous oil. Dissolution in CDCl3 followed by slow evaporation under a stream of N2 resulted in crystallization, affording the product 1p as a white solid; yield: 1.53 g (54%); mp 127–129 °C; Rf = 0.29 (silica gel, 60% EtOAc/hexanes, UV).
IR (neat): 2963, 1713, 1302, 1246, 1134, 1005, 812, 658 cm−1.
1H NMR (500 MHz, CDCl3): δ = 4.20 (dd, J = 9.3, 7.4 Hz, 2 H), 3.92−3.83 (m, 3 H), 2.86 (s, 4 H), 1.41 (s, 9 H).
13C NMR (126 MHz, CDCl3): δ = 177.0, 155.8, 80.1, 55.2, 38.1, 28.6, 28.3.
HRMS (ESI): m/z [M + Na]+ calcd for C12H18 N2O4SNa+: 309.0880; found: 309.0889.
1-(tert-Butylthio)pyrrolidine-2,5-dione (1q)
The compound was synthesized according to the literature procedure,9 employing 2-methylpropane-2-thiol (1.13 mL, 10.0 mmol, 1.0 equiv). Purification by silica gel chromatography (30% EtOAc in hexanes, then 50% EtOAc in hexanes) afforded the product 1q as a white solid; yield: 1.41 g (75%); mp 156–159 °C; Rf = 0.40 (silica gel, 50% EtOAc/hexanes, UV).
IR (neat): 2984, 1711, 1300, 1138, 820, 650 cm−1.
1H NMR (500 MHz, CDCl3): δ = 2.86 (s, 4 H), 1.31 (s, 9 H).
13C NMR (151 MHz, CDCl3): δ = 177.7, 51.3, 29.5, 28.5.
HRMS (ESI): m/z [M + H]+ calcd for C8H14NO2S+: 188.0740; found: 188.0745.
1-((3,5-Bis(trifluoromethyl)phenyl)thio)pyrrolidine-2,5-dione (1t)
The compound was synthesized according to the literature procedure,9 employing 3,5-bis(trifluoromethyl)benzenethiol (1.69 mL, 10.0 mmol, 1.0 equiv). Purification by silica gel chromatography (30% EtOAc in hexanes, then 50% EtOAc in hexanes) afforded the product 1t as a pale yellow solid; yield: 460 mg (13%); mp 94–96 °C; Rf = 0.58 (silica gel, 50% EtOAc/hexanes, UV).
IR (neat): 3094, 2965, 1726, 1354, 1281, 1115, 1005, 816, 681 cm−1.
1H NMR (600 MHz, CDCl3): δ = 7.87 (s, 2 H), 7.76 (s, 1 H), 2.86 (s, 4 H).
13C NMR (151 MHz, CDCl3): δ = 175.8, 137.2, 132.6 (q, 2JC,F = 34.0 Hz), 130.1 (q, 3JC,F = 3.8 Hz), 123.0 (hept, 3JC,F = 3.8 Hz), 122.5 (q, 1JC,F = 273.2 Hz), 28.6.
19F NMR (376 MHz, CDCl3): δ = −63.2.
HRMS (ESI): m/z [M + H]+ calcd for C12H8F6NO2S+: 344.0175; found: 344.0185.
1-((2,6-Dichlorophenyl)thio)pyrrolidine-2,5-dione (1u)
The compound was synthesized according to the literature procedure,9 employing 2,6-dichlorobenzenethiol (1.79 g, 10.0 mmol, 1.0 equiv). Purification by silica gel chromatography (10% EtOAc in hexanes, then 50% EtOAc in hexanes) afforded the product 1u as a white solid; yield: 893 mg (32%); mp 162–163 °C; Rf = 0.48 (silica gel, 50% EtOAc/hexanes, UV).
IR (neat): 3096, 2968, 1726, 1354, 1281, 1115, 1005, 816, 656 cm−1.
1H NMR (500 MHz, CDCl3): δ = 7.31 (d, J = 8.0 Hz, 2 H), 7.18 (t, J = 8.1 Hz, 1 H), 2.77 (s, 4 H).
13C NMR (126 MHz, CDCl3): δ = 175.3, 138.6, 131.0, 129.7, 129.1, 28.7.
HRMS (ESI): m/z [M + H]+ calcd for C10H8Cl2NO2S+: 275.9647; found: 275.9656.
1-(Pyridin-2-ylthio)pyrrolidine-2,5-dione (1v)
The compound was synthesized according to the literature procedure,9 employing pyridine-2-thiol (1.11 g, 10.0 mmol, 1.0 equiv). After purification by silica gel chromatography (50% EtOAc in hexanes, then 75% EtOAc in hexanes) and concentration of the appropriate fractions, the product was diluted with CH2Cl2 and extracted with sat.aq NaHCO3 to remove a co-polar succinimide by-product. Concentration of the organic layer then provided product 1v as a yellow solid; yield: 334 mg (16%); mp 84–85 °C; Rf = 0.24 (silica gel, 60% EtOAc/hexanes, UV).
IR (neat): 2965, 1713, 1574, 1416, 1302, 1138, 756, 660 cm−1.
