

Abstract
Clinical trials seeking type 1 diabetes prevention are challenging in terms of identifying patient populations likely to progress to type 1 diabetes within limited (i.e., short‐term) trial durations. Hence, we sought to improve such efforts by developing a quantitative disease progression model for type 1 diabetes. Individual‐level data obtained from the TrialNet Pathway to Prevention and The Environmental Determinants of Diabetes in the Young natural history studies were used to develop a joint model that links the longitudinal glycemic measure to the timing of type 1 diabetes diagnosis. Baseline covariates were assessed using a stepwise covariate modeling approach. Our study focused on individuals at risk of developing type 1 diabetes with the presence of two or more diabetes‐related autoantibodies (AAbs). The developed model successfully quantified how patient features measured at baseline, including HbA1c and the presence of different AAbs, alter the timing of type 1 diabetes diagnosis with reasonable accuracy and precision (<30% RSE). In addition, selected covariates were statistically significant (p < 0.0001 Wald test). The Weibull model best captured the timing to type 1 diabetes diagnosis. The 2‐h oral glucose tolerance values assessed at each visit were included as a time‐varying biomarker, which was best quantified using the sigmoid maximum effect function. This model provides a framework to quantitatively predict and simulate the time to type 1 diabetes diagnosis in individuals at risk of developing the disease and thus, aligns with the needs of pharmaceutical companies and scientists seeking to advance therapies aimed at interdicting the disease process.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Type 1 diabetes (T1D) prevention clinical trials are particularly challenging in terms of identifying patient populations with a high likelihood of progression to clinical T1D over a narrow window of time. Clinical trials in the T1D prevention space must be optimized to account for the presence of different antibody numbers, types, titers, and additional patient features.
WHAT QUESTION DID THIS STUDY ADDRESS?
The present work developed a quantitative T1D disease progression joint model using individual‐level data obtained from natural history studies to inform T1D prevention trial designs.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
The developed disease progression model can accurately predict the time‐to‐T1D diagnosis of individuals at risk of T1D by combining different subjects' characteristics defined at the derived baseline.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
The use of the developed model has the potential to optimize T1D prevention study designs; for instance, it could reduce the time of the trial and/or the number of enrolled individuals needed.
INTRODUCTION
Type 1 diabetes (T1D) is a chronic autoimmune disease characterized by immune‐mediated destruction of insulin‐producing beta cells and a lifelong dependence on exogenous insulin to avoid diabetic ketoacidosis, hyperglycemia, and long‐term microvascular and macrovascular diabetes complications. The incidence and prevalence of T1D are increasing worldwide 1 ; with projections showing that the number of younger individuals with T1D in the United States will be nearly tripled by 2050 in comparison with 2010. 2 , 3 At the same time, over half of people with T1D are adults underscoring the need to recognize this disease at any age. 4
Over the past several decades, substantial progress has been made in the effort to better understand T1D pathogenesis. For example, a new disease progression staging paradigm that differentiates the development of T1D into three phases was proposed in 2015 5 and is widely accepted by the American Diabetes Association, International Society for Pediatric and Adolescent Diabetes, and other major diabetes organizations worldwide. 6 , 7 , 8 In this paradigm, stage 1 is characterized by the presence of two or more islet autoantibodies (AAbs) with normoglycemia and no clinical symptoms. Progression to stage 2 is characterized by progression from normoglycemia to dysglycemia in the context of ongoing autoimmunity and carries a 5‐year risk of symptomatic disease of ~75%. 9 Finally, stage 3 is defined as the clinical diagnosis of the disease.
Despite many advances in the study of the pathophysiology and progression of T1D, there is currently just one recently approved therapy to delay the onset of the disease, 10 and none to prevent it. In fact, insulin replacement therapy, which was discovered over a century ago and transformed T1D from an acutely fatal disease into a chronic one (because it is not a cure), remains the primary treatment for T1D. 11 Drug development for T1D prevention is active, and, consequently, several clinical trials are being conducted to foster therapies intended to halt or prevent the disease. Subject inclusion criteria for T1D prevention trials frequently, but not always, include: (i) presence of multiple AAbs and (ii) having a first‐degree relative with T1D. However, there is a clear need to better identify patients likely to respond to prophylactic treatments prior to a T1D diagnosis. Specifically, Warshauer and collaborators 12 have pointed out on the need to identify biomarkers for the purpose of enriching T1D prevention trials for potential treatment responders. Therefore, in the context of drug development, existing and emerging biomarkers could be better leveraged to develop a quantitative understanding of when T1D onset will likely occur, but there is still much room for improvement in this area.
Disease‐drug‐trial models are uniquely positioned to meet the before‐mentioned need of T1D prevention trials because they integrate information on the disease processes, treatment effects, and variability therein in a strictly quantitative fashion. The use of such models has become standard in drug development and regulatory evaluation and is referred to as model‐informed drug development. 13 , 14 The application of these models in quantitative model‐based clinical trial simulation (CTS) tools holds great potential for T1D drug development because it is neither time‐ nor cost‐efficient to evaluate all possible scenarios in head‐to‐head clinical trials. CTS tools have proven particularly valuable for optimizing clinical trial design by informing the selection of biomarkers or surrogate end points, inclusion/exclusion criteria, optimal number of participants, trial duration, and frequency of observations. 15 They also allow drug developers to simulate possible trial design scenarios prior to trial execution enabling sponsors to optimize trial design characteristics, reducing the overall costs of drug development, and increasing the ability to detect efficacy. Some of these tools have been generated in public‐private partnerships that include industry, academia, and regulatory agencies. 16 , 17 Once developed and verified, these tools have undergone review through formal regulatory pathways at the US Food and Drug Administration and the European Medicines Agency.
The objective of this study was to inform future T1D prevention trial designs by developing a T1D disease progression model. The model aims to enable optimization of study designs by accounting for relevant sources of variability including patient demographics (e.g., age, sex, and first‐degree relative status) and biomarkers (e.g., presence of AAbs, glycemic measures, and genetics) to predict the time to T1D diagnosis. A user‐friendly T1D prevention CTS tool will be developed utilizing this disease progression joint model in order to make it available to the non‐modeler audience.
