

Abstract
Interferons (IFNs) are antiviral cytokines that play a key role in the innate immune response to viral infections. In response to viral stimuli, cells produce and release interferons, which then act on neighboring cells to induce the transcription of hundreds of genes. Many of these gene products either combat the viral infection directly, e.g., by interfering with viral replication, or help shape the following immune response. Here, we review how viral recognition leads to the production of different types of IFNs and how this production differs in spatial and temporal manners. We then continue to describe how these IFNs play different roles in the ensuing immune response depending on when and where they are produced or act during an infection.
Keywords: Interferon, Interferon induction, Interferon Regulatory Factor 3 (IRF3), viral recognition
Subject Categories: Immunology
This review discusses the regulation of IFNs and their role in viral recognition and immune responses.
Introduction
An effective immune response starts with the recognition of an invading pathogen by the innate immune system, which is mediated by a set of receptors called pattern recognition receptors (PRRs) (Mogensen, 2009; Chow et al, 2015). Once the PRRs detect a virus, they initiate a series of signaling events, leading to the establishment of an immune response towards the intruding pathogen. The key mammalian PRRs responsible for the induction of IFN production are shown in Fig 1. This concept is conserved throughout metazoan evolution (Holleufer et al, 2021; Slavik et al, 2021) and early components of this system are found in bacteria as well (Morehouse et al, 2020). In this review, we focus on the mammalian response to viral infections but with a particular focus on interferon (IFN), a group of cytokines that acts as signaling molecules and triggers antiviral defenses in cells. IFNs are divided into three types, depending upon their receptor usage: Type I IFNs encompass multiple subtypes but here we will only be discussing IFN‐α and IFN‐β. Type II IFN only has one member, IFN‐γ, which does not play a major role in the induction of an antiviral state, and it is not discussed further here. Finally, there are type III IFNs, which are also called IFN‐λ. Table 1 lists the different subtypes of IFN and classifies them into types I, II, or III IFNs.
Figure 1. Recognition of viral infection by pattern recognition receptors leads to interferon production.
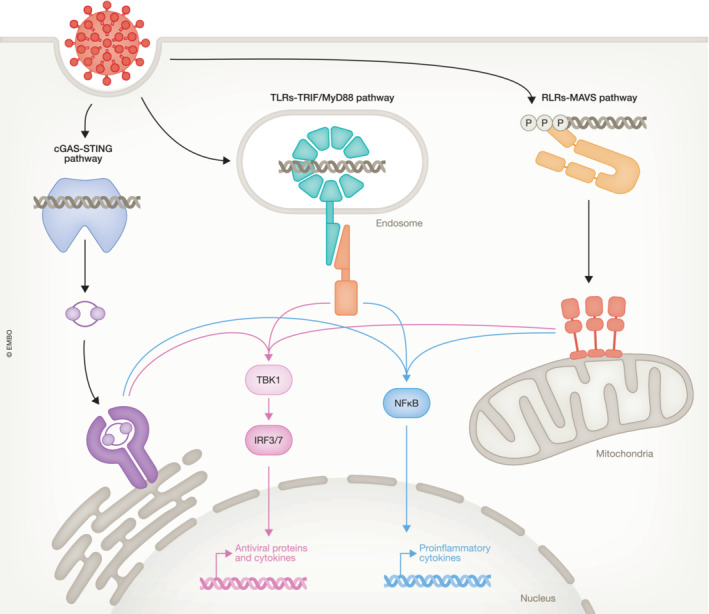
Viral recognition leads to interferon induction via three principal pathways that are characterized by the adaptor proteins used to connect the upstream pattern recognition receptors with the downstream signaling machinery. Signaling via those pathways is orchestrated by the adaptor proteins STING, MAVS and either TRIF or MyD88. All the three pathways activate the kinase TBK1, which in turn phosphorylates the transcription factors IRF3 and IRF7. Notably, IRF7 is constitutively expressed in pDCs but needs to be induced by IFN signaling in other cells. In addition, these pathways also activate the NF–κB family of transcription factors, yet the molecular mechanism behind this activation remains poorly characterized.
Table 1.
Provides an overview of the different subtypes of IFN found in humans, key references and main characteristic is listed.
| Type | Subtypes | Receptor | Comments | References |
|---|---|---|---|---|
| Type I IFNs | ||||
| IFN‐α | IFN‐α1, ‐α2, ‐α4, ‐α5, ‐α6, ‐α7, ‐α8, ‐α10, ‐α13, ‐α14, ‐α16, ‐α17, ‐α21 | IFN‐αR1/IFN‐αR2 | IFN‐α is primarily produced by pDCs | Cella et al (1999), Barchet et al (2002), Dai et al (2004), Hardy et al (2004), Jaks et al (2007) |
| IFN‐β | IFN‐αR1/IFN‐αR2 | IFN‐β is produced by most infected cells | Platanias (2005), Khaitov et al (2009), Ioannidis et al (2013) | |
| IFN‐ε | IFN‐αR1/IFN‐αR2 | IFN‐ε is associated with the female reproductive tract | Fung et al (2013), Marks et al (2019) | |
| IFN‐κ | IFN‐αR1/IFN‐αR2 | IFN‐κ is selectively expressed in keratinocytes | LaFleur et al (2001) | |
| IFN‐ω | IFN‐αR1/IFN‐αR2 | One of the least studied IFNs but the presence of neutralizing auto‐antibodies against it in severe COVID‐19 patients suggests its role in antiviral immunity may be underappreciated | Hauptmann and Swetly (1985), Bastard et al (2020) | |
| Type II IFNs | ||||
| IFN‐γ | IFN‐γR1/ IFN‐γR2 | Not discussed here | ||
| Type III IFNs | ||||
| IFN‐λ | IFN‐λ1, ‐λ2, ‐λ3, ‐λ4 | IFN‐λR1/IL‐10R2 | IFN‐λ is produced by infected cells at barrier tissues such as epithelial cells in the respiratory tract | Meager et al (2005), Sommereyns et al (2008), Jewell et al (2010), Mordstein et al (2010), Crotta et al (2013), Wack et al (2015), Ye et al (2019) |
Mammals are complex animals and viral recognition can lead to widely different outcomes depending on the kind of tissue in which it happens. The implications of this on the spatial/temporal distribution of IFN production and the ensuing consequences will be discussed in detail throughout this review, as we suggest the following working model. Recognition of a viral infection in barrier tissues leads to local IFN responses driven by the local production of IFN‐β and IFN‐λ, whereas recognition of viruses that have penetrated the barrier tissue leads to large‐scale production of IFN‐α and the result is a systemic IFN response. In the first case, IFN production generally originates from infected cells whereas in the latter case, immune cells, such as plasmacytoid dendritic cells (pDCs), can contribute with large amounts of IFN. Notably, pDCs do not need to be productively infected (defined as an infection that leads to viral progeny) themselves in order to initiate IFN production (Swiecki & Colonna, 2015). Finally, microbiota found at mucosal surfaces also drives a tonic IFN response (Bradley et al, 2019).
