

Abstract
Rare diseases collectively exact a high toll on society due to their sheer number and overall prevalence. Their heterogeneity, diversity, and nature pose daunting clinical challenges for both management and treatment. In this review, we discuss recent advances in clinical applications of gene therapy for rare diseases, focusing on a variety of viral and non‐viral strategies. The use of adeno‐associated virus (AAV) vectors is discussed in the context of Luxturna, licenced for the treatment of RPE65 deficiency in the retinal epithelium. Imlygic, a herpes virus vector licenced for the treatment of refractory metastatic melanoma, will be an example of oncolytic vectors developed against rare cancers. Yescarta and Kymriah will showcase the use of retrovirus and lentivirus vectors in the autologous ex vivo production of chimeric antigen receptor T cells (CAR‐T), licenced for the treatment of refractory leukaemias and lymphomas. Similar retroviral and lentiviral technology can be applied to autologous haematopoietic stem cells, exemplified by Strimvelis and Zynteglo, licenced treatments for adenosine deaminase‐severe combined immunodeficiency (ADA‐SCID) and β‐thalassaemia respectively. Antisense oligonucleotide technologies will be highlighted through Onpattro and Tegsedi, RNA interference drugs licenced for familial transthyretin (TTR) amyloidosis, and Spinraza, a splice‐switching treatment for spinal muscular atrophy (SMA). An initial comparison of the effectiveness of AAV and oligonucleotide therapies in SMA is possible with Zolgensma, an AAV serotype 9 vector, and Spinraza. Through these examples of marketed gene therapies and gene cell therapies, we will discuss the expanding applications of such novel technologies to previously intractable rare diseases.
Keywords: adeno‐associated virus vectors, antisense oligonucleotides, CAR‐T cells, retrovirus vectors
1. INTRODUCTION
This review outlines current gene therapy strategies to treat rare diseases (RDs). In‐depth analysis or a full overview of the RD field is beyond our scope, but other reviews are available. 1 , 2 , 3 Though definitions for RDs vary, the defining factor is low prevalence, typically <0.05%. 1 There are nearly 10,000 RDs that cumulatively affect over 5% of the global population, about 400 million people, thus exacting a high global health burden. 4 Their phenotype spectrum is extremely diverse, ranging from mild, for example, Inherited Macroglossia, 5 , 6 to severe, for example, Huntington's chorea 7 , 8 or adenosine deaminase—severe combined immunodeficiency (ADA‐SCID). 9 , 10 Approx. 80% of RDs involve genetic alterations, 11 and typically for each disease, there exist multiple different causative mutations with important implications for disease management. RDs also include some infectious diseases, such as Babesiosis, 12 , 13 a tick‐borne infection.
The healthcare cost for RDs is high; they can be chronic, often have devastating consequences and effective treatments are lacking, typically translating into extensive and expensive symptomatic management, including hospitalization. Undiagnosed RDs compound the problem. Without underlying cause identification, managing patient symptoms is inefficient and ineffective, worsening outcomes and increasing healthcare resource consumption. 14 , 15 , 16 Cumulatively, in developed countries RDs account for ~10% of total direct healthcare spending for a patient population of 5%–7%. 17 , 18
Research and development for disease treatments are expensive and protracted, regardless of patient numbers. For common diseases with a patient base of millions, they deliver value and can be funded largely by the patients themselves directly or indirectly. This is not the case for RDs whose patient base typically ranges from a few thousands to a few hundred thousand but can be as low as a single patient. Treatment development for RDs may not be commercially viable, but the suffering and high healthcare costs imposed by RDs, make it worthwhile for governments to step in. Initiatives such as the orphan drug designation status 19 , 20 , 21 have been instrumental in incentivizing pharmaceutical companies and spurring innovation.
As a group, the nature of RDs largely precludes small molecule therapeutics; functions of aberrant or missing genes are not readily replaced by other molecules. Successes, such as Imatinib for acute lymphoblastic leukaemia (ALL) are exceptions. 22 Biologic therapeutics such as protein supplementation can offer solutions but often fail to fully restore homeostatic balance, offering only partial symptom relief. Moreover, the development of one biologic agent benefits only modestly from work done on previous agents and their applicability is not universal. Haemophilia A, 23 , 24 affects just one protein and is effectively treated with recombinant Factor VIII. This is not the case for 47XXY (occasionally also 48XXYY) Klinefelter syndrome, 25 , 26 a congenital X and Y chromosome duplication, which profoundly impacts global gene expression patterns. Its correction is beyond current technological capabilities leaving symptomatic management as the only option.
By contrast, nucleic‐acid‐based therapies are exceptionally well‐suited to treat RDs, because (i) the nucleic‐acid payload is interchangeable, so platform and delivery developments can benefit many different disease areas; and (ii) the internal homeostatic balance is more effectively restored, either permanently or transiently depending on the technology used, to confer greater protection against the disease‐inflicted damage. In brief, gene therapy promises more effective treatments and a much more efficient therapeutic discovery process.
Here, we discuss current clinical development and practice of gene therapy for RDs. We focus almost exclusively on treatments that have received regulatory approval and are being used in people affected, contrasting them to conventional therapeutics and illustrating the wider applicability of their platforms.
Originally envisaged as alleviating or curing disease by correcting defective genes, gene therapy has evolved to encompass several therapeutic interventions (Figure 1). Genetic defects can cause disease by abolishing, reducing or increasing the expression of one or more proteins, or by creating novel proteins with altered functions (gain‐of‐function). The scale of these defects varies widely from point mutations to multi‐nucleotide deletions or insertions, gene copy number variation and karyotypic alterations. Current technology limits gene therapy to individual gene defects, but recent advances have the potential to correct larger scale abnormalities. To facilitate our study of the subject, we will divide our discussion of successful clinical applications of gene therapy into the following broad categories:
Direct modification of somatic cell DNA in vivo.
Modification of DNA in differentiated somatic cells, prior to reimplantation.
Modification of DNA in stem cells, prior to reimplantation.
Manipulation of post‐transcriptional RNA processing and translation with nucleic acid technology.
FIGURE 1.

The expanding Gene Therapy field. Originally gene therapy was envisioned as the in situ modification of genetic information of cells within tissues. The field has evolved beyond that encompassing more aspects of nucleic acid technology, particularly oligonucleotide technology, which aims to modify gene expression, without necessarily changing the cell's genetic information. The modification of a patient's cells ex vivo, outside the body prior to reimplantation has proven to be a successful clinical strategy. Although recent technological advancements have now enabled mitochondrial and germ line or embryonic cell gene therapy, these approaches are not yet being used due to safety and ethical issues.
2. DIRECT MODIFICATION OF SOMATIC CELL DNA IN VIVO
2.1. Gene supplementation in somatic and post‐mitotic tissues: Luxturna AAV‐based gene supplementation treatment for LCA2
The absence of a functional copy of a gene, key to the function of a highly differentiated tissue (e.g. lung or eye) is a common cause of RDs. Such cases lend themselves to direct addition of a functional gene copy to cells of the target tissue. This is gene supplementation: delivery of DNA containing the gene of interest to the nucleus, while ensuring its expression and persistence therein (Figure 2). Viruses can be engineered into powerful gene supplementation platforms. The basic premise is to create a custom viral genome with the gene of interest replacing viral genes and artificially package it into virions. These virions can transduce cells and deliver the target gene but cannot replicate or cause disease. As an example, we shall look at the recent successful clinical use of viral vector technology in inherited retinal dystrophies.
FIGURE 2.

Common gene supplementation strategies. (A) A gene of interest (GoI) can be incorporated into a chromosomal break, which may disrupt an existing gene, as the insertion point may be random. Examples include integrating viral vectors (RV, retrovirus vector; LV, lentivirus vector) and transposons. (B) Persistence of the new genetic material as an extrachromosomal element. Adeno‐associated virus (AAV), adenovirus (Ad) and integration‐deficient lentivirus vectors (IDLVs) are common examples. Without matrix‐attachment region and sequences directing replication, the extrachromosomal element will be diluted out through cell division. (C) Homology‐dependent repair (HDR) involves the targeted replacement of a host sequence. It is the safest method, yet also the hardest to harness.
The retinal pigment epithelium‐specific 65‐kDa protein (RPE65) is an enzyme critical for the regeneration of 11‐cis‐retinal during the visual cycle 27 , 28 (Figure 3). Without RPE65, 11‐cis‐retinal is depleted, leaving the photoreceptors unable to operate, while other intermediates in the metabolic pathway build up to potentially toxic levels. RPE65 mutations cause a spectrum of inherited retinal dystrophies, which result in blindness at birth or very early childhood. 27 , 28 The most common phenotypes are Leber's Congenital Amaurosis 29 , 30 and Retinitis Pigmentosa, 31 but other rarer phenotypes are also possible depending on the RPE65 genetic defect. 28
FIGURE 3.

The visual cycle in rod cells. The optical signal is generated by the opsin proteins with the help of 11‐cis retinal (RAL), which absorbs light, changes to all‐trans retinal and in the process activates opsin. All‐trans retinal is no longer photosensitive and needs to be converted back to 11‐cis RAL. This conversion is not carried out by the rod cells themselves but by the supporting retinal pigment epithelium (RPE). Trans‐RAL is released from opsin and since it is membrane permeable it is transported with the help of special carrier proteins (interphotoreceptor retinoid‐binding protein, IRBP) in the extracellular matrix to the RPE, where a series of specialized enzymes catalyse the conversion. The 11‐cis‐retinal product is transported back to the rod cells. Metabolic defects in the RPE enzymes block the conversion of 11‐cis retinal and lead to accumulation of various intermediates such as retinol (ROL) esters, which can reach toxic levels. External supply of 11‐cis retinal and removal of the intermediates via the blood is not sufficient to maintain vision.
Replacement of RPE65 function in the patient's eye cannot currently be achieved by means other than gene therapy and is an attractive gene therapy target (see Figure 3). An intense research and development effort culminated in the development of Voretigene neparvovec, an adeno‐associated virus (AAV) RPE65 gene replacement platform. 32 , 33 , 34 , 35 , 36 , 37 In 2017, it was approved by the US Food and Drug Administration (FDA), under the commercial name Luxturna for the treatment of type 2 Leber's Congenital Amaurosis (LCA2). 38
Adeno‐associated virus is a non‐pathogenic commensal Parvovirus, 39 , 40 , 41 whose biology makes it suitable as a gene therapy platform (Figure 4). It is unable to exit its latent stage and begin its lytic cycle spontaneously, without superinfection by another virus. 40 , 41 The natural AAV genome is capable of preferential site‐specific integration into the host genome at chromosome 19, but it can also maintain itself for long periods of time in the cell nucleus episomally (as an extra‐chromosomal element). 42 , 43 , 44 , 45 , 46 There are 12 different natural AAV variants in humans (referred to as serotypes), each with a unique type of capsid which controls their tropism, 40 , 47 that is, the type of cells it can infect. Collectively, these variants confer upon AAV a very wide tropism, which can be further extended using non‐human and genetically engineered variants. 40
FIGURE 4.
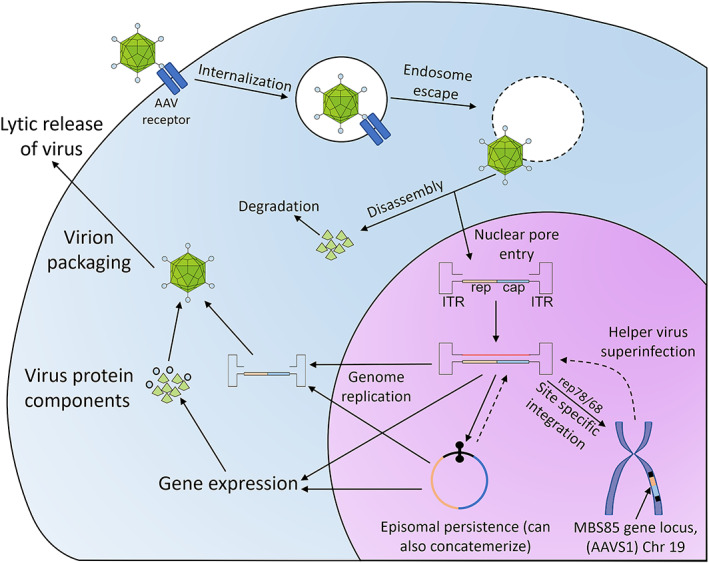
The lifecycle of adeno‐associated virus (AAV). AAV enters the cells via receptor‐mediated endocytosis and then disrupts the endosome to escape into the cytoplasm. The capsid disassembles and simultaneously passes the viral genome onto nuclear pores facilitating nuclear entry. In the nucleus, the AAV genome, which is single‐stranded, makes use of the inverted terminal repeats (ITRs) to become double‐stranded. In the absence of a concomitant helper virus, AAV goes dormant, preferentially integrating into the MBS85 locus on Chromosome 19 in a site‐specific manner that requires the AAVS1 genomic sequence. Superinfection with a helper virus reactivates AAV, allowing it to reproduce its genome and express its lytic stage proteins. Lysis of the cell by the helper virus helps AAV escape the cell.