1H NMR (500 MHz, C6D6): δ = 8.05−8.02 (m, 1 H), 6.72−6.67 (m, 1 H), 6.49 (d, J = 8.1 Hz, 1 H), 6.27 (ddd, J = 7.4, 4.9, 1.1 Hz, 1 H), 1.97 (s, 4 H).
13C NMR (126 MHz, C6D6): δ = 174.9, 155.7, 149.4, 136.1, 120.4, 117.9, 28.3.
HRMS (ESI): m/z [M + H]+ calcd for C9H9N2O2S+: 209.0379; found: 209.0386.
Sulfenamides 3a–l; General Procedure A
To a flame-dried 100 mL round- bottomed flask under N2 and equipped with a stir bar was added the indicated amide (1.1 mmol, 1.1 equiv). Then THF (0.1 M, 10 mL) was added, and the solution was cooled to 0 °C. A 60% dispersion of NaH (100 mg, 2.50 mmol, 2.5 equiv) was added, and the resulting mixture was warmed to rt and stirred for 30 min. The mixture was cooled to 0 °C and 2-(phenylthio)isoindoline-1,3-dione (1a; 255 mg, 1.0 mmol, 1.0 equiv) was added. The mixture was warmed to rt and stirred until reaction completion as assessed by TLC. The mixture was then slowly diluted with sat. aq NH4Cl with stirring until white precipitates dissolved, and the resulting solution was extracted with EtOAc (3 ×). The combined organic layers were washed with brine, dried (Na2SO4), and concentrated. The crude residue was purified by the indicated chromatographic method to afford the desired product.
N-(Phenylthio)pivalamide (3a)
General procedure A was followed, employing pivalamide (111 mg, 1.10 mmol, 1.1 equiv) as the nucleophile. The reaction was run for 20 min. Purification by silica gel chromatography (10% EtOAc in hexanes, then 20% EtOAc in hexanes) afforded the product 3a as a white solid; yield: 185 mg (88%); mp 101–105 °C; Rf = 0.38 (silica gel, 20% EtOAc/hexanes, UV).
IR (neat): 3264, 2970, 1674, 1585, 1474, 1408, 1153, 1022, 729, 687 cm−1.
1H NMR (600 MHz, CDCl3): δ = 7.48 (s, 1 H), 7.20 (t, J = 8.0 Hz, 2 H), 7.13−7.07 (m, 3 H), 1.19 (s, 9 H).
13C NMR (151 MHz, CDCl3): δ = 180.5, 139.3, 128.8, 126.2, 124.4, 39.9, 27.6.
HRMS (ESI): m/z [M + H]+ calcd for C11H16NOS+: 210.0947; found: 210.0956.
N-(Phenylthio)acetamide (3b)
General procedure A was followed, employing acetamide (65.0 mg, 1.10 mmol, 1.1 equiv) as the nucleophile. The reaction was run for 45 min. Purification by silica gel chromatography (20% EtOAc in hexanes) afforded the product 3b as a white solid; yield: 167 mg (88%); mp 106–107 °C; Rf = 0.10 (silica gel, 20% EtOAc/hexanes, UV).
IR (neat): 3356, 2967, 2430, 2372, 1670, 1477, 733, 689 cm−1.
1H NMR (600 MHz, CD3OD): δ = 7.30 (t, J = 7.7 Hz, 2 H), 7.22 (d, J = 7.1 Hz, 2 H), 7.17 (t, J = 7.4 Hz, 1 H), 2.13 (s, 3 H).
13C NMR (151 MHz, CD3OD): δ = 174.1, 139.0, 128.7, 126.1, 124.2, 21.5.
HRMS (ESI): m/z [M + H]+ calcd for C8H10NOS+: 168.0478; found: 168.0482.
tert-Butyl 4-((Phenylthio)carbamoyl)piperidine-1-carboxylate (3c)
General procedure A was followed, employing tert-butyl 4-carbamoylpiperidine-1-carboxylate (251 mg, 1.10 mmol, 1.1 equiv) as the nucleophile. The reaction was run for 20 min. Purification by silica gel chromatography (20% EtOAc in hexanes, then 50% EtOAc in hexanes) afforded the product 3c as a white solid; yield: 304 mg (90%); mp 49–54 °C; Rf = 0.48 (silica gel, 50% EtOAc/hexanes, UV).
IR (neat): 2930, 2859, 2407, 1657, 1225, 1157, 1125, 737, 689 cm−1.
1H NMR (500 MHz, CD3OD): δ = 7.29 (t, J = 7.7 Hz, 2 H), 7.22−7.13 (m, 3 H), 4.11−4.03 (m, 2 H), 2.78 (br s, 2 H), 2.60 (tt, J = 11.6, 3.8 Hz, 1 H), 1.83−1.76 (m, 2 H), 1.59 (qd, J = 8.2, 4.1 Hz, 2 H), 1.45 (s, 9 H).
13C NMR (126 MHz, CD3OD): δ = 178.2, 154.9, 139.1, 128.8, 126.2, 124.2, 79.8, 43.3, 42.8, 42.7, 28.4, 27.5.
