

Abstract
Moraxella catarrhalis is a symbiotic as well as mucosal infection-causing bacterium unique to humans. Currently, it is considered as one of the leading factors of acute middle ear infection in children. As M. catarrhalis is resistant to multiple drugs, the treatment is unsuccessful; therefore, innovative and forward-thinking approaches are required to combat the problem of antimicrobial resistance (AMR). To better comprehend the numerous processes that lead to antibiotic resistance in M. catarrhalis, we have adopted a computational method in this study. From the NCBI-Genome database, we investigated 12 strains of M. catarrhalis. We explored the interaction network comprising 74 antimicrobial-resistant genes found by analyzing M. catarrhalis bacterial strains. Moreover, to elucidate the molecular mechanism of the AMR system, clustering and the functional enrichment analysis were assessed employing AMR gene interactions networks. According to the findings of our assessment, the majority of the genes in the network were involved in antibiotic inactivation; antibiotic target replacement, alteration and antibiotic efflux pump processes. They exhibit resistance to several antibiotics, such as isoniazid, ethionamide, cycloserine, fosfomycin, triclosan, etc. Additionally, rpoB, atpA, fusA, groEL and rpoL have the highest frequency of relevant interactors in the interaction network and are therefore regarded as the hub nodes. These genes can be exploited to create novel medications by serving as possible therapeutic targets. Finally, we believe that our findings could be useful to advance knowledge of the AMR system present in M. catarrhalis.
Keywords: Moraxella catarrhalis, clustering analysis, functional enrichment analysis, antimicrobial resistance system, AMR genes, gene ontology
Introduction
Antimicrobial resistance (AMR) in pathogenic bacterial strains is currently a serious problem causing a lot of death and suffering to mankind around the globe. It has a negative impact on diagnostic, therapeutic and financial consequences, with implications varying from a patient’s failure to retaliate to treatment. Moreover, the rising incidence of multidrug-resistant (MDR) bacterial pathogens causing clinical and community-acquired diseases is restricting antibiotic treatment choices [1]. There are several pathogens found in the intensive care unit that can develop antibiotic resistance, but gram-negative strains of bacteria are the most prone to developing barriers to various kinds of antibiotics [2]. The most frequent methods of resistance to β-lactam in gram-negative bacteria are antimicrobials obliteration via beta-lactamases; insulative properties, which include the shutdown of gene encoding channels in the bacterial cell membrane; and antibiotic deformation by efflux pumps [3]. To better understand the numerous antibiotic resistance systems that lead to AMR in Moraxella catarrhalis, we employed a computational method in our current study (Figure 1).
Figure 1.

This workflow illustrates the whole exploration of how we predicted antibiotic resistance in M. catarrhalis by applying multiple computational techniques, depending on complete-genome sequencing and systems biology approaches.
Moraxella catarrhalis is a gram-negative, gamma-proteobacterium and oxygenated ubiquitous bacteria which generates acute otitis media (AOM) in youngsters and reduced respiratory tract problems in adults, putting a strain on medical infrastructures across the world [4]. Previously, this microorganism was referred to as Neisseria catarrhalis or Micrococcus catarrhalis [5]. It is commonly encountered as a top lung system pathogen in humans [6] and also a prevalent source of otitis media (OM) in newborns and children, contributing to 15–20% of severe OM episodes [7]. Moreover, M. catarrhalis-induced OM is thought to be moderate in compared with pneumococcal illness, multiple potential pathogens have already been discovered and it has been demonstrated that some epidermal constituents of M. catarrhalis produce inflammatory responses [8]. This bacterium can easily attach to the epithelium of a variety of nasal surfaces, including the lungs as well as the nasopharynx and elicits a powerful chronic inflammation defined by incursions of macrophages, lymphocytes and neutrophils into diseased tissue following infection, that is thought to be the etiology of OM and COPD relapses [9]. OM is common and widespread in developing nations and is a prominent reason of illness and death in children below the age of five [10]. It can cause major abnormalities in children’s linguistic, intellectual, academic and psychosocial development [11]. Additionally, M. cattarhalis is currently a good cause of around 10% of acute inflammatory comorbid conditions in individuals, the chronic obstructive pulmonary disease (COPD) [9]. In the United States, this bacterium is expected to trigger 2–4 million cases of chronic degenerative pulmonary disease in people annually [7].
In Bangladesh, several studies found substantial pervasiveness of OM in rural, urban and elementary school youngsters, with frequency rates of 43.2/1000, 32.6/1000 and 16.3/1000, correspondingly [11]. The detection of M. catarrhalis by several fatalities receptors (TLRs) including TLR4 and TLR9 induces the generation of erythrogenic mediators IL-6 and TNF- by the host defenses, according to several research studies [12, 13]. Also severe and recurring AOM infections have long been believed to include bacterial survival inside a biofilm due to their extremely resistant feature [14, 15]. Direct detection of bacterial biofilms in inpatient clinical specimens and the chinchilla model system of OM provide medical confirmation of bacterial biofilms [16, 17]. Several processes, along with epigenetic modification heterogeneity and slower spread of microorganisms inside this biofilm, postponed antibiotic infiltration through composite content, and the existence of viable cells [18] can dramatically increase antibiotic susceptibility in microbes within a biofilm congregation [19].
Moreover, some other preliminary research stated that M. catarrhalis exhibits a variety of antibiotic susceptibility strategies, including membrane porosity, active efflux mechanisms and alterations in antibacterial sites [20]. This bacteria shows that these strategies of susceptibility to beta-lactam antibiotics are usually related to type A serine β-lactamase enzyme [20, 21]. Beta-lactamases are enzymes generated by the preponderance of strains isolated from M. catarrhalis that allow them to withstand beta-lactam medications such as penicillin, amoxicillin and cephalosporins [22, 23]. In addition to this, the exterior layer porin M35 of M. catarrhalis is the factor that determines whether or not the bacteria are susceptible to aminopenicillins [24]. The resistance frequency of M. catarrhalis identified in infants to Beta-lactam antibiotics has attained 99% in China as a result of therapeutic experimental usage of antibiotics [25]. Furthermore, according to reports, M. catarrhalis appeared extremely susceptible to macrolide antibiotics, erythromycin and rokitamycin [25, 26]. In addition, some investigations have demonstrated that M. catarrhalis is sensitive to the antibiotics cefaclor, clarithromycin, azithromycin, doxycycline, co-trimoxazole, cefuroxime, cefixime and ceftriaxone, as well as ofloxacin and ciprofloxacin [27]. The percentage of resistance of M. catarrhalis to tetracycline has attained 65.7%, representing a remarkable rise in resistance [28].