METHODS
Data
Individual‐level data from the T1D TrialNet Pathway to Prevention (PTP, TN01) and The Environmental Determinants of Diabetes in the Young (TEDDY) natural history studies were used for model development. TrialNet is an international network dedicated to studying and preventing T1D, with centers located throughout the United States, Canada, Finland, the United Kingdom, Italy, Germany, Sweden, Australia, and New Zealand. 18 , 19 TrialNet PTP screened relatives of individuals with T1D for the presence of circulating AAbs, and those identified at risk for developing T1D received close monitoring until the diagnosis of the disease. During the follow‐up (annual or semi‐annual depending on risk level), individuals had regular assessment visits where blood samples were collected for continued evaluation of AAb status. In addition, the collection of longitudinal samples, the determination of metabolic status (e.g., dysglycemia), and the comprehensive collection of metadata were performed during the visits. 20 On the other hand, TEDDY is a birth cohort and includes centers in the United States, Finland, Sweden, and Germany, where 424,788 newborns were screened for HLA‐DR‐DQ genotypes giving increased T1D risk. 21 , 22 , 23 Participating children completed their initial study visit by 4 months of age, and this closed cohort (n = 8676) was followed for the appearance of AAbs and/or T1D diagnosis. Clinic visits occur quarterly until age 4 years, and then every 6 months through age 15 years. Children with one or more persistent AAbs were followed quarterly, and an Oral Glucose Tolerance Test (OGTT) was performed on these children every 6 months. In addition, the participants completed additional questionnaires and anthropometric measurements at each visit. 22 Data from both PTP and TEDDY studies were combined in this study to create a unique database for modeling. In addition, placebo data from the phase II randomized controlled TrialNet TN10 Anti‐CD3 (Teplizumab) Prevention Trial 24 was further used for additional validation of the final disease progression model.
Modeling variables, time metric, and covariates
The 2‐h OGTT (GLU120) measures the body's ability to control the blood glucose level after drinking a standardized glucose drink (1.75 g/kg of body weight up to a maximum of 75 g 25 ). Being used to classify glucose tolerance status as either normal or impaired, the GLU120 is one of the diagnostic criteria for diabetes. 26 Additionally, one of the typical primary outcomes in T1D prevention clinical trials is the measurement of time‐to‐T1D diagnosis. 24 , 27 Because GLU120 and time‐to‐T1D diagnosis were among the variables available in the database, they were both selected for model fitting.
The time metric used for the disease progression model was time in years after a derived baseline visit, which was defined as the earliest timepoint from study entry at which the subject had multiple (≥2) AAbs and non‐missing information for the covariates evaluated (see Table 1 for covariates tested). Specifically, the derived baseline was identified after a retrospective analysis of the data based on two key findings; (1) in both TEDDY and PTP, data were frequently missing for glycemic measures (OGTT and HbA1c); and (2) non‐missing information for glycemic measures was recorded when subjects began experiencing dysglycemia. Under this definition, the profile of a subject became much more homogenous between both studies, despite TEDDY being a birth cohort and PTP being a cross‐sectional study. In both studies, longitudinal assessments of AAb status were generally non‐missing. Therefore, a data curation process was done using the combined database of PTP and TEDDY natural history studies (Figure 1), where individuals must contain all features/covariates to be considered at the derived baseline. Individuals whose measures at the derived baseline indicate diabetes were further excluded (i.e., GLU120 ≥ 200 mg/dL, fasting glucose ≥126 mg/dL, or HbA1c ≥ 6.5%). 28 Presence of greater than or equal to two AAbs at the derived baseline was requested for individuals to be included in the analysis. Additionally, from the time of the derived baseline, the duration of follow‐up was truncated at 6 years after which data became sparse. The derived baseline results in a population of individuals who are likely to enter T1D prevention trials, because they are probable to start showing dysglycemia. Such individuals are potentially to progress to T1D within a typical trial duration (<2 years). 5
TABLE 1.
Summary of the curated dataset.
| Variable description | Units | TEDDY | TrialNet | Total |
|---|---|---|---|---|
| Numerical variables, n | ||||
| Number of individuals | 259 | 1006 | 1265 | |
| Continuous variables, mean (SD) | ||||
| Age at derived baseline | Years | 7.3 (2.1) | 14.1 (10.4) | 12.7 (9.7) |
| Stimulated glucose measurement (120 min of OGTT) at baseline | mg/dL | 107 (24.6) | 118.6 (28.6) | 116.3 (28.2) |
| Fasting glucose measurement (0 min of OGTT) at baseline | mg/dL | 87.4 (8.7) | 89.1 (9.4) | 88.8 (9.2) |
| Glycated hemoglobin result at baseline | % | 5.2 (0.2) | 5.1 (0.3) | 5.1 (0.3) |
| C peptide level at baseline (0 min of OGTT). | ng/mL | 1.1 (0.5) | 1.6 (0.8) | 1.5 (0.8) |
| C peptide level at baseline (120 min of OGTT). | ng/mL | 3.9 (1.8) | 6.1 (3.1) | 5.7 (3) |
| GLU120 – GLU0 at baseline | mg/dL | 19.7 (25.4) | 29.5 (29.5) | 27.5 (29) |
| PEP120 – PEP0 at baseline | ng/mL | 2.8 (1.6) | 4.6 (2.6) | 4.2 (2.5) |
| GLU120/GLU0 at baseline | – | 1.2 (0.3) | 1.3 (0.4) | 1.3 (0.4) |