Viral recognition
As the name implies, PRRs recognize specific molecular patterns called pathogen‐associated molecular patterns (PAMPs) originating from the pathogen. The primary PAMP for viruses is nucleic acids, including foreign nucleic acids structures, like 5′‐phosphorylated RNA and dsRNAs that are specifically associated with viral infections, and generic nucleic acids that may be perceived as foreign when located in a cellular compartment where they are not normally found. In mammals, there are three major pathways for the recognition of a viral infection (see Fig 1). (i) The endosomal toll‐like receptors (TLRs), which can recognize either dsRNA (TLR3) (Alexopoulou et al, 2001) or a variety of single‐stranded nucleic acids present within endosomes or phagosomes (TLR7, 8 or 9; Lund et al, 2003; Diebold et al, 2004; Heil et al, 2004; Akira et al, 2006). (ii) The cytosolic RIG‐I‐like receptors (RLR), which recognize dsRNA, 5′‐phosphorylated RNAs or a combination thereof (Hornung et al, 2006; Kato et al, 2006; Pichlmair et al, 2006). (iii) Cytosolic cyclic GMP–AMP synthase (cGAS), which recognizes dsDNA present in the cytoplasm and perceives it as a sign of viral infection (Civril et al, 2013; Sun et al, 2013; Gao et al, 2013a). Upon binding of the PAMP, the receptor conveys a signal to a distinct downstream adaptor protein (Fig 1). The TLRs signal via the TIR‐domain‐containing adapter‐inducing IFN‐β (TRIF) or the myeloid differentiation factor 88 (MyD88) (Liu & Ding, 2016), the RLRs signal via mammalian mitochondrial antiviral signaling protein (MAVS; Yoneyama et al, 2015), and cGAS produces the secondary messenger cyclic GMP–AMP (cGAMP) to signal via stimulator of interferon genes (STING; Ishikawa & Barber, 2008; Wu et al, 2013; Gao et al, 2013b). Once activated, the adaptor proteins recruit and activate several cellular kinases, like the TANK‐binding kinase 1 (TBK1), which leads to the activation of the transcription factors interferon regulatory factor (IRF) 3 and 7 (IRF7). The abovementioned PRRs also activate signaling through the NF‐κB pathway but our understanding of the underlying molecular mechanisms remains incomplete and will therefore not be discussed any further here. Thus, activation of PRRs ultimately leads to a transcriptional response creating an antiviral state in the infected cell. IRF3 is central in this response as it initiates two defensive mechanisms. Firstly, activation of IRF3 leads to the production of IFNs, in particular, IFN‐β and IFN‐λ1 (Osterlund et al, 2007), and other cytokines working in a paracrine manner to warn surrounding cells of the infection. Secondly, the IRF3 transcriptional response leads to the production of antiviral proteins that help combat the virus within the affected cell (Grandvaux et al, 2002). These responses enable cells to limit viral infection within the affected cell and simultaneously establish a strong defensive state in neighboring cells.
Molecular mechanisms of IFN production
Activation of IRF3
In this section, we focus on how STING activates IFN production as this is our research area, but we believe that the mechanism is largely conserved among the different adaptor molecules. In the model of IRF3 activation proposed by Chen and colleagues, the activation of IRF3 is initiated by it docking to a phosphorylated pLxIS motif on an adaptor molecule (Liu et al, 2015). This docking is mediated by electrostatic interactions between the positively charged surface on IRF3 and the phosphorylated pLxIS motif (Fig 2). The positively charged surface of IRF3 can be divided into 5 patches consisting of residues R211/R213, R255/R262/H263, R285/H288/H290, K313/K315, and K360/R361, and mutation of each of these patches abolishes activation of IRF3, thereby supporting the current model of IRF3 activation (Takahasi et al, 2003; Liu et al, 2015). Structural work using peptides representing the adaptors suggested that IRF3 docks to the adaptor proteins by forming a direct contact between R285 of IRF3 and the phosphorylated serine in the pLxIS motif (Zhao et al, 2016). The importance of residue R285 in IRF3 activation was further supported by the finding of a R285Q mutation in a patient suffering from viral encephalitis as well as by our detailed analysis of IRF3 docking to the adaptor (Andersen et al, 2015; Dalskov et al, 2020). The docking to an adaptor molecule positions IRF3 for phosphorylation by TBK1 on S386 (Liu et al, 2015). The importance of phosphorylation at either S386 or S396 has been debated in the literature. On one hand, the phosphorylation of S386 has been shown to be critical for IRF3 activity in several publications (Mori et al, 2004; Takahasi et al, 2010; Dalskov et al, 2020), but mutation of S396 and surrounding serine residues to the phosphomimetic aspartic acid was shown to cause a constitutively active phenotype, which suggests that S396 also plays a role in the activation of IRF3 (Lin et al, 1998; Servant et al, 2003). However, it is now clear that mutation of S396 to aspartic acid leads to a conformational change within IRF3, which facilitates the phosphorylation of S386. Thus, phosphorylation of S386 is required for the ability of IRF3 to dimerize and become transcriptionally active. Furthermore, mutation of S396 to alanine does not impact the activity of IRF3 (Dalskov et al, 2020) but a role for phosphorylation of S396 in facilitating S386 phosphorylation and thereby activation cannot be excluded.
Figure 2. The activation mechanism of IRF3.
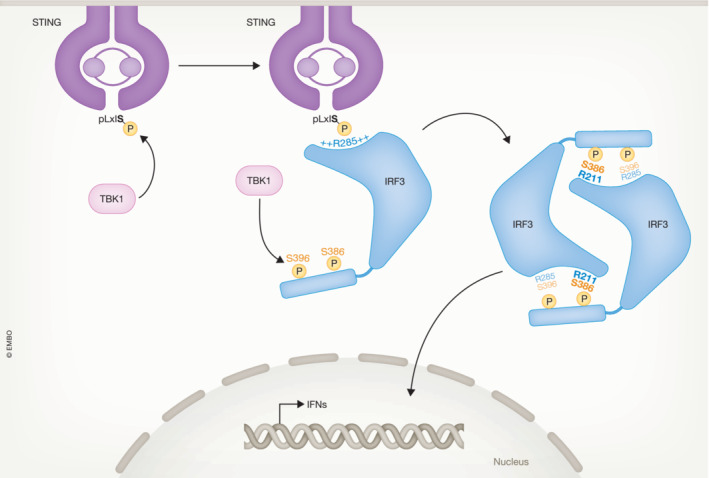
IRF3 docks to the phosphorylated form of the adaptor proteins illustrated here by STING. Once docked at the adaptor proteins, TBK1 phosphorylates IRF3 at key serine residues where residue 386 is particularly critical for IRF3 activation. Upon phosphorylation, IRF3 dimerizes and forms the transcriptionally active dimer, which then translocates to the nucleus where it drives transcription of IFNs and certain antiviral genes. The available data suggest that IRF7 is activated through a similar mechanism.
Once IRF3 is phosphorylated, it forms the transcriptionally active dimer, and this dimerization is driven by the interaction between the phosphorylated residue S386 and R211. This view is supported by structural evidence (Qin et al, 2003; Takahasi et al, 2003) and was confirmed by mutational studies. Furthermore, the fact that mutation of R211 still resulted in IRF3 phosphorylation but no dimer formation also supports this model (Dalskov et al, 2020). (Dalskov et al, 2020). S396 is found in a position, where if phosphorylated, it can potentially interact with R385, as shown in Fig 2. However, S396 is dispensable for the activity of IRF3. Analysis using dimeric IRF3 with phosphomimic substitutions at position S386 and S396 and the structure of murine dimeric IRF3 with a natural phosphoserine at position S386 (murine S379) provided further support for this model (Zhao et al, 2016; Jing et al, 2020). Finally, the manner upon which phosphorylation of S386 drives dimerization of IRF3 also offers a putative mechanism for avoiding aberrant activation of IRF3 by unspecific phosphorylation. Once it is phosphorylated, IRF3 will leave STING but the phosphoserine 386 is vulnerable to dephosphorylating enzymes as long as IRF3 is in the monomeric state. A characteristic of adaptor proteins, like STING, is the formation of higher order oligomers (Zhang et al, 2019). This means that the local concentration of phosphorylated IRF3 monomers is higher around those structures and thus IRF3 can rapidly find an equally phosphorylated partner to dimerize with. In the dimeric form, the phosphoserine is buried and is not accessible to phosphatases.
