

Abstract
One of the unsolved mysteries of medicine is how do volatile anesthetics (VAs) cause a patient to reversibly lose consciousness. In addition, identifying mechanisms for the collateral effects of VAs, including anesthetic-induced neurotoxicity (AiN) and anesthetic preconditioning (AP), has proven challenging. Multiple classes of molecules (lipids, proteins, and water) have been considered as potential VA targets, but recently proteins have received the most attention. Studies targeting neuronal receptors or ion channels had limited success in identifying the critical targets of VAs mediating either the phenotype of “anesthesia” or their collateral effects. Recent studies in both nematodes and fruit flies may provide a paradigm shift by suggesting that mitochondria may harbor the upstream molecular switch activating both primary and collateral effects. The disruption of a specific step of electron transfer within the mitochondrion causes hypersensitivity to VAs, from nematodes to Drosophila and to humans, while also modulating the sensitivity to collateral effects. The downstream effects from mitochondrial inhibition are potentially legion, but inhibition of presynaptic neurotransmitter cycling appears to be specifically sensitive to the mitochondrial effects. These findings are perhaps of even broader interest since two recent reports indicate that mitochondrial damage may well underlie neurotoxic and neuroprotective effects of VAs in the central nervous system (CNS). It is, therefore, important to understand how anesthetics interact with mitochondria to affect CNS function, not just for the desired facets of general anesthesia but also for significant collateral effects, both harmful and beneficial. A tantalizing possibility exists that both the primary (anesthesia) and secondary (AiN, AP) mechanisms may at least partially overlap in the mitochondrial electron transport chain (ETC).
Keywords: Anesthetics, genetics, sensitivity, neurotoxicity, pharmacogenetics, mitochondria
Impact Statement
Understanding the mechanisms of action of volatile anesthetics (VAs) will not only reveal information about their roles as agents used worldwide to induce unconsciousness and analgesia but also the causes of the desirable and undesirable collateral effects of these agents. In addition to satisfying intellectual curiosity about the nature of consciousness, an understanding of the metabolic pathways mediating the complex effects of VAs could reveal molecular targets for therapeutic exploitation as neuroprotectants. Genetic studies in invertebrates have provided a paradigm shift by suggesting that mitochondria may harbor the molecular switch activating both primary and collateral effects. The disruption of a specific step of electron transfer within the mitochondrion causes hypersensitivity to VAs, from nematodes and fruit flies to humans while also modulating the sensitivity to collateral effects. The striking evolutionary consistency raises the possibility that an ancient mechanism is at hand, linking metabolism in prokaryotes to neuronal silencing in mammals.
Introduction
The first proposal linking anesthesia directly to the effects of respiratory enzymes was formulated in 1914 by Otto Warburg 1 within the larger framework of hypotheses of asphyxia as a mechanism of volatile anesthetic (VA) action. The asphyxic framework of anesthesia lost favor in subsequent years but the “Warburg apparatus” continued to be used in the studies of tissue respiration. In the 1960s, as the chemiosmotic hypothesis of the general mechanism of oxidative and photosynthetic phosphorylation was formulated by Peter D. Mitchell, halothane was noted to suppress tissue oxygen consumption.2,3
In subsequent years, better understanding of oxidative phosphorylation and refinements in experimental techniques allowed the conclusion that VAs had a direct effect on mitochondrial respiration.4–6 Furthermore, the data suggested a specific interaction between halothane and complex I (Figure 1) of the mitochondrial electron transport chain (ETC). 5 Entry of electrons via complex II of the ETC was relatively unaffected by halothane compared to the flow of electrons via complex I. More recent data with improved techniques have confirmed that complex I-dependent respiration is indeed sensitive to VAs in the “clinical range,” whereas the other complexes of the ETC are not sensitive despite exposure to much higher VA concentrations.7,8
Figure 1.

Complex I in the mitochondrial electron transport chain (mETC). Electrons (e−) are donated to complex I (green) by NADH and to complex II (gray) by FADH2. Both complexes I and II then donate electrons (dashed line) to co-enzyme Q (CoQ) which transfers them complex III (turquoise) and then, via cytochrome C, to complex IV (red). Complexes I, III, and IV of the mETC pump protons to serve as a proton-motive force to drive production of ATP by complex V (blue). Embedded in the inner mitochondrial membrane, complex I is a large (~46 subunits) structure that transfers electrons from NADH to ubiquinone (CoQ). In the mETC, complex I is uniquely inhibited by VAs (red line) and contains NDUFS2, NDUFS4, NDUFS8, and ND2, all proteins discussed in this review.
The overall conclusion about anesthetics and mitochondrial function was that a variety of anesthetics suppressed tissue oxygen consumption and that this effect was observed with complex I-specific substrates but not with a complex II substrate.9,10 However, correlative studies of anesthetized tissues showed that “energy reserves” (ATP, glycogen phosphocreatine etc.) were maintained or increased 11 compared to the unanesthetized state. Further work from Michenfelder also found that ATP levels in the cortex were not decreased at high VA concentrations and after prolonged exposure. 12 Therefore, the degree to which interference with mitochondrial function was a significant contributor to anesthesia remained, at best, controversial. However, recent genetic studies have rekindled the interest in mitochondrial inhibition as a primary effect of VAs.
