

Abstract
There is strong interest in developing predictive models to better understand individual heterogeneity and disease progression in Alzheimer's disease (AD). We have built upon previous longitudinal AD progression models, using a nonlinear, mixed‐effect modeling approach to predict Clinical Dementia Rating Scale – Sum of Boxes (CDR‐SB) progression. Data from the Alzheimer's Disease Neuroimaging Initiative (observational study) and placebo arms from four interventional trials (N = 1093) were used for model building. The placebo arms from two additional interventional trials (N = 805) were used for external model validation. In this modeling framework, CDR‐SB progression over the disease trajectory timescale was obtained for each participant by estimating disease onset time (DOT). Disease progression following DOT was described by both global progression rate (RATE) and individual progression rate (α). Baseline Mini‐Mental State Examination and CDR‐SB scores described the interindividual variabilities in DOT and α well. This model successfully predicted outcomes in the external validation datasets, supporting its suitability for prospective prediction and use in design of future trials. By predicting individual participants' disease progression trajectories using baseline characteristics and comparing these against the observed responses to new agents, the model can help assess treatment effects and support decision making for future trials.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Disease progression models in Alzheimer's Disease (AD) have previously been developed for the Alzheimer's Disease Assessment Scale‐Cognitive Subscale and Clinical Dementia Rating Scale – Sum of Boxes (CDR‐SB) score.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study assessed the longitudinal progression of CDR‐SB score for prodromal‐to‐moderate AD using a disease progression model developed with placebo arm data from multiple interventional clinical trials and Alzheimer's Disease Neuroimaging Initiative (ADNI).
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
The model adequately predicted longitudinal progression of CDR‐SB score, informed by the baseline Mini‐Mental State Examination score and baseline CDR‐SB score. We finalized a robust model using data from interventional studies and ADNI.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
The disease progression model could be used to predict longitudinal progression, and as a virtual control arm in an open‐label extension study. Prospective prediction of progression could also support the design of future clinical trials and be used to assess treatment effects for ongoing trials via cohort‐ or individual‐level analysis.
INTRODUCTION
Alzheimer's disease (AD) is a neurodegenerative disorder and the most common cause of dementia worldwide, with an estimated prevalence of greater than 150 million by 2050. 1 , 2 AD causes progressive cognitive and functional impairment, which lead to significant disability and healthcare burdens. 2 However, there is considerable interindividual variability (IIV) in the rate of disease progression. 2 Disease‐modifying therapies for AD are being investigated, but the failure rate of AD trials is high. 3 The anti–amyloid‐beta (Aβ) antibody aducanumab recently received accelerated approval for the treatment of AD from the US Food and Drug Administration (FDA) based on reductions in Aβ positron emission tomography signals, which have been hypothesized to be related to reductions in the rates of clinical decline in cognition and function. 4 , 5 Other available, approved treatments for AD target disease symptoms and do not alter disease progression; however, several other anti‐amyloid monoclonal antibodies are also under investigation. 2
Given the complex and heterogeneous nature of AD, there is strong interest in predictive disease models to better understand the disease trajectory. 6 Research and clinical diagnostic criteria have been developed by several organizations which state that people living with AD progress along a continuum, from preclinical, without any overt symptoms, to prodromal AD (also described as mild cognitive impairment [MCI] due to AD). This can then develop into mild, moderate, and severe forms of AD dementia, with the spectrum of these changes stretching over a period of 15–25 years. 7 Disease models are increasingly important in the context of model‐informed drug development, which has been encouraged by global regulatory agencies (e.g., the FDA Prescription Drug User Fee Act VI, 8 the European Medicines Agency 2025 strategy, 9 and the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use 10 ), as they have the potential to impact drug development and clinical trial design by enabling robust assessment of treatment effects.
Several groups have developed disease progression models in AD using a nonlinear, mixed‐effect modeling approach and the Alzheimer's Disease Neuroimaging Initiative (ADNI) database. 11 , 12 , 13 , 14 , 15 , 16 , 17 Previous disease progression modeling work in AD is summarized in Table S1. The majority of these models have focused on describing the longitudinal trajectory of cognitive scores as measured by the Alzheimer's Disease Assessment Scale – Cognitive Subscale (ADAS‐Cog), 11 , 13 , 15 with a few studies focusing on the Clinical Dementia Rating Scale – Sum of Boxes (CDR‐SB). 14 , 16 Although the ADAS‐Cog assesses cognitive impairment, the CDR‐SB evaluates both cognitive and functional impairment and has been proposed as a primary outcome for use in prodromal‐to‐mild AD trials. 16 In these models, subpopulations that differ in progression rates (e.g., progressors/nonprogressors 14 or slow/fast progressors) 16 can be defined as parameters that are estimated in the mixture model. Additionally, covariate analysis can help identify main drivers of disease progression, such as baseline score values (e.g., CDR‐SB, Mini‐Mental State Examination [MMSE], and ADAS‐Cog), apolipoprotein E ε4 allele (APOEε4) genotype, sex, fluid biomarkers, measures of brain volume (hippocampal and intracranial volumes), and education level. 15 , 18 , 19 , 20
The base structure for disease models in AD has evolved over time. Earlier models assumed a linear score progression from baseline (start of the trial). 11 Holford and Peace developed a population pharmacodynamics model of five clinical trials and characterized the response to treatment via an offset in disease progression curve. 21 , 22 Yang et al. 12 proposed the concept of disease onset time (DOT) by accounting for the fact that participants enrolled in a trial are at different stages of their disease, thus fitting the data to a theoretical curve of disease progression for ADAS‐Cog 13. The ADAS‐Cog 13 includes all ADAS‐Cog 11 items as well as a test of delayed word recall and a number cancellation or maze task. 23 ADAS‐Cog 13 scores range from 0–85. 23 Ueckert et al. combined item response theory and a pharmacometrics approach to describe progression of total ADAS‐Cog and item level scores. 24 Rogers et al. used a beta regression model to describe the longitudinal progression of ADAS‐Cog in people with AD in both natural history and randomized clinical trial settings. 25 Quartino et al. used a similar approach to describe progression of six items of the CDR scale, ADAS‐Cog 12 (ADAS‐Cog 12 includes all ADAS‐Cog 11 items and a delayed word recall task and scores range from 0 to 80 23 ), and hippocampal and ventricular volumes. 26 Delor et al. implemented the DOT concept via a logistic function, to describe change in CDR‐SB scores following DOT via a differential equation. 16
These models discussed have improved our quantitative understanding of the time course of the disease and important covariates affecting disease progression. Most models developed to date focus on describing progression of one score (e.g., CDR‐SB or ADAS‐Cog 11). Ultimately, developing joint models of such scores could be of interest as proposed by Quartino et al. 26 This study builds upon the previous works in disease modeling for AD to characterize longitudinal progression of CDR‐SB in the absence of active treatment (i.e., progression of participants in the placebo arm of clinical trials and progression of participants in the observational study, ADNI). Even though the model building in this study is focused on CDR‐SB score, we leverage learnings from previous modeling work on ADAS‐Cog (such as the concept of DOT). We began our model‐building effort based on the CDR‐SB disease model developed by Delor et al., 16 which was developed using the ADNI cohort. We used a similar base structural model to Delor et al. 16 and conducted model building and covariate selection. The strength of our model‐building effort is using data from the placebo arms of multiple interventional clinical trials in AD (SCarlet RoAD [SR], Marguerite RoAD [MR], ABBY, and BLAZE, as described in Table 1) and data from the ADNI. 27 , 28 , 29 , 30 Additionally, we used placebo data from the CREAD 31 and CREAD2 32 trials to assess performance of the model on studies that were not used for model building. The model‐building population included participants across the range of disease severity, from prodromal AD to moderate AD dementia.