An AAV vector is created by flanking a transgene expression cassette capable of producing the transcript of interest with viral sequences called inverted terminal repeats (ITRs). 40 The ITRs allow the viral structural proteins to package the transgene cassette into virions. These engineered virions are typically produced by the expression of the ITR‐transgene cassette in cells that are also made to express the viral structural genes, from an expression vector rather than the viral backbone. Supplying the structural genes in trans, with only the ITR‐transgene available as the genome, allows packaging of the ITR‐transgene, without including any of the structural genes in the virion and therefore with a much lower risk of producing live virus in the process. By removing the structural genes, AAV vectors can transduce to transmit the transgene, but cannot replicate and create new virions. Typically, the genes needed for site‐specific integration of the AAV genome are not supplied during packaging and are not present in the vector. AAV vectors, therefore, lack the capacity for site‐specific integration and rely on episomal maintenance, substantially reducing their potential genotoxicity. The downside of episomal maintenance is rapid loss of the viral genome in replicating cells, limiting the utility of AAV to somatic post‐mitotic cells. 40 An important constraint of AAV vectors is packaging capacity. AAV packaging has a size limit of approximately 5 kbp, this being at the low end of what viral vector systems can offer (i.e. lentiviral vectors can carry 8 kb inserts, and high‐capacity adenoviral vectors can include 37 kb). Considering all the elements (e.g. promoters, enhancers, regulatory domains) that need to be included, this is an important limitation. This size limitation is particularly salient for a second generation of AAV vectors that use the self‐complementary strategy, but we will discuss that along with an example of a self‐complementary AAV vector in Section 5.3. A key advantage of AAV vector systems is serotype switching, to alter vector tropism. 40 , 48 , 49 Serotype switching involves packaging the vector with the capsid of the AAV variant most efficient at transducing the target cell population.
Until very recently LCA2 was both incurable and untreatable. The approval of Luxturna has brought new hope, not just for LCA2 but also for a host of other previously incurable retinopathies. Luxturna is an AAV2‐based recombinant, non‐integrating vector designed to deliver the RPE65 gene (Figure 5). In clinical trials, Luxturna was administered via subretinal injection into both eyes with a gap of 6–18 days. 33 , 35 , 36 , 37 Patients treated with Luxturna showed a strong and durable improvement in visual acuity 1 year after treatment. The visual field and the ability to perceive light also showed substantial improvements. Remarkably, patients from earlier phase 1 and 2 trials retained most of this improvement for 3 years or more. Such changes can have enormous effects on the quality of life of affected people, taking them from near blindness to partial sight.
FIGURE 5.

Luxturna vector design and treatment protocol. Luxturna is produced by packaging an expression cassette for RPE65 into an AAV2 capsid. The RPE65 vector virions are harvested from cells transfected with the relevant plasmids, purified and a high‐titre vector preparation is injected directly into the sub‐retinal space. The vector will diffuse outwards and insert the gene into a large region of the retinal pigment epithelium, creating a new source of 11‐cis‐retinal. The transcellular nature of the 11‐cis‐retinal cycle allows the effects to spread more widely throughout the retina. The cassette design contains the genomic RPE65 sequence with all three exons and two introns. Efficient high‐level expression is ensured by an artificial promoter/enhancer pair created by joining the chicken β‐Actin promoter with the cytomegalovirus (CMV) enhancer and an artificial poly‐adenylation sequence. The cassette is flanked by the AAV2 ITR sequences.
The success of Luxturna has validated an entirely new therapeutic avenue for congenital retinopathies and other genetic afflictions of the eyes. Indeed, RPE65 mutations account for only a small proportion of inherited retinopathies. Luxturna inspired an explosion in clinical development for gene therapy products targeting the eye. 34 Outcomes should start filtering through in the mid to late 2020s.
2.2. Gene therapy for solid tumours: Imlygic and HSV gene supplementation for melanoma
Cancer is a genetic disease, whose extreme heterogeneity makes it a virtual microcosm for the RD field. Collectively cancer is common, but with so many different cancers, individual types can be rare. The highly variable ontogenesis and resistance to conventional treatments means personalized medicine is very challenging and yet also a key priority. Gene therapy offers an attractive proposition: taking advantage of specific defects within a particular cancer to create engineered viral vectors selectively toxic to that cancer. The technology can then be readily repurposed to target other cancer types.
Melanoma is the fifth most common cancer in the United Kingdom, with an incidence of approximately 25 per 100,000. 50 It is very aggressive, with in situ melanoma quickly progressing to metastatic disease, at which point survival rates drop precipitously. 50 Talimogene laherparepvec (Imlygic) is a licenced herpes simplex virus‐1 (HSV1) gene therapy treatment for melanoma. 51 , 52 The lifecycle of herpes viruses is illustrated in Figure 6. Herpes viruses rely on key virulence factors that disrupt the interferon I pathway 53 and antigen presentation to evade immunity 54 and cause disease (Figure 6). In cancer, particularly melanoma, the same processes are often defective. Imlygic lacks these virulence factors, crippling its ability to infect normal cells, but leaving cancer cells highly vulnerable (Figure 7). 51 , 55 Two additional modifications enhance Imlygic's anti‐cancer potency: the virus expresses granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) during replication, plus is unable to undergo lysogeny, immediately activating the lethal lytic cycle (Figure 7). 51 , 55
FIGURE 6.

Herpes virus lifecycle. Herpes enter the cell by receptor‐mediated fusion of their envelope with the plasma membrane. The viral genome is injected into the nucleus, where it circularizes to facilitate simultaneous replication (through a rolling circle mechanism) and viral gene transcription. The linear viral genome copies are exported to the cytoplasm and interact with newly synthesized capsid proteins. The complete virion is internalized into vesicles within the endoplasmic reticulum (ER) guided by the viral envelope protein that is inserted into the ER membrane. The enveloped virus is trafficked within the ER into the endosomes and eventually released by exocytosis. Two viral proteins are important in blocking host responses against the virus. ICP47 blocks the loading of virion peptides onto MHC class I, hindering cytotoxic T‐cell recognition. ICP34.5 blocks protein kinase R (PKR), crippling the interferon pathway response to the virus.
FIGURE 7.

Talimogene laherparepvec (Imlygic) overall design. The herpes genome has two long inverted repeats (terminal and internal, TRL and IRL) and two inverted short repeats (IRS and TRS). It also has a long unique segment (UL) and short unique segment (US). In Imlygic, the promoters of the lytic cycle genes are altered for immediate activation, preventing lysogeny, ICP34.5 is replaced with GM‐CSF and ICP47 is deleted. Imlygic can only replicate effectively in cancer cells, where the interferon pathway and antigen presentation are compromised. Replication lyses the cancer cells and produces GM‐CSF, enhancing immune destruction of the tumour.
Imlygic performed well in clinical trials against stage III‐IV melanoma, refractory to surgery. 56 , 57 , 58 , 59 It increased the proportion of patients achieving durable disease remission, increasing disease‐free survival at 60 months by 50%. Although Imlygic was ineffective against late‐stage IV melanoma, it more than doubled overall survival in stage IIIB/B and IIIB‐IVM1. Remarkably, almost all patients achieving complete remission remained disease‐free at the 5‐year follow‐up. In addition, it was found that Imlygic shows substantial synergy with checkpoint inhibitors. 60 , 61
3. MODIFICATION OF DNA IN DIFFERENTIATED SOMATIC CELLS, PRIOR TO REIMPLANTATION
3.1. Ex vivo gene therapy benefits and challenges
Ex vivo gene therapy is the genetic modification of cells outside the body, followed by transplantation. These cells could be differentiated somatic cells or stem/progenitor cells. 62 The main advantages of this ex vivo approach include the selective targeting of the cell population of interest, the avoidance of immune defences and the implementation of quality control systems before the genetically modified cells are reimplanted. In this section, we will focus on differentiated somatic cells that retain sufficient replicative capacity to allow extraction, modification outside the body and re‐implantation.
3.2. Retroviral vectors for ex vivo gene therapy: chimeric antigen receptor (CAR) T cells
Retroviruses are enveloped single‐stranded RNA viruses, whose life cycle involves converting their RNA genome into double‐stranded DNA and stably integrating it into the host genome. 63 , 64 , 65 Their RNA‐containing capsid is surrounded by a lipid bilayer derived from the host cell plasma membrane and containing the envelope protein, a transmembrane host cell invasion factor. Gammaretroviruses 66 and Lentiviruses 67 are most used as viral vectors. Figure 8 shows a brief summary of the retroviral/lentiviral life cycle. Retroviruses use special sequences called long terminal repeats (LTRs) to direct packaging of their genome into virions. 66 , 67 The LTRs contain signals facilitating several steps in the virus life cycle and act as powerful promoters. Retroviral vectors are made from an LTR‐flanked transgene cassette by supplying the virus structural proteins in trans.
FIGURE 8.
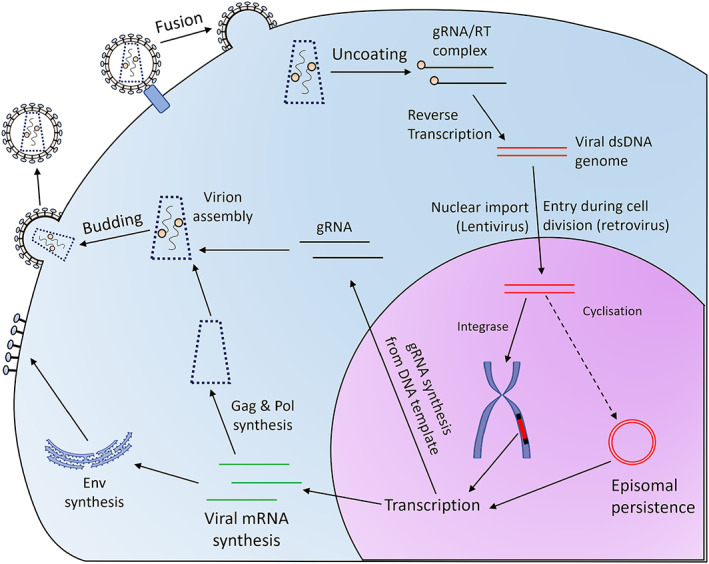
Retrovirus life cycle. Retroviruses and lentiviruses enter the cell via receptor‐mediated fusion of their envelope with the cell membrane. In the cytoplasm, the viral capsid disassembles and genomic RNA (gRNA) is converted to a double‐stranded (dsDNA) genome by reverse transcriptase (RT). The dsDNA genome is either transported through the nuclear pores by viral proteins (lentivirus) or enters the nucleus during cell division (gamma‐retrovirus). In the nucleus, cyclization and episomal persistence or direct integration into the host chromosomes occur. Integrated dsDNA produces both viral transcripts and the gRNA genome. The viral particles are assembled in the cytoplasm. The envelope protein, which is inserted in the plasma membrane, directs budding of the assembled nucleocapsid out of the cell and acquisition of the viral membrane envelope.
Retrovirus vectors are thus used to directly insert transgenes into the host genome. Although integration is promiscuous, it is not random and integration patterns have been reported. For example, HIV prefers active transcriptional units but integrates at similar frequencies across the gene. 68 Although the promiscuity of the integration is potentially problematic, it also ensures that the payload will be inserted in an area of active chromatin. Lentiviruses offer an additional advantage over other retroviruses. They can translocate their genome across the intact nuclear membrane and do not need to wait for cell division to access the host genome for integration. Lentivirus vectors can thus transduce quiescent cells. 67
Acute lymphoblastic leukaemia (ALL) is a malignancy of the lymphocyte progenitor cells that primarily affects paediatric patients. 69 It is associated with a range of chromosomal translocations in key oncogenes, such as PAX5 and TCF3 and, in some cases, gain of function mutants, such as the BCR‐ABL1 (Philadelphia translocation) and ETV6–RUNX1 rearrangements. 70 , 71 , 72 The incidence of ALL is estimated at 10–20 cases per million. 69 Unlike many other inherited cancers, ALL develops early in life making it one of the most common paediatric cancers. B‐cell precursor malignancies account for the bulk of ALL. Advances in chemotherapy and the understanding of the signalling networks that are disrupted by mutations in ALL, have led to the development of relatively effective chemotherapy regimens, 73 , 74 , 75 but unfortunately, when patients relapse after treatment or even worse when initial responses are poor, the prognosis is typically bleak. 76 , 77
Non‐Hodgkin lymphoma (NHL) is one of the more common lymphoid tissue‐derived cancers affecting the head and neck. 78 , 79 It includes a highly heterogeneous group of malignancies that collectively have an incidence of approximately 1 in 10,000 in the European Union. Diffuse Large B‐cell Lymphoma (DLBCL) accounts for between a quarter and a third of NHL incidence and it is highly aggressive. 80 , 81 Familial forms of NHL have been characterized. 82 , 83 Like ALL, effective treatment options are generally available for NHL, but refractory DLBCL has a very poor prognosis. 80
The development of artificial T‐cell receptors (TCRs) which target CD19, a surface marker of B‐cell precursors that is strongly expressed in most B‐cell lymphomas, 84 , 85 was a revolutionary advance (Figure 9). These chimeric receptors are constructed by joining together the signalling apparatus of the TCR with parts taken from costimulatory molecules and a binding site against the target protein, in this case CD19. 86 , 87 , 88 , 89 , 90 Anti‐CD19 CARs target T‐cells artificially to B cells. The CAR signalling steps are distinct from actual TCR signalling, 91 , 92 , 93 , 94 , 95 vary depending on the type of costimulatory molecule used, and the immunological synapse formed is atypical. Nevertheless, the signals produced are sufficient to emulate T‐cell receptor signalling, enabling the modified T‐cells to attack and destroy targets expressing CD19 while bypassing the human leucocyte antigen (HLA) restriction.