Genetic studies
Mitochondrial complex I and anesthetic sensitivity – invertebrate genetic models
Early work in the roundworm (Caenorhabditis elegans) identified several mutations which increased VA sensitivity. A mutation in one gene, gas-1(fc21), increased sensitivity to all VAs but the animals moved normally in air. 13 gas-1(fc21) reduced the EC50s of multiple VAs (isoflurane by over 80%) and shifted the dose-response curves for all VAs far to the left of wildtype. gas-1 overrides the effects of other genes that affect VA sensitivity in C. elegans and as such was postulated to be the closest gene to a putative anesthetic target.13,14
gas-1 was cloned and found to encode the highly conserved 49-kDa subunit (aka NDUFS2) of complex I, an entry point into the mitochondrial ETC (Figure 1).15,16 In contrast, animals with defects in complexes II, III, and IV of the ETC shared many phenotypes with gas-1, but, remarkably, were NOT hypersensitive to VAs.17–20 Complex I (NADH-ubiquinone oxidoreductase) catalyzes the reduction of ubiquinone (CoQ) by transferring electrons from NADH to complex III. 16 Another mutation, clk-1, is defective in CoQ synthesis and increases sensitivity to VAs.21,22 NDUFS2 is part of the CoQ binding site for complex I23–26 pinpointing this interaction as a potential determinant of anesthetic sensitivity. An RNAi-based screen indicated that subunits residing within subcomplex Iλ, close to the CoQ binding site, most directly influenced whole-worm anesthetic behavior. 27
The work in worms prompted investigations of mitochondrial defect-linked hypersensitivity to VAs in fruit flies (Drosophila melanogaster). Fruit flies have a complex CNS and roughly 70% of disease-causing genes are conserved between humans and fruit flies, the percentage being even higher in the nervous system.28–30 In analogy to the findings in rodents and humans (see below), a model of Leigh syndrome (LS) in fruit flies was used to investigate anesthetic action. LS is a fatal neurodegenerative disease caused primarily (but not exclusively) by mutations in complex I of the ETC. The ND2360114 line used in these experiments was generated by ethyl methanesulfonate mutagenesis in previous screens for temperature-sensitive paralytic mutants. 31 Characterization of this line revealed that flies homozygous for the ND2360114 allele reproduce many features of LS: morphological signs of neurodegeneration as young adults, shortened lifespan, abnormal mitochondrial morphology, and reduced adenosine triphosphate (ATP) levels. The causal mutation is an SNP in the ND23 gene (mammalian Ndufs8), a highly conserved subunit of mitochondrial complex I. 32 Anesthetic sensitivity was determined for isoflurane and sevoflurane using a modified rapid iterative negative geotaxis (RING) assay 33 and compared to six strains of wildtype flies (including the Canton-S, the background strain for ND2360114). 34 The EC50s for suppression of locomotion by isoflurane and sevoflurane were reduced from 0.36% and 0.89%, respectively, in Canton-S to 0.20% and 0.47%, respectively in ND2360114 flies, 34 corroborating the findings of increased VA sensitivity in patients and other animal models with complex I defects. Notably, after determining the EC50s of wildtype and mutant flies at one to eight days of age, all flies recovered from the exposure without mortality. When exposed at 10 days of age, ND2360114 flies present a mortality phenotype discussed in Part III.
Mitochondrial complex I and anesthetic sensitivity – humans
In general, patients with mitochondrial dysfunction (documented by biochemical studies or by histology) tolerate general anesthesia well.35,36 However, there were reports in the literature of poor outcomes in patients after anesthetic exposure.37–39 These reports led to the question of whether there might be variations in anesthetic sensitivity in humans with mitochondrial defects. We questioned whether sensitivity to VAs might vary significantly in mitochondrial patients. Since sevoflurane was, and remains, the predominant VA in use in children, the concentration of sevoflurane necessary to induce loss of consciousness was correlated with mitochondrial function. Children presented with a common constellation of symptoms included hypotonia, developmental delay, failure to thrive, and lactic acidosis. The results in a small cohort of children indicated that some patients with complex I defects were extremely sensitive to sevoflurane. 40
These results led to a larger multicenter case series of 91 patients presenting for diagnosis of mitochondrial defects. 41 Anesthesia was again induced with sevoflurane only, titrating the dose to that which caused loss of consciousness prior to surgical incision. The concentrations used to achieve this endpoint were then compared to measured function of complexes I–IV in skeletal muscle biopsies. Underlying severity of disease did not predict who might be hypersensitive to sevoflurane. The results were remarkable in that all patients with complex I dysfunction showed an increase in sensitivity to sevoflurane. However, not all patients who were hypersensitive to sevoflurane had complex I dysfunction. The authors speculated that ETC supercomplexes, consisting of complexes I–III–IV, are thought to be the functioning mitochondrial respiratory complex, and that allosteric interactions may be important for its function. However, most astonishingly, as in both the nematode and Drosophila, changes in complex I function uniformly resulted in hypersensitivity to sevoflurane.
Mitochondrial complex I and anesthetic sensitivity – mice
Recently, the importance of mitochondria for VA sensitivity was extended to a rodent model, the mouse, in which further physiologic studies were possible. Ndufs4 encodes one of over 40 subunits of mammalian mitochondrial complex I.15,42,43 The knockout (KO) of the nuclear gene Ndufs4 exhibited large increases in behavioral sensitivity (resistance to tail clamp, loss of righting reflex) to halothane and isoflurane.44,45 For both endpoints, the EC50s of the KO for isoflurane and halothane were 1/3–1/2 those of the controls. 44 Clearly, the effects of complex I dysfunction on VA sensitivity are also profound in mice as in the nematode, fly, and human. Importantly, and frankly surprisingly, this mitochondrial defect causes a resistance to ketamine, indicating that the behavioral change is not simply the result of baseline non-specific decreases in CNS function. Since complex I dysfunction causes VA hypersensitivity in organisms from nematodes to man, the implication is that an ancient mechanism is at hand, linking movement of electrons within the ETC (Figure 1) to neuronal silencing in the presence of VAs. The data indicate that complex I function is central to VA sensitivity and is likely an important VA target.
There are many possible links between mitochondrial function and neurotransmission. 46 For example, mitochondria are the site of both glutamate synthesis and the degradation of GABA, which is synthesized by decarboxylation of glutamate. 47 In addition, complex I represents the rate-limiting step for energy production in the mitochondrion.48,49 Synaptic transmission is energetically demanding; excitatory neuronal activity has been estimated to account for 75% of the energy requirement for cerebellum and 50% of the cortex.50,51 Inhibition of excitatory neurotransmission (cholinergic or glutamatergic) has been postulated as a mechanism by which VAs work and could be most sensitive to defective mitochondrial function.46,52,53 This is especially true of glutamatergic neurotransmission, which has been shown to be the most energetically demanding neurotransmission in the CNS.50,54,55 However, in addition to energy production, the roles of mitochondria/complex I in other important phenomena, such as ROS signaling and calcium homeostasis, make it easy to implicate them in the most important facets of neuronal signaling, both desired and collateral.
Several laboratories have shown that presynaptic function of excitatory neurons is inhibited by VAs.56–61 Studies with wildtype and cell-specific Ndufs4(KO) mice showed that mitochondrial function in glutamatergic neurons determined VA sensitivity to tail clamp and loss of righting reflex.61,62 Furthermore, presynaptic frequencies of excitatory signaling were inhibited by VAs at concentrations approximating the whole animal EC50s of wildtype and Ndufs4(KO) mice.61,62 The sensitivity of presynaptic frequencies to isoflurane increased when neurons were made dependent on mitochondrial respiration.