The activated dimeric form of IRF3 described above is transcriptionally competent and can induce the transcription of a number of genes (Grandvaux et al, 2002; Schoggins et al, 2011). A key role for IRF3 is to induce different IFN genes, but IRF3 also induces a series of antiviral genes within the infected cells, thus there is a significant overlap between the gene set induced by IFN and the gene set induced by IRF3. Notably, IFNs cannot induce their own transcription. The ability of IRF3 to induce a variety of antiviral genes (which are also under the control of IFN) hint at an ancestral role of the IRF family of transcription factors. We believe that prior to the emergence of IFN, the role of IRF was likely to directly induce antiviral genes upon detection of the virus by the PRRs. However, the strong phenotype seen in different model systems with impaired or completely inactivated IFN signaling suggests that the direct induction of antiviral genes by IRF3 plays a lesser role in vertebrates. The overlap between IFN‐induced genes and IRF3‐induced genes is also important to keep in mind when using modern bioinformatics tools to analyze transcriptomic data, as the programs struggle to differentiate between the signatures left by IRF3 and IFN.
IRF7 is activated in a similar manner as IRF3
IRF7 plays a key role in the production of IFN‐α and is constitutively expressed in pDCs while its expression is inducible by IFN in other cells (Au et al, 1998; Marié et al, 1998; Sato et al, 1998; Ning et al, 2011). Our data suggests that the molecular mechanism of IRF7 activation is very similar to what we just discussed for IRF3, but the regulation of IRF7 seems stricter, and it is possible that IRF7 needs to be phosphorylated at multiple sites to gain full activity (Dalskov et al, 2020). We have attempted to induce IFN‐α expression in a series of cell lines and primary cells that express IRF7 after IFN priming, but with little success. Thus, it appears that phosphorylation of IRF7 is insufficient to activate IFN‐α production in many cell types and this suggests the existence of additional layers of regulation in IFN‐α production, which may possibly involve epigenetic modifications at the IFN‐α loci. In other words, substantial new research of the molecular mechanisms that drive IFN‐α production is needed.
IRF3 and IRF7 differ in their ability to induce specific IFN subtypes
The structure and function of IFN promoters have been characterized in detail by others, which will be summarized in the following. The IFN‐β promoter contains IRF binding sites, which can bind both IRF3 and ‐7. Furthermore, it also contains binding sites for factors of the NF–κB family. For full activation of the IFN–β promoter, binding of both IRF3 and NF–κB is required (Thanos & Maniatis, 1995; Sato et al, 2000; Panne et al, 2004, 2007). The IFN‐λ1 promoter appears to show much of the same characteristics as the IFN‐β promoter (Onoguchi et al, 2007; Osterlund et al, 2007; Thomson et al, 2009). The IFN‐α promoter can bind several different members of the IRF family, most prominently IRF7, but IRF3 can also bind to at least some IFN‐α promoters (Schafer et al, 1998; Andrilenas et al, 2018; Wittling et al, 2020). Likewise, IRF5 has also been suggested to play a role in induction of some IFN‐α genes (Andrilenas et al, 2018). In contrast to the IFN‐β promoter, IFN‐α promoters do not appear to rely upon the NF–κB pathway (Wittling et al, 2020).
Naturally, the differences in promoter composition among IFNs also result in different regulatory patterns. IFN‐λ1 and IFN‐β have a similar mode of regulation with both being primarily controlled by IRF3 in conjunction with the NF–κB pathway. While both IRF3 and IRF7 can bind the IFN‐β promoter, the rapid induction of those IFNs in virus infected cells primarily relies upon IRF3, as IRF7 is not constitutively expressed in most cells. Importantly, the relative amount of IFN‐λ1 and IFN‐β being produced by a given cell might vary dependent upon cell type. In terms of gene regulation, human IFN‐λ3 appears to share some characteristics with IFN‐α (Onoguchi et al, 2007; Osterlund et al, 2007; Kotenko, 2011; Wack et al, 2015), but current data on the regulation of IFN‐λ3 are limited and more work is needed in this area.
Role of other IRF family members in IFN production
Besides IRF3 and ‐7, other IRFs have also been investigated for their involvement in IFN induction. Although IRF1 was identified as a positive regulator of the IFN‐β gene, it was later shown that IRF1 is not needed for IFN induction since IRF1 deficient mice produced normal levels of IFN in response to Newcastle disease virus (Fujita et al, 1988; Reis et al, 1994; Feng et al, 2021). IRF5 is widely expressed by immune cells such as macrophages and pDCs where it has been shown that IRF5 increases IFN‐β production. Furthermore, IRF5 has been shown to bind to a subset of IFN‐α promoters (Schoenemeyer et al, 2005; Lazear et al, 2013; Yasuda et al, 2013; Andrilenas et al, 2018; Khoyratty & Udalova, 2018). IRF8 has also been implicated in enhancing IFN production, especially in the later phases of IFN induction in pDCs and monocytes, by increasing recruitment of the basal transcription machinery (Tailor et al, 2007; Li et al, 2011; Jefferies, 2019). Yet, detailed knowledge about the role of other IRFs in IFN production at the molecular level is currently lacking.
Temporal and spatial regulation of IFN production
Effect of IRF3 and IRF7 deficiency on susceptibility to viral infections
The biological importance of IRF3 versus IRF7 in IFN production has been studied primarily in mice but with the recent progress in human genetics, more and more data emerge from humans as well. IRF3 knockout (KO) mice exhibited lower IFN‐β expression in response to both DNA and RNA viruses (Steinberg et al, 2009; Hatesuer et al, 2017; Yanai et al, 2018) whereas IRF7 KO mice exhibited an almost complete loss of IFN‐α expression as well as a severe drop in IFN‐λ2/3 expression (Hatesuer et al, 2017). Here, it is important to note that mice do not have an equivalent to the human IFN‐λ1 gene. Thus, the in vivo data agrees with the molecular model for IFN expression discussed above. Both the IRF3 and IRF7 KO mice exhibited decreased survival upon infection with influenza A virus (IAV). However, the effect of IRF7 KO was marginally larger than for IRF3 KO (Hatesuer et al, 2017). A lack of IRF3 should lead to an initially lower IFN production by epithelial cells, which agrees with the observed decrease in IFN‐β production at day 1 in these mice. It is unclear whether IRF7 is needed due to its ability to produce IFN‐λ2/3, which is particularly important in respiratory infections, or due to its role in driving a systemic IFN‐α response. Thus, the role of IRF7 in the induction of IFN‐λ in respiratory infections merits further investigations. Another group investigated the role of IRF3 and IRF7 in infection with murine cytomegalovirus (CMV) and found that IRF7 was particularly important whereas KO of IRF3 had little effect (Steinberg et al, 2009). This is what we would expect from a systemic viral infection with broad tropism, like CMV, where the systemic IFN‐α response is expected to be important.
Turning toward humans, a patient, which was heterozygote for the IRF3 missense variant (p.Arg285Gln/WT), displayed high susceptibility toward DNA viruses, such as herpes simplex virus 1 (HSV‐1), but not to RNA viruses (Andersen et al, 2015). In vitro characterization of this IRF3 variant demonstrated a selective failure in STING‐mediated activation, explaining the high susceptibility toward herpes infection. Several IRF7 variants have been identified in patients with severe IAV infection. Common for all of these is a reduced IFN production and defective IFN priming (Ciancanelli et al, 2015; Thomsen et al, 2019; Zhang et al, 2020). More specifically, it was observed that the IRF7 variants affected IFN‐α and IFN‐λ production, but to a lesser extent IFN‐β production (Ciancanelli et al, 2015; Campbell et al, 2022). These observations correlate with the findings in the IRF3/7 KO mice, where IRF7 plays a role not only in IFN‐α production but also in IFN‐λ production.