FIGURE 9.

The chimeric antigen receptor (CAR) design. The artificial T‐cell receptor (TCR) is made in a modular manner by combining the following parts: the CD3ζ signalling domain from the TCR, a transmembrane and hinge region and co‐stimulatory domains from receptors that are needed for TCR signalling, such as CD8, CD28 and 4‐1BB (CD137), and a single chain variable fragment (scFv) targeted against the protein of interest. This creates a receptor capable of generating a full TCR signal upon binding of the scFv target. The CAR is thought to become activated by dimerization or multimerization allowing cross‐interaction between the signalling domain of one CAR molecule with the co‐stimulatory domain of another. This drives CAR activation, creating a CD3 signal and activating other accessory receptors, particularly the IL2 receptor.
Autologous CD19‐CAR T‐cells turned out to be highly effective in treating refractory NHL and ALL. The first two products approved for this purpose were tisagenlecleucel (Kymriah) and axicabtagene ciloleucel (Yescarta). Kymriah 88 , 89 is a preparation of CD19‐CAR engineered T‐cells using the 4‐1BB co‐stimulatory domain and the CD8alpha transmembrane and hinge region. CD3‐positive cells (CD4 and CD8 T‐cells) isolated from autologous blood are transduced, using a lentiviral vector, with a CD19‐CAR transgene driven by the Elongation Factor‐1 promoter. 88 After selection and expansion, the CAR‐T cells are reinfused into patients that have undergone lymphocyte depletion treatment. 89 Yescarta 96 , 97 , 98 , 99 is prepared in a similar manner, except it uses the CD28 transmembrane/hinge region and co‐stimulatory domain instead of CD8alpha/4‐1BB. The vector is Retroviral and driven by the mouse stem cell virus (MSCV) promoter. 99
Kymriah is licenced for the treatment of refractory ALL and NHL/DLBCL, while Yescarta is only licenced for refractory NHL/DLBCL. 100 , 101 Both help drive substantial remission rates in excess of 50%, whereas conventional treatments are largely ineffective. 89 , 96 They can also have serious side‐effects. 102 , 103 , 104 , 105 , 106 Cytokine release syndrome is a potentially life‐threatening complication capable of harming several major organs, including the nervous system via the IEC (immune effector cell) associated neurological syndrome or ICANS. A second distinct complication of CD19 cell elimination is predictably beta‐cell aplasia and a collapse in blood immunoglobulin levels. This can potentially predispose the patient to infections and must be managed to mitigate the risks.
The success of CAR‐T technology in blood cancers has spearheaded substantial research on possible applications to solid tumours, a more challenging environment. Applications of gene therapy to cancer now account for around half the gene therapy treatments under development. 107
4. MODIFICATION OF DNA IN STEM CELLS, PRIOR TO REIMPLANTATION
An alternative ex vivo approach targets stem/progenitor cells, 62 which underpin the natural maintenance of organs. This strategy is particularly suited to correction of genetic defects of the blood, which builds on the clinical experience of bone marrow transplantation. 108 , 109 , 110 , 111
To illustrate the effectiveness of this approach, we will look at two clinically approved treatments for genetic diseases affecting blood cells, Strimvelis for ADA‐SCID and Zynteglo for β‐thalassaemia.
4.1. Severe combined immunodeficiency: Strimvelis HSC gene therapy
Severe combined immunodeficiency is a heterogenous group of genetic disorders that cause complete or nearly complete impairment of T‐lymphocyte function, combined with primary or secondary dysfunction in other immune cell types. 112 The SCID spectrum is very rare, with a prevalence of approximately 1 in 60,000 live births. 113 The complexity of T‐lymphocyte ontogenesis explains the extensive genetic heterogeneity of SCID. There are currently 16 known causative genes and over 20 separate defects. 114 , 115 The most common mutations are in X‐linked IL2 receptor components (SCID‐X1).
Adenosine deaminase‐severe combined immunodeficiency is another common form and one of the most damaging. 116 The Adenosine Deaminase enzyme is essential for the purine salvage pathway that regulates the purine nucleotide balance. ADA activity is important in preventing adenine nucleotide accumulation. Lack of ADA results in a marked imbalance in the dNTP pool, compromising DNA polymerase function. 117 , 118 , 119 In rapidly or continuously proliferating cells the result is genotoxic shock and apoptosis, 120 , 121 and the lymphoid cell differentiation pathway is particularly sensitive to ADA deficiency. 116 , 122 , 123 ADA deficiency also impacts cAMP synthesis, disrupting general cell signalling and giving rise to a more diffuse pathology, in most other tissues including the brain. 124
Adenosine deaminase‐severe combined immunodeficiency can be treated by allogeneic haematopoietic stem cell (HSC) transplantation 10 , 125 and PEGylated ADA (PEG‐ADA). 10 , 126 PEG‐ADA has a long plasma half‐life and can help reduce intracellular adenine build‐up, by keeping extracellular levels low and facilitating transporter‐mediated efflux, alleviating some of the worst symptoms. 10 Bone marrow transplantation is limited by the availability of HLA‐matched donors and by the risk of graft‐versus‐host disease (GVHD) with allogeneic donors. 127
A gene therapy option for ADA‐SCID has been licenced by the European Medicines Agency (EMA) in the European Union (EU). Strimvelis is a preparation of autologous HSCs, engineered to express functional ADA. 128 , 129 , 130 , 131 CD34‐positive HSC cells are isolated from the person affected and expanded using a cocktail of soluble mediators: FLT3L, KITL/SCF, THPO, IL3, and IL6 (FKT36). In this proliferative state, the cells are transduced with a functional ADA copy using an amphotropic Murine Moloney Leukaemia virus vector, 132 whose 4070A envelope gene, targets Pit‐2 and mediates efficient transduction of HSCs. 133 The transduced HSC pool is re‐infused after non‐myeloablative conditioning with anti‐proliferative agents such as busulfan to suppress the proliferation of endogenous HSCs.
Strimvelis has performed exceptionally well in clinical trials. 131 , 134 , 135 A follow‐up of 18 people treated with Strimvelis at a very early age revealed that all of them survived (follow‐up 2–13 years, median 7 years), and they were well enough to resume normal social interactions. Several of the patients were able to return to school. In those who could be evaluated ADA expression reached or exceeded 10% normal and remained stable in all myeloid and lymphoid cells, immune function was successfully reconstituted and a response to antigen challenge could be observed. The rate of infection decreased dramatically, and the recipients managed to resolve infections in most cases. In almost all cases PEG‐ADA treatment could be discontinued. Intervention‐free survival remained above 80%. These results match autologous HSC transplantation and compete very favourably with all other treatments.
Successful reconstitution of the T‐cell population does not eliminate the complete health impact of ADA‐SCID, since it does not replace ADA function in cells of a non‐haematopoietic linage, but it effectively provides (via expression in red blood cells) a ready pool of plasma ADA that can serve the same function as PEG‐ADA. At the same time, it eliminates the supply issues with donor‐matched HSC transplantation and the risks associated with allogeneic transplantation (e.g. GvHD).
4.2. β‐Thalassaemia gene therapy with Zynteglo HSCs
Beta‐thalassaemia is one of the most common genetic anaemias. 136 , 137 It is autosomal recessive, with a highly variable distribution. Its prevalence approaches 1 in 1000 live births in areas where malaria is currently endemic or was endemic in the recent past but is very rare elsewhere. Globally, prevalence is close to 1:100,000 live births.
The disease phenotype depends on the exact genetic defect in the HBB (adult β‐globin) gene, a subunit of haemoglobin. Homozygous inheritance of an allele that produces no functional protein causes β‐thalassaemia major and severe life‐threatening anaemia. 136 , 137 Homozygous inheritance of a partial loss of function mutant leads to β‐thalassaemia intermedia and milder disease.
Beta‐thalassaemia has all the typical hallmarks of anaemia 136 , 137 including fatigue, weakness, and palpitations. The major disease also leads to muscle cachexia, skeletal and cartilage deformities, osteoporosis and splenomegaly. Regular blood transfusions can address most of these symptoms, but they generally also cause iron overload, leading to heart, liver and endocrine complications.
Recently an ex vivo gene therapy approach, Zynteglo 138 , 139 , 140 , 141 was licenced for the treatment of severe, transfusion‐dependent, β‐thalassaemia. The patient is treated with G‐CSF and a CXCR4/SDF‐1 antagonist, which leads to substantial proliferation and mobilization of HSCs from the bone marrow into the blood. CD34‐positive HSCs are collected from the blood and transduced in the laboratory with the BB305 lentiviral vector, which contains a mutated HBB (T87Q). 142 , 143 The BB305 lentiviral vector has a self‐inactivating design, which removes the transcriptional activity of the LTR. It includes the entire HBB coding sequence with its native control elements: the β‐globin promoter, 144 its 3′ enhancer 145 and selected fragments from the upstream locus control region, 146 facilitating high‐level expression. The T87Q variant confers enhanced anti‐sickling activity and can be differentiated chromatographically, serving as a biomarker. 147 The patient is conditioned with myelosuppressive drugs to facilitate donor cell engraftment prior to infusion of the corrected HSCs.
Like Strimvelis, Zynteglo was highly successful in clinical trials. 138 , 139 , 140 , 141 The majority of patients showed long‐lasting improvement in haemoglobin levels and were able to stop blood transfusions. Most patients that could be evaluated for over a year, achieved near normalization of haemoglobin levels and blood transfusion independence.
5. GENE THERAPY THROUGH MANIPULATION OF POST‐TRANSCRIPTIONAL RNA PROCESSING AND TRANSLATION
Gene expression levels can be modulated after transcription using synthetic nucleic acid molecules able to interfere with splicing, translation or RNA degradation, without directly altering the cell's genetic material (Figure 10). 148 Diseases resulting from gain‐of‐function mutants are particularly amenable to this intervention method.
FIGURE 10.

Post‐transcriptional control of gene expression in eukaryotic cells. The first control point is during splicing. Splicing in eukaryotic cells is controlled by a series of splicing site elements and factors, and more than one product can be produced from the same gene. The splicing factors expressed by the cell determine the splice variant balance. The mature RNA is further regulated by degradation. A special protein complex recognizes double‐stranded RNA and then uses it as a template to degrade matching mRNA molecules. This mechanism (RNA interference) allows the cell to fine‐tune gene expression, by producing special RNA molecules (micro‐RNAs or miRNA). Splicing and RNA interference can be controlled using artificial oligonucleotides. The final control point is binding to the ribosome and translation initiation.
Control of RNA levels within the cell occurs through RNA interference, 148 , 149 , 150 which uses endogenous (e.g. miRNA) or exogenous (e.g. siRNA) double‐stranded RNA templates, to target specific mRNA sequences for degradation. Artificially produced RNA molecules (short hairpin RNAs, which are artificial miRNA mimics 151 , 152 ) can be used to hijack this system and selectively target mRNA molecules for degradation 153 , 154 (Figure 11).
FIGURE 11.

Oligonucleotide control of gene expression. (A) siRNAs are duplex RNA molecules made from two complementary 20–21 nt strands designed to leave 1–2 nt overhangs on the 3′ side after annealing. The Dicer protein complex processes siRNAs and incorporates one of the strands and degrades the other based on their physical properties. Any mRNA sequence that can base pair with the chosen strand is degraded. Artificial siRNA molecules are designed to force selection of the non‐coding strand. (B) Another design is gapmer antisense oligonucleotides (gASOs), which consist of a DNA core flanked by artificial nucleotides. The artificial nucleotides are nuclease resistant and have a high affinity for RNA. When the gapmer ASO anneals to its target RNA, the DNA core forms a DNA/RNA heteroduplex, thus recruiting RNaseH and marking the target RNA for degradation. The resistance of gASOs to nucleases allows for cytoplasmic persistence and durable responses.
Antisense oligonucleotides (ASO) are short nucleic acid sequences designed to base pair with a specific RNA target within the cell. 148 Typically, ASOs consist of modified nucleotides with increased stability, and frequently include artificial nucleotide analogues, such as morpholinos 155 and locked/bridged nucleic acids. 156 ASOs can manipulate the post‐transcriptional fate of mRNA in various ways, 157 but here we will mostly focus on RNase H targeting (Figure 11). Splice‐switching oligonucleotides (SSO) are designed to base pair with splicing sites, or splicing enhancers/suppressors within a pre‐mRNA sequence and direct alternative splicing of the target gene. 158 RNA ASOs designed to base pair with sequences within the 5′ or 3′ untranslated region (UTR) can suppress or enhance mRNA translation. 159 , 160
Here, we will discuss some key examples of RNA interference and SSO‐based therapeutics that have recently cleared clinical trials and are now being used to treat rare diseases. In particular, we will look at familial transthyretin amyloidosis (FTA) and spinal muscular atrophy (SMA).