Synaptic neurotransmitter recycling, in particular endocytosis, is uniquely dependent on ATP availability.63,64 Since VAs are known potent inhibitors of mitochondrial complex I,5,6,8,11 its inhibition may underlie the presynaptic changes caused by VAs. Recent work in neuronal cultures demonstrated that isoflurane specifically inhibited neurotransmitter endocytosis and ATP production at a concentration approximating the whole animal EC95 of wildtype mice. In addition, a similar inhibition was seen in Ndufs4(KO) at lower concentrations of isoflurane matching the EC95 of the mutant. 65 When ATP levels were supported by increasing glucose (to increase glycolysis) or use of a complex II-specific substrate to bypass inhibition of complex I, endocytosis and ATP levels were not affected by isoflurane exposure in either wildtype or mutant cultures. The results indicated that isoflurane inhibition of complex I leads to ATP depletion at the presynapse resulting in a failure of neurotransmitter cycling. 65
To establish that complex I is a primary component controlling VA sensitivity, a change in complex I leading to resistance would be invaluable. The recent availability of a genetic model that can bypass complex I (NDi1)66,67 as an electron donor allowed for direct analysis of the mitochondrial effects of isoflurane on both whole animal behavior and endocytosis. The presence of NDi1 in the CNS of mice led to a ~25% resistance to isoflurane and halothane in loss of righting reflex (LORR) and tail clamp assays, while rescuing both the ATP decrease and failure of endocytosis in the presence of isoflurane. 65 The behavioral effects of NDi1 on VA resistance were reproducible in flies using a locomotion assay (RING) (MP, unpublished data).
VAs and metabolism
In addition to the effects of VAs on the mitochondrial ETC,5,6,8,11 other effects of these drugs on metabolism have been also noted. Work from the Klein laboratory noted increased lactate levels in CNS tissue from animals anesthetized with VAs but not those anesthetized with parenteral agents.68,69 Follow-up work from this group recently showed that the increase in lactate was in turn related to complex I inhibition by the VAs, specifically by halothane and isoflurane and, as they note, to a lesser extent by sevoflurane. 7
Preliminary work from the Johnson laboratory has shown that neonates may carry an additional metabolic risk from VA exposure.70,71 Unexpectedly, they found that in neonates blood, β-hydroxybutyrate was rapidly depleted by VAs at concentrations well below those necessary for anesthesia, while adult levels were unaffected. Depletion of β-HB was accompanied by an increase in lactate, citrate accumulation, malonyl-CoA production by acetyl-CoA carboxylase, and inhibition of fatty acid oxidation. Acylcarnitines were also depleted but the decrease was not dependent on complex I inhibition but was most consistent with an effect on carnitine-palmitoyltransferase-1 resulting in a defect in transport of fatty acids across the mitochondrial membrane. Broad changes in metabolism have also been hinted at in other studies of the isoflurane-induced changes in a metabolome. 72 While it is not yet possible to specifically identify the pathways most affected by VAs, they have broad effects; the specific pathways affected may change during development.
An extensively studied VA, isoflurane, has long been known to be effective in preconditioning tissue from exposure to hypoxia. The mechanism, not fully understood, is felt to involve metabolic inhibition (probably of mitochondrial complexes I and III)73,74 with resulting release of reactive oxygen species (ROS).73,75 Recent work from the AN laboratory indicated that this may involve an indirect effect of isoflurane on complex II. 76 If so, this widens the range of important effects of VAs on the ETC.
Genetic determinants of collateral anesthetic pharmacodynamics
Exposure to VAs carries the risk of anesthetic-induced neurotoxicity (AiN). Situations in which a “fragile” (developing, diseased, injured) brain is exposed to VAs increase the AiN risk. 77 Understanding of risk factors and mechanisms of toxicity is still rudimentary, and progress is slowed by cost, animal welfare concerns, and limited throughput due in part to frequently ambiguous behavioral phenotypes of mammalian models.77,78 An important question is whether AiN is purely a xenotoxic phenomenon (anyone can be affected by an exposure to an appropriate dose of the toxin) or whether biological factors other than age determine the individual risk (i.e. could individuals at high risk be identified and protected?). Phrased differently, do genetic and environmental backgrounds determine the risk of an individual to be negatively affected by AiN? Genetic determination of risk from exposure to VAs is not unknown: due to its fulminant phenotype and Mendelian inheritance pattern, Malignant Hyperthermia Syndrome remains the best known pharmacogenetic disorder in perioperative medicine. 79 The core anesthetic phenotype of VA sensitivity, using inhibition of movement in response to a noxious stimulus, is controlled by genetic background.80–84 It is therefore plausible that AiN is also a quantitative genetic trait. Mitochondria are extremely genetically complex organelles with a unique inheritance pattern (contributions from the nuclear genome collaborate with purely maternally inherited genes of the mitochondrial genome). High rates of mutations and mitochondrial damage have been documented in the context of AiN85,86 and numerous neurodegenerative diseases. 87
Due to its experimental flexibility, advanced and well-characterized neurocircuitry, and the availability of an unmatched genetic toolbox, the fruit fly is widely used as (1) a model for neurodegenerative diseases; (2) for toxicologic studies; and (3) for genetic studies and can be arguably considered the ideal model to test the hypothesis that AiN is a quantitative trait. Two models were used to explore the pharmacogenomics of the interactions of isoflurane with “fragile” brains: (1) the brain affected by neurodegeneration using a fly model of LS and (2) the injured brain using a fly Traumatic Brain Injury (TBI) model.
Mitochondrial complex I mutations predispose to AiN
Exposure of neonate and young “wildtype” (i.e. not harboring known mutations) rodents to general anesthetics causes morphological changes in mitochondria. 88 In a seven-day-old rat pups, a 6-h exposure to commonly used sedatives and anesthetics (midazolam, nitrous oxide, isoflurane) resulted in a disturbed balance between mitochondrial fission and fusion with excessive fission. 86 This impaired mitochondrial homeostasis, attributed to excessive production of ROS, was linked to the neurotoxic consequences of anesthesia.89,90 The injectable anesthetic propofol, while less extensively studied than isoflurane, also has toxic mitochondrial effects.91,92 Long-term infusions of propofol can cause the propofol infusion syndrome possibly unmasking latent mitochondrial disease. 93 Short administrations have also been associated with alterations of mitochondrial dynamics in vitro. 85
To examine the interaction of VAs with mitochondria, we used a fly model of LS caused by a hypomorphic mutation in the ND23 subunit (Ndufs8 in mice) of mitochondrial complex I. In addition to behavioral hypersensitivity, carriers of the ND2360114 allele present an age- and O2-dependent anesthetic toxicity phenotype. Wildtype fruit flies recover from 4%h isoflurane (concentration anesthetic × duration of exposure), 34 with no mortality and 10–20%h isoflurane causes less than 3% mortality in the wildtype. In contrast, exposure of ND2360114 flies to 4%h isoflurane in room air results in ~50% mortality within 24 h after emergence from anesthesia. 34 Notable features of the mortality phenotype in ND2360114 flies are: (1) age-dependence (no mortality until 10 days of adult life); (2) O2 concentration-dependence: 75% O2 and 5% O2 during isoflurane exposure increases and suppresses 24 h mortality, respectively; (3) mortality can be rescued by overexpression of wildtype ND23 in neurons; (4) heterozygous carriers of ND2360114 develop a mortality phenotype after 4%h of isoflurane at an advanced age and only under hyperoxic conditions. Preliminary data indicate that mutations in certain other subunits of complex I also sensitize to AiN to different degrees (Borchert submitted). We conclude that flies used as models of LS offer an easily scored and unambiguous phenotype that in combination with the available genetic tools can be used to effectively investigate collateral effects of VAs.