In our current study, we gathered entire genomic sequences of M. catarrhalis strains from the NCBI Genome resource and built a phylogenetic tree to better comprehend the biological and developmental connection among the M. catarrhalis strains. We additionally obtained antibiotic resistance genes (AMR) of M. catarrhalis strains from many databases, including Comprehensive Antibiotic Resistance Database (CARD), Pathway-systems Resource Investigation Center (PATRIC) and ResFinder, as well as built a gene interaction network to analyze the multidrug susceptibility systems by utilizing those AMR genes. The application of gene interaction-based network is to identify whether the influence of genomic outcomes on biological activities is becoming progressively pertinent [29]. On the contrary, researchers have increasingly been interested in gene interaction networking investigations, which are thought to be valuable in understanding multidrug susceptibility in infectious and exploitative microorganisms, as well as other biological disorders [30, 31]. Currently, one of the most promising approaches to research the roles of genes and proteins, as well as their related collaborators, is to use gene interactions. It aids in the discovery of relevant biological information about AMR processes, which in turn assists in the identification of critical candidate genes or proteins in the cycle, as well as the development of innovative medications to combat ailments triggered by AMR virulent strains [32, 33]. The molecular linkages and processes of the AMR genes have been explored in this work, which will be crucial in the development of innovative and effective medications for the disease’s therapy. Moreover, we performed protein–protein interactions (PPIs) among the antibiotic-resistant genes, cluster investigation, recognition of hub proteins and pathway assessment to unveil the complicated biological framework, as well as gene linkage with resistance mechanisms and drug class. We employed a combination of clustering and topological techniques to uncover the physiologically significant genes involved in drug susceptibility pathways. Thus, we were able to pinpoint the origin of the antimicrobial-resistant genes as well as these gene interactions that occurred among the M. catarrhalis strains. All of these things were carried out to establish an affiliation in gene expression patterns in M. catarrhalis. However, the genes identified as prospective pharmacological targets can be employed to create novel molecules with pharmaceutical uses to reduce M. catarrhalis outbreaks. We anticipate that our findings will improve the knowledge about the molecular underpinnings of multidrug resistance pathways in M. catarrhalis bacterial strains.
Materials and methods
Genome information of M. Catarrhalis
From the NCBI Genome database (https://www.ncbi.nlm.nih.gov/genome), we have analyzed 215 strains of M. catarrhalis, but for the further analysis, we chose only the complete genome sequences of 12 strains of M. catarrhalis. The genome database is a comprehensive resource of NCBI that includes genome sequences and assembly metadata as well as mapping enrichment data such as variants, and indicators, including epigenomics data [34]. During the selection of genome sequences, chromosomes, scaffolds and contigs were not evaluated; only complete genome sequences were considered. Those strains were available from 1980 to 2022. Furthermore, most of the strains were from human middle ear and sputum sample.
Identification of AMR genes
After retrieving the complete genomes of M. Catarrhalis, we extracted AMR genes from those complete genomes using repositories including the CARD, PATRIC as well as ARDB and investigated them. Here, CARD or Comprehensive Antibiotic Resistance Database (https://card.mcmaster.ca/) is an elevated data source concerning the underlying mechanism of antibiotic resistance genes. It is a vetted platform that provides standard DNA and protein sequences, identification models and computational tools in a regulated ontology that is the Antibiotic Resistance Ontology, which is established by CARD’s biocuration group for program configuration [35, 36]. Another side, the PATRIC [https://www.patricbrc.org/] provides a collection of potential pathogens information kinds that have been combined from various data sources. PATRIC is a collaborative initiative between the Bioinformatics Resource Center and the National Institute of Allergy and Infectious Diseases [37]. ResFinder (https://cge.cbs.dtu.dk/services/ResFinder/) is a repository that indexes antibacterial resistant genes discovered in the whole genome of bacteria. This is accomplished through the usage of BLAST [38]. In the end, we compiled the resistance mechanisms and drug classes associated with these AMR genes. Then, we employed Venny 2.1 (https://bioinfogp.cnb.csic.es/tools/venny/) to gather only the unique AMR gene. Venny 2.1 is a tool for mapping and comparing gene lists that may be used interactively [39, 40]. Then, we used these unique AMR genes to build gene interaction networks and for other further analysis.
Phylogenetic tree construction
Most biological research requires an understanding of evolutionary interconnections between species. A reliable phylogenetic tree is essential for presuming the provenance of novel genes, identifying biochemical transformation, comprehension of morphological feature progression as well as recreating psychographic trends in diverged species [41]. We previously noted that we obtained a total of 12 complete-genome sequences of M. catarrhalis. The reference sequence was from the CCRI-195ME strain of M. catarrhalis. We employed Mega v11 software (https://www.megasoftware.net/) to do the phylogenetic investigation to determine the developmental and evolutionary connection between the M. catarrhalis strains. MEGA (Molecular Evolutionary Genetics Analysis) is a computer-based program for statistically analyzing molecular development, determining evolutionary process length and building phylogenetic relationships. It is open concerning safety and offers GDPR (General Data Protection Regulation) insurance to people all around the world [42]. However, the evolutionary history was estimated using the Neighbor-Joining statistical approach using 1000 bootstraps and then exported into iTOL (v. 6) (https://itol.embl.de/) for improved display. We also provided the length of each branch from the root. Here, the nodes in the phylogenetic tree indicate isolated strains, and the edges indicate the hamming distance between two strains.
PPI network construction and visualization
PPIs regulate a vast variety of biological activities, and physiological activities notably tissue connectivity as well as developmental management [43]. To build the PPI network and identify the associated genes or protein databases, we utilized a well-known search program STRING (http://string-db.org). The STRING database plays an important role in assembling, evaluating and disseminating PPI data in a user-friendly and extensive way [44]. As a starting point, we provided STRING with a list of unique AMR genes so that it could look for their neighboring interactors. The extracted PPI network was generated with medium confidence (>0.40) in STRING. Finally, we employed Cytoscape_v3.9.1 to create a visual representation of the target network. Cytoscape (https://cytoscape.org/) is a prominent bioinformatics program for visualizing biological interactions and integrating data.