| PEP120/PEP0 at baseline | – | 4 (2.2) | 4.3 (2.1) | 4.3 (2.1) |
| Body mass index at baseline | kg/m2 | 16.8 (2.8) | 20.9 (9.2) | 20.1 (8.4) |
| Categorical variables, n [it represents the number of individuals in each category indicator = 1.] (%) [percentage w.r.t. corresponding dataset] | ||||
| GADA AAb presence at baseline |
0 = no, 1 = yes |
223 (86.1) | 911 (90.6) | 1134 (89.6) |
| IA2A AAb presence at baseline |
0 = no, 1 = yes |
161 (62.2) | 622 (61.8) | 783 (61.9) |
| IAA AAb presence at baseline |
0 = no, 1 = yes |
155 (59.8) | 511 (50.8) | 666 (52.6) |
| ZnT8 AAb presence at baseline |
0 = no, 1 = yes |
172 (66.4) | 725 (72.1) | 897 (70.9) |
| Presence of at least 2 AAbs at baseline |
0 = no, 1 = yes |
125 (48.3) | 443 (44.0) | 568 (44.9) |
| Presence of at least 3 AAbs at baseline |
0 = no, 1 = yes |
75 (29.0) | 369 (36.7) | 444 (35.1) |
| Presence of at least 4 AAbs at baseline |
0 = no, 1 = yes |
59 (22.8) | 194 (19.3) | 253 (20.0) |
| Subject has FDR with T1D |
0 = no, 1 = yes |
40 (15.4) | 1006 (100) | 1046 (82.7) |
| Subject's sex |
0 = female, 1 = male |
156 (60.2) | 554 (55.1) | 710 (56.1) |
| HLA category as determined by TEDDY's increased risk groups. The high‐risk genotypes for participants screened from the general population were as follows: DR3‐DQ2.5/4‐DQ8, DR4/4, DR4/8, and DR3/3. An additional six genotypes were included for FDR to a subject with T1D: DR4/4, DR4/1, DR4/13, DR4/4, DR4/9, and DR3/9 |
0 = not increased risk, 1 = increased risk |
259 (100) | 364 (36.2) | 623 (49.2) |
| Presence of GADA and IAA AAbs at baseline |
0 = no, 1 = yes |
39 (15.1) | 137 (13.6) | 176 (13.9) |
| Presence of GADA and IA2A AAbs at baseline |
0 = no, 1 = yes |
26 (10.0) | 92 (9.1) | 118 (9.3) |
| Presence of GADA and ZNT8 AAbs at baseline |
0 = no, 1 = yes |
37 (14.3) | 149 (14.8) | 186 (14.7) |
| Presence of IA2A and IAA AAbs at baseline |
0 = no, 1 = yes |
5 (1.9) | 6 (0.6) | 11 (0.9) |
| Presence of IA2A and ZNT8 AAbs at baseline |
0 = no, 1 = yes |
13 (5.0) | 42 (4.2) | 55 (4.3) |
| Presence of IAA and ZNT8 AAbs at baseline |
0 = no, 1 = yes |
5 (1.9) | 17 (1.7) | 22 (1.7) |
| Presence of GADA, IA2A, and IAA AAbs at baseline |
0 = no, 1 = yes |
17 (6.6) | 46 (4.6) | 63 (5.0) |
| Presence of GADA, IAA, and ZNT8 AAbs at baseline |
0 = no, 1 = yes |
17 (6.6) | 81 (8.1) | 98 (7.7) |
| Presence of GADA, IA2A, and ZNT8 AAbs at baseline |
0 = no, 1 = yes |
28 (10.8) | 212 (21.1) | 240 (19.0) |
| Presence of IA2A, IAA, and ZNT8 AAbs at baseline |
0 = no, 1 = yes |
13 (5.0) | 30 (3.0) | 43 (3.4) |
| Presence of GADA, IA2A, IAA, and ZNT8 AAbs at baseline |
0 = no, 1 = yes |
59 (22.8) | 194 (19.3) | 253 (20.0) |
Abbreviations: AAb, autoantibody; FDR, first degree relative; GADA, glutamic acid decarboxylase autoantibody; GLU0, Fasting glucose measurement (0 min of OGTT); GLU120, stimulated glucose measurement (120 min of OGTT); HLA, human leukocyte antigen; IA2A, islet antigen‐2 autoantibody; IAA, insulin autoantibody; OGTT, oral glucose tolerance test; PEP0, C peptide level at 0 min of OGTT; PEP120, C peptide level at 120 min of OGTT; SD, standard deviation; T1D, type 1 diabetes; T1D, type 1 diabetes; TEDDY, The Environmental Determinants of Diabetes in the Young; ZnT8, zinc transporter‐8 autoantibody.
FIGURE 1.
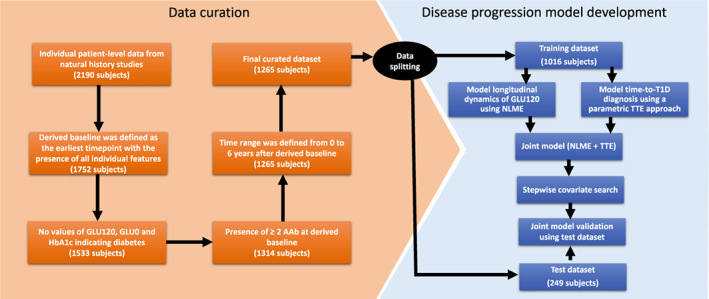
Data curation and disease progression model development workflow. AAb, autoantibody; GLU120, 120 min of oral glucose tolerance test; GLU0, fasting glucose measurement (0 min of oral glucose tolerance test); HbA1c, glycated hemoglobin; NLME, nonlinear mixed effects; T1D, type 1 diabetes; TTE, time to event.
The curated combined database was divided randomly into training and test datasets, whereas considering the balanced distribution of the covariates. Approximately 80% of the individuals were assigned to the training dataset for model development, whereas the remaining individuals were set apart for the test dataset for model validation (i.e., 80:20 train‐test split; Figure 1).
Disease progression model
Structural model development
In joint models created to track disease progression, changes are typically determined by repeatedly measuring a disease‐relevant biomarker (i.e., GLU120) and establishing the effect of the biomarker on the time‐to‐event (TTE) of interest (i.e., T1D diagnosis). The joint modeling framework proposes that the relative risk model for the TTE outcome is dependent upon the true underlying value of the longitudinal outcome, which is modeled using a nonlinear mixed effect (NLME) model, and where estimations are based on the joint distribution of both outcomes. 29 Therefore, in this study, the individual‐level longitudinal GLU120 values were incorporated as a time‐varying biomarker affecting one of the TTE model parameters. The longitudinal changes in GLU120 were quantified using a model equation, and all joint model parameters were simultaneously estimated.
Specifically, the NLME modeling approach was used to model the dynamics of the longitudinal time‐varying biomarker, GLU120, which allows quantifying random effects from between‐subject variability (BSV) and residual variability (RV). Several mathematical functions were tested as a structural model, including linear, quadratic, maximum effect (E max), and sigmoid E max functions. Structural model parameter distributions were considered lognormal. Several residual error model structures were tested, such as additive, proportional, and combined. Correlation between model parameters was also tested.