TABLE 1.
Data sources and inclusion criteria for model building and validation.
| Study | Role(s) | Primary end point(s) | MMSE scores for inclusion | CDR‐GS for inclusion | Disease category | Number of participants used for modeling (n) | Age range for inclusion (years) | Pathology inclusion criteria | Duration of study | |
|---|---|---|---|---|---|---|---|---|---|---|
| Real‐world data | ADNI | Model building | 1. Serial MRI, PET, other biological markers, and clinical and neuropsychological assessments can be combined to measure the progression of MCI and early AD |
Normal: 24–30 MCI: 24–30 Mild AD: 20–26 |
Normal: 0 MCI: 0.5 Mild AD: 0.5 or 1 |
Normal‐ to mild AD | 2033 total, (459 amyloid‐positive, prodromal AD) | 55–90 | Yes (for inclusion in model‐building dataset), CSF Aβ42 or Aβ PET | Initiated 2003, data extracted February 2, 2020 |
| Gantenerumab | SCarlet RoAD a | Model building, internal validation |
1. Mean change from baseline in CDR‐SB total score 2. AEs/SAEs (OLE phase) |
24–30 | 0.5 | Prodromal AD | 266 | 50–85 | Yes, CSF Aβ42 | 104 weeks |
| Marguerite RoAD | Model building, internal validation |
1. Mean change from baseline in ADAS‐Cog 13 score 2. Mean change from baseline in ADCS‐ADL score 3. AEs/SAEs |
20–26 | 0.5–1.0 | Mild AD | 195 | 50–89 | CSF consistent with amyloid pathology | 104 weeks | |
| Crenezumab | ABBY | Model building, internal validation |
1. Change in ADAS‐Cog score. 2. Change in CDR‐SB |
18–26 | >0.5 | Mild–moderate AD | 144 | 50–80 | Participants were assumed to be amyloid‐positive | 68 weeks |
| BLAZE | Model building, internal validation | 1. Change in brain amyloid load, as assessed by amyloid PET imaging | 18–26 | >0.5 | Mild–moderate AD | 29 | 50–80 | Yes, Aβ PET | 68 weeks | |
| CREAD | External validation | 1. Change from baseline in CDR‐SB | ≥22 | 0.5 or 1.0 | Prodromal‐mild AD | 407 | 50–85 | Yes, CSF Aβ42 | 100 weeks | |
| CREAD2 | External validation | 1. Change from baseline in CDR‐SB | ≥22 | 0.5 or 1.0 | Prodromal‐mild AD | 398 | 50–85 | Yes, CSF Aβ42 | 100 weeks |
Abbreviations: AD, Alzheimer's disease; ADAS‐Cog 13; Alzheimer's Disease Activity Scale – Cognitive subscale 13; ADCS‐ADL, Alzheimer's Disease Cooperative Study – Activities of Daily Living; ADNI, Alzheimer's Disease Neuroimaging Initiative; AE, adverse event; Aβ, amyloid‐beta; CDR‐GS, Clinical Dementia Rating – Global Score; CDR‐SB, Clinical Dementia Rating – Sum of Boxes; CSF, cerebrospinal fluid; MCI, mild cognitive impairment; MMSE, Mini‐Mental State Examination; MRI, magnetic resonance imaging; OLE, open‐label extension; PET, positron emission tomography; SAE, serious adverse event.
Participants in the SCarlet RoAD trial were not receiving memantine or cholinesterase inhibitors.
METHODS
Data for model building and validation
Model‐building dataset
The model‐building datasets included participants across the following disease stages: prodromal AD, mild AD, and moderate AD. Data were extracted from the placebo arms of clinical trials of crenezumab (ABBY [NCT01343966, n = 144, phase II, mild‐to‐moderate AD] and BLAZE [NCT01397578, n = 29, phase II, mild‐to‐moderate AD]) 27 , 33 and gantenerumab (SR [NCT01224106, n = 266, phase III, prodromal AD] and MR [NCT02051608, n = 195, phase III, mild AD] 30 ; Table 1). Participant data from the ADNI database (adni.loni.usc.edu) were also used for model building. 34 The ADNI study was launched in 2003 as a public–private partnership, led by Principal Investigator Michael W. Weiner. The primary goal of the ADNI has been to test whether biomarkers and clinical and neuropsychological assessments can be combined to measure the progression of MCI and AD. For up‐to‐date information, see www.adni‐info.org. For ADNI data (data extracted on February 2, 2020), only participants who were amyloid‐positive with a baseline diagnosis of late MCI or AD were included in the analysis (N = 459); in order to mirror inclusion criteria for the most recent AD clinical trials (e.g., SR, MR, BLAZE, CREAD, and CREAD2). Demographics and characteristics for participants included in the analysis are summarized in Table 2. Populations were similar in age and sex ratios. In SR, about 13% of participants were APOEε4‐positive; in the other studies, this percentage ranged from 67% to 79%. Participants did not receive any concomitant medications in the SR study, but the use of symptomatic therapies (i.e., rivastigmine, donepezil, galantamine, and memantine) was permitted in the other studies. The MR, ABBY, and BLAZE studies, which enrolled only participants with mild or mild‐to‐moderate AD, had slightly higher baseline CDR‐SB scores than other study populations. SR enrolled participants with prodromal AD, which resulted in lower baseline CDR‐SB scores.
TABLE 2.
Participant demographics.