HRMS (ESI): m/z [M + Na]+ calcd for C17H24N2O3SNa+: 359.1400; found: 359.1387.
N-(Phenylthio)benzamide (3d)
General procedure A was followed, employing benzamide (133 mg, 1.10 mmol, 1.1 equiv) as the nucleophile. The reaction was run for 30 min. Purification by silica gel chromatography (10% EtOAc in hexanes, then 20% EtOAc in hexanes) afforded the product 3d as a white solid; yield: 211 mg (92%); mp 122–125 °C; Rf = 0.25 (20% EtOAc/hexanes,UV).
IR (neat): 3260, 2970, 1670, 1582, 1420, 1261, 1157, 1022, 733, 687 cm−1.
1H NMR (600 MHz, CDCl3): δ = 8.05 (s, 1 H), 7.84 (d, J = 7.0 Hz, 2 H), 7.48 (t, J = 7.5 Hz, 1 H), 7.34 (t, J = 7.8 Hz, 2 H), 7.27−7.21 (m, 4 H), 7.14 (t, J = 7.0 Hz, 1 H).
13C NMR (151 MHz, CDCl3): δ = 169.9, 138.8, 133.0, 132.3, 129.0, 128.6, 127.9, 126.6, 125.1.
HRMS (ESI): m/z [M + H]+ calcd for C13H12NOS+: 230.0634; found: 230.0639.
4-Methoxy-N-(phenylthio)benzamide (3e)
General procedure A was followed, employing 4-methoxybenzamide (166 mg, 1.10 mmol, 1.1 equiv) as the nucleophile. The reaction was run for 30 min. Purification by reverse phase chromatography (10–100% MeCN in H2O with 0.1% Et3N) afforded the product 3e as a white solid; yield: 224 mg (86%); mp 140–143 °C.
IR (neat): 3186, 2392, 1601, 1432, 1250, 1177, 733, 675 cm−1.
1H NMR (600 MHz, acetone-d6): δ = 9.19 (s, 1 H), 8.02 (d, J = 8.9 Hz, 2 H), 7.32−7.25 (m, 4 H), 7.16 (t, J = 7.1 Hz, 1 H), 7.01 (d, J = 8.9 Hz, 2 H), 3.86 (s, 3 H).
13C NMR (151 MHz, acetone-d6): δ = 167.7, 162.9, 140.3, 129.9, 128.9, 125.9, 125.7, 123.9, 113.8, 55.0.
HRMS (ESI): m/z [M + H]+ calcd for C14H14NO2S+: 260.0740; found: 260.0746.
N-(Phenylthio)-4-(trifluoromethyl)benzamide (3f)
General procedure A was followed, employing 4-(trifluoromethyl)benzamide (208 mg, 1.10 mmol, 1.1 equiv) as the nucleophile. The reaction was run for 40 min. Purification by reverse phase chromatography (10–100% MeCN in H2O with 0.1% Et3N) afforded the product 3f as a white solid; yield: 272 mg (92%); mp 152 °C (dec.).
IR (neat): 3213, 1655, 1431, 1254, 1165, 1107, 853, 737, 677, 474 cm−1.
1H NMR (600 MHz, acetone-d6): δ = 9.56 (s, 1 H), 8.21 (d, J = 8.0 Hz, 2 H), 7.85 (d, J = 8.2 Hz, 2 H), 7.35−7.31 (m, 4 H), 7.21−7.17 (m, 1 H).
13C NMR (151 MHz, acetone-d6): δ = 167.5, 139.4, 137.3, 132.9 (q, 2JC,F = 32.4 Hz), 129.0, 128.7, 126.3, 125.6 (q, 3JC,F = 3.7 Hz), 124.4, 124.0 (q, 1JC,F = 272.2 Hz).
19F NMR (376 MHz, acetone-d6): δ = −63.5.
HRMS (ESI): m/z [M + H]+ calcd for C14H11F3NOS+: 298.0508; found: 298.0515.
2-Chloro-N-(phenylthio)benzamide (3g)
General procedure A was followed, employing 2-chlorobenzamide (171 mg, 1.10 mmol, 1.1 equiv) as the nucleophile. The reaction was run for 40 min. Purification by silica gel chromatography (10% EtOAc in hexanes, then 20% EtOAc in hexanes) afforded the product 3g as a white solid; yield: 246 mg (93%); mp 120–122 °C; Rf = 0.22 (silica gel, 20% EtOAc/hexanes, UV).
IR (neat): 3271, 1682, 1400, 1231, 1107, 1045, 741, 460 cm−1.
1H NMR (500 MHz, CDCl3): δ = 7.61 (d, J = 6.8 Hz, 1 H), 7.56 (s, 1 H), 7.42−7.33 (m, 4 H), 7.34−7.24 (m, 3 H), 7.21 (t, J = 7.3 Hz, 1 H).
13C NMR (126 MHz, CDCl3): δ = 168.5, 138.0, 134.0, 132.1, 130.9, 130.4, 130.3, 129.1, 127.2, 127.2, 126.0.