The time‐to‐T1D diagnosis was captured by a parametric TTE modeling approach. Several hazard functions were tested as a structural model, such as exponential, Weibull, log‐logistic, uniform, Gompertz, gamma, and generalized gamma functions.
After each of the GLU120 and time‐to‐T1D was modeled using the NLME and TTE approaches, respectively, these two components combined to a joint model. To quantify the association, we tested connections between parameters from each model of the two components by considering clinical plausibility as well as statistical metrics comparing the prediction and empirical data.
Covariate model development
Covariates measured at the derived baseline were assessed in both components using a classic stepwise covariate modeling (SCM) method where a set of iterations of forward selection were followed by a set of iterations of backward removal. The thresholds for the p values for the covariate forward addition and backward elimination were 0.05 and 0.01, respectively. 30 Finally, a rational criterion for covariate inclusion was applied based on previous knowledge of the disease. Covariates were incorporated into the models using a power function (see Equations S1 and S2), and, in the case of continuous covariates, they were centered by their weighted mean values (see Supplementary Material S2. Model Project File).
Model evaluation and selection
The model evaluation and selection were based on several criteria, such as the visual inspection of the goodness of fit (GOF) plots based on predictions and residuals, the Akaike Information Criterion (AIC), precision of parameter estimates, and successful minimization of the model, visual predictive checks (VPCs), and reasonable clinical explanation of the association.
The final disease progression covariate joint model was validated using internal hold‐out among the combined dataset of the PTP and TEDDY databases and further validated using TN10 database. The simulation‐based VPC plots were obtained based on 200 simulations. A workflow summarizing the disease progression model development process is shown in Figure 1.
Software
Monolix software (version 2021R1; Lixoft, Antony, France) was used for model development and R programming language (version 3.6.1) 31 was used for data processing and visualization.
RESULTS
Data summary
The combined and curated dataset included 1265 individuals with greater than or equal to two T1D‐related AAbs and 8341 observations for GLU120 measurements (age at baseline, 12.71 ± 9.69 years; age range at baseline, 1–56 years). The final dataset used for model development is summarized in Table 1. The training and test dataset included 1016 and 249 individuals, respectively. Figure 2 visualizes the maintained distributions of the covariates in both datasets.
FIGURE 2.
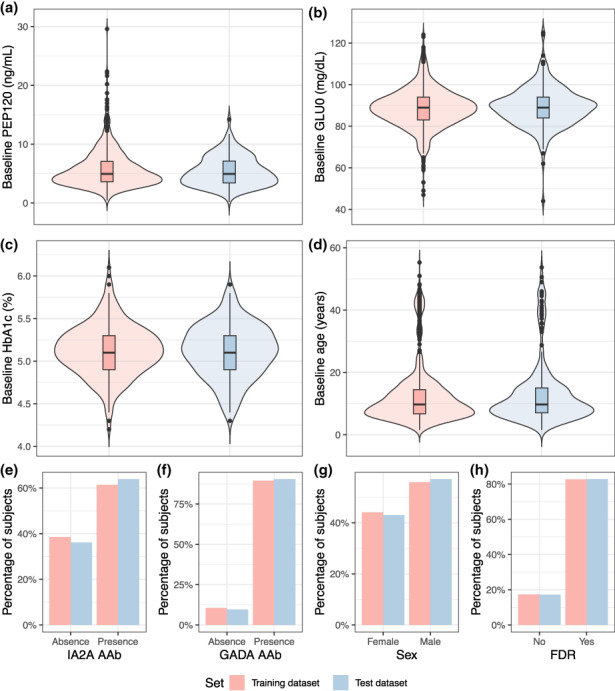
Distribution of covariates in the training and test datasets at derived baseline. FDR, first degree relative with type 1 diabetes; GADA AAb, glutamic acid decarboxylase autoantibody; GLU0, fasting glucose measurement (0 min of oral glucose tolerance test); HbA1c, glycated hemoglobin; IA2A AAb, islet antigen‐2 autoantibody; PEP120, C peptide level (120 min of oral glucose tolerance test).
Disease progression joint model
The sigmoid E max model was selected as the base model structure that best captured the longitudinal dynamics of GLU120 NLME model (Equation 1) based on the AIC criteria (see Table S1).
| (1) |
where t is the years from the derived baseline when GLU120 is measured for the individual i, and DP 0i , DP maxi , DP 50i , and represent the baseline score of GLU120, the maximum GLU120 change, the time producing 50% of DP max , and the steepness of the curve of the individual i, respectively.
The selection of the TTE model based on the AIC criteria was a Weibull model, given that there were no significant differences among Weibull, gamma, and log‐logistic models (see Table S2). The Weibull model was chosen as the base model structure for its simplicity (Equation 2).
| (2) |
where scale (Te) and shape (p) are the structural parameters of the TTE model. The scale parameter represents the time at which the survival value is ~0.4. The hazard function h(t) is the instantaneous rate of an event, given that it has not already occurred. This function is linked with the survival function S(t), which is the probability that the event happens after time t, by the following equation:
| (3) |
The final joint base model structure was defined in Equation 4, where the output of the longitudinal NLME model, GLU120(t)i, was incorporated into the TTE model to modify the Weibull scale parameter, Te. This incorporation was made in a way that, at a given time t, an increment on the result of the GLU120 model decreases the scale parameter Te (Figure 3), which translates into an augmentation of the hazard rate of the TTE part of the developed joint model, and therefore, the risk of T1D diagnosis increases. Conceptually, this implies that dysglycemia modifies the risk of a T1D diagnosis in a time‐dependent manner.
| (4) |
FIGURE 3.