The role of pDCs in IFN production
The abovedescribed division of labor between IRF3 and IRF7 results in temporal differences in IFN production. During early stages of a viral infection, the infected cells will activate IRF3 and drive an IFN‐β and IFN‐λ based response, which will be largely local in nature. However, as the infection progresses, a subset of dendritic cells, namely the pDCs, becomes major producers of IFN‐α (Cella et al, 1999; Barchet et al, 2002). In agreement with this, depletion of pDCs in a mouse model and/or in humans reduces IFN‐α production (Rowland et al, 2014; Karnell et al, 2021). Furthermore, in vitro pDCs will produce large amounts of IFN‐α after stimulation with a variety of viruses (Dai et al, 2004; Yin et al, 2012). There is no strict definition of a systemic response, but in our opinion, this occurs when pDCs produce sufficient IFN‐α to raise the serum concentration above the critical concentration needed for a systemic response. Thus, it is the response to IFN that is systemic, not the production of it. It is possible that IFN‐β also contributes to the systemic response whereas IFN‐λ acts in a more targeted fashion due to the limited distribution of its receptor. The IFN‐producing pDCs are most likely localized in the lymphatic tissue around the site of infection. It is possible that induction of IRF7 by IFN in non‐pDCs can lead to production of IFN‐α, which may then contribute to the systemic response. However, at present the role of non‐pDCs as IFN‐α producers is not clear. Importantly, we need a better understanding of the molecular mechanisms driving IFN‐α production and enabling pDCs to produce large amount of IFN‐α in order to understand how IFN‐α production is regulated.
Physiological effects of IFN and their therapeutic use
IFN signaling at a cellular level
The primary role of IFN is to warn uninfected cells of an approaching virus and thereby allow them to establish a defensive state before being infected. Figure 3 summarizes the discussion above and seeks to illustrate the primary functions of an IFN response. However, IFN also has secondary roles in promoting inflammatory reactions occurring upon detection of viral infection and increasing antigen presentation in an MHC I context. For type I IFNs, signaling occurs after IFN binds a heterodimeric receptor complex consisting of the IFN‐α receptor (IFN‐αR) 1 and IFN‐αR2 (Schreiber, 2017). The type III IFNs signal through a separate receptor complex formed by the IFN‐λ receptor 1 (IFN‐λR1) and the interleukin‐10 receptor 2 (IL‐10R2), the latter also used by the cytokines IL‐10 and IL‐22 (Kotenko et al, 2003; Sheppard et al, 2003). Upon binding of the ligand to the IFN receptor complexes, the receptor associated kinases, tyrosine kinase 2 (TYK2), and Janus kinase 1 (JAK1), are auto‐phosphorylated and then phosphorylate specific tyrosines in the cytoplasmic part of the receptor. This then leads to the recruitment and phosphorylation of signal transducer and activator of transcription 1 (STAT1) and STAT2 proteins. Despite using two different receptor complexes, this process is rather similar for both type I and III receptor complexes (Qureshi et al, 1995; Zhou et al, 2007). Following their activation, STAT1 and STAT2 join with IRF9 to form the IFN‐stimulated gene (ISG) factor 3 (ISGF3), which drives the expression of ISGs. Activation of the type I IFN receptor, but not the type III receptor, is thought to also lead to production of a phosphorylated STAT1 homodimer, which is transcriptionally competent. This might lead to physiologically significant gene expression in immune cells (Zhou et al, 2007; Forero et al, 2019) but more work is needed in this area.
Figure 3. Interferon signaling leads to the establishment of an antiviral state in uninfected cells.
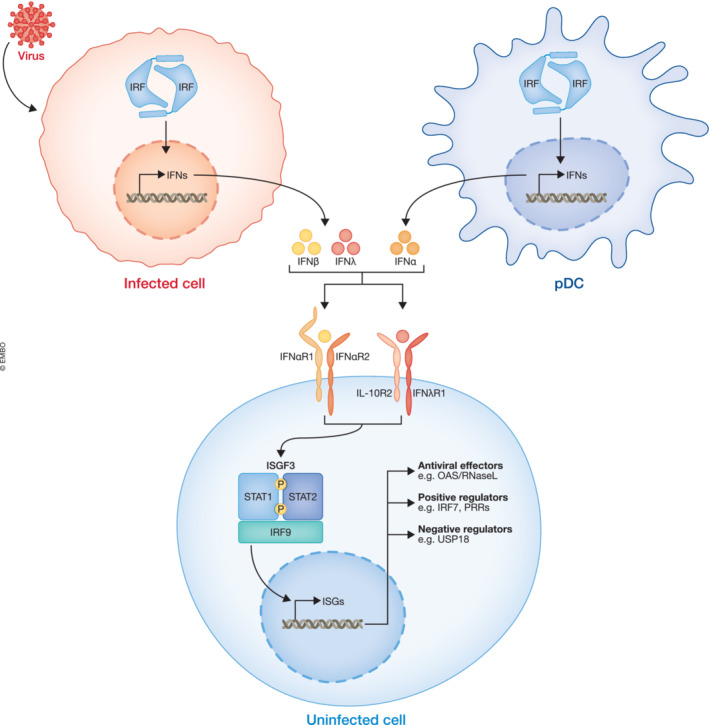
Recognition of a viral infection in infected cells leads to production of IFN‐β and IFN‐λ, whereas recognition of viruses by pDCs leads to production of large amount of IFN‐α. The IFNs signal through their respective receptor complexes to induce the expression of ISGs in uninfected cells and thereby establish an antiviral state.
The genes induced by IFN signaling are referred to as interferon‐stimulated genes (ISGs) and can be divided into three functional groups: antiviral effectors, positive regulators, and negative regulators (Schneider et al, 2014). The antiviral effectors control and combat an infection by directly targeting the replicating virus. The positive regulators aid in the induction of the immune response by enhancing recognition or innate immune signaling and many PRRs and signaling proteins are thus ISGs. At last, the negative regulators, such as USP18, help with terminating signaling to keep the immune system tightly controlled (Veer et al, 2001; Schneider et al, 2014).
The key difference, however, between type I and type III IFNs lies in the distribution of their receptor complexes. Whereas the type I receptor is found on all nucleated cells, the IFN‐λR1 chain is expressed in a highly tissue specific manner, which restricts the response toward type III IFNs (Sommereyns et al, 2008). IFN‐λR1 is expressed in all epithelial cells in both mice and humans, but whereas human hepatocytes do express IFN‐λR1, mice hepatocytes do not (Hermant et al, 2014). The responsiveness of immune cells to type III IFNs is actively being debated, but it is clear that only a subset of immune cells responds to type III IFNs. However, the identity of those cells and the effect that type III IFNs has upon them is still being worked out. We refer the reader to the review by Wack et al (2015) focusing upon type III IFNs for an in‐depth discussion of this topic (Wack et al, 2015).
Systemic IFN responses versus local IFN responses
As used here, the phrasing “IFN responses” covers the entirety of effects caused by the IFN system with the range of these responses being determined by a combination of how much IFN is produced and how specifically/broadly the receptor for the given subtype of IFN is expressed. Since the receptor for type I IFN is found on all nucleated cells, this means that if enough type I IFN is produced to raise the plasma level above a certain concentration, a systemic response will follow. In contrast to this, the type III IFN receptor is only expressed in specific cell types and thus even in the event of massive type III IFN production, the response will be restricted by its tissue tropism. As described above, the primary cause of systemic IFN responses is thought to be large‐scale production of IFN‐α by pDCs.