HRMS (ESI): m/z [M + H]+ calcd for C13H11ClNOS+: 264.0244; found: 264.0251.
tert-Butyl (Phenylthio)carbamate (3h)
General procedure A was followed, employing tert-butyl carbamate (129 mg, 1.10 mmol, 1.1 equiv) as the nucleophile. The reaction was run for 20 min. Purification by silica gel chromatography (10% EtOAc in hexanes) afforded the product 3h as a white solid; yield: 201 mg (89%); mp 65–67 °C; Rf = 0.53 (20% EtOAc/hexanes, UV).
IR (neat): 3271, 2970, 1670, 1477, 1416, 1153, 733, 656 cm−1.
1H NMR (600 MHz, CDCl3): δ = 7.30 (t, J = 8.2 Hz, 2 H), 7.24 (d, J = 7.0 Hz, 2 H), 7.17 (t, J = 7.3 Hz, 1 H), 5.87 (s, 1 H), 1.46 (s, 9 H).
13C NMR (151 MHz, CDCl3): δ = 156.0, 139.7, 128.9, 126.5, 124.5, 82.0, 28.1.
HRMS (EI): m/z [M]+ calcd for C11H15NO2S: 225.0824; found: 225.0819.
Benzyl (Phenylthio)carbamate (3i)
General procedure A was followed, employing benzyl carbamate (166 mg, 1.10 mmol, 1.1 equiv) as the nucleophile. The reaction was run for 20 min. Purification by silica gel chromatography (10% EtOAc in hexanes, then 20% EtOAc in hexanes) afforded the product 3i as a white solid; yield: 235 mg (91%); mp 83–85 °C; Rf = 0.40 (silica gel, 20% EtOAc/hexanes, UV).
IR (neat): 3256, 2970, 1670, 1427, 1153, 737, 687, 660 cm−1.
1H NMR (600 MHz, CDCl3): δ = 7.40−7.24 (m, 9 H), 7.21 (t, J = 6.9 Hz, 1 H), 6.33 (s, 1 H), 5.19 (s, 2 H).
13C NMR (151 MHz, CDCl3): δ = 157.3, 139.0, 135.6, 129.1, 128.6, 128.4, 128.3, 126.9, 125.2, 68.5.
HRMS (ESI): m/z [M + H]+ calcd for C14H14NO2S+: 260.0740; found: 260.0746.
4-Methyl-N-(phenylthio)benzenesulfonamide (3j)
General procedure A was followed, employing 4-methylbenzenesulfonamide (188 mg, 1.10 mmol, 1.1 equiv) as the nucleophile. The reaction was run for 80 min. Purification by silica gel chromatography (10% EtOAc in hexanes, then 20% EtOAc in hexanes) afforded the product 3j as a white solid; yield: 223 mg (80%); mp 101–103 °C; Rf = 0.24 (silica gel, 20% EtOAc/hexanes, UV).
IR (neat): 3248, 2967, 1670, 1474, 1366, 1153, 1088, 864, 733, 660, 544 cm−1.
1H NMR (500 MHz, CDCl3): δ = 7.76 (d, J = 8.1 Hz, 2 H), 7.32 (d, J = 7.2 Hz, 2 H), 7.27−7.15 (m, 5 H), 6.59 (s, 1 H), 2.38 (s, 3 H).
13C NMR (126 MHz, CDCl3): δ = 144.2, 137.3, 136.0, 129.6, 128.9, 127.7, 127.4, 126.3, 21.6.
HRMS (CI): m/z [M]+ calcd for C13H13NO2S2: 279.0388; found: 279.0392.
(S)-2-Methyl-N-(phenylthio)propane-2-sulfinamide (3k)
General procedure A was followed, employing (S)-2-methylpropane-2-sulfinamide (133 mg, 1.10 mmol, 1.1 equiv) as the nucleophile. The reaction was run for 20 min. Purification by silica gel chromatography (10% EtOAc in hexanes, then 33% EtOAc in hexanes) afforded the product 3k as an off-white solid; yield: 167 mg (73%); mp 108–112 °C; [α]D20 +54.38 (c 0.1, CHCl3); Rf = 0.14 (silica gel, 20% EtOAc/hexanes, UV).
HPLC: Chiralpak AD-H, 90:10 hexanes:i-PrOH, 1 mL/min, 254 nm, tR (minor) = 6.72 min, tR (major) = 7.64 min; 98:2 er.
IR (neat): 3078, 1578, 1439, 1049, 1022, 737, 691 cm−1.
1H NMR (400 MHz, CDCl3): δ = 7.38 (d, J = 7.0 Hz, 2 H), 7.29 (t, J = 7.8 Hz, 2 H), 7.17 (t, J = 7.3 Hz, 1 H), 5.35 (s, 1 H), 1.21 (s, 9 H).
13C NMR (151 MHz, CDCl3): δ = 139.8, 128.9, 126.9, 125.4, 58.0, 22.4.
HRMS (ESI): m/z [M + H]+ calcd for C10H16NOS2+: 230.0668; found: 230.0674.