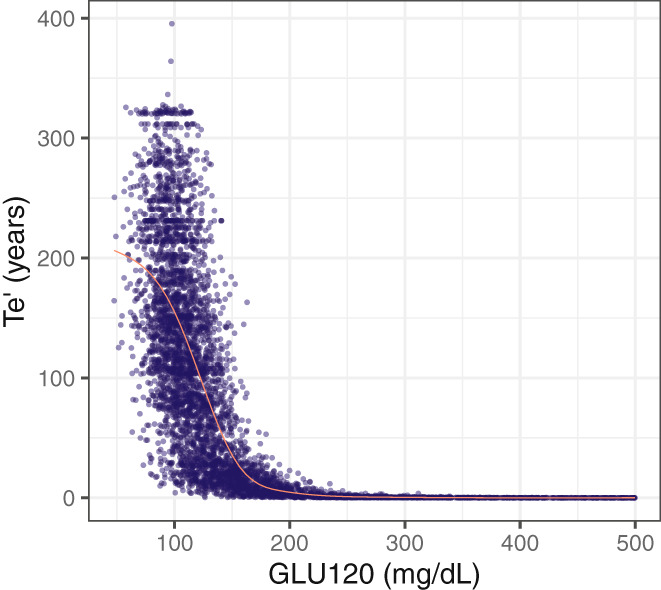
Effect of the time‐varying biomarker GLU120 on the Weibull scale parameter Te′. The data shown in this figure was obtained from 100 simulations for one subject. The red line represents the smooth line obtained using the generalized additive model smoothing method. GLU120, 120 min of oral glucose tolerance test; Te′, scale parameter of Weibull function.
Five covariates were added to the joint model through the SCM analysis, among the investigated covariates listed in Table 1. The baseline presence of glutamic acid decarboxylase 65 AAb (GADA) and the baseline value of HbA1c were associated with DPmax; the baseline presence of insulinoma associated protein‐2 AAb (IA2A) was associated with DP50; and GLU0 as well as GLUratio were associated with DP0. Table 2 summarizes the final parameter estimates of the joint model with the selected covariates, along with the interpretation of the parameters. The final model codes written in Monolix are available in Supplementary Materials S2 and S3.
TABLE 2.
Final parameter estimates of the joint model.
| Parameter | Value [%CV, %Shrinkage] | %RSE | Interpretation of parameter |
|---|---|---|---|
| Fixed effects | |||
| Typical value of DP50 | 6.09 years | 12.2 | Time producing 50% of DPmax |
| Typical value of DPmax | 208.72 mg/dL | 23.9 | Maximum GLU120 change |
| Typical value of γ (gamma) | 1.75 | 21.5 | Define the steepness of the curve |
| Typical value of DP0 | 107.31 mg/dL | 0.414 | Baseline GLU120 |
| Te | 91.81 years | 4.33 | Weibull scale parameter |
| p | 1.20 | 0.683 | Weibull shape parameter |
| β_Te | 6.13 | 2.29 | Parameter measuring the influence of longitudinal GLU120 in Te |
| β_DPmax_BSCORE_GADA_1 † | −0.73 | 29.5 | Presence of GADA AAb at the derived baseline decreases the risk of T1D diagnosis |
| β_DPmax_logtBSCORE_HbA1c † | 8.92 | 13.2 | A higher glycated hemoglobin level at the derived baseline increases the risk of T1D diagnosis |
| β_DP50_BSCORE_IA2A_1 † | −0.71 | 16.5 | Presence of IA2A AAb at the derived baseline increases the risk of T1D diagnosis |
| β_DP0_logtBSCORE_GLU0 † | 0.88 | 4.39 | A higher oral glucose tolerance test (0 min) level at derived baseline increases the risk of T1D diagnosis |
| β_DP0_logtBSCORE_GLURATIO † | 0.79 | 1.71 | A higher GLU120/GLU0 ratio at the derived baseline increases the risk of T1D diagnosis |
| Random effects | |||
| BSV DP50 | 0.72 [82, 77.4] | 10.1 | Random effect parameters representing standard deviations of inter‐individual random variables for the parameters [the coefficient of variation (%) and the shrinkage (%)] |
| BSV DPmax | 1.69 [405, 54.4] | 8.58 | |
| BSV γ (gamma) | 1.76 [460, 51.5] | 10.3 | |
| BSV DP0 | 0.043 [4, 68] | 8.099 | |
| RV proportional error | 0.16 | 1.23 | A proportional error model parameter added to describe residual variability |
Abbreviations: AAb, autoantibody; BSCORE, baseline value; BSV, between‐subject variability; GADA, glutamic acid decarboxylase autoantibody; GLU0, 0 min of oral glucose tolerance test; GLU120, 120 min of oral glucose tolerance test; IA2A, islet antigen‐2 autoantibody; RSE, relative standard error; RV, random variability; T1D, type 1 diabetes.
p < 0.0001 (Wald test).
For internal validation, visual inspection of standard GOF (see Figure S1) and VPC plots (see Figure 4a,b) on the training dataset revealed no visible bias and show good agreement between observed and predicted data. Additionally, the VPC plots for the longitudinal and TTE parts were generated for the test dataset and show good predictive performance of the final model (see Figures 4c,d).
FIGURE 4.
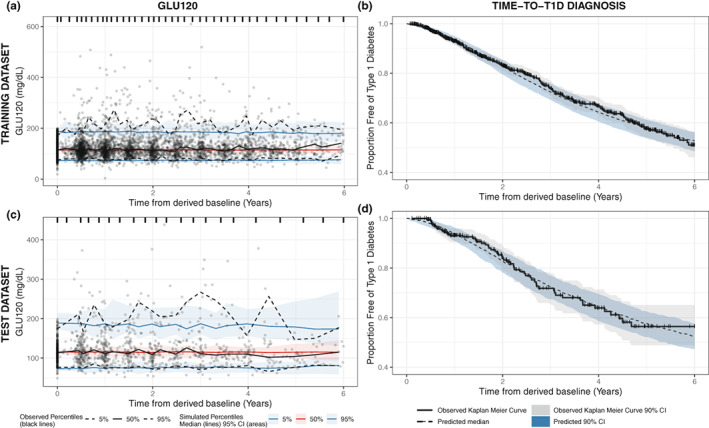
Visual predictive check model diagnostic plots of the final T1D disease progression joint model using training dataset (top panel) and test dataset (bottom panel), for the longitudinal glucose measure (left panel) and time to T1D diagnosis (right panel). The visual predictive check plots (a) and (c) show the median (black solid line) and the 5th and 95th percentiles (lower and upper black dashed lines, respectively) of the observed data. The red shaded areas indicate the 95% CIs of the model prediction of the median, and the blue shaded areas show 95% CIs of the model prediction for the 5th and 95th percentiles. The solid lines – red for the median and blue for the 5th and 95th percentiles – represent the model prediction. The visual predictive check plots (b) and (d) show the Kaplan Meier curve (black solid line) of the observed data. The gray shaded area indicates the 90% CIs of the observed Kaplan Meier curve. The black dashed line represents the median of the model prediction. The blue shaded areas indicate the 90% CIs of the model prediction of the median. CI, confidence interval; GLU120, 120 min of oral glucose tolerance test; T1D, type 1 diabetes.