Genetic background influences the extent of AiN in a fly TBI model
The availability of inbred collections of fully sequenced fly lines (e.g. the Drosophila Genetic Reference Panel [DGRP]) 94 presents an opportunity to directly test the hypothesis that VA pharmacodynamics for AiN are shaped by naturally occurring genetic polymorphisms. Segregating variation in the DGRP collection mimics genetic variation in human populations, allowing inferences on the involvement of conserved pathways.94–96 Schiffman et al., 97 subjected young adult flies from 146 DGRP strains to a standard TBI protocol, followed by exposure to 1 h of 2% isoflurane in 75% O2. Compared to paired strains in room air, treatment resulted in variable degrees of increased 24 h mortality across the lines (see supplementary figure in reference for an illustrative example). Genome-wide, five SNPs in three biologically plausible genes (Prip, Drip, and Gyc88E) were associated with variability in mortality at a p-value threshold < 107. Prip and Drip are orthologous to mammalian water permeable channels (aquaporins) and Gyc88E is orthologous to the oxygen sensor GUCYB1. Variants in the aquaporin-4 channel are associated with alterations in outcomes from various types of brain injury in rodents98–100 and humans.101,102 The identification of these genes using genome-wide association study (GWAS) analysis of a Drosophila collection attests to evolutionary conservation of key physiological pathways and to the potential for translatability of experimentally flexible model organisms. GUCYB1 has not yet been associated with outcome variability in mammalian TBI models. However, the experimental paradigm used includes hyperoxia and therefore makes a cellular oxygen sensor a plausible potential candidate gene for modulating outcome of TBI. In summary, the analysis of post-TBI toxicity using tools available in the fruit fly provides proof of principle that AiN is a quantitative trait and that identification of genes contributing to AiN can be accelerated by including fruit flies into research strategies.
As a counterpoint to AiN, VAs are also known to robustly protect the brain and other tissues from damage when administered prior to ischemic injury, a phenomenon referred to as anesthetic preconditioning (AP). A detailed understanding of AP mechanisms would yield valuable targets for cerebral protection. However, because anesthetics directly modulate injury, their use in experimental brain injury deprives the experimenter of a valid (i.e. unanesthetized) control and no feasible protective strategies against TBI have been proposed to date. Genetic variability, however, is a largely unexplored variable present in the human population but seldom considered in model animal studies because of cost.103–105 Similarly, throughput is low because of the time and effort needed to study each animal across its lifespan, which is desirable as pharmacodynamics, may be shaped by age-related changes that affect mitochondria. 97
AP can be reproduced in a Drosophila closed-head TBI model caused by blunt trauma. Using mortality at 24 h after TBI (MI24) as read-out, AP with the VAs isoflurane and sevoflurane substantially reduced the MI24. 34 AP in the fly TBI model replicated findings obtained in mammals in that in old flies and in flies rendered obese by starvation selection over generations, 106 the effectiveness of preconditioning was thwarted.97,107 Ongoing research suggests that, analogously to AiN, the protective effect conferred by AP in TBI varies in magnitude among DGRP lines, indicating that susceptibility to prophylactic pharmacologic brain protection is a quantitative trait. (Figure 2, unpublished observation). Once these experiments are completed, a GWAS analysis will identify genes and pathways associated with effective AP informing strategies aimed at identifying drug targets for prophylactic pharmacological brain protection.
Figure 2.
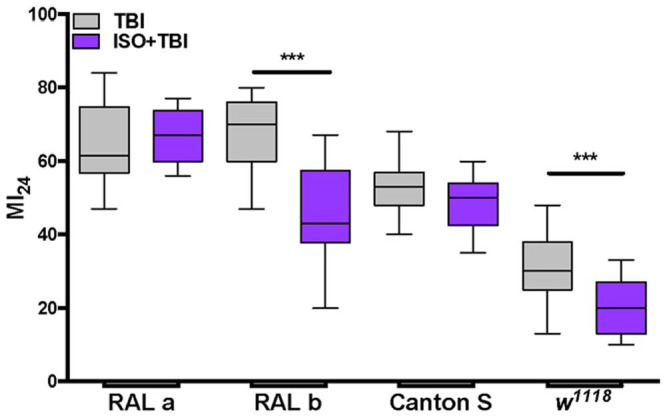
Genetic background influences the effectiveness of AP. Comparable mortality at 24 h after TBI (MI24) in two inbred fly lines (RAL a, RAL b) and different mortality in two standard laboratory fly lines (Canton-S and w1118) are shown in gray. Preconditioning (15 min with 2% isoflurane prior to TBI) suppressed the MI24 in RAL b and in w1118 but not in RAL a and Canton-S (purple). For each line and condition N = 10–20 vials with 20–30 flies each. Box 25th–75th percentiles, line indicates median.
***p < 0.0005.
Summary
The important targets for VAs have remained a point of debate for over a century. While neuronal receptors and gated ion channels have recently been favored proposed targets, proof of their behavioral importance has been elusive. Mitochondria are also known to contain molecular targets of VAs and have long been known to be inhibited by these drugs. However, it was unclear whether interaction of VAs with mitochondrial targets contributes to the conventional, clinically observable phenotypes of anesthesia and to the collateral phenotypes of exposure to VAs referred to as AiN and AP. Recently, genetic studies have indicated that metabolic interactions may be of more importance than previously appreciated. Because mitochondria are central to both normal cell function (both as energy generators and signaling relays) and stress response, both primary and collateral outcomes from exposure to VAs can result from their mitochondrial effects. Mitochondrial function is shaped by biological (age, genetic background, concurrent inflammation, and degenerative conditions) and environmental factors (stress intensity, duration of exposure to VAs, co-exposure to other agents, and injury) all of which contribute to the varied response to VAs between individuals. When seen within this framework, interaction between VAs and mitochondria can result in a spectrum of outcomes, ranging from anesthesia with rapid, complete recovery to long-term cognitive impairment and from tissue damage to organ protection.