5.1. Familial transthyretin amyloidosis: Onpattro RNAi and Tegsedi ASO
Familial transthyretin amyloidosis is a rare genetic disease of the Transthyretin (TTR) gene that causes the protein to misfold. 161 , 162 , 163 The misfolded protein forms amyloids that deposit into and damage tissues. This is a slow gradual process, so the symptoms typically begin in adulthood. The exact onset age is variable and correlates with disease progression. The peripheral nervous system is particularly vulnerable, so neuropathies are among the earlier symptoms, but as the disease progresses, eyes, kidney, heart, and CNS typically become involved. FTA is eventually fatal on average 10 years after the onset of symptoms, with a younger onset being associated with more aggressive disease. The prognosis in patients presenting with early cardiac involvement is extremely poor. Few patients survive for longer than 5 years.
Genetically, FTA mutations are autosomal dominant, but progression and penetrance vary depending on the exact genetic defect. 161 Most patients are heterozygotes. The global prevalence of FTA is of the order of 1 in 10,000, 164 though clusters have been observed within certain ethnic groups or populations, such as in certain areas of Portugal, Sweden, Japan, and West Africa. 162 , 164
The current gold standard treatment for FTA is liver transplantation 161 , 162 , 163 since the liver is a major source of TTR. Liver transplantation arrests the development of polyneuropathies and slows but does not prevent progressive degeneration of the eyes, heart, and kidney.
In the last few years, the FDA has approved two oligonucleotide‐based therapeutics for FTA: Patisiran (Onpattro) 165 and Inotersen (Tegsedi). 166 Onpattro 167 , 168 is a stable nucleic acid lipid particle (SNALP) formulation containing short interfering RNA (siRNA—Figure 11) against TTR. Onpattro is a new generation of siRNA, using DNA overhangs (dTdT), instead of RNA (UU), increasing RNA resistance for a longer lasting effect. In addition, Onpattro has most of the U and C residues methylated in the sense strand, to promote incorporation of the siRNA correct strand into the RISC (RNA‐induced silencing complex) assembly. Onpattro is designed to target the 3′ UTR of the TTR transcript and will suppress expression of both mutant and wild‐type forms. This is desirable because once misfolded aggregates are formed, they can induce misfolding and deposition of even the wild‐type protein.
The Onpattro SNALP's formulation 169 consists of a 1:1 mixture of cholesterol and phospholipids. The phospholipids have a strong positive charge (4:1 ratio of cationic to neutral) to facilitate complex formation with the siRNA. 5% of the cationic phospholipids used have a PEG2000 (polyethylene glycol 2000 MW) polymer attached to them, creating a sheath that greatly prolongs SNALP plasma half‐life. Lipid nanoparticles in the plasma are typically decorated by ApoE, despite the PEG sheath, and only extravasate effectively in tissues with fenestrated endothelium 170 , 171 ; thus, they naturally target the liver. After ApoE‐directed internalization, charged interaction between the positive SNALP and negatively charged endosomal membrane, mediates endosome escape, delivering the siRNA to the cytoplasm. The long SNALP half‐life, and the longevity of the primed RISC assembly, allow the effect to persist over several days.
Onpattro is highly effective at suppressing TTR expression. During clinical trials, 167 , 168 , 172 it was found that expression is reduced by >70% within 5 days and remains below that threshold for at least 20 days. Infusion of Patisiran every 3 weeks over 18 months halted progression in virtually all patients that achieved sustained TTR suppression. Small but significant improvements were also seen in the polyneuropathy and cardiomyopathy aspects of the disease. Adverse reactions to Onpattro are primarily related to infusion of the liposomal formulation. Serious adverse effects were rare.
Tegsedi 173 , 174 is an RNaseH‐dependent GAPmer ASO formulation (Figure 11) also targeting the TTR 3′UTR. The inner DNA core has phosphorothioate linkages 175 that make it strongly resistant to degradation but can also cause toxicity. 176 , 177 , 178 To mask the phosphorothioate toxicity, the central core is flanked on either side by five 2′‐O‐methoxyethyl ribonucleotide residues, which are also resistant to degradation. 174 The entire 20‐mer oligonucleotide is complementary to the target sequence in the TTR 3′UTR and stable enough to deliver via intramuscular injection of a preparation in saline, without a liposomal formulation. The ASO makes its way into the circulation and is actively taken up by cells in various tissues, with the liver being a primary site. During clinical trials, 173 , 174 patients received three injections of Inotersen in the first week, followed by weekly injections for a period of 64 weeks. At the end of the first‐week plasma TTR levels reduced by ~70% and remained at that level for the entire 64‐week period. Like Onpattro, Tegsedi effectively halted disease progression over the entire treatment period, albeit with variations between different patient groups.
The ability to control gene expression levels is vital in shutting down gain‐of‐function mutants. Like other nucleic‐based therapies, once the delivery method is optimized, it can be repurposed for any payload. For example, research into liposomal siRNA delivery paved the way for the SARS‐CoV2 mRNA vaccines, by BioNTech and Moderna. 179 , 180
5.2. Spinal muscular atrophy: Spinraza splice‐switching oligonucleotide
Spinal muscular atrophy is the most common cause of genetic death in childhood with a prevalence of approximately 1 in 10,000 live births. 181 , 182 It is caused by loss‐of‐function mutations in the survival motor neuron 1 (SMN1) gene. 183 It is autosomal recessive and the severity of the phenotype inversely correlates with the copy number and expression level of the highly related, but only partially functional, SMN2 gene. 184 , 185 , 186 SMA is a systemic disease, due to SMN being ubiquitously expressed, but the lower motor neurons are particularly sensitive to loss of SMN function. 187
SMN2 differs by a few nucleotides from SMN1. 181 , 184 , 188 Crucially, it is spliced differently, with 85%–90% of the transcripts typically skipping exon 7. The truncated protein isoform is unstable and rapidly degraded. SMN2 proved to be pivotal in developing an oligonucleotide‐based therapeutic for SMA. Nusinersen (Spinraza) is an SSO designed to prevent the excision of exon 7 from the SMN2 gene product, increasing the production of full‐length, stable SMN protein from it. 189 The 20‐mer SSO oligo is made from 2′‐O‐methoxyethyl (2MOE) ribonucleotides, 190 which resist degradation and base‐pair more efficiently, 191 enhancing intronic splicing silencer‐N1 (ISS‐N1) inhibition. The oligo is injected directly into the spinal cord via lumbar puncture. 192 Initially, a small number of frequent injections are given to quickly establish a steady state, followed by maintenance doses that are more infrequent. In the most recent clinical trial 192 the conditioning regime is three bi‐weekly doses, followed by a maintenance regime with 4 months between doses. Spinraza has proven highly efficacious in infantile‐onset SMA (type I), 193 , 194 , 195 producing dramatic improvements in survival and motor milestone achievement, with some infants developing skills never seen in the natural history of this disease. The incidence of adverse events was high, but mostly related to the complex spinal injection procedure in this vulnerable patient population.
Onpattro, Tegsedi, Spinraza and other examples represent important milestones for the oligo therapeutics field, by demonstrating that it is possible to exert sustained, effective, direct control over gene expression without stable genetic modification.
5.3. A gene therapy comparison of oligonucleotides versus viral vectors: Spinraza versus Zolgensma
The existence of a viral vector gene therapy alternative to Spinraza offers a unique opportunity to directly compare the effectiveness of the two approaches. Zolgensma (Onasemnogene Abeparvovec) 196 , 197 , 198 , 199 , 200 is an AAV‐based gene supplementation treatment aimed at directly and permanently restoring SMN1 expression with a single dose. The design of the Zolgensma expression cassette is similar to Luxturna (Figure 5), in using the hybrid CMV–Chicken beta actin promoter to drive the expression of SMN1 cDNA. To enhance expression, the design incorporates an artificial intron (from SV40) and codon optimization. The sequence of AVXS‐101 (the vector for Zolgensma) is proprietary and the exact optimizations are not in the public domain, but the effectiveness of this approach was documented by using a similar AAV9 platform. 201 , 202 , 203 A self‐complementary design (Figure 12) was employed, where one of the flanking ITRs was a specially engineered variant to synthesize genome dimers, rather than monomers. 204 This design is advantageous in that it can speed up transgenic expression without the need for DNA synthesis, a possible rate‐limiting step for single‐stranded AAV vectors.
FIGURE 12.

Generation of self‐complementary AAV vectors. AAV variants with faster expression kinetics can be produced through the use of a mutated ITR (blue, right), to frustrate terminal resolution. As a result, the two strands fail to separate, leaving them joined through the mutant ITR. The complete viral genome is still flanked by normal ITRs, therefore it can be replicated and packaged. Once released in the host cell the two strands can reanneal to form the structure marked with (*). This structure does not rely on second‐strand synthesis to stabilize it within the cell and initiate transgenic expression.
Zolgensma performed very well in clinical trials, 196 , 197 , 198 , 199 , 200 , 205 showing both a high response rate and substantial symptom alleviation. In the SPR1NT phase 3 trial, all 14 participants achieved the primary endpoint of sitting unaided, while 11 and 10 out of 14 managed to stand and walk respectively. None of the 23 untreated SMA patients achieved these developmental milestones. Impressively, a substantial number of children (40%–80% depending on the endpoint), reached the milestones within the regular developmental time. Motor assessment showed that all children improved rapidly after administration and reached at least 80% of the normal score. Typically, SMA children achieve on average 40% of the normal score and their scores decline, rather than improve with age. All children managed to avoid the need for mechanical ventilation during the study and 13 children were spared the need for assisted feeding. This was confirmed by other studies, which also found that the gains were durable. These results compare favourably with Spinraza. Indeed, Bitetti et al. 205 investigated children previously treated with Spinraza and found that Zolgensma helped the children make further gains, with the greatest benefits in children that had responded less well to Spinraza. An important factor in this improved response is likely to be the systemic treatment provided by Zolgensma, while Spinraza is delivered by intrathecal injection, with direct beneficial effects expected to be restricted to the CNS.
Although the evidence so far points to a higher efficacy and response rate for Zolgensma compared with Spinraza, safety concerns with the use of AAV9 use have emerged. Two common serious adverse events have been observed, hepatotoxicity and thrombocytopenia. Although these adverse effects proved self‐limiting in the clinical trials, a subsequent meta‐analysis 206 of the clinical data confirmed that the majority of patients show evidence of liver damage, though this responded well to steroid treatment.
In 2021, Thomsen et al. 207 reported the expression of SMN1 in two infants that received Zolgensma but had died due to reasons unrelated to treatment. SMN1 expression was readily observed in the central nervous system, but also in several peripheral organs, particularly the liver where expression was 2–3 orders of magnitude higher compared to the CNS. Although the reasons for AAV9 vector hepatotoxicity are not fully understood, the observation that it responded to steroid treatment suggests that it might be related to an immune response to the vector. Sadly, two patients recently treated with Zolgensma have died of acute liver failure. Both deaths occurred several weeks post‐treatment, shortly after corticosteroid taper was initiated. 208 Clinical trials with AAV vectors for other indications have also been marred by similar severe adverse events to those described for Zolgensma, including death at high vector doses. While Zolgensma has been used on more than 2300 people so far, these findings reiterate the need for better understanding and control of viral vector tropism and the associated immune response, to develop even safer treatments.
Just as the lessons learned from Luxturna are expected to greatly reduce the effort required to target retinopathies, so will Zolgensma aid the development of other gene therapy solutions targeting the central nervous system, 209 thereby offering further evidence for the suitability of gene therapy to treat RDs. Moreover, as development and production costs fall with the adoption of these methods into mainstream clinical practice, we can expect treatment costs to reduce significantly from their current high price tags.
6. CONCLUDING REMARKS AND FUTURE PROSPECTS
In this review, we have used specific examples of successful clinical implementation to showcase what gene therapy can achieve and how it is already helping address the challenges of treating RDs. Around 50 years have passed since gene therapy was first mooted as a possible therapeutic avenue, illustrating how complicated it can be to implement a novel concept into clinical practice. Some of the licenced therapies fit the original mould (gene supplementation to rescue a genetic defect), but many do not as they involve mechanistic pathways discovered and adapted more recently, such as dsRNA interference, splicing modulation and exon skipping. Significant initial successes in treating SCID with ex vivo gene therapies built on the accumulated experience of allogeneic bone marrow transplantation, showed how existing clinical practice can be instrumental for implementation of novel technologies. Application of gene therapies to some forms of SCID paved the way to develop clinical gene therapies for other immunodeficiencies, and also additional disorders of the haematopoietic system like β‐thalassaemia, thus demonstrating how therapeutic strategies can be adapted relatively quickly to different diseases of the same tissue. Moreover, advances in ex vivo modification of haematopoietic cells have also led to unforeseen successes such as CAR‐T cells. A similar expansion is underway with AAV9 vectors, which can cross the blood–brain barrier via intravascular delivery to treat inherited diseases of the CNS, as first demonstrated with Zolgensma for SMA. 196 , 197 , 198 , 199 , 200 For those interested in tracking marketed gene (and cell) therapies across the world, the International Society for Stem Cell Research maintains an up‐to‐date map. 210 Note, however, that this resource does not include oligonucleotide therapies, as they are not technically considered gene therapies by FDA or Advanced Therapeutic Medicinal Products by EMA.