The validation results using TN10 dataset were visualized in Figure S2. It was performed using the complete (n = 32; see Figure S2a,b) and reduced (n = 24; see Figure S2c,d) TN10 trial placebo group data. For the reduced validation dataset, eight individuals were excluded as they were diagnosed with T1D before 4 months since randomization. Although the GLU120 model was able to make decent predictions in spite of the small sample size, the TTE model showed some discrepancies.
Subject features influencing the likelihood of eventual T1D diagnosis
In order to understand the influence of the covariates incorporated into the final joint disease progression model, survival curves were simulated for virtual individuals with different features. Figure 5 visualizes the predicted survival curves that were stratified by the selected covariates. Four different datasets were created where the only characteristic, measured at the derived baseline, that changed were the presence of GADA (Figure 5a), the presence of IA2A (Figure 5b), and HbA1c (Figure 5c), respectively. The presence of IA2A and a high HbA1c at the derived baseline increased the risk of T1D diagnosis, as it is visualized in the survival curves of Figure 5b,c. On the other hand, Figure 5a shows how the presence of GADA at the derived baseline affects the Kaplan–Meier curves, decreasing the risk of T1D diagnosis.
FIGURE 5.
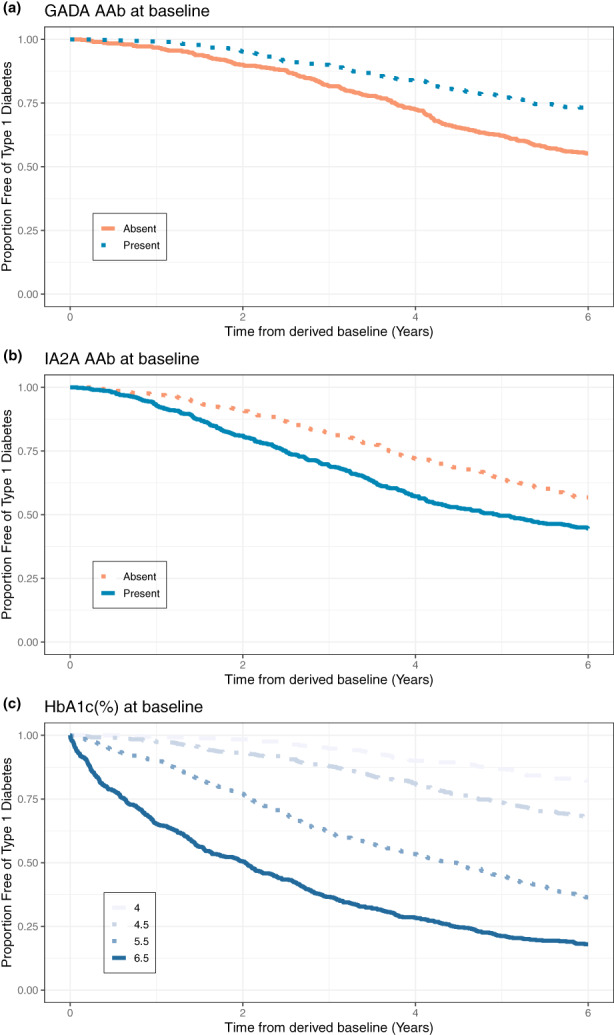
Predicated influence of selected covariates to the time to T1D diagnosis. GADA, glutamic acid decarboxylase autoantibody; HbA1c, glycated hemoglobin; IA2A, islet antigen‐2 autoantibody.
DISCUSSION
T1D prevention clinical trials are challenging especially in terms of identifying patient subgroups which are likely to reach a T1D diagnosis during the trial. Although it has long been established that AAbs are pathologic predictors of T1D, the presence of different AAb combinations is associated with highly variable durations until diagnosis. As a result, there is a need to optimize clinical trials in order to simultaneously account for patient factors (i.e., age) and disease factors (i.e., presence of AAbs) that drive the progression of the disease. The use of available patient‐level data which captures such features offers the potential to define quantitative descriptions of disease progression. This work utilized data from two T1D natural history studies (PTP and TEDDY) to develop a T1D disease progression model that can be used to inform T1D prevention trial designs. A joint disease progression model was developed that linked a longitudinal glycemic measure (GLU120) to timing of T1D diagnosis through longitudinal and TTE models with other patient features measured at the derived baseline, including HbA1c and the presence of different AAbs. The use of a joint model approach instead of separate longitudinal and TTE analysis can potentially reduce parameter estimate bias and additionally permitted the inclusion of a relevant T1D disease progression longitudinal biomarker (GLU120) measured with random effects (i.e., BSV and RV) into the TTE model. 32 Essentially, this innovative model provides the ability to quantitatively predict and simulate the time to T1D diagnosis in individuals at risk of developing the disease.
The combined effect of multiple covariates on the time to T1D diagnosis was quantified. Of the five covariates added to the model, one of them decreased the risk of T1D diagnosis, and four of them increased that risk. The presence of GADA at the derived baseline was the selected covariate that decreased the risk of T1D diagnosis. It is important to remark that the analyzed population dataset was ensembled with individuals that had greater than or equal to two T1D‐related AAbs. In this context, lower risk due to the presence of GADA at baseline does not contradict the evidence that GADA is a factor that increases the risk of T1D in the general population. 33 In addition, this finding is consistent with published previous works, where the presence of GADA as a primary AAb (i.e., the first islet AAb detected) is associated with a slower disease progression relative to the presence of insulin as the primary AAb, for example. 33 , 34 , 35 The presence of IA2A, higher HbA1c, higher GLU0, and higher GLUratio at derived baseline were the covariates added that increased the risk of T1D diagnosis. This is in concordance with the hypothesis that IA2A positivity is identified in subjects at more advanced phases in the process of beta‐cell damage and, therefore, closer to clinical diagnosis. 35 HbA1c, GLU0, and GLUratio at the derived baseline are each related to blood glucose control impairment. Consequently, these covariates make sense in the advancing of T1D disease progression.