1,1-Dimethyl-3-(phenylthio)urea (3l)
General procedure A was followed with slight modification, employing 1,1-dimethylurea (96.9 mg, 1.10 mmol, 1.1 equiv) as the nucleophile. The initial addition of NaH was performed in a pre-heated oil bath set for 50 °C. After 30 min, the mixture was cooled to rt, and the 2-(phenylthio)isoindoline-1,3-dione was then added at 0 °C after cooling the mixture in an ice bath. The reaction was then run for 30 min at 50 °C in a preheated oil bath. Purification by silica gel chromatography (60% EtOAc in hexanes, then 80% EtOAc in hexanes) afforded the product 3l as a white solid; yield: 109 mg (56%); mp 112–114 °C; Rf = 0.21 (silica gel, 60% EtOAc/hexanes, UV).
IR (neat): 3075, 2654, 1582, 1435, 1173, 1022, 737, 691 cm−1.
1H NMR (600 MHz, CDCl3): δ = 7.22 (t, J = 8.2 Hz, 2 H), 7.17 (d, J = 7.0 Hz, 2 H), 7.08 (t, J = 7.3 Hz, 1 H), 6.47 (s, 1 H), 2.88 (s, 6 H).
13C NMR (151 MHz, CDCl3): δ = 158.0, 140.8, 128.7, 125.9, 123.9, 37.0.
HRMS (ESI): m/z [M + H]+ calcd for C9H13N2OS+: 197.0743; found: 197.0748.
Sulfenamides 3m–w; General Procedure B
To a flame-dried 100 mL round-bottomed flask under N2 and equipped with a stir bar was added pivalamide (2a; 111 mg, 1.1 mmol, 1.1 equiv). Then THF (0.1 M, 10 mL) was added, and the solution was cooled to 0 °C. A 60% dispersion of NaH (100 mg, 2.5 mmol, 2.5 equiv) was added, then the resulting mixture was warmed to rt and stirred for 30 min. The mixture was cooled to 0 °C and the indicated N-thiosuccinimide (1.0 mmol, 1.0 equiv) was added. The mixture was warmed to rt and stirred until reaction completion as assessed by TLC. The crude mixture was then slowly diluted with sat. aq NH4Cl with stirring until white precipitates dissolved, and the resulting solution was extracted with EtOAc (3 ×). The combined organic layers were washed with brine, dried (Na2SO4), and concentrated. The crude residue was purified by the indicated chromatographic method to afford the desired product.
N-(Butylthio)pivalamide (3m)
General procedure B was followed, employing 1-(butylthio)pyrrolidine-2,5-dione (187 mg, 1.00 mmol, 1.0 equiv) as the electrophile. The reaction was run for 20 min. Purification by silica gel chromatography (15% EtOAc in hexanes) afforded the product 3m as a white solid; yield: 166 mg (88%); mp 50–52 °C; Rf = 0.34 (silica gel, 20% EtOAc/hexanes, UV).
IR (neat): 3258, 2961, 1663, 1476, 1412, 1157, 1026, 824, 681 cm−1.
1H NMR (500 MHz, CDCl3): δ = 6.67 (s, 1 H), 2.63 (t, J = 7.4 Hz, 2 H), 1.44 (p, J = 7.2 Hz, 2 H), 1.31 (hept, J = 7.3 Hz, 2 H), 1.13 (s, 9 H), 0.80 (t, J = 7.4 Hz, 3 H).
13C NMR (126 MHz, CDCl3): δ = 180.4, 39.7, 38.0, 29.6, 27.6, 21.6, 13.6.
HRMS (ESI): m/z [M + H]+ calcd for C9H20NOS+: 190.1260; found: 190.1268.
Methyl 3-(Pivalamidothio)propanoate (3n)
General procedure B was followed with slight modification, employing methyl 3-((2,5-dioxopyrrolidin-1-yl)thio)propanoate (217 mg, 1.00 mmol, 1.0 equiv) as the electrophile. The reaction mixture was left on ice after the addition of methyl 3-((2,5-dioxopyrrolidin-1yl)thio)propanoate. The reaction was run for 15 min at 0 °C. Purification by silica gel chromatography (20% EtOAc in hexanes, then 40% EtOAc in hexanes) afforded the product 3n as a white solid; yield: 138 mg (63%); mp 36–40 °C; Rf = 0.31 (silica gel, 40% EtOAc/hexanes, UV).
IR (neat): 3283, 2967, 1730, 1665, 1402, 1159, 667 cm−1.
1H NMR (500 MHz, CDCl3): δ = 6.63 (s, 1 H), 3.66 (s, 3 H), 2.93 (t, J = 7.0 Hz, 2 H), 2.63 (t, J = 7.0 Hz, 2 H), 1.19 (s, 9 H).
13C NMR (126 MHz, CDCl3): δ = 180.4, 172.3, 51.9, 39.8, 33.9, 33.4, 27.6.
HRMS (ESI): m/z [M + H]+ calcd for C9H18NO3S+: 220.1002; found: 220.0989.