Observing patients receiving systemic IFN treatment (this will typically be pegylated IFN‐α) provides a good idea of the global effect of IFN on the human body. Generally, patients report “flu like” symptoms, including fatigue, headache and nausea, after treatment with type I IFN. One phase II clinical trial compared pegylated IFN‐α to pegylated IFN‐λ for the treatment of hepatitis C virus (HCV) infection. The two IFNs had similar antiviral properties but the abovedescribed side effects were substantially lower for IFN‐λ (Muir et al, 2014). This illustrates our current view of why two different IFN systems that regulate the same set of genes have proved to be an advantage throughout evolution. In essence, type III IFNs provide efficient control of viral replication in high‐risk tissues but avoid some of the negative effect of an IFN response due to the targeted nature of the system as compared to type I IFNs.
IFN responses in the respiratory tract
As alluded to above, type III IFNs act in a much more targeted manner than type I IFNs, and here, we will discuss the IFN response at barrier tissues in more details. Our focus will be on IFN responses in the respiratory tract since this is our particular area of expertise. Since barrier tissues suffer from a particular high risk of viral infection, they have evolved specific defense mechanisms. This includes the type III IFNs, which are largely specialized in defending the mucosal surfaces in our body (Wack et al, 2015), but also the IFN‐ε, a member of the type I IFN family, which is produced only in the female reproductive tract and act there to protect from viral infection (Fung et al, 2013; Marks et al, 2019). Primary human bronchiolar epithelial cells as well as human airway type II epithelial cells were shown to primarily produce IFN‐λs (IFN‐λ1 and IFN‐λ2/3) and IFN‐β and to a lesser extent IFN‐αs following infection with respiratory syncytial virus (RSV) or IAV (Khaitov et al, 2009; Wang et al, 2009; Ioannidis et al, 2013). Similar observations have been made in murine alveolar epithelial cells where IFN‐λ was mostly produced followed by IFN‐β and at last IFN‐α, which was only produced in small amounts (Ioannidis et al, 2013). Combining the data, we arrive at a model where production of IFN‐λ provides efficient protection of the lung epithelium supplemented by limited production of IFN‐β, which provides local protection of underlying tissues. Clearly, type I IFNs are critically important, as they prevent the virus from penetrating deeper into our body and offer the strength of a systemic IFN response should the virus penetrate the epithelium. Yet, their effect comes with a substantial cost caused by inflammation and therefore it is beneficial for the host only to deploy systemic type I IFN responses when absolutely needed.
In some of the important respiratory infections, such as IAV and severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2), the inflammatory response poses more of a threat than direct cytotoxicity caused by the virus. The absence of the IFN‐λR1 receptor chain on most professional immune cells means that type III IFNs have much weaker pro‐inflammatory activity than type I IFNs. In contrast, the IFN‐λR1 chain is expressed by all epithelial cells and therefore type III IFNs can exert the same antiviral effect as type I IFNs during respiratory infections while causing less inflammation. Furthermore, treatment with type I IFN led to successful inhibition of IAV replication in mice, although with increased production of pro‐inflammatory cytokines and increased mortality of the infected mice. In contrast, treatment with type III IFN also led to successful inhibition of IAV replication but was accompanied by a reduced inflammatory response as well as increased survival of infected mice (Davidson et al, 2016; Galani et al, 2017). Type I IFN has been used extensively as an antiviral therapy against HCV but is being replaced with modern direct acting antiviral drugs, which have substantially fewer side effects and higher success rates. Based upon the abovedescribed mice data, type III IFN was suggested as a potential therapeutic against both IAV and SARS‐CoV‐2 (Davidson et al, 2016; O'Brien et al, 2020; Prokunina‐Olsson et al, 2020) with less inflammatory damage than type I IFN. Recent clinical trials using pegylated IFN‐λ in SARS‐CoV‐2 infected patients, show a benefit if patients are treated with IFN‐λ during the early phase of the infection (Reis et al, 2023). In turn, this then poses a clinical challenge to identify and treat patients early enough.
At higher doses, IFNs also exert an anti‐proliferative effect and prolonged IFN responses can harm highly proliferative tissues, like the bone marrow or epithelial tissues (Parker et al, 2016). In lung epithelial cells, both type I and type III IFNs induce an anti‐proliferative effect via a mechanism involving induction of p53 (Broggi et al, 2020; Major et al, 2020). Thus, at early stages of a respiratory infection, the IFN response is critical to limit viral replication but at later stages, a prolonged IFN response can prevent proper repair of the lung epithelial due to its anti‐proliferative effect and thereby expose patients to secondary bacterial infections (Planet et al, 2016; Rich et al, 2019).
We would like to finish this section by alluding to some of the important unanswered questions in this area. Which cells are the primary source of IFN during viral infections? IFN production can originate both from infected cells that we presume recognize the virus through one of the cytosolic sensing pathways (STING or MAVS) or from non‐infected immune cells that acquire virally derived PAMPs by phagocytosis or related mechanisms and recognize those PAMPS via TLR receptors (Bruni et al, 2015). Many studies address the ability of individual cell types to produce various subtypes of IFN in vitro, but at present we have little information on the in vivo importance of different cell types during an ongoing viral infection. Specifically, we need to address the role of infected versus non‐infected cells as IFN producers, the balance between production of IFN‐λ versus IFN‐β during early stages of infection and potential differences between the mouse model and humans while keeping in mind that mice lack IFN‐λ1 and ‐4.
Author contributions
Louise Dalskov: Conceptualization; data curation; writing – original draft; writing – review and editing. Hans Henrik Gad: Conceptualization; data curation; writing – original draft; writing – review and editing. Rune Hartmann: Conceptualization; data curation; writing – original draft; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Acknowledgements
Research in the RH laboratory are supported by the following agencies: The Novo Nordisk foundation (NNF) grant No. 0071969; Independent research found Denmark, Medical science grant No. 2034‐00225B and Independent research found Denmark, Natural science, grant No. 0135‐00338B.