Cluster formation and hub proteins extraction
Cluster analysis is a comprehensive method for combining expression profiles with protein–protein-interacting networks. In systems biology, it has a vital role in identifying regulatory components and estimating protein expression. We utilized the MCODE (https://baderlab.org/Software/MCODE) plug-in in Cytoscape to form the clusters. The MCODE plugin is intended to find densely connected zone also known as clusters in a biological network. In our current study, cluster formation was carried out applying the default parameter including degree score cutoff of 2, node score cutoff of 0.2 and K-Core of 2, and the maximum depth of 100 in the MCODE to verify the efficacy of interactive collaborators in the context of AMR gene expression. On the other hand, hub proteins, also known as key proteins, are characterized as proteins that have a significant degree of association on a wide range throughout the PPI network. In our ongoing study, we utilized the Cytoscape plug-in cytohubba (http://apps.cytoscape.org/apps/cytohubba) to find highly interconnected protein nodes as well as to investigate the network topology. The cytoHubba plugin is employed to obtain the protein nodes that are largely attributable inside the PPI network. Eleven topology analytical techniques are accessible in cytoHubba [45]. Our study included six analytical techniques from the Cytohubba plugin, including three locally ranked methodologies: degree, maximum neighborhood component (MNC) and maximum clique centrality (MCC), as well as three globally ranked methodologies: closeness centrality, betweenness and also the stress method. In the following step, the collected genes from the cytoHubba were submitted to jvenn (an interactive Venn diagram analyzer) (http://jvenn.toulouse.inra.fr/) for more analysis and the genes that were intersected among the six approaches of cytohubba were designated as significant hub proteins.
Assessment of gene enrichment
Gene enrichment is a process of analyzing collections of genes using the gene ontology categorization system, whereby genes are classified into preset groups based on their operational features. Gene Ontology is categorized into three distinct activities: biological activities, cellular activities and molecular activities. Here, the term biological activities refer to the major cellular or metabolic significance of genes in coordination with other genes, cellular activities refer to the role of gene products within the cell, whereas molecular activities refer to the specific molecular function (MF) of a gene [46]. On the other hand, Kyoto Encyclopedia of Genes and Genome (KEGG) is also a biological explanatory scientific route database. KEGG pathway analysis aids in the discovery of linkages between core activities of critical genes, as well as in gaining a thorough understanding of the fundamental activities of genes [26, 47]. In this work, we retrieved GO keywords and KEGG pathway data from the STRING database and then utilized SRplot—Science and Research online plot (http://www.bioinformatics.com.cn/en) to display and further analyze them.
Genes correlation with antibiotic resistance mechanism and drug class
Microbes develop methods to defend themselves against antimicrobial compounds, which are known as AMR mechanisms. These mechanisms have developed in bacteria due to a variety of reasons. A few of them include modifications of the permeability in the bacterial cell that constrain bacterial direct exposure to target areas, alternations of the enzyme’s catalytic activity, oversaturation of the intended enzymes, antimicrobial drugs modification and deterioration, development of metabolic processes other than those blocked by the medication, active efflux pump and so on [48]. On the other hand, penicillin and beta-lactam were the first antibacterial compounds identified [49]. These antibiotics were successful in treating bacterial infectious diseases. Other antibiotics, such as macrolides, aminoglycosides, chloramphenicol, tetracycline and streptothricin, as well as sulfonamide and trimethoprim, perform a significant function in the diagnosis and therapies of microbial pathogens [49]. These agents can repress the antimicrobial protein production while also interfering with DNA and RNA production, negatively affect the microbial cell wall production and prevent microbial cell energy biosynthesis [50]. In our ongoing study, during the process of collecting AMR genes from the CARD and PATRIC databases, we also put together a list of the resistance mechanisms and drug classes. Afterward, we reorganized all of the information, which included distinct genes, drug classes as well as resistance mechanisms, to execute the sunbursts plot using python. Python is a programming language that is employed to develop computer programs. Additionally, it is frequently utilized to perform automated operations, as well as statistical exploration [51]. However, this analysis helps to dig out the causes behind susceptibility, the enhanced strategy of detecting resistance when it emerges, alternative therapeutic choices for diseases triggered by resistant organisms, as well as attempts to mitigate and regulate the formation [52, 53].
Results
Collection of AMR gene
We retrieved a total of 288 AMR genes from these 12 strains of bacteria (Figure 2). Among these AMR genes, 18 were from the card, 264 were from PATRIC and 6 were from ResFinder. Out of 288 collected resistance genes, 74 entries were found to be unique (Table S1 available online at http://bib.oxfordjournals.org/). However, these unique AMR genes were implemented to conduct additional exploration of this current study. The detailed information of bacterial genome size, genome coverage and gene number was provided in Table 1.
Figure 2.

The number of AMR genes found in M. catarrhalis. The X-axis indicates the number of AMR genes, while the Y-axis indicates the name of the M. catarrhalis genomes.
Table 1.