Footnotes
Authors’ Contributions: MP and PGM participated in writing, review, and editing; MMS participated in review and editing; DJ-S contributed in experimental data collection, review, and editing.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: MP: NIGMS R01GM134107 and R&D fund of the Department of Anesthesiology. University of Wisconsin–Madison; PGM: NIGMS R35GM139556 and Northwest Mitochondrial Research Guild; MMS, NIGMS R01GM105696; NIGMS R01GM133865, and Northwest Mitochondrial Research Guild.
ORCID iD: Philip G Morgan  https://orcid.org/0000-0003-4857-2756
https://orcid.org/0000-0003-4857-2756
References
- 1.Warburg O. Uber die empfindlichkeit der sauerstoffarmung gegenüber indifferenten narkotika. Pflügers Archiv 1914;158:19–28 [Google Scholar]
- 2.Hoech GP, Jr, Matteo RS, Fink BR. Effect of halothane on oxygen consumption of rat brain, liver and heart and anaerobic glycolysis of rat brain. Anesthesiology 1966;27:770–7 [DOI] [PubMed] [Google Scholar]
- 3.Matteo RS, Hoech GP, Jr, Hoskin FC. The effects of cyclopropane and diethyl ether on tissue oxygen consumption and anaerobic glycolysis of brain in vitro. Anesthesiology 1969;30:156–63 [DOI] [PubMed] [Google Scholar]
- 4.Miller RN, Hunter FE., Jr.The effect of halothane on electron transport, oxidative phosphorylation, and swelling in rat liver mitochondria. Mol Pharmacol 1970;6:67–77 [PubMed] [Google Scholar]
- 5.Harris RA, Munroe J, Farmer B, Kim KC, Jenkins P. Action of halothane upon mitochondrial respiration. Arch Biochem Biophys 1971;142: 435–44 [DOI] [PubMed] [Google Scholar]
- 6.Hanley PJ, Ray J, Brandt U, Daut J. Halothane, isoflurane and sevoflurane inhibit NADH: ubiquinone oxidoreductase (complex I) of cardiac mitochondria. J Physiol 2002;544:687–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fedorov A, Lehto A, Klein J. Inhibition of mitochondrial respiration by general anesthetic drugs. Naunyn Schmiedebergs Arch Pharmacol 2023;396:375–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kayser EB, Suthammarak W, Morgan PG, Sedensky MM. Isoflurane selectively inhibits distal mitochondrial complex I in Caenorhabditis elegans. Anesth Analg 2011;112:1321–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nahrwold ML, Cohen PJ. The effects of forane and fluroxene on mitochondrial respiration: correlation with lipid solubility and in-vivo potency. Anesthesiology 1973;38:437–44 [DOI] [PubMed] [Google Scholar]
- 10.Nahrwold ML, Cohen PJ. Anesthetics and mitochondrial respiration. Clin Anesth 1975;11:25–44 [PubMed] [Google Scholar]
- 11.Cohen PJ. Effect of anesthetics on mitochondrial function. Anesthesiology 1973;39:153–64 [DOI] [PubMed] [Google Scholar]
- 12.Newberg LA, Milde JH, Michenfelder JD. The cerebral metabolic effects of isoflurane at and above concentrations that suppress cortical electrical activity. Anesthesiology 1983;59:23–8 [DOI] [PubMed] [Google Scholar]
- 13.Morgan PG, Sedensky MM. Mutations conferring new patterns of sensitivity to volatile anesthetics in Caenorhabditis elegans. Anesthesiology 1994;81:888–98 [DOI] [PubMed] [Google Scholar]
- 14.Kayser EB, Morgan PG, Sedensky MM. GAS-1: a mitochondrial protein controls sensitivity to volatile anesthetics in the nematode Caenorhabditis elegans. Anesthesiology 1999;90:545–54 [DOI] [PubMed] [Google Scholar]
- 15.Hirst J, Carroll J, Fearnley IM, Shannon RJ, Walker JE. The nuclear encoded subunits of complex I from bovine heart mitochondria. Biochim Biophys Acta 2003;1604:135–50 [DOI] [PubMed] [Google Scholar]
- 16.Walker JE. The NADH:ubiquinone oxidoreductase (complex I) of respiratory chains. Q Rev Biophys 1992;25:253–324 [DOI] [PubMed] [Google Scholar]
- 17.Suthammarak W, Morgan PG, Sedensky MM. Mutations in mitochondrial complex III uniquely affect complex I in Caenorhabditis elegans. J Biol Chem 2010;285:40724–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suthammarak W, Yang YY, Morgan PG, Sedensky MM. Complex I function is defective in complex IV-deficient Caenorhabditis elegans. J Biol Chem 2009;284:6425–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo J, Lemire BD. The ubiquinone-binding site of the Saccharomyces cerevisiae succinate-ubiquinone oxidoreductase is a source of superoxide. J Biol Chem 2003;278:47629–35 [DOI] [PubMed] [Google Scholar]
- 20.Ishii N, Fujii M, Hartman PS, Tsuda M, Yasuda K, Senoo-Matsuda N, Yanase S, Ayusawa D, Suzuki K. A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature 1998;394:694–7 [DOI] [PubMed] [Google Scholar]
- 21.Falk MJ, Kayser EB, Morgan PG, Sedensky MM. Mitochondrial complex I function modulates volatile anesthetic sensitivity in C. Elegans. Curr Biol 2006;16:1641–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kayser EB, Sedensky MM, Morgan PG, Hoppel CL. Mitochondrial oxidative phosphorylation is defective in the long-lived mutant clk-1. J Biol Chem 2004;279:54479–86 [DOI] [PubMed] [Google Scholar]
- 23.Efremov RG, Sazanov LA. Structure of the membrane domain of respiratory complex I. Nature 2011;476:414–20 [DOI] [PubMed] [Google Scholar]
- 24.Sazanov LA. Respiratory complex I: mechanistic and structural insights provided by the crystal structure of the hydrophilic domain. Biochemistry 2007;46:2275–88 [DOI] [PubMed] [Google Scholar]
- 25.Zickermann V, Kerscher S, Zwicker K, Tocilescu MA, Radermacher M, Brandt U. Architecture of complex I and its implications for electron transfer and proton pumping. Biochim Biophys Acta 2009;1787:574–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chung I, Wright JJ, Bridges HR, Ivanov BS, Biner O, Pereira CS, Arantes GM, Hirst J. Cryo-EM structures define ubiquinone-10 binding to mitochondrial complex I and conformational transitions accompanying Q-site occupancy. Nat Commun 2022;13:2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Falk MJ, Rosenjack JR, Polyak E, Suthammarak W, Chen Z, Morgan PG, Sedensky MM. Subcomplex Ilambda specifically controls integrated mitochondrial functions in Caenorhabditis elegans. PLoS ONE 2009;4:e6607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reiter LT, Bier E. Using Drosophila melanogaster to uncover human disease gene function and potential drug target proteins. Expert Opin Ther Targets 2002;6:387–99 [DOI] [PubMed] [Google Scholar]