| Real‐world data | Gantenerumab | Crenezumab | |||||
|---|---|---|---|---|---|---|---|
| ADNI | SCarlet RoAD | Marguerite RoAD | ABBY | BLAZE | CREAD | CREAD2 | |
| n = 459 | n = 266 | n = 195 | n = 144 | n = 29 | n = 407 | n = 398 | |
| Age, mean (SD), years | 73.8 (7.6) | 69.5 (7.5) | 69.9 (8.6) | 70.1 (7.2) | 69.2 (7.8) | 70 (8.4) | 70.7 (7.9) |
| Sex, female, n (%) | 195 (42) | 149 (56) | 113 (58) | 76 (53) | 14 (48) | 247 (61) | 224 (56) |
| APOEε4 carrier, % positive | 72 | 13 | 67 | 67 | 79 | 71 | 67 |
| Treatment with symptomatic therapies, n (%) | |||||||
| Rivastigmine (Y) | 27 (6) | 0 | 34 (17) | 19 (13) | 2 (7) | 54 (13) | 54 (14) |
| Donepezil (Y) | 254 (55) | 0 | 98 (50) | 82 (57) | 19 (66) | 209 (33) | 179 (45) |
| Galantamine (Y) | 428 (93) | 0 | 17 (9) | 18 (13) | 2 (7) | 25 (4) | 17 (4) |
| Memantine (Y) | 145 (31) | 0 | 40 (21) | 41 (29) | 13 (45) | 20 (5) | 82 (20) |
| Diagnosis, n (%) | |||||||
| Prodromal AD | 244 (53) | 266 (100) | 0 | 0 | 0 | 265 (65) | 226 (57) |
| Mild or moderate AD dementia | 215 (47) | 0 | 195 (100) | 144 (100) | 29 (100) | 142 (35) | 172 (43) |
| CDR‐SB, mean (SD) | 3 (1.9) | 2.1 (1.0) | 4 (1.8) | 4.6 (2.2) | 4.9 (1.8) | 3.8 (1.6) | 3.8 (1.6) |
| MMSE, mean (SD) | 25.2 (2.7) | 25.7 (2.1) | 22.4 (2.9) | 21.8 (3.3) | 21 (3.3) | 23.4 (2.9) | 23.5 (2.9) |
| ADAS‐Cog 11, mean (SD) | 15.9 (7) | 14.1 (5.6) | 18.9 (6) | 19.5 (7.3) | 22.6 (9.5) | 20 (5.9) | 20.1 (5.5) |
Abbreviations: AD, Alzheimer's disease; ADAS‐Cog 11, Alzheimer's Disease Assessment Scale – Cognitive Subscale 11; ADNI, Alzheimer's Disease Neuroimaging Initiative; APOEε4, apolipoprotein E ε4 allele; CDR‐SB, Clinical Dementia Rating Scale – Sum of Boxes; MMSE, Mini‐Mental State Examination; SD, standard deviation.
Participants with only one CDR‐SB score value or a missing baseline CDR‐SB score were excluded from the analysis. The lengths of the treatment duration in interventional trials were generally up to 2 years, and data up to 4 years were included from the ADNI. For the covariate analysis, missing data were imputed to the population median from the study and the corresponding diagnosis group. For baseline scores, we had fewer than 5% missing. We did not have missing categorical covariates.
External validation dataset
Data from the placebo arms of the crenezumab clinical trials (CREAD [NCT02670083, phase III] and CREAD2 [NCT03114657, phase III]; pooled N = 805) were used for external model validation. The CREAD studies included participants with prodromal‐to‐mild AD.
Definition of CDR‐SB score
The CDR‐SB is a common clinical end point in interventional trials that assesses both cognitive and functional ability. 35 The score is bound between zero and 18 and increases as the disease advances.
Base structural model
The AD progression model for CDR‐SB was developed using a nonlinear, mixed‐effect modeling approach. The modeling was performed in the logit domain, resulting in the estimate of a score that was bound between zero and one; this was multiplied by 18, to ensure that the number stays within the bounds of the CDR‐SB score (zero and 18). To obtain the CDR‐SB score, an estimated term (baseline correction [BLC]; Table 3) repositioned the logit at the most probable level (i.e., 1–4.11 = −3.11; Equation 1).
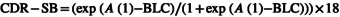 |
(1) |
TABLE 3.
Population parameter estimates.
| Estimate | Relative standard error, % | 95% CI | Shrinkage, % | |
|---|---|---|---|---|
| Objective function value | 5923.514 | – | – | |
| DOT, years | 16.7 (i.e., 3.3 before the start of the trial/study) | 1.5 | [16.3; 17.3] | – |
| RATE, per year | 0.305 | 5.4 | [0.274; 0.345] | – |
| α (range) | 0.0624 | 11.9 | [0.049; 0.079] | – |
| Baseline correction | 4.11 | 1.9 | [3.91; 4.25] | – |
| Covariate baseline MMSE on DOT | 0.246 | 15.5 | [0.169; 0.316] | – |
| Covariate baseline CDR‐SB on DOT | −0.076 | 10.1 | [−0.094; −0.060] | – |
| Covariate baseline CDR‐SB on RATE | 0.261 | 15.6 | [0.181; 0.351] | – |
| Covariate baseline MMSE on α | 1.9 | 30.2 | [0.576; 2.735] | – |
| IIV for DOT | 0.0016 | 13.57 | [0.001; 0.002] | 35 |
| IIV for α | 0.718 | 15.70 | [0.56; 1.00] | 32 |
| Additive error | 0.126 | 4 | [0.119; 0.136] | 11 |
Abbreviations: CDR‐SB, Clinical Dementia Rating Scale – Sum of Boxes; DOT, disease onset time; IIV, interindividual variability; MMSE, Mini‐Mental State Examination; RATE, population disease‐progression rate; α, individual progression rates.
An equation (Equation 2) describing the rate of progression of A (1) was fitted to the model‐building dataset and parameters of the equation describing progression were estimated.
| (2) |
Change in A (1) over time was described via a differential equation, similar to Delor et al. (Equation 2). 16 , 36 To account for the fact that different participants enter a trial at different stages of disease, a DOT was estimated prior to the start of the trial for each participant by fitting Equation 2 to the model‐building data; this DOT concept is illustrated in Figure 1. To ensure that DOT had a positive value, an arbitrary time shift (20 years) was imposed on the data such that the start of the trial was at year 20; DOT was then estimated to be between 0 and 20 years. A virtual dose was administered in NONMEM version 7.3 at time 0 with AMT = 1. The sigmoidal term, , ensured that the score only started increasing after DOT and did not change evidently between time 0 and DOT (exponent 30 is arbitrary and very high, to model the onset of the disease). In addition to DOT, the change in CDR‐SB score was further described by a global disease progression rate, linear with time (RATE), and a first order rate constant (α), describing exponential progression. IIV was implemented on DOT and α. It was not possible to estimate IIV on all three parameters (when IIV was implemented on RATE it was estimated to be close to the boundary of zero [0.00003]). Similar to Delor et al., 16 an additive residual error model was implemented.
FIGURE 1.