The application of gene therapy technologies to the vaccine field has provided resounding successes in the fight against COVID‐19, with mRNA‐based and adenovirus‐based formulations being developed in record time and used to immunize a large part of the world population. These platforms are now being explored for other applications in both vaccinology and RD therapy, with very promising prospects.
Our discussion has focused on some of the clinical successes of gene therapy. Consequently, we have not dwelt on other recent advances in gene therapy methods that are yet to reach full clinical implementation. However, to conclude our review we briefly cite some recent advances likely to drive the field forward. These include synthetic virology, 211 lipid nanoparticles 212 and membrane‐active peptides 213 as key areas of intense research development. Similarly, AAV capsid engineering to alter tropism has seen much resource investment and is starting to deliver optimized AAV serotypes for targeted therapies in vivo. However, to close this review it is clear that genome editing technologies are easily the most promising therapeutic strategies for the future. The accessible engineering of CRISPR/Cas enzymes, based on short synthetic RNAs, has facilitated enormously the introduction of defined genetic and epigenetic modifications in the genome 214 , 215 through a variety of approaches including indel‐mediated knockouts, homology‐dependent repair, prime editing, base editing, and epigenetic regulation of transcription. Indeed, recent clinical trials with CRISPR in Transthyretin Amyloidosis, 216 sickle cell disease and β‐thalassaemia 217 offer very promising demonstrations of the technology and its exciting potential for patient benefit.
CONFLICT OF INTEREST STATEMENT
The authors have no conflict of interest.
ACKNOWLEDGEMENTS
RJY‐M thanks Spinal Muscular Atrophy UK (formerly the SMA Trust) for generic funding to the UK SMA Research Consortium, and Action for A‐T for funding their research on ataxia telangiectasia.
Papaioannou I, Owen JS, Yáñez‐Muñoz RJ. Clinical applications of gene therapy for rare diseases: A review. Int J Exp Path. 2023;104:154‐176. doi: 10.1111/iep.12478
REFERENCES
- 1. Haendel M, Vasilevsky N, Unni D, et al. How many rare diseases are there? Nat Rev Drug Discov. 2020;19:77‐78. doi: 10.1038/d41573-019-00180-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tambuyzer E, Vandendriessche B, Austin CP, et al. Therapies for rare diseases: therapeutic modalities, progress and challenges ahead. Nat Rev Drug Discov. 2020;19:93‐111. doi: 10.1038/s41573-019-0049-9 [DOI] [PubMed] [Google Scholar]
- 3. Klimova B, Storek M, Valis M, Kuca K. Global view on rare diseases: a mini review. Curr Med Chem. 2017;24:3153‐3158. doi: 10.2174/0929867324666170511111803 [DOI] [PubMed] [Google Scholar]
- 4. Nguengang Wakap S, Lambert DM, Olry A, et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur J Hum Genet. 2020;28:165‐173. doi: 10.1038/s41431-019-0508-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reynoso MC, Hernández A, Lizcano‐Gil LA, et al. Autosomal dominant congenital macroglossia: further delineation of the syndrome. Genet Couns. 1994;5:151‐154. [PubMed] [Google Scholar]
- 6. Reynoso MC, Hernandez A, Soto F, et al. Autosomal dominant macroglossia in two unrelated families. Hum Genet. 1986;74:200‐202. doi: 10.1007/BF00282095 [DOI] [PubMed] [Google Scholar]
- 7. Tabrizi SJ, Flower MD, Ross CA, Wild EJ. Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nat Rev Neurol. 2020;16:529‐546. doi: 10.1038/s41582-020-0389-4 [DOI] [PubMed] [Google Scholar]
- 8. Bates GP, Dorsey R, Gusella JF, et al. Huntington disease. Nat Rev Dis Primers. 2015;1:15005. doi: 10.1038/nrdp.2015.5 [DOI] [PubMed] [Google Scholar]
- 9. Fischer A, Notarangelo LD, Neven B, Cavazzana M, Puck JM. Severe combined immunodeficiencies and related disorders. Nat Rev Dis Primers. 2015;1:15061. doi: 10.1038/nrdp.2015.61 [DOI] [PubMed] [Google Scholar]
- 10. Gaspar HB, Aiuti A, Porta F, Candotti F, Hershfield MS, Notarangelo LD. How I treat ADA deficiency. Blood. 2009;114:3524‐3532. doi: 10.1182/blood-2009-06-189209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rare diseases: understanding this Public Health Priority. EURORDIS. Accessed March 1, 2023. https://www.eurordis.org/sites/default/files/publications/princeps_document‐EN.pdf
- 12. Young KM, Corrin T, Wilhelm B, et al. Zoonotic Babesia: a scoping review of the global evidence. PLoS One. 2019;14:e0226781. doi: 10.1371/journal.pone.0226781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vannier E, Krause PJ. Human babesiosis. N Engl J Med. 2012;366:2397‐2407. doi: 10.1056/NEJMra1202018 [DOI] [PubMed] [Google Scholar]
- 14. A preliminary assessment of the potential impact of rare diseases on the NHS. Imperial College Health Partners. Accessed March 1, 2023. https://imperialcollegehealthpartners.com/wp‐content/uploads/2019/05/ICHP‐RD‐Report‐Nov‐2018‐APPROVED‐002.pdf
- 15. Spillmann RC, McConkie‐Rosell A, Pena L, et al. A window into living with an undiagnosed disease: illness narratives from the Undiagnosed Diseases Network. Orphanet J Rare Dis. 2017;12:71. doi: 10.1186/s13023-017-0623-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shashi V, McConkie‐Rosell A, Rosell B, et al. The utility of the traditional medical genetics diagnostic evaluation in the context of next‐generation sequencing for undiagnosed genetic disorders. Genet Med. 2014;16:176‐182. doi: 10.1038/gim.2013.99 [DOI] [PubMed] [Google Scholar]
- 17. Mestre‐Ferrandiz J, Palaska C, Kelly T, Hutchings A, Parnaby A. An analysis of orphan medicine expenditure in Europe: is it sustainable? Orphanet J Rare Dis. 2019;14:287. doi: 10.1186/s13023-019-1246-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang G, Cintina I, Pariser A, Oehrlein E, Sullivan J, Kennedy A. The national economic burden of rare disease in the United States in 2019. Orphanet J Rare Dis. 2022;17:163. doi: 10.1186/s13023-022-02299-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Designating an orphan product: drugs and biological products. US Food and Drug Administration. Accessed March 1, 2023. https://www.fda.gov/industry/developing‐products‐rare‐diseases‐conditions/designating‐orphan‐product‐drugs‐and‐biological‐products
- 20. Orphan designation: overview. The European Medicines Agency. Accessed March 1, 2023. https://www.ema.europa.eu/en/human‐regulatory/overview/orphan‐designation‐overview
- 21. Tambuyzer E. Rare diseases, orphan drugs and their regulation: questions and misconceptions. Nat Rev Drug Discov. 2010;9:921‐929. doi: 10.1038/nrd3275 [DOI] [PubMed] [Google Scholar]
- 22. Biondi A, Gandemer V, De Lorenzo P, et al. Imatinib treatment of paediatric Philadelphia chromosome‐positive acute lymphoblastic leukaemia (EsPhALL2010): a prospective, intergroup, open‐label, single‐arm clinical trial. Lancet Haematol. 2018;5:e641‐e652. doi: 10.1016/S2352-3026(18)30173-X [DOI] [PubMed] [Google Scholar]
- 23. Berntorp E, Fischer K, Hart DP, et al. Haemophilia. Nat Rev Dis Primers. 2021;7:45. doi: 10.1038/s41572-021-00278-x [DOI] [PubMed] [Google Scholar]
- 24. Marchesini E, Morfini M, Valentino L. Recent advances in the treatment of hemophilia: a review. Biol Theory. 2021;15:221‐235. doi: 10.2147/BTT.S252580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tartaglia N, Davis S, Hench A, et al. A new look at XXYY syndrome: medical and psychological features. Am J Med Genet A. 2008;146A:1509‐1522. doi: 10.1002/ajmg.a.32366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Simpson JL, de la Cruz F, Swerdloff RS, et al. Klinefelter syndrome: expanding the phenotype and identifying new research directions. Genet Med. 2003;5:460‐468. doi: 10.1097/01.gim.0000095626.54201.d0 [DOI] [PubMed] [Google Scholar]
- 27. Cai X, Conley SM, Naash MI. RPE65: role in the visual cycle, human retinal disease, and gene therapy. Ophthalmic Genet. 2009;30:57‐62. doi: 10.1080/13816810802626399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thompson DA, Gal A. Vitamin A metabolism in the retinal pigment epithelium: genes, mutations, and diseases. Prog Retin Eye Res. 2003;22:683‐703. doi: 10.1016/s1350-9462(03)00051-x [DOI] [PubMed] [Google Scholar]
- 29. Daich Varela M, Cabral de Guimaraes TA, Georgiou M, et al. Leber congenital amaurosis/early‐onset severe retinal dystrophy: current management and clinical trials. Br J Ophthalmol. 2022;106:445‐451. doi: 10.1136/bjophthalmol-2020-318483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Astuti GD, Bertelsen M, Preising MN, et al. Comprehensive genotyping reveals RPE65 as the most frequently mutated gene in Leber congenital amaurosis in Denmark. Eur J Hum Genet. 2016;24:1071‐1079. doi: 10.1038/ejhg.2015.241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bravo‐Gil N, Gonzalez‐Del Pozo M, Martin‐Sanchez M, et al. Unravelling the genetic basis of simplex retinitis pigmentosa cases. Sci Rep. 2017;7:41937. doi: 10.1038/srep41937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rodrigues GA, Shalaev E, Karami TK, Cunningham J, Slater NKH, Rivers HM. Pharmaceutical development of AAV‐based gene therapy products for the eye. Pharm Res. 2018;36:29. doi: 10.1007/s11095-018-2554-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Russell S, Bennett J, Wellman JA, et al. Efficacy and safety of voretigene neparvovec (AAV2‐hRPE65v2) in patients with RPE65‐mediated inherited retinal dystrophy: a randomised, controlled, open‐label, phase 3 trial. Lancet. 2017;390:849‐860. doi: 10.1016/S0140-6736(17)31868-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Boye SE, Boye SL, Lewin AS, Hauswirth WW. A comprehensive review of retinal gene therapy. Mol Ther. 2013;21:509‐519. doi: 10.1038/mt.2012.280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med. 2008;358:2240‐2248. doi: 10.1056/NEJMoa0802315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber's congenital amaurosis. N Engl J Med. 2008;358:2231‐2239. doi: 10.1056/NEJMoa0802268 [DOI] [PubMed] [Google Scholar]
- 37. Hauswirth WW, Aleman TS, Kaushal S, et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno‐associated virus gene vector: short‐term results of a phase I trial. Hum Gene Ther. 2008;19:979‐990. doi: 10.1089/hum.2008.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. LUXTURNA. US Food and Drug Administration. Accessed March 1, 2023. https://www.fda.gov/vaccines‐blood‐biologics/cellular‐gene‐therapy‐products/luxturna
- 39. Cotmore SF, Agbandje‐McKenna M, Chiorini JA, et al. The family Parvoviridae. Arch Virol. 2014;159:1239‐1247. doi: 10.1007/s00705-013-1914-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Daya S, Berns KI. Gene therapy using adeno‐associated virus vectors. Clin Microbiol Rev. 2008;21:583‐593. doi: 10.1128/CMR.00008-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ward P. Replication of adeno‐associated virus DNA. In: Kerr JR, Cotmore SF, Bloom ME, Linden RM, Parrish CR, eds. Parvoviruses. Hodder Arnold; 2006:189‐212. [Google Scholar]
- 42. Henckaerts E, Dutheil N, Zeltner N, et al. Site‐specific integration of adeno‐associated virus involves partial duplication of the target locus. Proc Natl Acad Sci USA. 2009;106:7571‐7576. doi: 10.1073/pnas.0806821106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Penaud‐Budloo M, Le Guiner C, Nowrouzi A, et al. Adeno‐associated virus vector genomes persist as episomal chromatin in primate muscle. J Virol. 2008;82:7875‐7885. doi: 10.1128/JVI.00649-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Duan D, Sharma P, Yang J, et al. Circular intermediates of recombinant adeno‐associated virus have defined structural characteristics responsible for long‐term episomal persistence in muscle tissue. J Virol. 1998;72:8568‐8577. doi: 10.1128/JVI.72.11.8568-8577.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Samulski RJ, Zhu X, Xiao X, et al. Targeted integration of adeno‐associated virus (AAV) into human chromosome 19. EMBO J. 1991;10:3941‐3950. doi: 10.1002/j.1460-2075.1991.tb04964.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kotin RM, Siniscalco M, Samulski RJ, et al. Site‐specific integration by adeno‐associated virus. Proc Natl Acad Sci USA. 1990;87:2211‐2215. doi: 10.1073/pnas.87.6.2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mietzsch M, Grasse S, Zurawski C, et al. OneBac: platform for scalable and high‐titer production of adeno‐associated virus serotype 1‐12 vectors for gene therapy. Hum Gene Ther. 2014;25:212‐222. doi: 10.1089/hum.2013.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kuzmin DA, Shutova MV, Johnston NR, et al. The clinical landscape for AAV gene therapies. Nat Rev Drug Discov. 2021;20:173‐174. doi: 10.1038/d41573-021-00017-7 [DOI] [PubMed] [Google Scholar]