- 29.Bilen J, Bonini NM. Drosophila as a model for human neurodegenerative disease. Annu Rev Genet 2005;39:153–71 [DOI] [PubMed] [Google Scholar]
- 30.Marsh JL, Thompson LM. Drosophila in the study of neurodegenerative disease. Neuron 2006;52:169–78 [DOI] [PubMed] [Google Scholar]
- 31.Palladino MJ, Hadley TJ, Ganetzky B. Temperature-sensitive paralytic mutants are enriched for those causing neurodegeneration in Drosophila. Genetics 2002;161:1197–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loewen CA, Ganetzky B. Mito-nuclear interactions affecting lifespan and neurodegeneration in a Drosophila model of Leigh Syndrome. Genetics 2018;208:1535–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gargano JW, Martin I, Bhandari P, Grotewiel MS. Rapid iterative negative geotaxis (RING): a new method for assessing age-related locomotor decline in Drosophila. Exp Gerontol 2005;40:386–95 [DOI] [PubMed] [Google Scholar]
- 34.Fischer JA, Olufs ZPG, Katzenberger RJ, Wassarman DA, Perouansky M. Anesthetics influence mortality in a Drosophila model of blunt trauma with traumatic brain injury. Anesth Analg 2018;126:1979–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Driessen J, Willems S, Dercksen S, Giele J, van der Staak F, Smeitink J. Anesthesia-related morbidity and mortality after surgery for muscle biopsy in children with mitochondrial defects. Paediatr Anaesth 2007;17:16–21 [DOI] [PubMed] [Google Scholar]
- 36.Footitt EJ, Sinha MD, Raiman JA, Dhawan A, Moganasundram S, Champion MP. Mitochondrial disorders and general anaesthesia: a case series and review. Br J Anaesth 2008;100:436–41 [DOI] [PubMed] [Google Scholar]
- 37.Casta A, Quackenbush EJ, Houck CS, Korson MS. Perioperative white matter degeneration and death in a patient with a defect in mitochondrial oxidative phosphorylation. Anesthesiology 1997;87:420–5 [DOI] [PubMed] [Google Scholar]
- 38.Cooper MA, Fox R. Anesthesia for corrective spinal surgery in a patient with Leigh’s disease. Anesth Analg 2003;97:1539–41 [DOI] [PubMed] [Google Scholar]
- 39.Grattan-Smith PJ, Shield LK, Hopkins IJ, Collins KJ. Acute respiratory failure precipitated by general anesthesia in Leigh’s syndrome. J Child Neurol 1990;5:137–41 [DOI] [PubMed] [Google Scholar]
- 40.Morgan PG, Hoppel CL, Sedensky MM. Mitochondrial defects and anesthetic sensitivity. Anesthesiology 2002;96:1268–70 [DOI] [PubMed] [Google Scholar]
- 41.Hsieh VC, Niezgoda J, Sedensky MM, Hoppel CL, Morgan PG. Anesthetic hypersensitivity in a case-controlled series of patients with mitochondrial disease. Anesth Analg 2021;133:924–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lenaz G, Genova ML. Supramolecular organisation of the mitochondrial respiratory chain: a new challenge for the mechanism and control of oxidative phosphorylation. Adv Exp Med Biol 2012;748:107–44 [DOI] [PubMed] [Google Scholar]
- 43.Agip AA, Blaza JN, Bridges HR, Viscomi C, Rawson S, Muench SP, Hirst J. Cryo-EM structures of complex I from mouse heart mitochondria in two biochemically defined states. Nat Struct Mol Biol 2018;25:548–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quintana A, Morgan PG, Kruse SE, Palmiter RD, Sedensky MM. Altered anesthetic sensitivity of mice lacking Ndufs4, a subunit of mitochondrial complex I. PLoS ONE 2012;7:e42904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramadasan-Nair R, Hui J, Zimin PI, Itsara LS, Morgan PG, Sedensky MM. Regional knockdown of NDUFS4 implicates a thalamocortical circuit mediating anesthetic sensitivity. PLoS ONE 2017;12:e0188087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Franks NP. General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci 2008;9:370–86 [DOI] [PubMed] [Google Scholar]
- 47.Buddhala C, Hsu CC, Wu JY. A novel mechanism for GABA synthesis and packaging into synaptic vesicles. Neurochem Int 2009;55:9–12 [DOI] [PubMed] [Google Scholar]
- 48.Lenaz G, D’Aurelio M, Merlo Pich M, Genova ML, Ventura B, Bovina C, Formiggini G, Parenti Castelli G. Mitochondrial bioenergetics in aging. Biochim Biophys Acta 2000;1459:397–404 [DOI] [PubMed] [Google Scholar]
- 49.Lenaz G, Fato R, Genova ML, Bergamini C, Bianchi C, Biondi A. Mitochondrial complex I: structural and functional aspects. Biochim Biophys Acta 2006;1757:1406–20 [DOI] [PubMed] [Google Scholar]
- 50.Howarth C, Gleeson P, Attwell D. Updated energy budgets for neural computation in the neocortex and cerebellum. J Cereb Blood Flow Metab 2012;32:1222–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Howarth C, Peppiatt-Wildman CM, Attwell D. The energy use associated with neural computation in the cerebellum. J Cereb Blood Flow Metab 2010;30:403–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Franks NP, Lieb WR. Molecular and cellular mechanisms of general anaesthesia. Nature 1994;367:607–14 [DOI] [PubMed] [Google Scholar]
- 53.Forman SA, Chin VA. General anesthetics and molecular mechanisms of unconsciousness. Int Anesthesiol Clin 2008;46:43–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 2001;21:1133–45 [DOI] [PubMed] [Google Scholar]
- 55.Schousboe A, Sickmann HM, Bak LK, Schousboe I, Jajo FS, Faek SA, Waagepetersen HS. Neuron-glia interactions in glutamatergic neurotransmission: roles of oxidative and glycolytic adenosine triphosphate as energy source. J Neurosci Res 2011;89:1926–34 [DOI] [PubMed] [Google Scholar]
- 56.Baumgart JP, Zhou ZY, Hara M, Cook DC, Hoppa MB, Ryan TA, Hemmings HC., Jr.Isoflurane inhibits synaptic vesicle exocytosis through reduced Ca2+ influx, not Ca2+-exocytosis coupling. Proc Natl Acad Sci U S A 2015;112:11959–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xie Z, McMillan K, Pike CM, Cahill AL, Herring BE, Wang Q, Fox AP. Interaction of anesthetics with neurotransmitter release machinery proteins. J Neurophysiol 2013;109:758–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hemmings HC, Jr, Yan W, Westphalen RI, Ryan TA. The general anesthetic isoflurane depresses synaptic vesicle exocytosis. Mol Pharmacol 2005;67:1591–9 [DOI] [PubMed] [Google Scholar]