Nevertheless, we acknowledge the limitations of our analysis. The joint disease progression model should be applied just to individuals at risk of developing T1D because that was the population used for model fitting. Any prediction on individuals that do not fulfill this requirement is an extrapolation, and there is a lack of confidence in the results. In addition, the validation results using TN10 dataset showed some discrepancies between observed and predicted measures. It is important to remark that the number of subjects in the validation dataset was limited (i.e., a total of 32 subjects). Twenty‐five percent of them (8 individuals) were diagnosed with T1D within 4 months since randomization, which was different from our curated dataset that was used to develop the joint disease progression model. In fact, in the training dataset, only 10 individuals (<1% of the training set) were diagnosed with T1D within 4 months since the derived baseline. In addition, the validation dataset is not fully independent of the data used for model development because participants of TrialNet TN10 Anti‐CD3 (Teplizumab) Prevention Trial could be part of the PTP population. Therefore, a more extensive external validation dataset is needed in order to be able to test the robustness of the predictions from the developed T1D disease progression model.
As a future direction, a model‐based CTS platform will be created. This tool will use the developed T1D disease progression joint model, and it could be used by drug developers to support the decision making of future T1D prevention trial designs through simulations. In addition to the disease progression model, the CTS tool will include: (1) a trial design section where the user can select, for instance, the trial duration and number of subjects per arm; (2) a trial population section where the user can choose the target population based on baseline covariate selection; and (3) a drug behavior section where the user can select a hypothetical treatment effect modifying the magnitudes of the final disease progression model parameters. Simulations of T1D prevention clinical trials, where users could define subjects' characteristics, trial design parameters, and a hypothetical drug effect for the treatment arm, could be performed. The power of the virtual trial could be computed as the ratio of the number of simulations in which the placebo and treatment arms are statistically different over the total number of simulations, providing a tangibly readable output for the drug development community.
In conclusion, the developed T1D disease progression model accurately reflects data from TEDDY and PTP natural history studies. The model was validated using the internal hold‐out approach, increasing its credibility. It was further validated using TN10 dataset, but more data are needed. The developed model quantifies the combined effect of multiple covariates on the time to T1D diagnosis. The model provides a framework to quantitatively predict and simulate the time to T1D diagnosis in individuals at risk of developing the disease and thus, aligns with the needs of drug developers and academic researchers seeking to advance therapies capable of delaying T1D disease progression. Finally, A user‐friendly T1D prevention CTS platform will be developed based on the T1D disease progression model. This graphical user interface app will have the advantage to expand the use of the outcome of this project to a broader audience.
AUTHOR CONTRIBUTIONS
J.F.M., R.M., J.P., J.B., S.D., S.S., K.R., I.O., F.M., M.C.‐T., M.J.H., M.A.A., and S.K. wrote the manuscript. J.P., J.B., K.R., I.O., and S.K. designed the research. J.F.M., R.M., and S.K. performed the research. J.F.M., R.M., J.P., J.B., S.D., P.L., S.S., K.R., I.O., F.M., M.C.‐T., M.J.H., M.A.A., and S.K. analyzed the data. J.F.M., R.M., J.P., J.B., and S.K. contributed new reagents/analytical tools.
FUNDING INFORMATION
This work was funded by the JDRF International (grant numbers: 2‐SRA‐2020‐903‐A‐N and 3‐SRA‐2022‐1157‐S‐B).
CONFLICT OF INTEREST STATEMENT
The authors declared no competing interests for this work.
Supporting information
Data S1
ACKNOWLEDGMENTS
The data from the TEDDY and TrialNet Study reported here were supplied by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Central Repositories. This paper does not necessarily reflect the opinions or views of the TEDDY, TrialNet Study, the NIDDK Central Repositories, or the NIDDK. This research was performed under the auspices of the TEDDY study group, a collaborative clinical study sponsored by the NIDDK, NIAID, NICHD, NIEHS, JDRF, and CDC. The Type 1 Diabetes TrialNet Study Group is a clinical trials network funded through a cooperative agreement by the NIH through the NIDDK, the NIAID, the NICHD, and the JDRF. The authors wish to thank Amanda Posgai (University of Florida) and Sara Williams (University of Florida) for their editorial assistance.
Morales JF, Muse R, Podichetty JT, et al. Disease progression joint model predicts time to type 1 diabetes onset: Optimizing future type 1 diabetes prevention studies. CPT Pharmacometrics Syst Pharmacol. 2023;12:1016‐1028. doi: 10.1002/psp4.12973
[Correction added on 17 May 2023, after first online publication: The ORCID ID for Jackson Burton has been added in this version.]