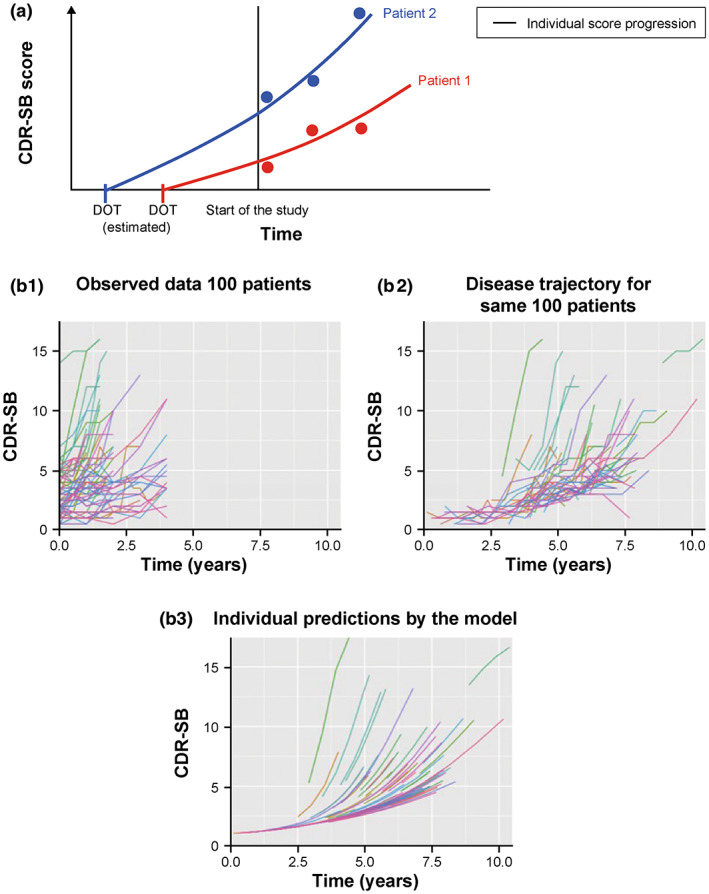
The concept of DOT (i.e., the time at which the score starts increasing before the start of the trial) (a) and observed longitudinal trajectories of CDR‐SB scores for 100 randomly selected participants from the model‐building dataset on the clinical trial time scale (b1), on the disease trajectory time scale (using DOT) (b2), and corresponding individual predictions from the model for those participants (b3). CDR‐SB, Clinical Dementia Rating Scale – Sum of Boxes; DOT, disease onset time.
Covariate analysis
Covariate analysis was conducted to identify factors that help explain IIV. Age, body mass index, education level (years), sex, APOEε4 status, disease diagnosis (AD vs. prodromal), baseline cognition (MMSE and ADAS‐Cog), baseline cognition/function (CDR‐SB), baseline functional score (Functional Activities Questionnaire [FAQ] or Alzheimer's Disease Cooperative Study – Activities of Daily Living Scale), and baseline Clinical Dementia Rating – Global Score (CDR‐GS) were selected based on understanding of the disease and graphical analysis of correlation between covariates and score, as well as parameters of IIV (ETAs) and covariates. Covariates were retained in the model based on statistical significance (Table S2 shows the drop in objective function after addition of each covariate). In addition to a drop in the objective function value, plots of ETAs versus covariates were examined after addition of covariates, to ensure that the effect was fully captured. The functional forms of the significant covariates are presented in Table S2.
Model evaluation and validation
In the first step, parameter estimates and goodness‐of‐fit plots were carefully examined on the population and individual levels, such as plot of observation (DV) versus population prediction (PRED), DV versus individual prediction (IPRED), individual weighted residuals (IWRES) versus TIME, IWRES versus IPRED, conditional weighted residuals (CWRES) versus TIME, CWRES versus PRED, and ETA distributions. Diagnostic plots of observed versus predicted changes from baseline were also examined, and the objective function value together with parameter uncertainty (posterior predictive check [PPC]) were used as a guide throughout model development. Internal validation via visual predictive checks (VPCs) was used to confirm that the model captured the central tendency and variability observed in the model‐building data for each study. In producing VPCs, 1000 simulated replications of the dataset were created by random sampling of parameter‐level random error (i.e., IIV) and observation‐level random error (residual error or unexplained differences between predicted and observed values). Simulations to obtain VPCs were conducted in NONMEM version 7.3 and plotted in R version 3.6.1. 37 External validation using VPCs was then performed with data from the placebo arms of the CREAD trials. In addition to VPCs, the PPC including parameter uncertainty was conducted to compare model‐predicted and observed mean changes from baseline for each study. For this analysis, similar to VPCs, 1000 replications of the dataset were simulated across various sources of variability, and predicted mean changes from baseline, and confidence intervals for the mean changes from baseline, were calculated at each timepoint and compared with observed changes. Thus, the model was developed using a dataset with participants ranging in disease severity from prodromal AD to moderate AD, and internal validation was conducted to confirm that the model can predict all different disease stages used for model building.
RESULTS
Model development
We were able to capture progression of CDR‐SB scores for the entire population using Equation 2, by including baseline CDR‐SB as a covariate on DOT and RATE (structural covariates). Including the baseline MMSE score as a covariate on DOT and α was significant in further explaining between‐subject variability (Table 4). The final model thus included baseline CDR‐SB as a covariate on RATE, baseline MMSE as a covariate on α, and baseline CDR‐SB and baseline MMSE as covariates on DOT. Baseline CDR‐SB was included as a structural covariate on DOT (Equation 3). Overall, model diagnostic plots did not show signs of systemic bias with respect to study, diagnostic group, time, or PRED magnitude (Figure S1 shows plots of DV vs. PRED and DV vs. IPRED; plot of observed and predicted changes from baseline at 12 months is shown in Figure S2). All parameters were well‐estimated (Table 3). DOT was estimated as 3.3 years before entering the trial, or start of study for the ADNI data (with a relative standard error [RSE] of 1.5%). The estimated IIV of α was large (84.7% [RSE 9.3%]). Despite the large range of DOT (up to 6.94 years before the start of the study in model‐building studies), the covariates largely explained the variability in DOT very well, and IIV was estimated at 4% (RSE 6.4%) (correlation plots of IIV on DOT and α are presented in Figure S3). Shrinkage of IIV on DOT and α were 35% and 32%, respectively. Fewer than 1% of participants had estimated DOTs after the start of the study (DOT <0); this occurred in participants whose score did not progress or decreased over time. Larger values of α were associated with fast progressors (DOTs close to the start of the trial/study, but scores progressed rapidly). Population RATE was estimated at 0.305 (RSE 4.6%) per year. The direction of the estimated covariate effects on DOT was in line with our expectation based on the directionality of these scores. For example, MMSE score decreases whereas CDR‐SB increases with advanced disease, and the corresponding covariate parameters are estimated as positive and negative values, respectively, which capture the nature of these scores (Figure S3A–C).
| (3) |
TABLE 4.
Distribution of individual parameter values for studies used in model building and external validation.
| ADNI | SR | MR | ABBY | BLAZE | CREAD | CREAD2 | Prodromal‐mild AD | Mild–moderate AD | |
|---|---|---|---|---|---|---|---|---|---|
| DOT (range), years | 3.36 (6.01, −1.13) | 2.92 (4.95, −0.64) | 4.47 (6.94, 2.2) | 4.67 (6.84, 2.07) | 4.81 (6.69, 3.57) | 4.28 (6.76, 1.56) | 4.25 (7.04, 1.33) | 2.69 (−1.13, 4.95) | 4.49 (2.07, 6.94) |
|
RATE (range), per year |
0.3 (0.2, 0.43) | 0.29 (0.2, 0.26) | 0.33 (0.20, 0.44) | 0.35 (0.24, 0.48) | 0.35 (0.29, 0.45) | 0.33 (0.24, 0.45) | 0.33 (0.24, 0.44) | 0.28 (0.20, 0.37) | 0.35 (0.20, 0.48) |
| α (range) | 0.066 (0.017, 0.929) | 0.057 (0.02, 0.8) | 0.055 (0.016, 0.65) | 0.047 (0.016, 0.3) | 0.044 (0.018, 0.22) | 0.066 (0.012, 0.19) | 0.068 (0.006, 0.21) | 0.09 (0.02, 0.93) | 0.07 (0.02, 0.65) |
Note: Mean and range of parameters. For CREAD and CREAD2 studies, parameter values are obtained using estimation mode, MaxEval = 0. Individual predictions for each study are presented in Figure S4.