N-(Cyclopentylthio)pivalamide (3o)
General procedure B was followed, employing 1-(cyclopentylthio)pyrrolidine-2,5-dione (199 mg, 1.00 mmol, 1.0 equiv) as the electrophile. The reaction was run for 30 min. Purification by silica gel chromatography (20% EtOAc in hexanes) afforded the product 3o as a white solid; yield: 172 mg (86%); mp 108–109 °C; Rf = 0.34 (silica gel, 20% EtOAc/hexanes, UV).
IR (neat): 3258, 2961, 1663, 1477, 1412, 1159, 1026, 675 cm−1.
1H NMR (500 MHz, CDCl3): δ = 6.82 (s, 1 H), 3.49−3.40 (m, 1 H), 1.78−1.68 (m, 2 H), 1.67−1.56 (m, 2 H), 1.51−1.42 (m, 2 H), 1.42−1.33 (m, 2 H), 1.13 (s, 9 H).
13C NMR (126 MHz, CDCl3): δ = 180.4, 48.8, 39.7, 30.5, 27.6, 24.7.
HRMS (ESI): m/z [M + H]+ calcd for C10H20NOS+: 202.1260; found: 202.1266.
tert-Butyl 3-(Pivalamidothio)azetidine-1-carboxylate (3p)
General procedure B was followed, employing tert-butyl 3-((2,5-dioxopyrrolidin-1-yl)thio)azetidine-1-carboxylate (286 mg, 1.00 mmol, 1.0 equiv) as the electrophile. The reaction was run for 45 min. Purification by silica gel chromatography (40% EtOAc in hexanes) afforded the product 3p as a white solid; yield: 200 mg (69%); mp 162–163 °C; Rf = 0.27 (silica gel, 40% EtOAc/hexanes, UV).
IR (neat): 3235, 2982, 2234, 1655, 1423, 1152, 910, 725, 644 cm−1.
1H NMR (500 MHz, CDCl3): δ = 6.95 (s, 1 H), 4.17−4.11 (m, 2 H), 3.87 (dd, J = 9.4, 5.6 Hz, 2 H), 3.68−3.62 (m, 1 H), 1.38 (s, 9 H), 1.22 (s, 9 H).
13C NMR (126 MHz, CDCl3): δ = 181.1, 156.0, 79.8, 54.5, 40.0, 38.3, 28.3, 27.7.
HRMS (ESI): m/z [M + Na]+ calcd for C13H24N2O3SNa+: 311.1400; found: 311.1410.
N-(tert-Butylthio)pivalamide (3q)
General procedure B was followed, employing 1-(tert-butylthio)pyrrolidine-2,5-dione (187 mg, 1.00 mmol, 1.0 equiv) as the electrophile. The reaction was run for 20 min. Purification by silica gel chromatography (15% EtOAc in hexanes) afforded the product 3q as a white solid; yield: 111 mg (59%); mp 138–140 °C; Rf = 0.31 (silica gel, 20% EtOAc/hexanes, UV).
IR (neat): 3256, 2963, 1661, 1476, 1402, 1157, 1026, 826, 638 cm−1.
1H NMR (600 MHz, CDCl3): δ = 6.51 (s, 1 H), 1.19 (s, 9 H), 1.19 (s, 9 H).
13C NMR (151 MHz, CDCl3): δ = 180.4, 48.5, 39.9, 28.5, 27.8.
HRMS (ESI): m/z [M + H]+ calcd for C9H20NOS+: 190.1260; found: 190.1267.
N-(Benzylthio)pivalamide (3r)
General procedure B was followed, employing 1-(benzylthio)pyrrolidine-2,5-dione (221 mg, 1.00 mmol, 1.0 equiv) as the electrophile. The reaction was run for 30 min. Purification by silica gel chromatography (15% EtOAc in hexanes) afforded the product 3r as a white solid; yield: 183 mg (82%); mp 85–87 °C; Rf = 0.30 (silica gel, 20% EtOAc/hexanes, UV).
IR (neat): 3258, 2961, 1663, 1476, 1414, 1157, 1026, 826, 694 cm−1.
1H NMR (500 MHz, CDCl3): δ = 7.27 (t, J = 7.1 Hz, 2 H), 7.23−7.18 (m, 3 H), 6.29 (s, 1 H), 3.89 (s, 2 H), 1.10 (s, 9 H).
13C NMR (126 MHz, CDCl3): δ = 180.1, 136.2, 129.4, 128.5, 127.3, 42.0, 39.7, 27.6.
HRMS (ESI): m/z [M + H]+ calcd for C12H18NOS+: 224.1104; found: 224.1111.
N-((4-Methoxyphenyl)thio)pivalamide (3s)
General procedure B was followed, employing 1-((4-methoxyphenyl)thio)pyrrolidine-2,5-dione (237 mg, 1.00 mmol, 1.0 equiv) as the electrophile. The reaction was run for 30 min. Purification by silica gel chromatography (20% EtOAc in hexanes, then 30% EtOAc in hexanes) afforded the product 3s as a white solid; yield: 214 mg (89%); mp 115 °C (dec.); Rf = 0.20 (silica gel, 20% EtOAc/hexanes, UV).