REFERENCES
- 1. Mobasseri M, Shirmohammadi M, Amiri T, Vahed N, Hosseini Fard H, Ghojazadeh M. Prevalence and incidence of type 1 diabetes in the world: a systematic review and meta‐analysis. Health Promot Perspect. 2020;10:98‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dabelea D, Mayer‐Davis EJ, Saydah S, et al. Prevalence of type 1 and type 2 diabetes among children and adolescents from 2001 to 2009. JAMA. 2014;311:1778‐1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Imperatore G, Boyle JP, Thompson TJ, et al. Projections of type 1 and type 2 diabetes burden in the U.S. population aged <20 years through 2050. Diabetes Care. 2012;35:2515‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thomas NJ, Jones SE, Weedon MN, Shields BM, Oram RA, Hattersley AT. Frequency and phenotype of type 1 diabetes in the first six decades of life: a cross‐sectional, genetically stratified survival analysis from UK biobank. Lancet Diabetes Endocrinol. 2018;6:122‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Insel RA, Dunne JL, Atkinson MA, et al. Staging presymptomatic type 1 diabetes: a scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care. 2015;38:1964‐1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Podichetty JT, Lang P, O’Doherty IM, et al. Leveraging real‐world data for EMA qualification of a model‐based biomarker tool to optimize Type‐1 diabetes prevention studies. Clin Pharmacol Ther. 2022;111:1133‐1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Karpen SR, Dunne JL, Frohnert BI, et al. Consortium‐based approach to receiving an EMA qualification opinion on the use of islet autoantibodies as enrichment biomarkers in type 1 diabetes clinical studies. Diabetologia. 2022;1–10:415‐424. doi: 10.1007/s00125-022-05751-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Association, A. D . 2. Classification and diagnosis of diabetes: standards of medical Care in Diabetes—2019. Diabetes Care. 2019;42:S13‐S28. [DOI] [PubMed] [Google Scholar]
- 9. Dayan CM, Korah M, Tatovic D, Bundy BN, Herold KC. Changing the landscape for type 1 diabetes: the first step to prevention. Lancet. 2019;394:1286‐1296. [DOI] [PubMed] [Google Scholar]
- 10. FDA Approves First Drug That Can Delay Onset of Type 1 Diabetes | FDA. https://www.fda.gov/news‐events/press‐announcements/fda‐approves‐first‐drug‐can‐delay‐onset‐type‐1‐diabetes
- 11. Roep BO, Montero E, Van Tienhoven R, Atkinson MA, Schatz DA, Mathieu C. Defining a cure for type 1 diabetes: a call to action. Lancet Diabetes Endocrinol. 2021;9:553‐555. [DOI] [PubMed] [Google Scholar]
- 12. Warshauer JT, Bluestone JA, Anderson MS. New Frontiers in the treatment of type 1 diabetes. Cell Metab. 2020;31:46‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marshall S, Madabushi R, Manolis E, et al. Model‐informed drug discovery and development: current industry good practice and regulatory expectations and future perspectives. CPT Pharmacometrics Syst Pharmacol. 2019;8:87‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marshall SF, Burghaus R, Cosson V, et al. Good practices in model‐informed drug discovery and development: practice, application, and documentation. CPT Pharmacometrics Syst Pharmacol. 2016;5:93‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Conrado DJ, Larkindale J, Berg A, et al. Towards regulatory endorsement of drug development tools to promote the application of model‐informed drug development in Duchenne muscular dystrophy. J Pharmacokinet Pharmacodyn. 2019;46:441‐455. [DOI] [PubMed] [Google Scholar]
- 16. Maxfield KE, Buckman‐Garner S, Parekh A. The role of public–private partnerships in catalyzing the critical path. Clin Transl Sci. 2017;10:431‐442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shahin MH, Bhattacharya S, Silva D, et al. Open data revolution in clinical research: opportunities and challenges. Clin Transl Sci. 2020;13:665‐674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sims EK, Geyer S, Johnson SB, et al. Who is enrolling? The path to monitoring in type 1 diabetes TrialNet's pathway to prevention. Diabetes Care. 2019;42:2228‐2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mahon JL, Sosenko JM, Rafkin‐Mervis L, et al. The TrialNet natural history study of the development of type 1 diabetes: objectives, design, and initial results. Pediatr Diabetes. 2009;10:97‐104. [DOI] [PubMed] [Google Scholar]
- 20. Battaglia M, Anderson MS, Buckner JH, et al. Understanding and preventing type 1 diabetes through the unique working model of TrialNet. Diabetologia. 2017;60:2139‐2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hagopian WA, Erlich H, Lernmark Å, et al. The environmental determinants of diabetes in the young (TEDDY): genetic criteria and international diabetes risk screening of 421 000 infants. Pediatr Diabetes. 2011;12:733‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rewers M, Hyöty H, Lernmark Å, et al. The environmental determinants of diabetes in the young (TEDDY) study: 2018 update. Curr Diab Rep. 2018;18:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rewers M, TEDDY Study Group . The environmental determinants of diabetes in the young (TEDDY) study. Ann N Y Acad Sci. 2008;1150:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Herold KC, Bundy BN, Long SA, et al. An anti‐CD3 antibody, teplizumab, in relatives at risk for type 1 diabetes. N Engl J Med. 2019;381:603‐613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boudreau V, Reynaud Q, Denis A, et al. Impact of 1h oral glucose tolerance test on the clinical status of adult cystic fibrosis patients over a 4‐year period. PLoS One. 2021;16:e0246897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ismail HM, Xu P, Libman IM, et al. The shape of the glucose concentration curve during an oral glucose tolerance test predicts risk for type 1 diabetes. Diabetologia. 2018;61:84‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krischer JP, Schatz DA, Bundy B, Skyler JS, Greenbaum CJ, Writing Committee for the Type 1 Diabetes TrialNet Oral Insulin Study Group . Effect of Oral insulin on prevention of diabetes in relatives of patients with type 1 diabetes: a randomized clinical trial. JAMA. 2017;318:1891‐1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Association, A. D . Diagnosis and classification of diabetes mellitus. Diabetes Care. 2014;37:S81‐S90. [DOI] [PubMed] [Google Scholar]
- 29. Papageorgiou G, Mauff K, Tomer A, Rizopoulos D. An overview of joint modeling of time‐to‐event and longitudinal outcomes. Annual Rev. 2019;6:223‐240. doi: 10.1146/annurev-statistics-030718-105048 [DOI] [Google Scholar]
- 30. Ayral G, Si Abdallah JF, Magnard C, Chauvin J. A novel method based on unbiased correlations tests for covariate selection in nonlinear mixed effects models: The COSSAC approach. CPT Pharmacometrics Syst Pharmacol. 2021;10:318‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. R Core Team . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2019. https://www.r‐project.org/ [Google Scholar]
- 32. Sudell M, Kolamunnage‐Dona R, Tudur‐Smith C. Joint models for longitudinal and time‐to‐event data: a review of reporting quality with a view to meta‐analysis. BMC Med Res Methodol. 2016;16:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jacobsen LM, Bocchino L, Evans‐Molina C, et al. The risk of progression to type 1 diabetes is highly variable in individuals with multiple autoantibodies following screening. Diabetologia. 2020;63:588‐596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gorus FK, Balti EV, Messaaoui A, et al. Twenty‐year progression rate to clinical onset according to autoantibody profile, age, and HLA‐dq genotype in a registry‐based group of children and adults with a first‐degree relative with type 1 diabetes. Diabetes Care. 2017;40:1065‐1072. [DOI] [PubMed] [Google Scholar]
- 35. So M, Speake C, Steck AK, et al. Advances in type 1 diabetes prediction using islet autoantibodies: beyond a simple count. Endocr Rev. 2021;42:584‐604. doi: 10.1210/endrev/bnab013 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1