The EMBO Journal (2023) 42: e112907
References
- Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124: 783–801 [DOI] [PubMed] [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA (2001) Recognition of double‐stranded RNA and activation of NF‐kappaB by toll‐like receptor 3. Nature 413: 732–738 [DOI] [PubMed] [Google Scholar]
- Andersen LL, Mork N, Reinert LS, Kofod‐Olsen E, Narita R, Jorgensen SE, Skipper KA, Honing K, Gad HH, Ostergaard L et al (2015) Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J Exp Med 212: 1371–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrilenas KK, Ramlall V, Kurland J, Leung B, Harbaugh AG, Siggers T (2018) DNA‐binding landscape of IRF3, IRF5 and IRF7 dimers: implications for dimer‐specific gene regulation. Nucleic Acids Res 46: 2509–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au WC, Moore PA, LaFleur DW, Tombal B, Pitha PM (1998) Characterization of the interferon regulatory factor‐7 and its potential role in the transcription activation of interferon a genes. J Biol Chem 273: 29210–29217 [DOI] [PubMed] [Google Scholar]
- Barchet W, Cella M, Odermatt B, Asselin‐Paturel C, Colonna M, Kalinke U (2002) Virus‐induced interferon a production by a dendritic cell subset in the absence of feedback signaling in vivo . J Exp Med 195: 507–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann H-H, Zhang Y, Dorgham K, Philippot Q, Rosain J, Béziat V et al (2020) Autoantibodies against type I IFNs in patients with life–threatening COVID–19. Science 370: eabd4585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley KC, Finsterbusch K, Schnepf D, Crotta S, Llorian M, Davidson S, Fuchs SY, Staeheli P, Wack A (2019) Microbiota‐driven tonic interferon signals in lung stromal cells protect from influenza virus infection. Cell Rep 28: 245–256.e4 [DOI] [PubMed] [Google Scholar]
- Broggi A, Ghosh S, Sposito B, Spreafico R, Balzarini F, Lo Cascio A, Clementi N, De Santis M, Mancini N, Granucci F et al (2020) Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science 369: 706–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruni D, Chazal M, Sinigaglia L, Chauveau L, Schwartz O, Despres P, Jouvenet N (2015) Viral entry route determines how human plasmacytoid dendritic cells produce type I interferons. Sci Signal 8: ra25 [DOI] [PubMed] [Google Scholar]
- Campbell TM, Liu Z, Zhang Q, Moncada‐Velez M, Covill LE, Zhang P, Alavi Darazam I, Bastard P, Bizien L, Bucciol G et al (2022) Respiratory viral infections in otherwise healthy humans with inherited IRF7 deficiency. J Exp Med 219: e20220202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, Colonna M (1999) Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med 5: 919–923 [DOI] [PubMed] [Google Scholar]
- Chow J, Franz KM, Kagan JC (2015) PRRs are watching you: localization of innate sensing and signaling regulators. Virology 479‐480: 104–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciancanelli MJ, Huang SX, Luthra P, Garner H, Itan Y, Volpi S, Lafaille FG, Trouillet C, Schmolke M, Albrecht RA et al (2015) Infectious disease. Life‐threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 348: 448–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, Hornung V, Hopfner KP (2013) Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498: 332–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotta S, Davidson S, Mahlakoiv T, Desmet CJ, Buckwalter MR, Albert ML, Staeheli P, Wack A (2013) Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza‐infected airway epithelia. PLoS Pathog 9: e1003773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Megjugorac NJ, Amrute SB, Fitzgerald‐Bocarsly P (2004) Regulation of IFN regulatory factor‐7 and IFN‐alpha production by enveloped virus and lipopolysaccharide in human plasmacytoid dendritic cells. J Immunol 173: 1535–1548 [DOI] [PubMed] [Google Scholar]
- Dalskov L, Narita R, Andersen LL, Jensen N, Assil S, Kristensen KH, Mikkelsen JG, Fujita T, Mogensen TH, Paludan SR et al (2020) Characterization of distinct molecular interactions responsible for IRF3 and IRF7 phosphorylation and subsequent dimerization. Nucleic Acids Res 48: 11421–11433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson S, McCabe TM, Crotta S, Gad HH, Hessel EM, Beinke S, Hartmann R, Wack A (2016) IFNlambda is a potent anti‐influenza therapeutic without the inflammatory side effects of IFNalpha treatment. EMBO Mol Med 8: 1099–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C (2004) Innate antiviral responses by means of TLR7‐mediated recognition of single‐stranded RNA. Science 303: 1529–1531 [DOI] [PubMed] [Google Scholar]
- Feng H, Zhang YB, Gui JF, Lemon SM, Yamane D (2021) Interferon regulatory factor 1 (IRF1) and anti‐pathogen innate immune responses. PLoS Pathog 17: e1009220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forero A, Ozarkar S, Li H, Lee CH, Hemann EA, Nadjsombati MS, Hendricks MR, So L, Green R, Roy CN et al (2019) Differential activation of the transcription factor IRF1 underlies the distinct immune responses elicited by type I and type III interferons. Immunity 51: 451–464.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita T, Sakakibara J, Sudo Y, Miyamoto M, Kimura Y, Taniguchi T (1988) Evidence for a nuclear factor(s), IRF‐1, mediating induction and silencing properties to human IFN‐b gene regulatory elements. EMBO J 7: 3397–3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung KY, Mangan NE, Cumming H, Horvat JC, Mayall JR, Stifter SA, De Weerd N, Roisman LC, Rossjohn J, Robertson SA et al (2013) Interferon‐epsilon protects the female reproductive tract from viral and bacterial infection. Science 339: 1088–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galani IE, Triantafyllia V, Eleminiadou EE, Koltsida O, Stavropoulos A, Manioudaki M, Thanos D, Doyle SE, Kotenko SV, Thanopoulou K et al (2017) Interferon‐lambda mediates non‐redundant front‐line antiviral protection against influenza virus infection without compromising host fitness. Immunity 46: e876 [DOI] [PubMed] [Google Scholar]
- Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G et al (2013a) Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA‐activated cyclic GMP‐AMP synthase. Cell 153: 1094–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, Gaffney BL, Shuman S, Jones RA, Deng L et al (2013b) Structure‐function analysis of STING activation by c[G(2′,5′)pA(3′,5′)p] and targeting by antiviral DMXAA. Cell 154: 748–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandvaux N, Servant MJ, tenOever B, Sen GC, Balachandran S, Barber GN, Lin R, Hiscott J (2002) Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon‐stimulated genes. J Virol 76: 5532–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy MP, Owczarek CM, Jermiin LS, Ejdeback M, Hertzog PJ (2004) Characterization of the type I interferon locus and identification of novel genes. Genomics 84: 331–345 [DOI] [PubMed] [Google Scholar]
- Hatesuer B, Hoang HT, Riese P, Trittel S, Gerhauser I, Elbahesh H, Geffers R, Wilk E, Schughart K (2017) Deletion of Irf3 and Irf7 genes in mice results in altered interferon pathway activation and granulocyte‐dominated inflammatory responses to influenza a infection. J Innate Immun 9: 145–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauptmann R, Swetly P (1985) A novel class of human type I interferons. Nucleic Acids Res 13: 4739–4749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S (2004) Species‐specific recognition of single‐stranded RNA via toll‐like receptor 7 and 8. Science 303: 1526–1529 [DOI] [PubMed] [Google Scholar]
- Hermant P, Demarez C, Mahlakoiv T, Staeheli P, Meuleman P, Michiels T (2014) Human but not mouse hepatocytes respond to interferon‐lambda in vivo. PLoS One 9: e87906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holleufer A, Winther KG, Gad HH, Ai X, Chen Y, Li L, Wei Z, Deng H, Liu J, Frederiksen NA et al (2021) Two cGAS‐like receptors induce antiviral immunity in drosophila. Nature 597: 114–118 [DOI] [PubMed] [Google Scholar]
- Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M et al (2006) 5′‐triphosphate RNA is the ligand for RIG‐I. Science 314: 994–997 [DOI] [PubMed] [Google Scholar]