IR (neat): 3262, 2972, 1665, 1155, 1026, 824, 679, 525 cm−1.
1H NMR (600 MHz, CDCl3): δ = 7.32 (d, J = 8.8 Hz, 2 H), 7.16 (s, 1 H), 6.77 (d, J = 8.8 Hz, 2 H), 3.72 (s, 3 H), 1.17 (s, 9 H).
13C NMR (151 MHz, CDCl3): δ = 180.0, 159.6, 130.9, 129.6, 114.5, 55.3, 39.7, 27.6.
HRMS (ESI): m/z [M + H]+ calcd for C12H18NO2S+: 240.1053; found: 240.1061.
N-((3,5-Bis(trifluoromethyl)phenyl)thio)pivalamide (3t)
General procedure B was followed, employing 1-((3,5-bis(trifluoromethyl)phenyl)thio)pyrrolidine-2,5-dione (343 mg, 1.00 mmol, 1.0 equiv) as the electrophile. The reaction was run for 45 min. Purification by silica gel chromatography (10% EtOAc in hexanes, then 20% EtOAc in hexanes) afforded the product 3t as a white solid; yield: 214 mg (62%); mp 108–109 °C; Rf = 0.40 (silica gel, 20% EtOAc/hexanes,UV).
IR (neat): 3256, 2961, 1663, 1476, 1157, 1026, 824, 696 cm−1.
1H NMR (500 MHz, CDCl3): δ = 7.76 (s, 1 H), 7.54 (s, 1 H), 7.43 (s, 2 H), 1.24 (s, 9 H).
13C NMR (126 MHz, CDCl3): δ = 180.5, 143.1, 132.1 (q, 2JC,F = 33.6 Hz), 122.9 (q, 1JC,F = 273.0 Hz), 122.7 (q, 3JC,F = 3.9 Hz), 119.4 (hept, 3JC,F = 3.7 Hz), 40.0, 27.3.
19F NMR (376 MHz, CDCl3): δ = −63.5.
HRMS (ESI): m/z [M + H]+ calcd for C13H14F6NOS+: 346.0695; found: 346.0707.
N-((2,6-Dichlorophenyl)thio)pivalamide (3u)
General procedure B was followed, employing 1-((2,6-dichlorophenyl)thio)pyrrolidine-2,5-dione (276 mg, 1.00 mmol, 1.0 equiv) as the electrophile. The reaction was run for 30 min. Purification by silica gel chromatography (10% EtOAc in hexanes) afforded the product 3u as a white solid; yield: 245 mg (88%); mp 138–139 °C; Rf = 0.40 (silica gel, 20% EtOAc/hexanes, UV).
IR (neat): 3289, 2972, 1665, 1476, 1420, 1152, 1026, 781, 625 cm−1.
1H NMR (600 MHz, CDCl3): δ = 7.28 (d, J = 8.1 Hz, 2 H), 7.13 (t, J = 8.2 Hz, 1 H), 7.02 (s, 1 H), 1.14 (s, 9 H).
13C NMR (151 MHz, CDCl3): δ = 178.8, 138.6, 134.2, 130.5, 128.9, 40.0, 27.5.
HRMS (ESI): m/z [M + H]+ calcd for C11H14Cl2NOS+: 278.0168; found: 278.0176.
N-(Pyridin-2-ylthio)pivalamide (3v)
General procedure B was followed, employing 1-(pyridin-2-ylthio)pyrrolidine-2,5-dione (208 mg, 1.00 mmol, 1.0 equiv) as the electrophile. The reaction was run for 45 min. Purification by silica gel chromatography (30% EtOAc in hexanes, then 50% EtOAc in hexanes) afforded the product 3v as a viscous oil; yield: 178 mg (85%); Rf = 0.35 (silica gel, 50% EtOAc/hexanes, UV).
IR (neat): 3256, 2968, 1670, 1574, 1418, 1130, 754, 725 cm−1.
1H NMR (500 MHz, CDCl3): δ = 8.31 (d, J = 4.1 Hz, 1 H), 7.51−7.44 (m, 2 H), 6.96−6.91 (m, 2 H), 1.24 (s, 9 H).
13C NMR (126 MHz, CDCl3): δ = 180.2, 161.1, 149.2, 136.8, 120.1, 117.4, 40.0, 27.6.
HRMS (ESI): m/z [M + H]+ calcd for C10H15N2OS+: 211.0900; found: 211.0907.
N-(Thiophen-2-ylthio)pivalamide (3w)
General procedure B was followed, employing 1-(thiophen-2-ylthio)pyrrolidine-2,5-dione (213 mg, 1.00 mmol, 1.0 equiv) as the electrophile. The reaction was run for 30 min. Purification by silica gel chromatography (15% EtOAc in hexanes) afforded the product 3w as a white solid; yield: 178 mg (83%); mp 103–104 °C; Rf = 0.37 (silica gel, 20% EtOAc/hexanes, UV).
IR (neat): 3256, 2963, 1661, 1476, 1402, 1157, 1026, 826, 708, 637, 515 cm−1.