Abbreviations: ADNI, Alzheimer's Disease Neuroimaging Initiative; CDR‐SB, Clinical Dementia Rating Scale – Sum of Boxes; DOT, disease onset time; MR, Marguerite RoAD; RATE, population disease progression rate; SR, SCarlet RoAD; α, individual progression rates.
Model validation
Cohort level
Internal validation by VPC confirmed that the model performed adequately in capturing the observed data from the trials used for model building (Figure 2). The deviation observed in SR after 1 year could be attributed to the fact that there was a very high proportion of participants with minimal/no progression. For this study, the model also over predicted the 90th percentile. Similarly, external validation via VPC confirmed that the model performed adequately in capturing the observed data from the CREAD and CREAD2 trials. Overlap between the confidence intervals from the simulations and observed data for median, 10th percentile, and 90th percentile of the data demonstrates that the model adequately captures central tendency and variability in the trials used for model building (Figure 2a–e). The external validation, which included early AD, was also well‐predicted by this model (Figure 2f,g).
FIGURE 2.
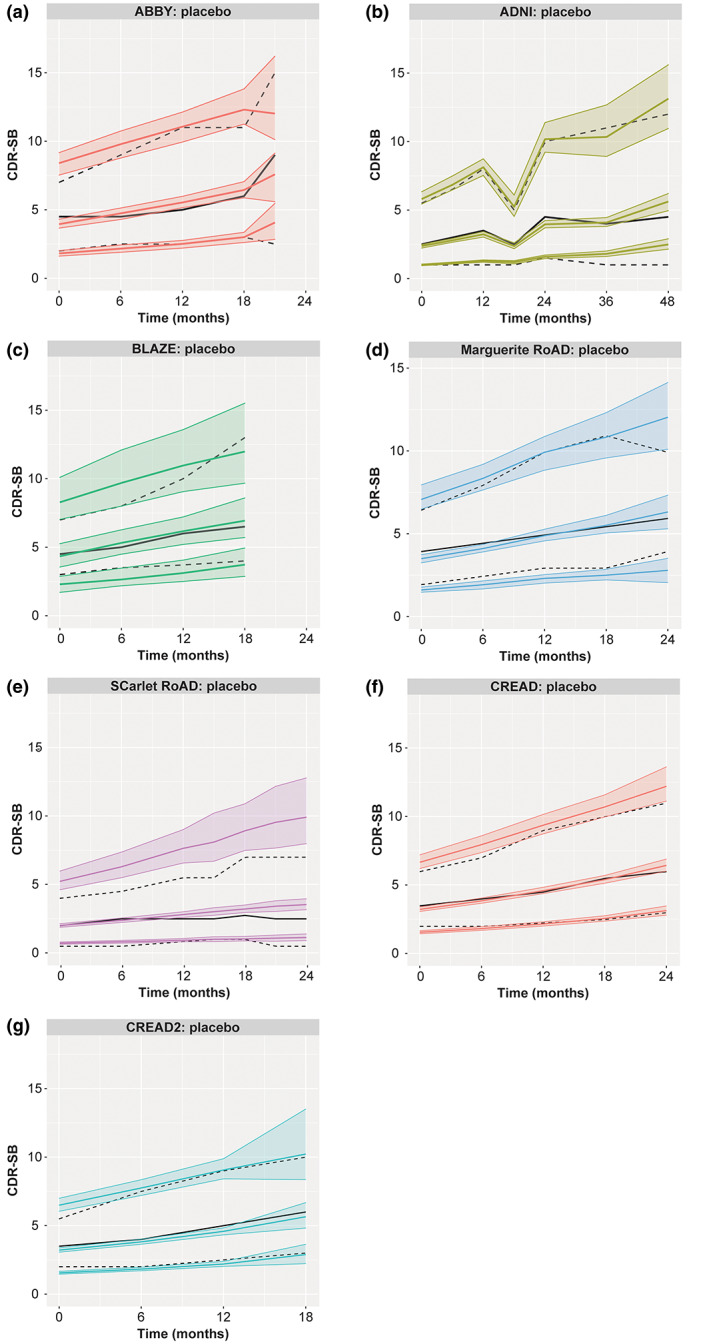
VPC internal validation of the model in studies used for model development (a–e) and external validation of the model in studies not used for model development (f, g). *ADNI, Alzheimer's Disease Neuroimaging Initiative; CDR‐SB, Clinical Dementia Rating Scale – Sum of Boxes; CI, confidence interval; VPC, visual predictive check. Solid and dashed black lines are the observed median and 10th and 90th percentiles, respectively. Three predicted percentiles are shown in color for each of the observed metrics (solid line is median, shaded areas are 10th, and 90th percentiles). *Obtained from 1000 simulated datasets using the final model.
Individual level
In addition to using simulations to obtain model predictions for an external dataset, the model can be used in estimation mode to obtain individual‐level parameter estimates to perform such prediction (post hoc Bayesian forecasting [MaxEval = 0]). In this setting, the R 2 for observed versus predicted changes from baseline for post‐baseline timepoints was 0.16.
Comparison of parameters describing disease progression across studies, disease stage, and APOEε4 status
Mean DOT was less than 3 years before the start of the study for the prodromal‐to‐mild AD population (summary statistics based on individual empirical Bayes estimates [EBEs] of DOT; Table 4), while the mild‐to‐moderate AD population had a mean DOT greater than 4 years prior to the start of the study. Global disease progression rate (RATE) varied between 0.29 and 0.35 across studies, with higher values in the mild‐to‐moderate AD population (it should be noted that variability in RATE is due to fixed effects [covariates]). In addition to study and disease stage, we also looked at summary statistics based on individual EBEs for DOT, and α based on APOEε4 status. Mean values for DOT, RATE, and α parameters were similar in APOEε4‐positive and APOEε4‐negative groups (Table S3).
Prediction of clinical end point (CDR‐SB mean change from baseline)
The final model can be used to simulate the clinical end point: here, mean change from baseline in CDR‐SB at each timepoint. We compared model‐predicted versus observed mean changes from baseline in CDR‐SB scores for the placebo arms from model‐building studies (Figure 3a–e) and external validation studies (Figure 3f,g). Apart from the CREAD2 study, mean change from baseline predicted by the model at 12 months was higher than observed mean change from baseline at this timepoint. Observed mean change from baseline was within 95% predicted range for mean changes from baseline in CDR‐SB scores (Table S3). For SR, the model predicted greater progression than what was observed in this study.
FIGURE 3.