- 49. Srivastava A. In vivo tissue‐tropism of adeno‐associated viral vectors. Curr Opin Virol. 2016;21:75‐80. doi: 10.1016/j.coviro.2016.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Melanoma skin cancer incidence statistics. Cancer Research UK. Accessed March 1, 2023. https://www.cancerresearchuk.org/health‐professional/cancer‐statistics/statistics‐by‐cancer‐type/melanoma‐skin‐cancer/incidence#heading‐Two
- 51. Conry RM, Westbrook B, McKee S, Norwood TG. Talimogene laherparepvec: first in class oncolytic virotherapy. Hum Vaccin Immunother. 2018;14:839‐846. doi: 10.1080/21645515.2017.1412896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kohlhapp FJ, Kaufman HL. Molecular pathways: mechanism of action for Talimogene Laherparepvec, a new oncolytic virus immunotherapy. Clin Cancer Res. 2016;22:1048‐1054. doi: 10.1158/1078-0432.CCR-15-2667 [DOI] [PubMed] [Google Scholar]
- 53. Orvedahl A, Alexander D, Talloczy Z, et al. HSV‐1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23‐35. doi: 10.1016/j.chom.2006.12.001 [DOI] [PubMed] [Google Scholar]
- 54. Goldsmith K, Chen W, Johnson DC, Hendricks RL. Infected cell protein (ICP)47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J Exp Med. 1998;187:341‐348. doi: 10.1084/jem.187.3.341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu BL, Robinson M, Han ZQ, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti‐tumour properties. Gene Ther. 2003;10:292‐303. doi: 10.1038/sj.gt.3301885 [DOI] [PubMed] [Google Scholar]
- 56. Franke V, Berger DMS, Klop WMC, et al. High response rates for T‐VEC in early metastatic melanoma (stage IIIB/C‐IVM1a). Int J Cancer. 2019;145:974‐978. doi: 10.1002/ijc.32172 [DOI] [PubMed] [Google Scholar]
- 57. Andtbacka RHI, Collichio F, Harrington KJ, et al. Final analyses of OPTiM: a randomized phase III trial of talimogene laherparepvec versus granulocyte‐macrophage colony‐stimulating factor in unresectable stage III‐IV melanoma. J Immunother Cancer. 2019;7:145. doi: 10.1186/s40425-019-0623-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kaufman HL, Andtbacka RHI, Collichio FA, et al. Durable response rate as an endpoint in cancer immunotherapy: insights from oncolytic virus clinical trials. J Immunother Cancer. 2017;5:72. doi: 10.1186/s40425-017-0276-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Andtbacka RH, Kaufman HL, Collichio F, et al. Talimogene Laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33:2780‐2788. doi: 10.1200/JCO.2014.58.3377 [DOI] [PubMed] [Google Scholar]
- 60. Chesney J, Puzanov I, Collichio F, et al. Patterns of response with talimogene laherparepvec in combination with ipilimumab or ipilimumab alone in metastatic unresectable melanoma. Br J Cancer. 2019;121:417‐420. doi: 10.1038/s41416-019-0530-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ribas A, Dummer R, Puzanov I, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti‐PD‐1 immunotherapy. Cell. 2017;170:1109‐1119.e10. doi: 10.1016/j.cell.2017.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zakrzewski W, Dobrzynski M, Szymonowicz M, et al. Stem cells: past, present, and future. Stem Cell Res Ther. 2019;10:68. doi: 10.1186/s13287-019-1165-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nisole S, Saib A. Early steps of retrovirus replicative cycle. Retrovirology. 2004;1:9. doi: 10.1186/1742-4690-1-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Voisset C, Andrawiss M. Retroviruses at a glance. Genome Biol. 2000;1:reports4015.4011. doi: 10.1186/gb-2000-1-3-reports4015 [DOI] [Google Scholar]
- 65. Griffiths AJF, Gelbart WM, Miller JH, Lewontin R. Modern Genetic Analysis. W. H. Freeman; 1999. [Google Scholar]
- 66. Maetzig T, Galla M, Baum C, Schambach A. Gammaretroviral vectors: biology, technology and application. Viruses. 2011;3:677‐713. doi: 10.3390/v3060677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Milone MC, O'Doherty U. Clinical use of lentiviral vectors. Leukemia. 2018;32:1529‐1541. doi: 10.1038/s41375-018-0106-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang GP, Ciuffi A, Leipzig J, Berry CC, Bushman FD. HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 2007;17:1186‐1194. doi: 10.1101/gr.6286907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Terwilliger T, Abdul‐Hay M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. 2017;7:e577. doi: 10.1038/bcj.2017.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mullighan CG, Collins‐Underwood JR, Phillips LA, et al. Rearrangement of CRLF2 in B‐progenitor‐ and Down syndrome‐associated acute lymphoblastic leukemia. Nat Genet. 2009;41:1243‐1246. doi: 10.1038/ng.469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mullighan CG, Goorha S, Radtke I, et al. Genome‐wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758‐764. doi: 10.1038/nature05690 [DOI] [PubMed] [Google Scholar]
- 72. Greaves MF, Wiemels J. Origins of chromosome translocations in childhood leukaemia. Nat Rev Cancer. 2003;3:639‐649. doi: 10.1038/nrc1164 [DOI] [PubMed] [Google Scholar]
- 73. Ravandi F, O'Brien SM, Cortes JE, et al. Long‐term follow‐up of a phase 2 study of chemotherapy plus dasatinib for the initial treatment of patients with Philadelphia chromosome‐positive acute lymphoblastic leukemia. Cancer. 2015;121:4158‐4164. doi: 10.1002/cncr.29646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rowe JM, Buck G, Burnett AK, et al. Induction therapy for adults with acute lymphoblastic leukemia: results of more than 1500 patients from the international ALL trial: MRC UKALL XII/ECOG E2993. Blood. 2005;106:3760‐3767. doi: 10.1182/blood-2005-04-1623 [DOI] [PubMed] [Google Scholar]
- 75. Kantarjian HM, O'Brien S, Smith TL, et al. Results of treatment with hyper‐CVAD, a dose‐intensive regimen, in adult acute lymphocytic leukemia. J Clin Oncol. 2000;18:547‐561. doi: 10.1200/JCO.2000.18.3.547 [DOI] [PubMed] [Google Scholar]
- 76. Topp MS, Gokbuget N, Stein AS, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B‐precursor acute lymphoblastic leukaemia: a multicentre, single‐arm, phase 2 study. Lancet Oncol. 2015;16:57‐66. doi: 10.1016/S1470-2045(14)71170-2 [DOI] [PubMed] [Google Scholar]
- 77. DeAngelo DJ, Yu D, Johnson JL, et al. Nelarabine induces complete remissions in adults with relapsed or refractory T‐lineage acute lymphoblastic leukemia or lymphoblastic lymphoma: Cancer and Leukemia Group B study 19801. Blood. 2007;109:5136‐5142. doi: 10.1182/blood-2006-11-056754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Connors JM, Cozen W, Steidl C, et al. Hodgkin lymphoma. Nat Rev Dis Primers. 2020;6:61. doi: 10.1038/s41572-020-0189-6 [DOI] [PubMed] [Google Scholar]
- 79. Singh R, Shaik S, Negi BS, et al. Non‐Hodgkin's lymphoma: a review. J Family Med Prim Care. 2020;9:1834‐1840. doi: 10.4103/jfmpc.jfmpc_1037_19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Miao Y, Medeiros LJ, Li Y, Li J, Young KH. Genetic alterations and their clinical implications in DLBCL. Nat Rev Clin Oncol. 2019;16:634‐652. doi: 10.1038/s41571-019-0225-1 [DOI] [PubMed] [Google Scholar]
- 81. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375‐2390. doi: 10.1182/blood-2016-01-643569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fallah M, Kharazmi E, Pukkala E, et al. Familial risk of non‐Hodgkin lymphoma by sex, relationship, age at diagnosis and histology: a joint study from five Nordic countries. Leukemia. 2016;30:373‐378. doi: 10.1038/leu.2015.272 [DOI] [PubMed] [Google Scholar]
- 83. Cerhan JR, Slager SL. Familial predisposition and genetic risk factors for lymphoma. Blood. 2015;126:2265‐2273. doi: 10.1182/blood-2015-04-537498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Feins S, Kong W, Williams EF, Milone MC, Fraietta JA. An introduction to chimeric antigen receptor (CAR) T‐cell immunotherapy for human cancer. Am J Hematol. 2019;94:S3‐S9. doi: 10.1002/ajh.25418 [DOI] [PubMed] [Google Scholar]
- 85. Park JH, Geyer MB, Brentjens RJ. CD19‐targeted CAR T‐cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood. 2016;127:3312‐3320. doi: 10.1182/blood-2016-02-629063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Guedan S, Calderon H, Posey AD Jr, Maus MV. Engineering and design of chimeric antigen receptors. Mol Ther Methods Clin Dev. 2019;12:145‐156. doi: 10.1016/j.omtm.2018.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sadelain M, Riviere I, Riddell S. Therapeutic T cell engineering. Nature. 2017;545:423‐431. doi: 10.1038/nature22395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Milone MC, Fish JD, Carpenito C, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17:1453‐1464. doi: 10.1038/mt.2009.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507‐1517. doi: 10.1056/NEJMoa1407222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody‐binding domains and the gamma or zeta subunits of the immunoglobulin and T‐cell receptors. Proc Natl Acad Sci USA. 1993;90:720‐724. doi: 10.1073/pnas.90.2.720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lindner SE, Johnson SM, Brown CE, Wang LD. Chimeric antigen receptor signaling: functional consequences and design implications. Sci Adv. 2020;6:eaaz3223. doi: 10.1126/sciadv.aaz3223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Davenport AJ, Cross RS, Watson KA, et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc Natl Acad Sci USA. 2018;115:E2068‐E2076. doi: 10.1073/pnas.1716266115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Ramello MC, Benzaid I, Kuenzi BM, et al. An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci Signal. 2019;12:eaap9777. doi: 10.1126/scisignal.aap9777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chang ZL, Lorenzini MH, Chen X, et al. Rewiring T‐cell responses to soluble factors with chimeric antigen receptors. Nat Chem Biol. 2018;14:317‐324. doi: 10.1038/nchembio.2565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Bridgeman JS, Ladell K, Sheard VE, et al. CD3zeta‐based chimeric antigen receptors mediate T cell activation via cis‐ and trans‐signalling mechanisms: implications for optimization of receptor structure for adoptive cell therapy. Clin Exp Immunol. 2014;175:258‐267. doi: 10.1111/cei.12216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T‐cell therapy in refractory large B‐cell lymphoma. N Engl J Med. 2017;377:2531‐2544. doi: 10.1056/NEJMoa1707447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 results of ZUMA‐1: a multicenter study of KTE‐C19 anti‐CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25:285‐295. doi: 10.1016/j.ymthe.2016.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kochenderfer JN, Somerville RPT, Lu T, et al. Lymphoma remissions caused by anti‐CD19 chimeric antigen receptor T cells are associated with high serum interleukin‐15 levels. J Clin Oncol. 2017;35:1803‐1813. doi: 10.1200/JCO.2016.71.3024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kochenderfer JN, Feldman SA, Zhao Y, et al. Construction and preclinical evaluation of an anti‐CD19 chimeric antigen receptor. J Immunother. 2009;32:689‐702. doi: 10.1097/CJI.0b013e3181ac6138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. YESCARTA (axicabtagene ciloleucel). US Food and Drug Administration. Accessed March 1, 2023. https://www.fda.gov/vaccines‐blood‐biologics/cellular‐gene‐therapy‐products/yescarta‐axicabtagene‐ciloleucel
- 101. KYMRIAH (tisagenlecleucel). US Food and Drug Administration. Accessed March 1, 2023. https://www.fda.gov/vaccines‐blood‐biologics/cellular‐gene‐therapy‐products/kymriah‐tisagenlecleucel
- 102. Rubin DB, Al Jarrah A, Li K, et al. Clinical predictors of neurotoxicity after chimeric antigen receptor T‐cell therapy. JAMA Neurol. 2020;77:1536‐1542. doi: 10.1001/jamaneurol.2020.2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Rubin DB, Danish HH, Ali AB, et al. Neurological toxicities associated with chimeric antigen receptor T‐cell therapy. Brain. 2019;142:1334‐1348. doi: 10.1093/brain/awz053 [DOI] [PubMed] [Google Scholar]
- 104. Santomasso B, Bachier C, Westin J, Rezvani K, Shpall EJ. The other side of CAR T‐cell therapy: cytokine release syndrome, neurologic toxicity, and financial burden. Am Soc Clin Oncol Educ Book. 2019;39:433‐444. doi: 10.1200/EDBK_238691 [DOI] [PubMed] [Google Scholar]