Details annotation information of bacterial (M. catarrhalis) genome
| Sl. No | Accession No. | Strain Name of M. catarrhalis | Genome Coverage | Genome size (Mb) | Host/source | Genes number | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | GCF_002080125.1 | CCRI-195ME (Reference) | Not provided | 1.9 | Homo sapiens/middle ear | 1901 | NCBI:txid480 |
| 2 | GCA_000740455.1 | 25 240 | 317× | 1.9 | Unknown | 1777 | NCBI:txid480 |
| 3 | GCA_000766665.1 | 25 239 | 211× | 1.8 | H. sapiens | 1752 | NCBI:txid480 |
| 4 | GCA_002073215.2 | FDAARGOS_213 | 1000.94× | 1.9 | H. sapiens | 1747 | NCBI:txid480 |
| 5 | GCA_002984125.1 | FDAARGOS_304 | 825.126× | 1.9 | H. sapiens/nose of a healthy pediatric carrier | 1788 | NCBI:txid480 |
| 6 | GCA_003971285.1 | 74P50B1 | 30× | 1.8 | H. sapiens/sputum | 1681 | NCBI:txid480 |
| 7 | GCA_003971305.1 | 142P87B1 | 26× | 1.9 | H. sapiens/sputum | 1769 | NCBI:txid480 |
| 8 | GCA_003971325.1 | 46P58B1 | 24× | 2.05 | H. sapiens/sputum | 1933 | NCBI:txid480 |
| 9 | GCA_003971345.1 | 74P58B1 | 31× | 1.8 | H. sapiens/sputum | 1681 | NCBI:txid480 |
| 10 | GCA_003971365.1 | 5P47B2 | 29× | 1.9 | H. sapiens/sputum | 1771 | NCBI:txid480 |
| 11 | GCA_900476075.1 | NCTC11020 | 100× | 1.9 | Unknown | 1748 | NCBI:txid480 |
| 12 | GCA_000092265.1 | BBH18 | Not provided | 1.863 | Unknown | 1722 | NCBI:txid1236608 |
Phylogenetic tree analysis
We have indicated that there were 215 strains of M. catarrhalis in the NCBI up to 5 October 2022, but we analyzed only 12 complete strains with genome coverage of ≥20×, and size of the analyzed genomes varied from 1.8 to 2.05 Mbp (Table 1). We constructed a phylogenetic tree relying on the 12 complete genomes of M. catarrhalis and revealed the ancestral connection among them. Figure 3A and B represents the rooted and circular view of the phylogenetic tree, respectively. Among 12 strains, the phylogenetic tree revealed two major clades and one outgroup. Clade 1 consists of two strains and clade 2 consists of 9 strains of M. catarrhalis. Moreover, the out group strain was M. catarrhalis FDAARGOS_304 which was less linked to other strains. The reference sequence of M. catarrhalis, strain CCRI-195ME, has been highlighted in red in both images.
Figure 3.

Phylogenetic tree (12 strains of M. catarrhalis). (A) Rooted view and (B) Circular view. The spreading arrangement of two views of phylogenetic tree indicates how bacterial strains emerged from a prevalent origin. This phylogenetic tree was built using the genomes of 12 different M. catarrhalis strains using the Neighbor-Joining method and 1000 bootstraps. The strains are divided into two separate clades by the tree network (Clade 1 and Clade 2). In both views, the reference sequence M. catarrhalis, strain CCRI-195ME is marked (red). The edges of the phylogenetic tree indicate the hamming distance between two strains, and each node indicates a single strain.
PPI network analysis
In our present study, STRING was used to generate the PPI network through the unique genes. In a PPI network, proteins or genes are denoted as nodes, while interconnections between these nodes are denoted as edges. The PPI network that we extracted in our current analysis contains 43 nodes and 288 edges. Moreover, the clustering coefficient of the network was 0.414. Figure 4A depicts the PPI network. It represents the connectivity of folE, FabF, argG, RocD, ung, fabG and GalE genes to other nodes within the network.
Figure 4.
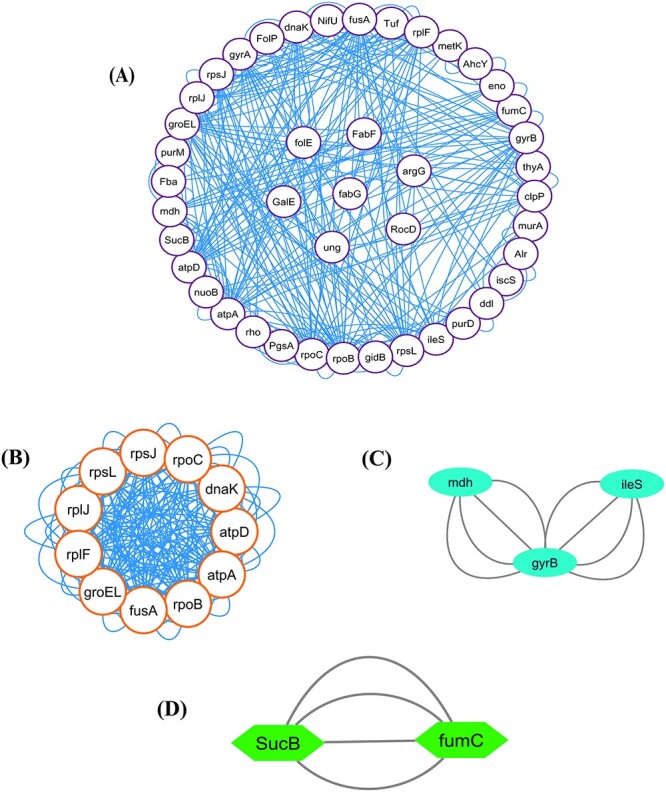
(A) PPI network. In this network, AMR genes are indicated as white nodes and gene connections are indicated by blue edges. Furthermore, cluster analysis was performed to produce relevant and persistent sets of comparable genes for biological identification and evaluation. (B) Cluster C1 (11 nodes, 192 edges), (C) Cluster C2 (3 nodes, 8 edges) and (D) Cluster C3 (2 nodes, 4 edges) show the highly connected proteins among the PPI network.
Cluster analysis and hub proteins identification
We detected three significant clusters in the PPI network employing the MCODE plugin of Cytoscape. Within the clusters, the 1st cluster (C1) comprised 11 nodes and 192 edges (score: 25.517), the 2nd cluster (C2) comprised 3 nodes and 8 edges (score 2) as well as the 3rd cluster (C3) comprised 2 nodes and 4 edges (score 2). The three clusters are shown in Figure 4B–D, respectively. In addition, Table 2 displays the genes involved within those three clusters. Furthermore, we discovered hub proteins by employing six Cytohubba plugin methodologies, comprising Stress, Betweenness, Closeness, Degree, MNC and MCC (Figure 5). Then, Jvenn analysis was performed on six classes of hub proteins obtained using the aforementioned methods. From the Jvenn analysis, we noticed that five proteins were shared by all methods namely rpoB, atpA, fusA, groEL and rpoL. These five proteins were identified as significant hub proteins. The Jvenn diagram is shown in Figure 6, and Table 3 shows the topological properties of significant hub proteins. On the contrary, these hub proteins were also detected in cluster 1, cluster 2 and cluster 3, indicating that they were the most crucial hub proteins. In Table 2, we have highlighted the hub genes which were presented in those three clusters.
Table 2.