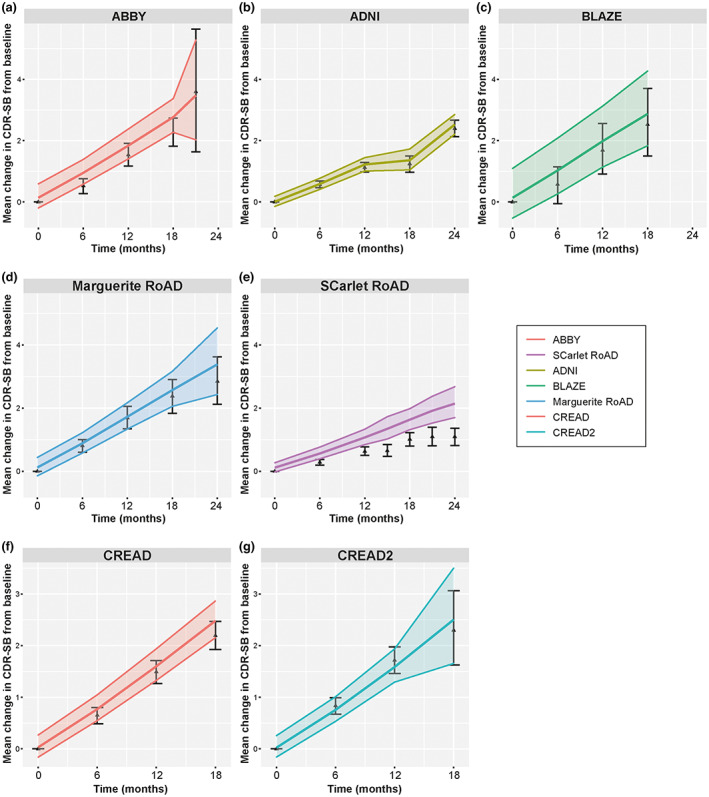
Model‐predicted and observed changes from baseline in CDR‐SB for participants in studies used for model building (a–e) and not used in model building (f, g). ADNI, Alzheimer's Disease Neuroimaging Initiative; CDR‐SB, Clinical Dementia Rating Scale – Sum of Boxes. Colored lines are mean and 95% prediction interval from the model. Black triangles and whiskers are mean and 95% confidence interval of the observed.
DISCUSSION
In this analysis, we built upon previous modeling by Delor et al. 16 to develop a disease progression model to describe longitudinal, clinical progression in AD (as measured by the CDR‐SB) using a nonlinear, mixed‐effect population modeling approach. The model was developed using a rich dataset, with participants ranging in disease severity from prodromal AD to moderate AD; internal validation confirmed that the model can predict all different disease stages used in model building. External validation, which included early AD, was also well‐predicted by this model. AD is a slowly progressive disease, and trials follow participants over relatively short periods of time (roughly 2 years); disease progression appears linear during trial duration (as seen in progression of scores such CDR‐SB over time), whereas there is an exponential trajectory when looking at such scores over a longer period of time (e.g., onset of disease). To account for this, in this model, change in CDR‐SB score over time was described via a differential equation, similar to Delor et al. 16 (Equation 2). We obtained the individual disease progression trajectory for each participant by estimating DOT prior to the start of the trial, thus enabling us to capture the nonlinear time course of the score. CDR‐SB score is bounded between zero and 18. In our model‐building dataset, fewer than 1% of participants had CDR‐SB greater or equal to 10. Due to nature of the score, the participants who start at a very high CDR‐SB score may show slower progression of score.
A previous model, developed by Delor et al., 16 used a mixture model to identify fast and slow progressors in the ADNI. They identified baseline CDR‐SB and ADAS‐Cog as significant covariates on DOT, baseline MMSE on α, and baseline CDR‐SB, FAQ, and RHPNM (hippocampal volumes normalized to that of a healthy subject with the same age and head size) as covariates on the modeled fraction of the population in the first (or second) subpopulation ($MIX). Our model uses the same base structural model as Delor et al. 16 In addition to data from the ADNI, we used data from the SR, MR, ABBY, and BLAZE trials for model building. We identified baseline CDR‐SB and baseline MMSE on DOT, baseline MMSE on α, and baseline CDR‐SB on RATE as significant covariates in our model. CDR‐SB assesses both cognition and function, whereas MMSE and ADAS‐Cog both focus on cognitive assessments. Thus, in both our model and the model by Delor et al., 16 we identified baseline CDR‐SB and a cognitive score as significant covariates of DOT. Not all covariates used by Delor et al. 16 were available (e.g., FAQ) or comparable (e.g., brain volume) for all studies we used in model building. A mixture model (mixture models allow for the estimation of two or more [sets of] parameters for different subpopulations of subjects without a specific variable to assign subjects to relevant groups) was also assessed during our model development; however, in this study, we were able to finalize a model without a mixture model, using data from interventional studies and the ADNI, and, thus, with an easier application in simulation mode. Indeed, differences between subpopulations identified by Delor et al. 16 could be characterized using baseline covariates.
Nevertheless, although the mixture model was not included in the final model, our model parameter estimates were in agreement with the estimates published by Delor et al., 16 where DOT was estimated 3.7 years prior to the start of the study for the ADNI. Our estimated rate of 0.305 falls between the slow progression rate (0.271) and the fast progression rate (0.383), as estimated by the mixture model by Delor et al. 16
We identified significant covariates to explain between‐individual variability in DOTs and nonlinear progression rates. Shrinkage of IIV on DOT and α were 35% and 32%, respectively. These shrinkages were higher than the 25%–30% range. In general, higher ETA shrinkage imposes a risk in selecting covariate based on EBEs. In our model‐building effort, we comprehensively examined the correlation between significant covariates and other parameters to ensure correct covariate selection. Baseline CDR‐SB score was a significant covariate on DOT and RATE, and baseline MMSE score was a significant covariate on DOT and α. Covariate estimates of DOT indicate earlier onset of the disease in trials with higher baseline CDR‐SB scores (estimated baseline CDR‐SB covariate on DOT = −0.076) and lower baseline MMSE scores (estimated baseline MMSE covariate on DOT = 0.246). This is expected, as higher baseline CDR‐SB and lower baseline MMSE correspond to more advanced disease and, thus, earlier DOT. Covariate estimate for α (estimated baseline MMSE covariate on α = 1.9) indicates higher α at higher baseline MMSE scores, but IIV on α is large. Higher α in Equation 1 corresponds to faster progression in CDR‐SB score. Covariate estimate for RATE (estimated baseline CDR‐SB covariate on RATE = 0.261) indicates higher RATE at higher baseline CDR‐SB scores. Higher RATE in Equation 1 corresponds to faster progression in CDR‐SB score. Even though we were not able to include brain volumes (due to lack of consistency across studies) or biomarkers (due to small sample size for participants with biomarker data) as covariates, these parameters seem to have impacts on disease progression and could be areas of further exploration. In our analysis of different subpopulations, we found that prodromal‐to‐mild AD is associated with a DOT closer to the start of the trial, as well as a lower global progression rate compared with the mild‐to‐moderate AD population. However, we did not find a difference in parameter estimates based on APOEε4 status. This is in agreement with Yang et al., where they found that calculating disease onset time in the model resulted in a model with fewer covariates compared to previous studies for ADAS‐Cog. 12
The model was successfully validated (via VPC and PPC) by assessing its ability to predict CDR‐SB progression for the model‐building dataset. The discrepancy in SR could be attributed to the high proportion of nonprogressors in this study. Assessing model predictive performance on data from independent clinical trials not seen by the model is highly preferable 38 ; to this end, the placebo arms of the two phase III CREAD trials were used for external validation. In addition to cohort‐level analysis, the model can be used to make individual‐level predictions. Such predictions could be used to prospectively predict progression of patients enrolled in an interventional study based on their score at the start of the trial. We tested the model performance for this individual level prediction, using only baseline score as the input. (Here, we used MaxEval = 0, taking observed baseline score as input and predicting for post‐baseline timepoints using the model, EVID = 0 for baseline, EVID = 2 for post‐baseline.) In this setting, R 2 for model‐predicted versus observed changes from baseline for post‐baseline timepoints was 0.16 (using MaxEval = 0). It should be noted that diagnostic plot for predicted and observed change from baseline during model building was observed to be reasonable (Figure S2). To our knowledge, previous disease modeling efforts did not explore performance and the application of such models on individual level predictions. Large between‐participant variability in AD could be a contributor to low R 2 using MaxEval = 0, as random effect plays a big role in capturing between‐participant variability. This is an active area of further exploration for us to identify adequate performance metrics for application of AD progression model for individual level predictions. The DV versus PRED plots did not show a major bias, a fact which supports this type of analysis could be used prospectively, to predict progression in participants enrolled in clinical trials and confirm placebo progression. In addition to application for the placebo arm, the model can be used to predict progression in the treatment arm as if enrollees had not received the treatment in the treatment arm. 22 , 39 By comparing the predicted individual trajectories to the observed, on‐treatment responses, the model can help assess a treatment effect of new molecules in development. 40 This assessment of treatment effect can be used to further confirm and support statistical analyses following study readout. The model can also be used for the design of future trials in AD, by selecting inclusion/exclusion criteria using clinical trial simulations.