- 59.Winegar BD, MacIver MB. Isoflurane depresses hippocampal CA1 glutamate nerve terminals without inhibiting fiber volleys. BMC Neurosci 2006;7:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.MacIver MB. Anesthetic agent-specific effects on synaptic inhibition. Anesth Analg 2014;119:558–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zimin PI, Woods CB, Quintana A, Ramirez JM, Morgan PG, Sedensky MM. Glutamatergic neurotransmission links sensitivity to volatile anesthetics with mitochondrial function. Curr Biol 2016; 26:2194–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zimin PI, Woods CB, Kayser EB, Ramirez JM, Morgan PG, Sedensky MM. Isoflurane disrupts excitatory neurotransmitter dynamics via inhibition of mitochondrial complex I. Br J Anaesth 2018;120:1019–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pathak D, Shields LY, Mendelsohn BA, Haddad D, Lin W, Gerencser AA, Kim H, Brand MD, Edwards RH, Nakamura K. The role of mitochondrially derived ATP in synaptic vesicle recycling. J Biol Chem 2015;290:22325–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rangaraju V, Calloway N, Ryan TA. Activity-driven local ATP synthesis is required for synaptic function. Cell 2014;156:825–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jung S, Zimin PI, Woods CB, Kayser EB, Haddad D, Reczek CR, Nakamura K, Ramirez JM, Sedensky MM, Morgan PG. Isoflurane inhibition of endocytosis is an anesthetic mechanism of action. Curr Biol 2022;32:3016–32.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.de Vries S, Grivell LA. Purification and characterization of a rotenone-insensitive NADH:Q6 oxidoreductase from mitochondria of Saccharomyces cerevisiae. Eur J Biochem 1988;176:377–84 [DOI] [PubMed] [Google Scholar]
- 67.McElroy GS, Reczek CR, Reyfman PA, Mithal DS, Horbinski CM, Chandel NS. NAD+ regeneration rescues lifespan, but not ataxia, in a mouse model of brain mitochondrial complex I dysfunction. Cell Metab 2020;32:301–8.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Horn T, Klein J. Lactate levels in the brain are elevated upon exposure to volatile anesthetics: a microdialysis study. Neurochem Int 2010;57:940–7 [DOI] [PubMed] [Google Scholar]
- 69.Schwarzkopf TM, Horn T, Lang D, Klein J. Blood gases and energy metabolites in mouse blood before and after cerebral ischemia: the effects of anesthetics. Exp Biol Med 2013;238:84–9 [DOI] [PubMed] [Google Scholar]
- 70.Stokes J, Freed A, Bornstein R, Su KN, Snell J, Pan A, Sun GX, Park KY, Jung S, Worstman H, Johnson BM, Morgan PG, Sedensky MM, Johnson SC. Mechanisms underlying neonate-specific metabolic effects of volatile anesthetics. Elife 2021;10:e65400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Johnson SC, Pan A, Sun GX, Freed A, Stokes JC, Bornstein R, Witkowski M, Li L, Ford JM, Howard CRA, Sedensky MM, Morgan PG. Relevance of experimental paradigms of anesthesia induced neurotoxicity in the mouse. PLoS ONE 2019;14:e0213543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Baer AG, Bourdon AK, Price JM, Campagna SR, Jacobson DA, Baghdoyan HA, Lydic R. Isoflurane anesthesia disrupts the cortical metabolome. J Neurophysiol 2020;124:2012–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen Q, Camara AK, Stowe DF, Hoppel CL, Lesnefsky EJ. Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. Am J Physiol Cell Physiol 2007;292:C137–47 [DOI] [PubMed] [Google Scholar]
- 74.Camara AK, Bienengraeber M, Stowe DF. Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury. Front Physiol 2011;2:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hirata N, Shim YH, Pravdic D, Lohr NL, Pratt PF, Jr, Weihrauch D, Kersten JR, Warltier DC, Bosnjak ZJ, Bienengraeber M. Isoflurane differentially modulates mitochondrial reactive oxygen species production via forward versus reverse electron transport flow: implications for preconditioning. Anesthesiology 2011;115:531–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang J, Sun J, Qiao S, Li H, Che T, Wang C, An J. Effects of isoflurane on complex II-associated mitochondrial respiration and reactive oxygen species production: roles of nitric oxide and mitochondrial KATP channels. Mol Med Rep 2019;20:4383–90 [DOI] [PubMed] [Google Scholar]
- 77.Vutskits L, Xie Z. Lasting impact of general anaesthesia on the brain: mechanisms and relevance. Nat Rev Neurosci 2016;17:705–17 [DOI] [PubMed] [Google Scholar]
- 78.Jevtovic-Todorovic V, Absalom AR, Blomgren K, Brambrink A, Crosby G, Culley DJ, Fiskum G, Giffard RG, Herold KF, Loepke AW, Ma D, Orser BA, Planel E, Slikker W, Jr, Soriano SG, Stratmann G, Vutskits L, Xie Z, Hemmings HC., Jr.Anaesthetic neurotoxicity and neuroplasticity: an expert group report and statement based on the BJA Salzburg Seminar. Br J Anaesth 2013;111:143–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Biesecker LG, Dirksen RT, Girard T, Hopkins PM, Riazi S, Rosenberg H, Stowell K, Weber J. Genomic screening for malignant hyperthermia susceptibility. Anesthesiology 2020;133:1277–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sonner JM, Gong D, Li J, Eger EI, II, Laster MJ. Mouse strain modestly influences minimum alveolar anesthetic concentration and convulsivity of inhaled compounds. Anesth Analg 1999;89:1030–4 [DOI] [PubMed] [Google Scholar]
- 81.Sonner JM, Gong D, Eger EI, II. Naturally occurring variability in anesthetic potency among inbred mouse strains. Anesth Analg 2000;91: 720–6 [DOI] [PubMed] [Google Scholar]