- Ioannidis I, Ye F, McNally B, Willette M, Flano E (2013) Toll‐like receptor expression and induction of type I and type III interferons in primary airway epithelial cells. J Virol 87: 3261–3270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN (2008) STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455: 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaks E, Gavutis M, Uze G, Martal J, Piehler J (2007) Differential receptor subunit affinities of type I interferons govern differential signal activation. J Mol Biol 366: 525–539 [DOI] [PubMed] [Google Scholar]
- Jefferies CA (2019) Regulating IRFs in IFN driven disease. Front Immunol 10: 325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewell NA, Cline T, Mertz SE, Smirnov SV, Flano E, Schindler C, Grieves JL, Durbin RK, Kotenko SV, Durbin JE (2010) Lambda interferon is the predominant interferon induced by influenza a virus infection in vivo . J Virol 84: 11515–11522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing T, Zhao B, Xu P, Gao X, Chi L, Han H, Sankaran B, Li P (2020) The structural basis of IRF‐3 activation upon phosphorylation. J Immunol 205: 1886–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnell JL, Wu Y, Mittereder N, Smith MA, Gunsior M, Yan L, Casey KA, Henault J, Riggs JM, Nicholson SM et al (2021) Depleting plasmacytoid dendritic cells reduces local type I interferon responses and disease activity in patients with cutaneous lupus. Sci Transl Med 13: eabf8442 [DOI] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ et al (2006) Differential roles of MDA5 and RIG‐I helicases in the recognition of RNA viruses. Nature 441: 101–105 [DOI] [PubMed] [Google Scholar]
- Khaitov MR, Laza‐Stanca V, Edwards MR, Walton RP, Rohde G, Contoli M, Papi A, Stanciu LA, Kotenko SV, Johnston SL (2009) Respiratory virus induction of alpha‐, beta‐ and lambda‐interferons in bronchial epithelial cells and peripheral blood mononuclear cells. Allergy 64: 375–386 [DOI] [PubMed] [Google Scholar]
- Khoyratty TE, Udalova IA (2018) Diverse mechanisms of IRF5 action in inflammatory responses. Int J Biochem Cell Biol 99: 38–42 [DOI] [PubMed] [Google Scholar]
- Kotenko SV (2011) IFN‐lambdas. Curr Opin Immunol 23: 583–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotenko SV, Gallagher G, Baurin VV, Lewis‐Antes A, Shen M, Shah NK, Langer JA, Sheikh F, Dickensheets H, Donnelly RP (2003) IFN‐lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol 4: 69–77 [DOI] [PubMed] [Google Scholar]
- LaFleur DW, Nardelli B, Tsareva T, Mather D, Feng P, Semenuk M, Taylor K, Buergin M, Chinchilla D, Roshke V et al (2001) Interferon–κ, a novel type I interferon expressed in human keratinocytes. J Biol Chem 276: 39765–39771 [DOI] [PubMed] [Google Scholar]
- Lazear HM, Lancaster A, Wilkins C, Suthar MS, Huang A, Vick SC, Clepper L, Thackray L, Brassil MM, Virgin HW et al (2013) IRF‐3, IRF‐5, and IRF‐7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog 9: e1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Wong JJ, Sum C, Sin WX, Ng KQ, Koh MB, Chin KC (2011) IRF8 and IRF3 cooperatively regulate rapid interferon‐beta induction in human blood monocytes. Blood 117: 2847–2854 [DOI] [PubMed] [Google Scholar]
- Lin R, Heylbroeck C, Pitha PM, Hiscott J (1998) Virus‐dependent phosphorylation of the IRF‐3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome‐mediated degradation. Mol Cell Biol 18: 2986–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Ding JL (2016) The molecular mechanisms of TLR‐signaling cooperation in cytokine regulation. Immunol Cell Biol 94: 538–542 [DOI] [PubMed] [Google Scholar]
- Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV et al (2015) Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347: aaa2630 [DOI] [PubMed] [Google Scholar]
- Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A (2003) Toll‐like receptor 9‐mediated recognition of herpes simplex virus‐2 by plasmacytoid dendritic cells. J Exp Med 198: 513–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major J, Crotta S, Llorian M, McCabe TM, Gad HH, Priestnall SL, Hartmann R, Wack A (2020) Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science 369: 712–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marié I, Durbin JE, Levy DE (1998) Differential viral induction of distinct interferon‐α genes by positive feedback through interferon regulatory factor‐7. EMBO J 17: 6660–6669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks ZRC, Campbell N, deWeerd NA, Lim SS, Gearing LJ, Bourke NM, Hertzog PJ (2019) Properties and functions of the novel type I interferon epsilon. Semin Immunol 43: 101328 [DOI] [PubMed] [Google Scholar]
- Meager A, Visvalingam K, Dilger P, Bryan D, Wadhwa M (2005) Biological activity of interleukins‐28 and ‐29: comparison with type I interferons. Cytokine 31: 109–118 [DOI] [PubMed] [Google Scholar]
- Mogensen TH (2009) Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22: 240–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordstein M, Neugebauer E, Ditt V, Jessen B, Rieger T, Falcone V, Sorgeloos F, Ehl S, Mayer D, Kochs G et al (2010) Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J Virol 84: 5670–5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morehouse BR, Govande AA, Millman A, Keszei AFA, Lowey B, Ofir G, Shao S, Sorek R, Kranzusch PJ (2020) STING cyclic dinucleotide sensing originated in bacteria. Nature 586: 429–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Yoneyama M, Ito T, Takahashi K, Inagaki F, Fujita T (2004) Identification of Ser‐386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J Biol Chem 279: 9698–9702 [DOI] [PubMed] [Google Scholar]
- Muir AJ, Arora S, Everson G, Flisiak R, George J, Ghalib R, Gordon SC, Gray T, Greenbloom S, Hassanein T et al (2014) A randomized phase 2b study of Peginterferon lambda‐1a for the treatment of chronic HCV infection. J Hepatol 61: 1238–1246 [DOI] [PubMed] [Google Scholar]
- Ning S, Pagano JS, Barber GN (2011) IRF7: activation, regulation, modification and function. Genes Immun 12: 399–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien TR, Thomas DL, Jackson SS, Prokunina‐Olsson L, Donnelly RP, Hartmann R (2020) Weak induction of interferon expression by severe acute respiratory syndrome coronavirus 2 supports clinical trials of interferon‐lambda to treat early coronavirus disease 2019. Clin Infect Dis 71: 1410–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onoguchi K, Yoneyama M, Takemura A, Akira S, Taniguchi T, Namiki H, Fujita T (2007) Viral infections activate types I and III interferon genes through a common mechanism. J Biol Chem 282: 7576–7581 [DOI] [PubMed] [Google Scholar]
- Osterlund PI, Pietila TE, Veckman V, Kotenko SV, Julkunen I (2007) IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN‐lambda) genes. J Immunol 179: 3434–3442 [DOI] [PubMed] [Google Scholar]
- Panne D, Maniatis T, Harrison SC (2004) Crystal structure of ATF‐2/c‐Jun and IRF‐3 bound to the interferon‐beta enhancer. EMBO J 23: 4365–4525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panne D, Maniatis T, Harrison SC (2007) An atomic model of the interferon‐beta enhanceosome. Cell 129: 1111–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker BS, Rautela J, Hertzog PJ (2016) Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer 16: 131–144 [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C (2006) RIG‐I‐mediated antiviral responses to single‐stranded RNA bearing 5′‐phosphates. Science 314: 997–1001 [DOI] [PubMed] [Google Scholar]
- Planet PJ, Parker D, Cohen TS, Smith H, Leon JD, Ryan C, Hammer TJ, Fierer N, Chen EI, Prince AS (2016) Lambda interferon restructures the nasal microbiome and increases susceptibility to Staphylococcus aureus superinfection. mBio 7: e01939‐15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platanias LC (2005) Mechanisms of type‐I‐ and type‐II‐interferon‐mediated signalling. Nat Rev Immunol 5: 375–386 [DOI] [PubMed] [Google Scholar]
- Prokunina‐Olsson L, Alphonse N, Dickenson RE, Durbin JE, Glenn JS, Hartmann R, Kotenko SV, Lazear HM, O'Brien TR, Odendall C et al (2020) COVID‐19 and emerging viral infections: the case for interferon lambda. J Exp Med 217: e20200653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin BY, Liu C, Lam SS, Srinath H, Delston R, Correia JJ, Derynck R, Lin K (2003) Crystal structure of IRF‐3 reveals mechanism of autoinhibition and virus‐induced phosphoactivation. Nat Struct Biol 10: 913–921 [DOI] [PubMed] [Google Scholar]