AUTHOR CONTRIBUTIONS
S.J., M.D., P.C., V.R., Y.F., K.W., P.M., E.T., J.Y.J., A.Q., and J.C.H., wrote the manuscript. S.J., M.D., P.C., V.R., J.Y.J., A.Q., and J.C.H, designed the research. S.J. performed the research and analyzed the data. S.J., Y.F., and P.C. contributed to new analytical tools.
FUNDING INFORMATION
This analysis was funded by Genentech, Inc., South San Francisco, California, USA, and F. Hoffmann‐La Roche Ltd, Basel, Switzerland. Data collection and sharing for this project were funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI; National Institutes of Health grant U01 AG024904) and Department of Defense (DOD) ADNI (DOD award number W81XWH‐12‐2‐0012). The ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie; Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; Biogen; Bristol Myers Squibb; CereSpir, Inc.; Clario; Cogstate; Eisai Co., Ltd.; Elan Pharmaceuticals, plc.; Eli Lilly and Company; EUROIMMUN; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx; Neurotrack; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
CONFLICT OF INTEREST STATEMENT
S.J., V.R., E.T., J.Y.J., and J.C.H. are all full‐time employees of Genentech, Inc. and may own company stock/stock options. M.D. is an employee of Roche Products Pty Ltd. and owns stock/stock options in F. Hoffmann‐La Roche Ltd. P.C. is an employee of Roche/Genentech and owns Roche stock options. Y.F. is a former employee of Genentech, Inc. K.W. is a former full‐time employee of Genentech, Inc. and may own company stock/stock options. P.M. is a former employee of Genentech, Inc. A.Q. was an employee of Genentech, Inc. at the time of this study (current affiliation: Clinical Pharmacology and Quantitative Pharmacology, AstraZeneca, Gothenburg, Sweden).
Supporting information
Data S1
Data S2
ACKNOWLEDGMENTS
The authors would like to acknowledge the following contributions: Balazs Toth and Christina Rabe of Genentech, Inc., and Tobias Bittner of F. Hoffmann‐La Roche Ltd., for reviewing the manuscript and data preparation. Michaela Mertes and Kellie Riley of F. Hoffmann‐La Roche Ltd., for data preparation. Medical writing support was provided by Adam Ruth, PhD, Sarah Engelberth, PhD, of Medical Expressions (Chicago, Illinois) and Ayesha Babar, MSc, of Chrysalis Medical Communications (United Kingdom), and funded by F. Hoffmann‐La Roche.
Jamalian S, Dolton M, Chanu P, et al. Modeling Alzheimer's disease progression utilizing clinical trial and ADNI data to predict longitudinal trajectory of CDR‐SB. CPT Pharmacometrics Syst Pharmacol. 2023;12:1029‐1042. doi: 10.1002/psp4.12974
REFERENCES
- 1. Alzheimer's Disease International (ADI) . World Alzheimer Report 2019: Attitudes to Dementia. Alzheimer’s Disease International (ADI); 2019. https://www.alz.co.uk/research/WorldAlzheimerReport2019.pdf. Accessed December 6, 2022. [Google Scholar]
- 2. Alzheimer's Association . 2020 Alzheimer's disease facts and figures. Alzheimers Dement. 2020;16:391‐460. [Google Scholar]
- 3. Cummings JL, Morstorf T, Zhong K. Alzheimer's disease drug‐development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014;6:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alzforum . Aduhelm. https://www.alzforum.org/therapeutics/aducanumab. Accessed December 6, 2022.
- 5. Tampi RR, Forester BP, Agronin M. Aducanumab: evidence from clinical trial data and controversies. Drugs Context. 2021;10:2021‐2027‐2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gavidia‐Bovadilla G, Kanaan‐Izquierdo S, Mataró‐Serrat M, Perera‐Lluna A. Alzheimer's disease neuroimaging initiative. Early prediction of Alzheimer's disease using null longitudinal model‐based classifiers. PLoS One. 2017;12:e0168011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jack CR Jr, Bennett DA, Blennow K, et al. NIA‐AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. US Food and Drug Administration (FDA) . PDUFA Reauthorization Performance Goals and Procedures Fiscal Years 2018 Through 2022. https://www.fda.gov/media/99140/download. Accessed December 6, 2022.
- 9. European Medicines Agency (EMA) . EMA Regulatory Science to 2025: Strategic reflection. https://www.ema.europa.eu/en/documents/regulatory‐procedural‐guideline/ema‐regulatory‐science‐2025‐strategic‐reflection_en.pdf. Accessed December 6, 2022.