Identification of gene clusters (C1–C3) of M. catarrhalis from the PPI network
| Cluster | Score (density) | Nodes | Edges | Gene name |
|---|---|---|---|---|
| C1 | 25.517 | 11 | 192 | rpsJ, rpoC, dnaK, atpD, atpA, atpA, fusA, groEL, rplF, rplJ, rpsL. |
| C2 | 2 | 3 | 8 | mdh, gyrB, ileS. |
| C3 | 2 | 2 | 4 | SucB, fumC. |
Note: Bold symbol indicates the potential hub genes.
Figure 5.

Hub gene identification using six Cytohubba plugins in Cytoscape. (A) Stress, (B) Betweenness, (C) Closeness, (D) Degree, (E) MNC and (F) MCC. These hub genes are referred to as highly linked fundamental nodes in a large-scale-free PPI network that includes diverse functional partners that combine various network components. Color gradients indicate the higher to lower value from red to yellow.
Figure 6.

Interpretation of JVENN analysis. The overlapped region comprises five genes (rpoB, atpA, fusA, groEL and rpoL) common among hub genes gathered using six Cytohubba approaches (Stress, Betweenness, Closeness, Degree, MNC and MCC).
Table 3.
Topological characteristics of unique hub genes
| Gene Name | MCODE Cluster | Stress | Betweenness | Closeness | Degree | MNC | MCC |
|---|---|---|---|---|---|---|---|
| rpoB | NIC | 8824 | 249.4637201 | 28.58333333 | 38 | 19 | 49 842 |
| dnaK | C1 | 8640 | 181.5186545 | 24.16666667 | 22 | 10 | 5769 |
| atpA | C1 | 5896 | 197.1844322 | 28.33333333 | 38 | 19 | 47 752 |
| clpP | NIC | 5584 | 188.6512432 | 22 | 14 | 5 | 12 |
| fusA | C1 | 5584 | 138.2553313 | 26.91666667 | 32 | 15 | 50 427 |
| mdh | C2 | 5320 | 153.4120443 | 23.3333 | 20 | 9 | 173 |
| atpD | C1 | 4904 | 115.8893319 | 26.08333333 | 30 | 15 | 6676 |
| groEL | C1 | 4880 | 106.0333377 | 26.66666667 | 30 | 15 | 47 654 |
| rpsL | C1 | 4512 | 122.0596986 | 25.66666667 | 30 | 14 | 43 939 |
| thyA | NIC | 4200 | 103.3791209 | 20.5 | 12 | 4 | 10 |
| rpoC | C1 | 3072 | 75.43951 | 25.91666667 | 30 | 15 | 49 706 |
| gyrB | C2 | 3512 | 66.69975 | 25.41666667 | 26 | 13 | 8070 |
| gyrA | NIC | 2476 | 54.03081 | 24.58333333 | 24 | 12 | 2168 |
| rpsJ | C1 | 400 | 8.7243 | 23.3333 | 22 | 11 | 41 784 |
| rplF | C1 | 224 | 3.08944 | 23 | 20 | 10 | 41 784 |
| rplJ | C1 | 184 | 3.00269 | 22.83333 | 20 | 10 | 41 760 |
Functional enrichment analysis
Functional enrichment was performed by employing unique AMR genes. In this present study, GO and KEGG pathway findings revealed some biological, cellular and molecular processes as well as some multidrug susceptible mechanisms that were strongly linked with AMR genes of M. catarrhalis. In this case, the outputs of GO enrichment provided a biological process (BP) that was significantly correlated with cellular metabolic process, cellular biosynthetic process, organic substance metabolic process, primary metabolic process, Cellular nitrogen compound biosynthetic, etc. MFs enriched with catalytic activity, organic cyclic compound binding, heterocyclic compound binding, anion binding and purine ribonucleoside triphosphate binding. Furthermore, intracellular, cytoplasm, cellular anatomical entity and tricarboxylic acid cycle enzyme complex principally involved with the cellular component. Meanwhile, the KEGG pathway enrichment study discovered that metabolic pathways, citrate cycle (TCA cycle), RNA degradation, carbon metabolism, biosynthesis of secondary metabolites, etc., were substantially related to the AMR genes (Figure 7). Furthermore, Tables 4 and 5 and Table S2, available online at http://bib.oxfordjournals.org/, provided the tabular representation of the GO and KEGG pathway.
Figure 7.

Functional Enrichment analysis (Gene Ontology and KEGG pathway) of AMR genes. The top 24 GO keywords and top 6 KEGG functional pathways are shown by the study of AMR genes. In the bubble plot, circular-shaped indicates BP; triangle-shaped represents cellular component (CP); plus sign indicates MF and square-shaped indicates KEEG pathway.
Table 4.
Most significant relevant pathway of each GO term of AMR genes
| Category | GO ID | GO Terms |
|---|---|---|
| BP | GO:0008152 | Metabolic process |
| GO:0044237 | Cellular metabolic process | |
| GO:0071704 | Organic substance metabolic process | |
| GO:0009987 | Cellular process | |
| GO:0044249 | Cellular biosynthetic process | |
| GO:1901576 | Organic substance biosynthetic process | |
| GO:0044238 | Primary metabolic process | |
| GO:0044281 | Small molecule metabolic process | |
| GO:0044271 | Cellular nitrogen compound biosynthetic process | |
| GO:0034641 | Cellular nitrogen compound metabolic process | |
| Molecular function | GO:0036094 | Small molecule binding |
| GO:0003824 | Catalytic activity | |
| GO:0097159 | Organic cyclic compound binding | |
| GO:1901363 | Heterocyclic compound binding | |
| GO:0043168 | Anion binding | |
| GO:0000166 | Nucleotide binding | |
| GO:0005488 | Binding | |
| GO:0017076 | Purine nucleotide binding | |
| GO:0043167 | Ion binding | |
| GO:0035639 | Purine ribonucleoside triphosphate binding | |
| Cellular function | GO:0005622 | Intracellular |
| GO:0005737 | Cytoplasm | |
| GO:0110165 | Cellular anatomical entity | |
| GO:0045239 | Tricarboxylic acid cycle enzyme complex |
Table 5.