1H NMR (500 MHz, CDCl3): δ = 7.39 (dd, J = 5.4, 1.3 Hz, 1 H), 7.30 (dd, J = 3.6, 1.3 Hz, 1 H), 7.15 (s, 1 H), 6.92 (dd, J = 5.3, 3.6 Hz, 1 H), 1.16 (s, 9 H).
13C NMR (126 MHz, CDCl3): δ = 179.6, 136.8, 134.9, 131.8, 127.2, 39.7, 27.5.
HRMS (ESI): m/z [M + H]+ calcd for C9H14NOS2+: 216.0511; found. 216.0519.
Supplementary Material
Funding Information
This work was supported by NIH Grant R35GM122473 (to J.A.E.). J.T.L. was funded by the Yale College First-Year Summer Research Fellowship in the Sciences and Engineering. D.S.B. acknowledges the Berson Graduate Research Fellowship in Chemistry for financial support. N.S.G. gratefully acknowledges the support of the National Science Foundation Graduate Research Fellowship Program.
Footnotes
This work is dedicated to the memory of David A. Evans for his commitment and lasting contributions to research and teaching.
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information
Supporting information for this article is available online at https://doi.org/10.1055/s-0041-1738430.
References
- (1).(a) Craine L; Raban M Chem. Rev 1989, 89, 689. [Google Scholar]; (b) Koval IV Russ. Chem. Rev 1996, 65, 421. [Google Scholar]; (c) Cao Y; Abdolmohammadi S; Ahmadi R; Issakhov A; Ebadi AG; Vessally E RSC Adv. 2021, 11, 32394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Greenwood NS; Champlin AT; Ellman JA J. Am. Chem. Soc 2022, 144, 17808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ma L; Bai L; Yu Z; Shen Q Chirality 2022, 34, 1191. [DOI] [PubMed] [Google Scholar]
- (4).(a) Behforouz M; Kerwood JE J. Org. Chem 1969, 34, 51. [Google Scholar]; (b) Harpp DN; Mullins DF; Steliou K; Triassi IJ Org. Chem 1979, 44, 4196. [Google Scholar]; (c) Koval IV; Tarasenko AI; Kremlev MM; Molchanova NR Zh. Org. Khim 1981, 17, 533. [Google Scholar]; (d) Miura Y; Shibata Y; Kinoshita M Bull. Chem. Soc. Jpn 1986, 59, 3291. [Google Scholar]; (e) Perrio S; Reboul V; Metzner P In Science of Synthesis., Vol. 31a; Ramsden CA, Ed.; Georg Thieme Verlag: Stuttgart, 2007, 1041. [Google Scholar]; (f) Ma L-J; Chen S-S; Li G-X; Zhu J; Wang Q-W; Tang Z ACS Catal. 2019, 9, 1525. [Google Scholar]
- (5).(a) Chen Q; Shen M; Tang Y; Li C Org. Lett 2005, 7, 1625. [DOI] [PubMed] [Google Scholar]; (b) Zhang X-S; Zhang X-H Phosphorus, Sulfur Silicon Relat. Elem 2016, 191, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Musiejuk M; Witt D Phosphorus, Sulfur Silicon Relat. Elem 2016, 191, 305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Bao M; Shimizu M; Shimada S; Tanaka M Tetrahedron 2003, 59, 303. [Google Scholar]; (b) Wang H; Xian M Angew. Chem. Int. Ed 2008, 47, 6598. [DOI] [PubMed] [Google Scholar]; (c) Nasab FAH; Fekri LZ; Monfared A; Hosseini A; Vessally E RSC Adv. 2018, 8, 18456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bai Z; Zhu S; Hu Y; Yang P; Chu X; He G; Wang H; Chen G Nat. Commun 2022, 13, 6445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dodds AC; Sutherland AJ Org. Chem 2021, 86, 5922. [DOI] [PubMed] [Google Scholar]
- (10).(a) Harpp DN; Back TG Tetrahedron Lett. 1971, 12, 4953. [Google Scholar]; (b) Harpp DN; Back TG J. Org. Chem 1976, 41, 2498. [Google Scholar]; (c) Liu Y; Xu Y; Zhang Y; Gao W-C; Shao X Org. Chem. Front 2022, 9, 6490. [Google Scholar]
- (11).Guarino VR; Olson RE; Everlof JG; Wang N; McDonald I; Haskell R; Clarke W; Lentz KA Bioorg. Med. Chem. Lett 2020, 30, 126856. [DOI] [PubMed] [Google Scholar]
- (12).Backes BJ; Dragoli DR; Ellman JA J. Org. Chem 1999, 64, 5472. [DOI] [PubMed] [Google Scholar]
- (13).Eitzinger A; Otevrel J; Haider V; Macchia A; Massa A; Faust K; Spingler B; Berkessel A; Waser M Adv. Synth. Catal 2021, 363, 1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kesavan A; Anbarasan P Chem. Commun 2022, 58, 282. [DOI] [PubMed] [Google Scholar]
- (15).Savarin C; Srogl J; Liebeskind LS Org. Lett 2002, 4, 4309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.