- 82.Hawasli AH, Saifee O, Liu C, Nonet ML, Crowder CM. Resistance to volatile anesthetics by mutations enhancing excitatory neurotransmitter release in Caenorhabditis elegans. Genetics 2004;168:831–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Morgan PG, Sedensky M, Meneely PM. Multiple sites of action of volatile anesthetics in Caenorhabditis elegans. Proc Natl Acad Sci U S A 1990;87:2965–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Olufs ZPG, Ganetzky B, Wassarman DA, Perouansky M. Mitochondrial complex I mutations predispose Drosophila to isoflurane neurotoxicity. Anesthesiology 2020;133:839–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Twaroski DM, Yan Y, Zaja I, Clark E, Bosnjak ZJ, Bai X. Altered mitochondrial dynamics contributes to propofol-induced cell death in human stem cell-derived neurons. Anesthesiology 2015;123:1067–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boscolo A, Milanovic D, Starr JA, Sanchez V, Oklopcic A, Moy L, Ori C C, Erisir A, Jevtovic-Todorovic V. Early exposure to general anesthesia disturbs mitochondrial fission and fusion in the developing rat brain. Anesthesiology 2013;118:1086–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kumar R, Harilal S, Parambi DGT, Kanthlal SK, Rahman MA, Alexiou A, Batiha GE, Mathew B. The role of mitochondrial genes in neurodegenerative disorders. Curr Neuropharmacol 2022;20:824–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sanchez V, Feinstein SD, Lunardi N, Joksovic PM, Boscolo A, Todorovic SM, Jevtovic-Todorovic V. General anesthesia causes long-term impairment of mitochondrial morphogenesis and synaptic transmission in developing rat brain. Anesthesiology 2011;115:992–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Boscolo A, Starr JA, Sanchez V, Lunardi N, DiGruccio MR, Ori C, Erisir A, Trimmer P, Bennett J, Jevtovic-Todorovic V. The abolishment of anesthesia-induced cognitive impairment by timely protection of mitochondria in the developing rat brain: the importance of free oxygen radicals and mitochondrial integrity. Neurobiol Dis 2012;45:1031–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Joksimovic SM, DiGruccio MR, Boscolo A, Jevtovic-Todorovic V, Todorovic SM. The role of free oxygen radicals in lasting hyperexcitability of rat subicular neurons after exposure to general anesthesia during brain development. Mol Neurobiol 2020;57:208–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barajas MB, Brunner SD, Wang A, Griffiths KK, Levy RJ. Propofol toxicity in the developing mouse heart mitochondria. Pediatr Res 2022;92:1341–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barajas MB, Wang A, Griffiths KK, Sun L, Yang G, Levy RJ. Modeling propofol-induced cardiotoxicity in the isolated-perfused newborn mouse heart. Physiol Rep 2022;10:e15402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Finsterer J, Frank M. Propofol is mitochondrion-toxic and may unmask a mitochondrial disorder. J Child Neurol 2016;31:1489–94 [DOI] [PubMed] [Google Scholar]
- 94.Chow CY, Reiter LT. Etiology of human genetic disease on the fly. Trends Genet 2017;33:391–8 [DOI] [PubMed] [Google Scholar]
- 95.Mackay TF, Richards S, Stone EA, Barbadilla A, Ayroles JF, Zhu D, Casillas S, Han Y, Magwire MM, Cridland JM, Richardson MF, Anholt RR, Barron M, Bess C, Blankenburg KP, Carbone MA, Castellano D, Chaboub L, Duncan L, Harris Z, Javaid M, Jayaseelan JC, Jhangiani SN, Jordan KW, Lara F, Lawrence F, Lee SL, Librado P, Linheiro RS, Lyman RF, Mackey AJ, Munidasa M, Muzny DM, Nazareth L, Newsham I, Perales L, Pu LL, Qu C, Ramia M, Reid JG, Rollmann SM, Rozas J, Saada N, Turlapati L, Worley KC, Wu YQ, Yamamoto A, Zhu Y, Bergman CM, Thornton KR, Mittelman D, Gibbs RA. The Drosophila melanogaster genetic reference panel. Nature 2012;482:173–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mackay TFC, Huang W. Charting the genotype-phenotype map: lessons from the Drosophila melanogaster genetic reference panel. Wiley Interdiscip Rev Dev Biol 2018;7:e289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schiffman HJ, Olufs ZPG, Lasarev MR, Wassarman DA, Perouansky M. Ageing and genetic background influence anaesthetic effects in a D. Melanogaster model of blunt trauma with brain injury. Br J Anaesth 2020;125:77–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ke C, Poon WS, Ng HK, Pang JC, Chan Y. Heterogeneous responses of aquaporin-4 in oedema formation in a replicated severe traumatic brain injury model in rats. Neurosci Lett 2001;301:21–4 [DOI] [PubMed] [Google Scholar]
- 99.Akdemir G, Ratelade J, Asavapanumas N, Verkman AS. Neuroprotective effect of aquaporin-4 deficiency in a mouse model of severe global cerebral ischemia produced by transient 4-vessel occlusion. Neurosci Lett 2014;574:70–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Katada R, Akdemir G, Asavapanumas N, Ratelade J, Zhang H, Verkman AS. Greatly improved survival and neuroprotection in aquaporin-4-knockout mice following global cerebral ischemia. FASEB J 2014;28:705–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Appelboom G, Bruce S, Duren A, Piazza M, Monahan A, Christophe B, Zoller S, LoPresti M, Connolly ES. Aquaporin-4 gene variant independently associated with oedema after intracerebral haemorrhage. Neurol Res 2015;37:657–61 [DOI] [PubMed] [Google Scholar]
- 102.Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol 2009;8:398–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fox GB, LeVasseur RA, Faden AI. Behavioral responses of C57BL/6, FVB/N, and 129/SvEMS mouse strains to traumatic brain injury: implications for gene targeting approaches to neurotrauma. J Neurotrauma 1999;16:377–89 [DOI] [PubMed] [Google Scholar]
- 104.Tan AA, Quigley A, Smith DC, Hoane MR. Strain differences in response to traumatic brain injury in Long-Evans compared to Sprague-Dawley rats. J Neurotrauma 2009;26:539–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Reid WM, Rolfe A, Register D, Levasseur JE, Churn SB, Sun D. Strain-related differences after experimental traumatic brain injury in rats. J Neurotrauma 2010;27:1243–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hardy CM, Birse RT, Wolf MJ, Yu L, Bodmer R, Gibbs AG. Obesity-associated cardiac dysfunction in starvation-selected Drosophila melanogaster. Am J Physiol Regul Integr Comp Physiol 2015;309: R658–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Johnson-Schlitz D, Fischer JA, Schiffman HJ, Scharenbrock AR, Olufs ZPG, Wassarman DA, Perouansky M. Anesthetic preconditioning of traumatic brain injury is ineffective in a Drosophila model of obesity. J Pharmacol Exp Ther 2022;381:229–35 [DOI] [PMC free article] [PubMed] [Google Scholar]