- Qureshi SA, Salditt‐Georgieff M, Darnell JE Jr (1995) Tyrosine‐phosphorylated Stat1 and Stat2 plus a 48‐kDa protein all contact DNA in forming interferon‐stimulated‐gene factor 3. Proc Natl Acad Sci USA 92: 3829–3833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis LFL, Ruffner H, Stark G, Aguet M, Weissmann C (1994) Mice devoid of interferon regulatory factor 1 (IRF‐1) show normal expression of type I interferon genes. EMBO J 13: 4798–4806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis G, Moreira Silva EAS, Medeiros Silva DC, Thabane L, Campos VHS, Ferreira TS, Santos CVQ, Nogueira AMR, Almeida A, Savassi LCM et al (2023) Early treatment with pegylated interferon lambda for Covid‐19. N Engl J Med 388: 518–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich HE, McCourt CC, Zheng WQ, McHugh KJ, Robinson KM, Wang J, Alcorn JF (2019) Interferon lambda inhibits bacterial uptake during influenza superinfection. Infect Immun 87: e00114‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland SL, Riggs JM, Gilfillan S, Bugatti M, Vermi W, Kolbeck R, Unanue ER, Sanjuan MA, Colonna M (2014) Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J Exp Med 211: 1977–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N (1998) Positive feedback regulation of type I IFN genes by the IFN inducible transcription factor IRF‐7. FEBS Lett 441: 106–110 [DOI] [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N et al (2000) Distinct and essential roles of transcription factors IRF‐3 and IRF‐7 in response to viruses for IFN‐alpha/beta gene induction. Immunity 13: 539–548 [DOI] [PubMed] [Google Scholar]
- Schafer SL, Lin R, Moore PA, Hiscott J, Pitha PM (1998) Regulation of type I interferon gene expression by interferon regulatory factor‐3. J Biol Chem 273: 2714–2720 [DOI] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, Rice CM (2014) Interferon‐stimulated genes: a complex web of host defenses. Annu Rev Immunol 32: 513–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenemeyer A, Barnes BJ, Mancl ME, Latz E, Goutagny N, Pitha PM, Fitzgerald KA, Golenbock DT (2005) The interferon regulatory factor, IRF5, is a central mediator of toll‐like receptor 7 signaling. J Biol Chem 280: 17005–17012 [DOI] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM (2011) A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472: 481–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber G (2017) The molecular basis for differential type I interferon signaling. J Biol Chem 292: 7285–7294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant MJ, Grandvaux N, tenOever BR, Duguay D, Lin R, Hiscott J (2003) Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double‐stranded RNA. J Biol Chem 278: 9441–9447 [DOI] [PubMed] [Google Scholar]
- Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, Kuestner R, Garrigues U, Birks C, Roraback J et al (2003) IL‐28, IL‐29 and their class II cytokine receptor IL‐28R. Nat Immunol 4: 63–68 [DOI] [PubMed] [Google Scholar]
- Slavik KM, Morehouse BR, Ragucci AE, Zhou W, Ai X, Chen Y, Li L, Wei Z, Bahre H, Konig M et al (2021) cGAS‐like receptors sense RNA and control 3′2'‐cGAMP signalling in Drosophila . Nature 597: 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommereyns C, Paul S, Staeheli P, Michiels T (2008) IFN‐lambda (IFN‐lambda) is expressed in a tissue‐dependent fashion and primarily acts on epithelial cells in vivo . PLoS Pathog 4: e1000017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg C, Eisenacher K, Gross O, Reindl W, Schmitz F, Ruland J, Krug A (2009) The IFN regulatory factor 7‐dependent type I IFN response is not essential for early resistance against murine cytomegalovirus infection. Eur J Immunol 39: 1007–1018 [DOI] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ (2013) Cyclic GMP‐AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiecki M, Colonna M (2015) The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol 15: 471–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tailor P, Tamura T, Kong HJ, Kubota T, Kubota M, Borghi P, Gabriele L, Ozato K (2007) The feedback phase of type I interferon induction in dendritic cells requires interferon regulatory factor 8. Immunity 27: 228–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahasi K, Suzuki NN, Horiuchi M, Mori M, Suhara W, Okabe Y, Fukuhara Y, Terasawa H, Akira S, Fujita T et al (2003) X‐ray crystal structure of IRF‐3 and its functional implications. Nat Struct Biol 10: 922–927 [DOI] [PubMed] [Google Scholar]
- Takahasi K, Horiuchi M, Fujii K, Nakamura S, Noda NN, Yoneyama M, Fujita T, Inagaki F (2010) Ser386 phosphorylation of transcription factor IRF‐3 induces dimerization and association with CBP/p300 without overall conformational change. Genes Cells 15: 901–910 [DOI] [PubMed] [Google Scholar]
- Thanos D, Maniatis T (1995) Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell 83: 1091–1100 [DOI] [PubMed] [Google Scholar]
- Thomsen MM, Jorgensen SE, Gad HH, Storgaard M, Gjedsted J, Christiansen M, Hartmann R, Mogensen TH (2019) Defective interferon priming and impaired antiviral responses in a patient with an IRF7 variant and severe influenza. Med Microbiol Immunol 208: 869–876 [DOI] [PubMed] [Google Scholar]
- Thomson SJ, Goh FG, Banks H, Krausgruber T, Kotenko SV, Foxwell BM, Udalova IA (2009) The role of transposable elements in the regulation of IFN‐lambda1 gene expression. Proc Natl Acad Sci USA 106: 11564–11569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, Silverman RH, Williams BRG (2001) Functional classification of interferon‐stimulated genes identified using microarrays. J Leukoc Biol 69: [PubMed] [Google Scholar]
- Wack A, Terczynska‐Dyla E, Hartmann R (2015) Guarding the frontiers: the biology of type III interferons. Nat Immunol 16: 802–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Oberley‐Deegan R, Wang S, Nikrad M, Funk CJ, Hartshorn KL, Mason RJ (2009) Differentiated human alveolar type II cells secrete antiviral IL‐29 (IFN‐λ1) in response to influenza a infection. J Immunol 182: 1296–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittling MC, Cahalan SR, Levenson EA, Rabin RL (2020) Shared and unique features of human Interferon‐Beta and Interferon‐alpha subtypes. Front Immunol 11: 605673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ (2013) Cyclic GMP‐AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339: 826–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai H, Chiba S, Hangai S, Kometani K, Inoue A, Kimura Y, Abe T, Kiyonari H, Nishio J, Taguchi‐Atarashi N et al (2018) Revisiting the role of IRF3 in inflammation and immunity by conditional and specifically targeted gene ablation in mice. Proc Natl Acad Sci USA 115: 5253–5258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda K, Nundel K, Watkins AA, Dhawan T, Bonegio RG, Ubellacker JM, Marshak‐Rothstein A, Rifkin IR (2013) Phenotype and function of B cells and dendritic cells from interferon regulatory factor 5‐deficient mice with and without a mutation in DOCK2. Int Immunol 25: 295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Schnepf D, Staeheli P (2019) Interferon‐lambda orchestrates innate and adaptive mucosal immune responses. Nat Rev Immunol 19: 614–625 [DOI] [PubMed] [Google Scholar]
- Yin Z, Dai J, Deng J, Sheikh F, Natalia M, Shih T, Lewis‐Antes A, Amrute SB, Garrigues U, Doyle S et al (2012) Type III IFNs are produced by and stimulate human plasmacytoid dendritic cells. J Immunol 189: 2735–2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, Onomoto K, Jogi M, Akaboshi T, Fujita T (2015) Viral RNA detection by RIG‐I‐like receptors. Curr Opin Immunol 32: 48–53 [DOI] [PubMed] [Google Scholar]
- Zhang C, Shang G, Gui X, Zhang X, Bai XC, Chen ZJ (2019) Structural basis of STING binding with and phosphorylation by TBK1. Nature 567: 394–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada‐Velez M, Chen J, Ogishi M, Sabli IKD, Hodeib S, Korol C et al (2020) Inborn errors of type I IFN immunity in patients with life‐threatening COVID‐19. Science 370: eabd4570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Shu C, Gao X, Sankaran B, Du F, Shelton CL, Herr AB, Ji J‐Y, Li P (2016) Structural basis for concerted recruitment and activation of IRF‐3 by innate immune adaptor proteins. Proc Natl Acad Sci USA 113: E3403‐12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hamming OJ, Ank N, Paludan SR, Nielsen AL, Hartmann R (2007) Type III interferon (IFN) induces a type I IFN‐like response in a restricted subset of cells through signaling pathways involving both the Jak‐STAT pathway and the mitogen‐activated protein kinases. J Virol 81: 7749–7758 [DOI] [PMC free article] [PubMed] [Google Scholar]