- 105. Neelapu SS, Tummala S, Kebriaei P, et al. Chimeric antigen receptor T‐cell therapy – assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15:47‐62. doi: 10.1038/nrclinonc.2017.148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127:3321‐3330. doi: 10.1182/blood-2016-04-703751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Picanco‐Castro V, Pereira CG, Covas DT, et al. Emerging patent landscape for non‐viral vectors used for gene therapy. Nat Biotechnol. 2020;38:151‐157. doi: 10.1038/s41587-019-0402-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Burt RK, Balabanov R, Burman J, et al. Effect of nonmyeloablative hematopoietic stem cell transplantation vs continued disease‐modifying therapy on disease progression in patients with relapsing‐remitting multiple sclerosis: a randomized clinical trial. JAMA. 2019;321:165‐174. doi: 10.1001/jama.2018.18743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Appelbaum FR. Hematopoietic‐cell transplantation at 50. N Engl J Med. 2007;357:1472‐1475. doi: 10.1056/NEJMp078166 [DOI] [PubMed] [Google Scholar]
- 110. Copelan EA. Hematopoietic stem‐cell transplantation. N Engl J Med. 2006;354:1813‐1826. doi: 10.1056/NEJMra052638 [DOI] [PubMed] [Google Scholar]
- 111. Ljungman P, Urbano‐Ispizua A, Cavazzana‐Calvo M, et al. Allogeneic and autologous transplantation for haematological diseases, solid tumours and immune disorders: definitions and current practice in Europe. Bone Marrow Transplant. 2006;37:439‐449. doi: 10.1038/sj.bmt.1705265 [DOI] [PubMed] [Google Scholar]
- 112. Al‐Herz W, Bousfiha A, Casanova JL, et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. Front Immunol. 2014;5:162. doi: 10.3389/fimmu.2014.00162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kwan A, Abraham RS, Currier R, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 2014;312:729‐738. doi: 10.1001/jama.2014.9132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Kumrah R, Vignesh P, Patra P, et al. Genetics of severe combined immunodeficiency. Genes Dis. 2020;7:52‐61. doi: 10.1016/j.gendis.2019.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Picard C, Al‐Herz W, Bousfiha A, et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency 2015. J Clin Immunol. 2015;35:696‐726. doi: 10.1007/s10875-015-0201-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Flinn AM, Gennery AR. Adenosine deaminase deficiency: a review. Orphanet J Rare Dis. 2018;13:65. doi: 10.1186/s13023-018-0807-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Hirschhorn R. Overview of biochemical abnormalities and molecular genetics of adenosine deaminase deficiency. Pediatr Res. 1993;33:S35‐S41. doi: 10.1203/00006450-199305001-00194 [DOI] [PubMed] [Google Scholar]
- 118. Seto S, Carrera CJ, Wasson DB, Carson DA. Inhibition of DNA repair by deoxyadenosine in resting human lymphocytes. J Immunol. 1986;136:2839‐2843. PMID 2870121. [PubMed] [Google Scholar]
- 119. Seto S, Carrera CJ, Kubota M, Wasson DB, Carson DA. Mechanism of deoxyadenosine and 2‐chlorodeoxyadenosine toxicity to nondividing human lymphocytes. J Clin Invest. 1985;75:377‐383. doi: 10.1172/JCI111710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Mathews CK. DNA precursor metabolism and genomic stability. FASEB J. 2006;20:1300‐1314. doi: 10.1096/fj.06-5730rev [DOI] [PubMed] [Google Scholar]
- 121. Apasov SG, Blackburn MR, Kellems RE, Smith PT, Sitkovsky MV. Adenosine deaminase deficiency increases thymic apoptosis and causes defective T cell receptor signaling. J Clin Invest. 2001;108:131‐141. doi: 10.1172/JCI10360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Gangi‐Peterson L, Sorscher DH, Reynolds JW, Kepler TB, Mitchell BS. Nucleotide pool imbalance and adenosine deaminase deficiency induce alterations of N‐region insertions during V(D)J recombination. J Clin Invest. 1999;103:833‐841. doi: 10.1172/JCI4320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Benveniste P, Zhu W, Cohen A. Interference with thymocyte differentiation by an inhibitor of S‐adenosylhomocysteine hydrolase. J Immunol. 1995;155:536‐544. [PubMed] [Google Scholar]
- 124. Whitmore KV, Gaspar HB. Adenosine deaminase deficiency – more than just an immunodeficiency. Front Immunol. 2016;7:314. doi: 10.3389/fimmu.2016.00314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Antoine C, Muller S, Cant A, et al. Long‐term survival and transplantation of haemopoietic stem cells for immunodeficiencies: report of the European experience 1968‐99. Lancet. 2003;361:553‐560. doi: 10.1016/s0140-6736(03)12513-5 [DOI] [PubMed] [Google Scholar]
- 126. Hershfield MS, Buckley RH, Greenberg ML, et al. Treatment of adenosine deaminase deficiency with polyethylene glycol‐modified adenosine deaminase. N Engl J Med. 1987;316:589‐596. doi: 10.1056/NEJM198703053161005 [DOI] [PubMed] [Google Scholar]
- 127. Kohn DB, Hershfield MS, Puck JM, et al. Consensus approach for the management of severe combined immune deficiency caused by adenosine deaminase deficiency. J Allergy Clin Immunol. 2019;143:852‐863. doi: 10.1016/j.jaci.2018.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Aiuti A, Roncarolo MG, Naldini L. Gene therapy for ADA‐SCID, the first marketing approval of an ex vivo gene therapy in Europe: paving the road for the next generation of advanced therapy medicinal products. EMBO Mol Med. 2017;9:737‐740. doi: 10.15252/emmm.201707573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Aiuti A, Slavin S, Aker M, et al. Correction of ADA‐SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296:2410‐2413. doi: 10.1126/science.1070104 [DOI] [PubMed] [Google Scholar]
- 130. Aiuti A, Vai S, Mortellaro A, et al. Immune reconstitution in ADA‐SCID after PBL gene therapy and discontinuation of enzyme replacement. Nat Med. 2002;8:423‐425. doi: 10.1038/nm0502-423 [DOI] [PubMed] [Google Scholar]
- 131. Cicalese MP, Ferrua F, Castagnaro L, et al. Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood. 2016;128:45‐54. doi: 10.1182/blood-2016-01-688226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Markowitz D, Goff S, Bank A. Construction and use of a safe and efficient amphotropic packaging cell line. Virology. 1988;167:400‐406. doi: 10.1016/s0042-6822(88)90101-8 [DOI] [PubMed] [Google Scholar]
- 133. Lu CW, O'Reilly L, Roth MJ. G100R mutation within 4070A murine leukemia virus Env increases virus receptor binding, kinetics of entry, and viral transduction efficiency. J Virol. 2003;77:739‐743. doi: 10.1128/jvi.77.1.739-743.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Gaspar HB, Cooray S, Gilmour KC, et al. Hematopoietic stem cell gene therapy for adenosine deaminase‐deficient severe combined immunodeficiency leads to long‐term immunological recovery and metabolic correction. Sci Transl Med. 2011;3:97ra80. doi: 10.1126/scitranslmed.3002716 [DOI] [PubMed] [Google Scholar]
- 135. Aiuti A, Cattaneo F, Galimberti S, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360:447‐458. doi: 10.1056/NEJMoa0805817 [DOI] [PubMed] [Google Scholar]
- 136. Galanello R, Origa R. Beta‐thalassemia. Orphanet J Rare Dis. 2010;5:11. doi: 10.1186/1750-1172-5-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Origa R. β‐Thalassemia. Genet Med. 2017;19:609‐619. doi: 10.1038/gim.2016.173 [DOI] [PubMed] [Google Scholar]
- 138. Lal A, Locatelli F, Kwiatkowski JL, et al. Northstar‐3: interim results from a phase 3 study evaluating Lentiglobin gene therapy in patients with transfusion‐dependent β‐thalassemia and either a β0 or IVS‐I‐110 mutation at both alleles of the HBB gene. Blood. 2019;134:815. doi: 10.1182/blood-2019-128482 [DOI] [Google Scholar]
- 139. Locatelli F, Walters MC, Kwiatkowski JL, et al. Lentiglobin gene therapy for patients with transfusion‐dependent β‐thalassemia (TDT): results from the phase 3 Northstar‐2 and Northstar‐3 studies. Blood. 2018;132:1025. doi: 10.1182/blood-2018-99-112667 [DOI] [Google Scholar]
- 140. Thompson AA, Walters MC, Kwiatkowski J, et al. Gene therapy in patients with transfusion‐dependent beta‐thalassemia. N Engl J Med. 2018;378:1479‐1493. doi: 10.1056/NEJMoa1705342 [DOI] [PubMed] [Google Scholar]
- 141. Schneiderman J, Thompson AA, Walters MC, et al. Interim results from the phase 3 Hgb‐207 (Northstar‐2) and Hgb‐212 (Northstar‐3) studies of betibeglogene autotemcel gene therapy (LentiGlobin) for the treatment of transfusion‐dependent β‐thalassemia. Biol Blood Marrow Transplant. 2020;26:S87‐S88. doi: 10.1016/j.bbmt.2019.12.588 [DOI] [Google Scholar]
- 142. Negre O, Bartholomae C, Beuzard Y, et al. Preclinical evaluation of efficacy and safety of an improved lentiviral vector for the treatment of beta‐thalassemia and sickle cell disease. Curr Gene Ther. 2015;15:64‐81. doi: 10.2174/1566523214666141127095336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Cavazzana‐Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human beta‐thalassaemia. Nature. 2010;467:318‐322. doi: 10.1038/nature09328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Leach KM, Vieira KF, Kang SH, et al. Characterization of the human beta‐globin downstream promoter region. Nucleic Acids Res. 2003;31:1292‐1301. doi: 10.1093/nar/gkg209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Wall L, deBoer E, Grosveld F. The human beta‐globin gene 3′ enhancer contains multiple binding sites for an erythroid‐specific protein. Genes Dev. 1988;2:1089‐1100. doi: 10.1101/gad.2.9.1089 [DOI] [PubMed] [Google Scholar]
- 146. Sawado T, Halow J, Bender MA, Groudine M. The beta‐globin locus control region (LCR) functions primarily by enhancing the transition from transcription initiation to elongation. Genes Dev. 2003;17:1009‐1018. doi: 10.1101/gad.1072303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Negre O, Eggimann AV, Beuzard Y, et al. Gene therapy of the beta‐hemoglobinopathies by lentiviral transfer of the beta(A(T87Q))‐globin gene. Hum Gene Ther. 2016;27:148‐165. doi: 10.1089/hum.2016.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nat Rev Drug Discov. 2020;19:673‐694. doi: 10.1038/s41573-020-0075-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Hammond SM, Bernstein E, Beach D, Hannon GJ. An RNA‐directed nuclease mediates post‐transcriptional gene silencing in Drosophila cells. Nature. 2000;16(404):293‐296. doi: 10.1038/35005107 [DOI] [PubMed] [Google Scholar]
- 150. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double‐stranded RNA in Caenorhabditis elegans . Nature. 1998;391:806‐811. doi: 10.1038/35888 [DOI] [PubMed] [Google Scholar]
- 151. Fellmann C, Lowe SW. Stable RNA interference rules for silencing. Nat Cell Biol. 2014;16:10‐18. doi: 10.1038/ncb2895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Maniataki E, Mourelatos Z. A human, ATP‐independent, RISC assembly machine fueled by pre‐miRNA. Genes Dev. 2005;19:2979‐2990. doi: 10.1101/gad.1384005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550‐553. doi: 10.1126/science.1068999 [DOI] [PubMed] [Google Scholar]
- 154. Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS. Short hairpin RNAs (shRNAs) induce sequence‐specific silencing in mammalian cells. Genes Dev. 2002;16:948‐958. doi: 10.1101/gad.981002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Nasevicius A, Ekker SC. Effective targeted gene ‘knockdown’ in zebrafish. Nat Genet. 2000;26:216‐220. doi: 10.1038/79951 [DOI] [PubMed] [Google Scholar]
- 156. Chery J, Petri A, Wagschal A, et al. Development of locked nucleic acid antisense oligonucleotides targeting Ebola viral proteins and host factor Niemann‐Pick C1. Nucleic Acid Ther. 2018;28:273‐284. doi: 10.1089/nat.2018.0722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Goodchild J. Therapeutic oligonucleotides. In: Goodchild J, ed. Therapeutic Oligonucleotides Methods and Protocols. Humana Press; 2011:1‐15. [Google Scholar]
- 158. Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. 2012;11:125‐140. doi: 10.1038/nrd3625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Liang XH, Sun H, Shen W, et al. Antisense oligonucleotides targeting translation inhibitory elements in 5' UTRs can selectively increase protein levels. Nucleic Acids Res. 2017;45:9528‐9546. doi: 10.1093/nar/gkx632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Marsollier AC, Ciszewski L, Mariot V, et al. Antisense targeting of 3' end elements involved in DUX4 mRNA processing is an efficient therapeutic strategy for facioscapulohumeral dystrophy: a new gene‐silencing approach. Hum Mol Genet. 2016;25:1468‐1478. doi: 10.1093/hmg/ddw015 [DOI] [PubMed] [Google Scholar]