Most significant relevant pathway KEGG pathway analysis along with term ID and description
| Pathway | Term ID | Term description |
|---|---|---|
| KEGG | mct01100 | Metabolic pathways |
| mct00020 | Citrate cycle (TCA cycle) | |
| mct03018 | RNA degradation | |
| mct01200 | Carbon metabolism | |
| mct01110 | Biosynthesis of secondary metabolites | |
| mct03010 | Ribosome |
Genes correlation with resistance mechanisms and drug class analysis
We used a sunburst plot to identify the genes associated with resistance mechanisms and drug classes in M. catarrhalis. According to the plot, various M. catarrhalis strains contain many genes and employ various mechanisms and techniques to increase their resistance capacities against various multiple medications. Antibiotic inactivation, antibiotic target replacement and alteration, reduced permeability to antibiotics and antibiotic efflux pump are some of the resistance mechanisms employed by M. catarrhalis. Figure 8A and B indicates the different types of resistance mechanisms and drug classes exploited by M. catarrhalis, as well as their numbers. Antibiotic target replacement mechanism is regulated by fabG-1 gene, antibiotic target alteration is regulated by ICR-Mc and antibiotic efflux pumps mechanism regulated by eptA, pgsA and OxyR genes. Furthermore, we observed that M. catarrhalis has resistance activity in a variety of antibiotics, including beta-lactam, peptide, penam, tetracyclines, rifamycins, aminoglycosides, fosfomycin and others, according to the CARD and PATRIC databases (Table S3 available online at http://bib.oxfordjournals.org/). Figure 9A–C represents the whole relationship between the genes with resistance mechanisms and drug classes.
Figure 8.
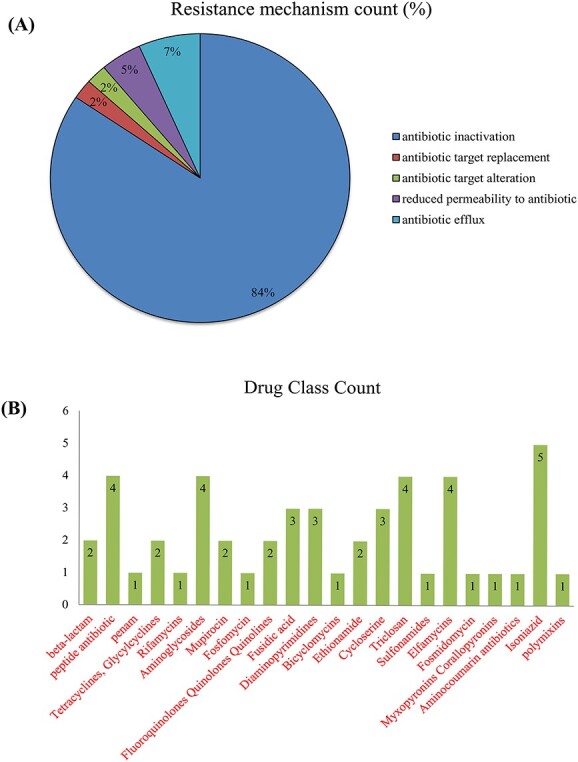
Moraxella catarrhalis significantly utilize several kinds of AMR mechanisms and drug classes. (A) Resistance mechanisms count and (B) Drug Class count.
Figure 9.

Sunburst plots were used to build interactions between different gene classes, drug classes and resistance mechanisms in networks. (A) The relationship between resistance mechanisms corresponding to their genes. (B) The relationship between drug classes corresponding to their genes. (C) The relationship between resistance mechanism and drug class.
Discussion
AMR is the inability of microbes to adapt to various antibiotics, which makes it more difficult to prevent infectious diseases and also raises the overall probability of disease transmission, serious morbidity and fatality. Several AMR genes perform a vital function in developing this resilience to several anti-disease medications in bacterial pathogens. Currently, it has become a persistent danger to our capacity to cure prevalent diseases due to the formation and transmission of drug-resistant bacteria. In the current work, we have explored the AMR system in the pathogenic bacterial strain M. catarrhalis. Moraxella catarrhalis is typically detected in humans as a symbiotic and mucosal parasite that might cause OM, chronic obstructive pulmonary disease (COPD), ocular infection, sinusitis as well as infrequently laryngitis in humans. This bacterium has shown that it can block the passage of drugs into the cell by a variety of distinct pathways, demonstrating that it has various strategies that it has adapted to do so. As previously mentioned, we gathered 288 AMR genes from the strains of M. catarrhalis via several databases; then, we conducted phylogenetic analysis, protein interaction network analysis, clustering analysis, hub protein identification and functional enrichment analysis of AMR genes, to determine the drug resistance mechanisms. In addition to this, we made an approach to identify how AMR genes are linked to diverse antibiotic resistance strategies as well as different categories of antibiotics.
Environmental microorganisms, including other species on the earth, are vulnerable to the processes of molecular diversity. Genome sequencing provides information on the molecular diversity of organisms through the analysis of those sequences. More than that, significant perspectives on microbiological as well as pragmatic applications such as disease identification might be gained by integrating the DNA sequencing of microorganisms and developmental modeling with phylogenetic studies [54]. The phylogenetic analysis of the data from our current investigation indicated the link between the M. catarrhalis strains. The strain CCRI-195ME of M. catarrhalis, which was considered a reference sequence, was highly correlated with other strains found in GenBank such as M. catarrhalis FDAARGOS 304, M. catarrhalis NCTC11020, M. catarrhalis 142P87B1, M. catarrhalis 74P58B1, M. catarrhalis 46P58B1 and M. catarrhalis BBH18. Among these strains, M. catarrhalis CCRI-195ME was the first fully sequenced genome with the modM3 allele, derived from the inner ear of a 16-month aged infant predisposed to OM [55]. About 80% of the total children have experienced a minimum of one episode of OM by the time they are 3 years old, resulting in the most frequent pediatric illnesses [56]. Additionally, this bacterium is currently regarded to be the 2nd highest prevalent reason for the deterioration in COPD [57]. Unfortunately, the specific function of bacteria is not well known and is a contentious topic. Although the importance of developing a vaccine that is effective against M. catarrhalis, none of the contenders have advanced to the medical testing stage. Because of this, we must interpret the diversity of M. catarrhalis to develop an improved insight of the epidemiological data as well as the propagation of genes involved in pathogenicity aspects, which will assist in the development of vaccines [57].