- 10. Eglovitch J. Regulatory affairs professionals society, regulatory news. ICH: MIDD Concept Paper Coming by year's End [News Article]. May 20, 2021. https://www.raps.org/news‐and‐articles/news‐articles/2021/5/ich‐midd‐concept‐paper‐coming‐by‐years‐end. Accessed December 6, 2022
- 11. Ito K, Corrigan B, Zhao Q, et al. Disease progression model for cognitive deterioration from Alzheimer's Disease Neuroimaging Initiative database. Alzheimers Dement. 2011;7:151‐160. [DOI] [PubMed] [Google Scholar]
- 12. Yang E, Farnum M, Lobanov V, et al. Quantifying the pathophysiological timeline of Alzheimer's disease. J Alzheimers Dis. 2011;26:745‐753. [DOI] [PubMed] [Google Scholar]
- 13. Samtani MN, Farnum M, Lobanov V, et al. An improved model for disease progression in patients from the Alzheimer's disease neuroimaging initiative. J Clin Pharmacol. 2012;52:629‐644. [DOI] [PubMed] [Google Scholar]
- 14. Samtani MN, Raghavan N, Novak G, Nandy P, Narayan VA. Disease progression model for clinical dementia rating–sum of boxes in mild cognitive impairment and Alzheimer's subjects from the Alzheimer's disease neuroimaging initiative. Neuropsychiatr Dis Treat. 2014;10:929‐952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Samtani MN, Raghavan N, Shi Y, et al. Disease progression model in subjects with mild cognitive impairment from the Alzheimer's disease neuroimaging initiative: CSF biomarkers predict population subtypes. Br J Clin Pharmacol. 2013;75:146‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Delor I, Charoin JE, Gieschke R, Retout S, Jacqmin P. Modeling Alzheimer's disease progression using disease onset time and disease trajectory concepts applied to CDR‐SOB scores from ADNI. CPT Pharmacometrics Syst Pharmacol. 2013;2:e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Conrado DJ, Denney WS, Chen D, Ito K. An updated Alzheimer's disease progression model: incorporating non‐linearity, beta regression, and a third‐level random effect in NONMEM. J Pharmacokinet Pharmacodyn. 2014;41:581‐598. [DOI] [PubMed] [Google Scholar]
- 18. Cho SH, Woo S, Kim C, et al. Disease progression modelling from preclinical Alzheimer's disease (AD) to AD dementia. Sci Rep. 2021;11:4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim KW, Woo SY, Kim S, et al. Disease progression modeling of Alzheimer's disease according to education level. Sci Rep. 2020;10:16808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Raket LL. Statistical disease progression modeling in Alzheimer disease. Front Big Data. 2020;3:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Holford NH, Peace K. The effect of tacrine and lecithin in Alzheimer's disease. A population pharmacodynamic analysis of five clinical trials. Eur J Clin Pharmacol. 1994;47:17‐23. [DOI] [PubMed] [Google Scholar]
- 22. Holford NH, Peace KE. Results and validation of a population pharmacodynamic model for cognitive effects in Alzheimer patients treated with tacrine. Proc Natl Acad Sci USA. 1992;89:11471‐11475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kueper JK, Speechley M, Montero‐Odasso M. The Alzheimer's Disease Assessment Scale‐Cognitive Subscale (ADAS‐cog): modifications and responsiveness in pre‐dementia populations. A narrative review. J Alzheimers Dis. 2018;63:423‐444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ueckert S, Plan EL, Ito K, et al. Improved utilization of ADAS‐cog assessment data through item response theory based pharmacometric modeling. Pharm Res. 2014;31:2152‐2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rogers JA, Polhamus D, Gillespie WR, et al. Combining patient‐level and summary‐level data for Alzheimer's disease modeling and simulation: a β regression meta‐analysis. J Pharmacokinet Pharmacodyn. 2012;39:479‐498. [DOI] [PubMed] [Google Scholar]
- 26. Quartino A, Polhamus D, Rogers J, Jin J. An integrated natural disease progression model of nine cognitive and biomarker endpoints in patients with Alzheimer's disease. Presentation at the Population Approach Group Europe (PAGE) Annual Meeting 2014; Alicante, Spain; Abstract 3187. https://www.page‐meeting.org/?abstract=3187. Accessed December 6, 2022
- 27. Cummings J, Cohen S, Van Dyck CH, et al. ABBY: a phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology. 2018;90:e1889‐e1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Salloway S, Cho W, Clayton D, et al. Amyloid PET imaging results from a study to evaluate the impact of crenezumab on fibrillar amyloid in patients with mild‐to‐moderate Alzheimer's disease. J Prev Alz. 2014;1:214‐296. [Google Scholar]
- 29. Klein G, Delmar P, Voyle N, et al. Gantenerumab reduces amyloid‐β plaques in patients with prodromal to moderate Alzheimer's disease: a PET substudy interim analysis. Alzheimers Res Ther. 2019;11:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ostrowitzki S, Lasser RA, Dorflinger E, et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer's disease. Alzheimers Res Ther. 2017;9:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. ClinicalTrials.gov . A study evaluating the efficacy and safety of crenezumab versus placebo in participants with prodromal to mild Alzheimer's disease (AD). (CREAD). ClinicalTrials.gov Identifier: NCT02670083. https://clinicaltrials.gov/ct2/show/NCT02670083. Accessed December 6, 2022
- 32. ClinicalTrials.gov . A study of crenezumab versus placebo to evaluate the efficacy and safety in participants with prodromal to mild Alzheimer's disease (AD) (CREAD 2). ClinicalTrials.gov Identifier: NCT03114657. https://clinicaltrials.gov/ct2/show/NCT03114657. Accessed December 6, 2022.
- 33. Salloway S, Honigberg LA, Cho W, et al. Amyloid positron emission tomography and cerebrospinal fluid results from a crenezumab anti‐amyloid‐beta antibody double‐blind, placebo‐controlled, randomized phase II study in mild‐to‐moderate Alzheimer's disease (BLAZE). Alzheimers Res Ther. 2018;10:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weiner MW, Aisen PS, Jack CR Jr, et al. The Alzheimer's disease neuroimaging initiative: progress report and future plans. Alzheimers Dement. 2010;6:202‐211.e207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McDougall F, Edgar C, Mertes M, et al. Psychometric properties of the Clinical Dementia Rating – Sum of Boxes and other cognitive and functional outcomes in a prodromal Alzheimer's disease population. J Prev Alzheimers Dis. 2021;8:151‐160. [DOI] [PubMed] [Google Scholar]
- 36. Retout S, Gieschke R, Weber C, et al. The importance of understanding the variable rate of progression among Alzheimer's disease patients: data from the gantenerumab program. J Prev Alzheimers Dis. 2015;2:LB13. [Google Scholar]
- 37. Keizer RJ, Karlsson MO, Hooker A. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometrics Syst Pharmacol. 2013;2:e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Owen JS, Fiedler‐Kelly J. PK/PD models. Introduction to Population Pharmacokinetic/Pharmacodynamic Analysis with Nonlinear Mixed Effects Models. John Wiley & Sons, Inc.; 2014:250‐264. [Google Scholar]
- 39. Nguyen TH, Mouksassi MS, Holford N, et al. Model evaluation of continuous data pharmacometric models: metrics and graphics. CPT Pharmacometrics Syst Pharmacol. 2017;6:87‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chanu P, Marchand M, Vadhavkar S, et al. A disease progression model for geographic atrophy. Presentation at the Population Approach Group Europe (PAGE) Annual Meeting 2019; Stockholm, Sweden; Abstract 9184. https://www.page‐meeting.org/default.asp?abstract=9184. Accessed December 6, 2022.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Data S2