- 161. Gertz MA, Mauermann ML, Grogan M, Coelho T. Advances in the treatment of hereditary transthyretin amyloidosis: a review. Brain Behav. 2019;9:e01371. doi: 10.1002/brb3.1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Gertz MA, Benson MD, Dyck PJ, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66:2451‐2466. doi: 10.1016/j.jacc.2015.09.075 [DOI] [PubMed] [Google Scholar]
- 163. Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin‐related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31. doi: 10.1186/1750-1172-8-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Schmidt HH, Waddington‐Cruz M, Botteman MF, et al. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2018;57:829‐837. doi: 10.1002/mus.26034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Drug approval package: Onpattro (patisiran). US Food and Drug Administration. Accessed March 1, 2023. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210922orig1s000toc.cfm
- 166. Drug approval package: Tegsedi (inotersen). US Food and Drug Administration. Accessed March 1, 2023. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/211172Orig1s000TOC.cfm
- 167. Adams D, Gonzalez‐Duarte A, O'Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379:11‐21. doi: 10.1056/NEJMoa1716153 [DOI] [PubMed] [Google Scholar]
- 168. Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369:819‐829. doi: 10.1056/NEJMoa1208760 [DOI] [PubMed] [Google Scholar]
- 169. Assessment report: Onpattro. European Medicines Agency. Accessed March 1, 2023. https://www.ema.europa.eu/en/documents/assessment‐report/onpattro‐epar‐public‐assessment‐report_pdf
- 170. Woitok MM, Zoubek ME, Doleschel D, et al. Lipid‐encapsulated siRNA for hepatocyte‐directed treatment of advanced liver disease. Cell Death Dis. 2020;11:343. doi: 10.1038/s41419-020-2571-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Akinc A, Querbes W, De S, et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand‐based mechanisms. Mol Ther. 2010;18:1357‐1364. doi: 10.1038/mt.2010.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Suhr OB, Coelho T, Buades J, et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi‐dose study. Orphanet J Rare Dis. 2015;10:109. doi: 10.1186/s13023-015-0326-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Benson MD, Waddington‐Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379:22‐31. doi: 10.1056/NEJMoa1716793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Ackermann EJ, Guo S, Benson MD, et al. Suppressing transthyretin production in mice, monkeys and humans using 2nd‐generation antisense oligonucleotides. Amyloid. 2016;23:148‐157. doi: 10.1080/13506129.2016.1191458 [DOI] [PubMed] [Google Scholar]
- 175. Stein CA, Subasinghe C, Shinozuka K, Cohen JS. Physicochemical properties of phosphorothioate oligodeoxynucleotides. Nucleic Acids Res. 1988;16:3209‐3221. doi: 10.1093/nar/16.8.3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Iannitti T, Morales‐Medina JC, Palmieri B. Phosphorothioate oligonucleotides: effectiveness and toxicity. Curr Drug Targets. 2014;15:663‐673. doi: 10.2174/1389450115666140321100304 [DOI] [PubMed] [Google Scholar]
- 177. Papaioannou I, Disterer P, Owen JS. Use of internally nuclease‐protected single‐strand DNA oligonucleotides and silencing of the mismatch repair protein, MSH2, enhances the replication of corrected cells following gene editing. J Gene Med. 2009;11:267‐274. doi: 10.1002/jgm.1296 [DOI] [PubMed] [Google Scholar]
- 178. Castro JE, Prada CE, Aguillon RA, et al. Thymidine‐phosphorothioate oligonucleotides induce activation and apoptosis of CLL cells independently of CpG motifs or BCL‐2 gene interference. Leukemia. 2006;20:680‐688. doi: 10.1038/sj.leu.2404144 [DOI] [PubMed] [Google Scholar]
- 179. El Sahly HM, Baden LR, Essink B, et al. Efficacy of the mRNA‐1273 SARS‐CoV‐2 vaccine at completion of blinded phase. N Engl J Med. 2021;385:1774‐1785. doi: 10.1056/NEJMoa2113017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Polack FP, Thomas SJ, Kitchin N, et al. Safety and efficacy of the BNT162b2 mRNA Covid‐19 vaccine. N Engl J Med. 2020;383:2603‐2615. doi: 10.1056/NEJMoa2034577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Kolb SJ, Kissel JT. Spinal muscular atrophy: a timely review. Arch Neurol. 2011;68:979‐984. doi: 10.1001/archneurol.2011.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Cusin V, Clermont O, Gerard B, et al. Prevalence of SMN1 deletion and duplication in carrier and normal populations: implication for genetic counselling. J Med Genet. 2003;40:e39. doi: 10.1136/jmg.40.4.e39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy‐determining gene. Cell. 1995;80:155‐165. doi: 10.1016/0092-8674(95)90460-3 [DOI] [PubMed] [Google Scholar]
- 184. Calucho M, Bernal S, Alias L, et al. Correlation between SMA type and SMN2 copy number revisited: an analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul Disord. 2018;28:208‐215. doi: 10.1016/j.nmd.2018.01.003 [DOI] [PubMed] [Google Scholar]
- 185. Prior TW, Krainer AR, Hua Y, et al. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Genet. 2009;85:408‐413. doi: 10.1016/j.ajhg.2009.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Cartegni L, Krainer AR. Disruption of an SF2/ASF‐dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002;30:377‐384. doi: 10.1038/ng854 [DOI] [PubMed] [Google Scholar]
- 187. Chaytow H, Huang YT, Gillingwater TH, Faller KME. The role of survival motor neuron protein (SMN) in protein homeostasis. Cell Mol Life Sci. 2018;75:3877‐3894. doi: 10.1007/s00018-018-2849-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Monani UR, Lorson CL, Parsons DW, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8:1177‐1183. doi: 10.1093/hmg/8.7.1177 [DOI] [PubMed] [Google Scholar]
- 189. Claborn MK, Stevens DL, Walker CK, Gildon BL. Nusinersen: a treatment for spinal muscular atrophy. Ann Pharmacother. 2019;53:61‐69. doi: 10.1177/1060028018789956 [DOI] [PubMed] [Google Scholar]
- 190. Hua Y, Sahashi K, Hung G, et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010;24:1634‐1644. doi: 10.1101/gad.1941310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Majlessi M, Nelson NC, Becker MM. Advantages of 2'‐O‐methyl oligoribonucleotide probes for detecting RNA targets. Nucleic Acids Res. 1998;26:2224‐2229. doi: 10.1093/nar/26.9.2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Konersman CG, Ewing E, Yaszay B, Naheedy J, Murphy S, Skalsky A. Nusinersen treatment of older children and adults with spinal muscular atrophy. Neuromuscul Disord. 2021;31:183‐193. doi: 10.1016/j.nmd.2020.12.006 [DOI] [PubMed] [Google Scholar]
- 193. Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile‐onset spinal muscular atrophy. N Engl J Med. 2017;377:1723‐1732. doi: 10.1056/NEJMoa1702752 [DOI] [PubMed] [Google Scholar]
- 194. Finkel RS, Chiriboga CA, Vajsar J, et al. Treatment of infantile‐onset spinal muscular atrophy with nusinersen: a phase 2, open‐label, dose‐escalation study. Lancet. 2016;388:3017‐3026. doi: 10.1016/S0140-6736(16)31408-8 [DOI] [PubMed] [Google Scholar]
- 195. Chiriboga CA, Swoboda KJ, Darras BT, et al. Results from a phase 1 study of nusinersen (ISIS‐SMN(Rx)) in children with spinal muscular atrophy. Neurology. 2016;86:890‐897. doi: 10.1212/WNL.0000000000002445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Mendell JR, Al‐Zaidy S, Shell R, et al. Single‐dose gene‐replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377:1713‐1722. doi: 10.1056/NEJMoa1706198 [DOI] [PubMed] [Google Scholar]
- 197. Al‐Zaidy SA, Kolb SJ, Lowes L, et al. AVXS‐101 (Onasemnogene Abeparvovec) for SMA1: comparative study with a prospective natural history cohort. JNeuromuscul Dis. 2019;6:307‐317. doi: 10.3233/JND-190403 [DOI] [PubMed] [Google Scholar]
- 198. Mendell JR, Al‐Zaidy SA, Lehman KJ, et al. Five‐year extension results of the phase 1 START trial of onasemnogene abeparvovec in spinal muscular atrophy. JAMA Neurol. 2021;78:834‐841. doi: 10.1001/jamaneurol.2021.1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Day JW, Finkel RS, Chiriboga CA, et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile‐onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): an open‐label, single‐arm, multicentre, phase 3 trial. Lancet Neurol. 2021;20:284‐293. doi: 10.1016/S1474-4422(21)00001-6 [DOI] [PubMed] [Google Scholar]
- 200. Strauss KA, Farrar MA, Muntoni F, et al. Onasemnogene abeparvovec for presymptomatic infants with two copies of SMN2 at risk for spinal muscular atrophy type 1: the phase III SPR1NT trial. Nat Med. 2022;28:1381‐1389. doi: 10.1038/s41591-022-01866-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Valori CF, Ning K, Wyles M, et al. Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci Transl Med. 2010;2:35ra42. doi: 10.1126/scitranslmed.3000830 [DOI] [PubMed] [Google Scholar]
- 202. Dominguez E, Marais T, Chatauret N, et al. Intravenous scAAV9 delivery of a codon‐optimized SMN1 sequence rescues SMA mice. Hum Mol Genet. 2011;20:681‐693. doi: 10.1093/hmg/ddq514 [DOI] [PubMed] [Google Scholar]
- 203. Benkhelifa‐Ziyyat S, Besse A, Roda M, et al. Intramuscular scAAV9‐SMN injection mediates widespread gene delivery to the spinal cord and decreases disease severity in SMA mice. Mol Ther. 2013;21:282‐290. doi: 10.1038/mt.2012.261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. McCarty DM. Self‐complementary AAV vectors; advances and applications. Mol Ther. 2008;16:1648‐1656. doi: 10.1038/mt.2008.171 [DOI] [PubMed] [Google Scholar]
- 205. Bitetti I, Lanzara V, Margiotta G, Varone A. Onasemnogene abeparvovec gene replacement therapy for the treatment of spinal muscular atrophy: a real‐world observational study. Gene Ther. 2022. doi: 10.1038/s41434-022-00341-6. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Chand D, Mohr F, McMillan H, et al. Hepatotoxicity following administration of onasemnogene abeparvovec (AVXS‐101) for the treatment of spinal muscular atrophy. J Hepatol. 2021;74:560‐566. doi: 10.1016/j.jhep.2020.11.001 [DOI] [PubMed] [Google Scholar]
- 207. Thomsen G, Burghes AHM, Hsieh C, et al. Biodistribution of onasemnogene abeparvovec DNA, mRNA and SMN protein in human tissue. Nat Med. 2021;27:1701‐1711. doi: 10.1038/s41591-021-01483-7 [DOI] [PubMed] [Google Scholar]
- 208. Philippidis A. Novartis confirms deaths of two patients treated with gene therapy Zolgensma. Hum Gene Ther. 2022;33:842‐844. doi: 10.1089/hum.2022.29216.bfs [DOI] [PubMed] [Google Scholar]
- 209. McMillan HJ, Proud CM, Farrar MA, et al. Onasemnogene abeparvovec for the treatment of spinal muscular atrophy. Expert Opin Biol Ther. 2022;22:1075‐1090. doi: 10.1080/14712598.2022.2066471 [DOI] [PubMed] [Google Scholar]
- 210. Patient resources. The International Society for Cell & Gene Therapy Committee on the Ethics of Cell and Gene Therapy. Accessed March 1, 2023. http://www.isct‐unprovencellulartherapies.org/patient‐resources/
- 211. Guenther CM, Kuypers BE, Lam MT, Robinson TM, Zhao J, Suh J. Synthetic virology: engineering viruses for gene delivery. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2014;6:548‐558. doi: 10.1002/wnan.1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Kulkarni JA, Cullis PR, van der Meel R. Lipid nanoparticles enabling gene therapies: from concepts to clinical utility. Nucleic Acid Ther. 2018;28:146‐157. doi: 10.1089/nat.2018.0721 [DOI] [PubMed] [Google Scholar]
- 213. Brand GD, Ramada MHS, Genaro‐Mattos TC, et al. Towards an experimental classification system for membrane active peptides. Sci Rep. 2018;8:1194. doi: 10.1038/s41598-018-19566-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Pickar‐Oliver A, Gersbach CA. The next generation of CRISPR‐Cas technologies and applications. Nat Rev Mol Cell Biol. 2019;20:490‐507. doi: 10.1038/s41580-019-0131-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun. 2018;9:1911. doi: 10.1038/s41467-018-04252-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Gillmore JD, Gane E, Taubel J, et al. CRISPR‐Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med. 2021;385:493‐502. doi: 10.1056/NEJMoa2107454 [DOI] [PubMed] [Google Scholar]
- 217. Frangoul H, Altshuler D, Cappellini MD, et al. CRISPR‐Cas9 gene editing for sickle cell disease and beta‐thalassemia. N Engl J Med. 2021;384:252‐260. doi: 10.1056/NEJMoa2031054 [DOI] [PubMed] [Google Scholar]