Furthermore, the PPI network that was analyzed in this study showed that the genes folE, FabF, argG, RocD and fabG are all connected to neighboring nodes in the network. Previous findings have suggested that the folE motif might act as a potent riboswitch for the structural intergenic RNAs found in microbial noncoding regions [58]. This folE motif, which is typically located on the proximal of folE genes, follows a straightforward construction [59]. Furthermore, these genes encode enzyme that catalyzes the initial stage of the de novo folate manufacturing process in bacteria [58]. FabF and fabG perform a critical part in the bacterial process of fatty acid production. For the discovery of novel antibacterial drugs, the fatty acid production route is substantially underutilized. Initially, FabH catalyzes the condensation process between acetyl CoA and malonyl-acetyl carrying molecule to create acetoacetyl-ACP, which is crucial for the commencement of fatty acid biosynthesis [60]. In this cycle, FabF generates -ketoacyl-ACP, which boosts the rate of fatty acid biosynthesis of different lengths for activation by the microorganism [61, 62]. But by producing butyryl-ACP, fabG reduces the rate of fatty acid biosynthesis [60]. Other side, argG gene, which encodes argininosuccinate synthetase enzyme, is required in arginine biosynthesis in bacteria [63]. Arginine performs a significant function in the metabolic process of M. catarrhalis [64]. In Bacillus subtilis, a gene named rocD was discovered adjacent the rocR gene which was also present in M. catarrhalis [65]. The rocD gene produces an enzyme that is structurally analogous to eukaryotic ornithine aminotransferases [66]. Therefore, these genes perform many crucial roles in the metabolic, cellular and BPs that take place in microorganisms. In our investigation, GO keywords relating to antibiotic-resistant processes including BPs, cellular components and molecular activities were significantly elevated. BPs were mostly related to the cellular metabolic process (GO:0044237), organic substance metabolic process (GO:0071704), cellular biosynthetic process (GO:0044249), organic substance cellular biosynthetic process (GO:0044249), organic substance biosynthetic process (GO:1901576), primary metabolic process (GO:0044238), small molecule metabolic process (GO:0044281), cellular nitrogen compound biosynthetic process (GO:0044271) and cellular nitrogen compound metabolic process (GO:0034641). Typically, bacteria’s cellular metabolic process determines their resistance to antibiotics, so drug potency could be increased by modifying bacteria’s metabolic condition [67]. Additionally, disruptions in bacterial metabolic equilibrium have substantial side effects on therapy concerning drugs [67]. On the other hand, the primary metabolic process entails biochemical events and mechanisms which generate substances during regular anabolic and catabolic events, and the organic substance metabolic process entails the series of biochemical events that an organic matter, which can be thought of as like any molecule or other unit that contains carbon, is involved in [68, 69].
Remarkably, both the cellular nitrogen compound biosynthetic process and the cellular nitrogen compound metabolic process are implicated in the formation of nitrogen, a crucial component for the production of, amino acids, proteins, different types of enzymes, DNA and RNA in all microorganisms [70]. Among the cellular function terms cellular anatomical entity (GO:0110165), tricarboxylic acid cycle enzyme complex (GO:0045239), cytoplasm (GO:0005737), etc., were mostly enriched. Similarly, the following MFs were found to be accumulated: small molecule binding (GO:0036094), catalytic activity (GO:0003824), organic cyclic compound binding (GO:1901363), nucleotide binding (GO:0000166), organic cyclic compound binding (GO:0097159), heterocyclic compound binding (GO:1901363) anion binding (GO:0043168), purine nucleotide binding (GO:0017076), purine ribonucleoside triphosphate binding (GO:0035639) and ion binding (GO:0043167). Currently, nanomaterial-based therapeutics are intriguing methods for combating complicated bacterial infestations, as they can circumvent established processes linked with accumulated antibiotic resistance [71]. Moreover, heterocyclic compounds, such as thiazole, benzothiazole and thiazolidinone, have been produced over the previous decades in an effort to acquire novel antibiotics capable of treating conditions triggered by antimicrobial resistant bacterial strains [72]. We also uncovered KEGG pathways associated with the citrate cycle (TCA cycle), RNA degradation, carbon metabolism and secondary metabolite production. In microorganisms, RNA can be degraded via multiple processes. Controlling gene expression relies heavily on RNA synthesis and degradation, which plays an important part in the molecular process [73]. Additionally, carbon metabolism is essential for bacterial proliferation [74]. Since bacteria cannot manufacture their food, they must rely on the source of carbon for the synthesis of energy and metabolic substances [75]. These substances are required for the production of anabolic subunits, which are then transformed into polymers, including organic molecules (proteins, nucleotides), as well as elements of the complicated cell membrane [75, 76]. Mainly the citrate cycle also referred to as the Krebs cycle, is the fundamental route through which cells obtain their supply of energy and is an essential component of cellular breathing. This cycle is also employed as the foundation for secondary metabolite synthesis because its byproducts are utilized as substrates in the production of metabolites; numerous molecular systems are linked to the CAC, forming a biochemical circuit [77].
Aforementioned, we identified three important clusters, which are referred to as C1, C2 and C3, and also detected hub proteins using six Cytohubba plugin approaches. In addition, we detected a total of 11 hub genes that were unique among six approaches including rpoB, dnaK, atpA, clpP, fusA, mdh, atpD, groEL, rpsL, thyA, rpoC, gyrB, gyrA, rpsJ, rplF and rplJ. Among these genes, dnaK, atpA, fusA, atpD, groEL, rpsL, rpoC, rpsJ, rplF and rplJ were present in C1 as well as mdh and gyrB were present in C2. We found that these hub genes significantly correlated with various types of drug resistance mechanisms along with drug classes. These mechanisms are carried on by changes to the drug, changes to the antimicrobial targets, restricted access to the target, or even a mixture of these processes [78–80]. Because of its capacity to manufacture BRO-lactamase, M. catarrhalis is reported to be resistant to penicillin as well as the foundation follows cephalosporins; nevertheless, it is normally sensitive to additional drugs, notably fluoroquinolones [81, 82]. However, in our analysis, we found some significant resistance mechanisms such as antibiotic inactivation, antibiotic target replacement, drug target alteration, reduced permeability to antibiotics, and antibiotic efflux pump. Antibiotic inactivation was carried out by 36 genes (fabI, inhA, gyrB, kasA, rpoB, rpsJ, rpoC, folA, S10p, fabF, gidB, gyrA, ileS, murA, rho, rplF, fusA, rpsL, tuf, tufA_1, tufA_2, folP, BRO-1, Ddl, Dfr, EF-G, EF-Tu, fabG, Iso-tRNA, OxyR, S12p, alr, bro-2, ddlB, dfrA and dxr). These genes inactivated the antibiotics including isoniazid, triclosan, diaminopyrimidines, aminoglycosides, ethionamide, peptide antibiotics, tetracyclines, fluoroquinolones, fosfomycin, penam, rifamycins, sulfonamide, beta-lactam antibiotics, etc. Antibiotic target replacement and alternation were performed by fabG_1 and ICR-Mc, respectively. They showed resistance to triclosan and peptide antibiotic. Moreover, PgsA and rsmG diminished the permeability to antibiotics. As a result, they exhibit resistance to peptide and aminoglycoside antibiotics. Then, PgsA, OxyR and eptA possessed antibiotic efflux pumps that were resistant to the drugs isoniazid and polymixins. Various studies concluded that the presence of efflux pumps, which potentially provide resistance to macrolides, b-lactams, macrolides, tetracyclines, aminoglycosides and fluoroquinolones, is common in MDR microorganisms [83, 84].
To prevent the propagation of the infection, prompt detection with appropriate management is crucial. Singpanomchai et al. [85] and Guitor [86] have indicated in their studies that infections that are prominent in respiratory regions cause significant rpoB gene alterations in RNA polymerase during RNA production, which is associated with rifamycin susceptibility. Mujawar et al. [87] revealed that DnaK is essential for antibiotic resistance through a variety of assessments using microbial extracts. The nucleotide alterations from cytosine to adenine on DnaK gene were identified by the SNPs discovered in the individual samples that potentially caused protein denaturing [87]. In the same way, the DnaK-GroEL interaction may play a significant contribution to antibiotic resistance in pathogenic microorganisms [88]. The GyrA and GyrB genes encode DNA gyrase enzyme that contributes to the key methods of quinolone (QN) susceptibility and comprises a reduction in interaction propensity to QNs caused by amino acid change in the QRDR (quinolone resistance-determining region) [89]. On the other hand, through the evaluation of the gene’s genomic structure, many researchers demonstrated in their paper that the rpsL, ClpP, rpsJ and rplJ function as potential antibiotic resistance genes [90–94].
In contrast, the major cause of antibiotic resistance may also be mutations. The terminology ‘mutation rate’ is used in investigations of biological evolvement to provide estimates of the percentage of genetic mutation for every nucleotide, for every region or subsequently for the entire genotype. Here, preferentially beneficial, detrimental or neutrality changes are all taken into consideration. The probability in which identifiable mutations appear in a microbial species that exposed to a specific therapeutic dose shows how the evolution rate is usually characterized in the perspective of antimicrobial sensitivity. Numerous investigations have demonstrated that even an one amino acid change can result in the development of beta-lactam resistance in isolates that are extremely sensitive to penicillin have additional penicillin-binding proteins changes than bacteria that are intermediately resistant [95]. Furthermore, Jacobs and Micheael R showed in their study the activation of an erythromycin ribosomal methylation gene leads in transcription factors alteration of 23S ribosomal RNA, which prevents the macrolide from attaching to the ribosome [96]. They discovered that it is the source of the majority of pneumococcal macrolide susceptibility. However, many clinical studies have revealed that M. catarrhalis isolates are extremely sensitive to macrolides. Even several studies have found that a mutation in the TonB-dependent receptor expressing gene MCR 0492 may be linked to macrolides susceptibility in M. catarrhalis strains [97]. Kasai et al. [98] used naturally occurring erythromycin-resistant variants to study the impact of alterations in the 23S rRNA gene and also the L4 and L22 ribosomal subunits. Additionally, the development of rising macrolide-susceptibility M. catarrhalis may be attributed to the A2330T Mutation in the 23S rRNA gene [99]. Therefore, we may conclude that alterations in 23S rRNA are the major cause of macrolide resistance. On the other hand, fluoroquinolone resistance is induced by significant mutations in the DNA gyrase enzymes, which can potentially be induced by the emergence of efflux pumps within the bacterium [100]. Warner et al. [101] attempted to demonstrate in their work that therapeutically significant mutations that induce derepression of the Neisseria gonorrhoeae MtrC-MtrD-MtrE Efflux pump system provide various rates of antimicrobial sensitivity. Likewise, mutation in Mycobacterium tuberculosis may be the cause of the individual’s inherent resistance to numerous antibiotics, which reduces the amount of drugs that are accessible for therapy [102]. Antibiotic resistance mutations may have cytoprotective impacts, resulting in a decrease in bacterial viability, as measured, for example, by a decrease in in laboratory multiplication efficiency.
Thus, our findings provide a comprehensive portrayal of antibiotic resistance mechanisms as well as the relevant entities participating in the interaction network. We believe that these observations will offer clarity on the underlying process that leads to AMR in M. catarrhalis. In near future, we will be validated these antibiotic susceptibility patterns in wet lab experiments. Firstly, we will collect sample from patients or pure culture in hospitals or clinic. Secondly, we will analyze bacterial growth curve for the test of antibiorgram susceptibility patterns and then validate the gene expression patterns (potential hub gene) through Polymerase Chain Reaction analysis. Finally, we will sequence the potential bacteria based on 16sRNA analysis for the identification of possible new bacterial strains.
Conclusion
Antibiotic resistance in virulent bacteria is a prominent cause for concern all around the world. It is a persistent issue in the medical sector. In this work, we investigated the AMR system in the virulent strain of M. catarrhalis using genomic interaction system biology. The AMR genes, in conjunction with their functional interacting partners, mostly manifest their antibiotic resistance through the inactivation of antibiotics, target replacement of antibiotics, target alteration, reduced permeability to antibiotics and also antibiotic efflux pump. Various antibiotics, notably beta-lactam, tetracyclines, glycylcyclines, rifamycins, aminoglycosides and fosfomycin are sensitive to these mechanisms. In addition, the clustering approach uncovered gene sets that are intricately linked to one another. The genes rpoB, atpA, fusA, groEL and rpoL have quite a significant contact as well as can be more important for figuring out how the AMR genes work together at the genomic level. Therefore, we believe that the findings that we have provided in this study will provide researchers with a solid foundation upon which to build their investigations into novel therapeutic approaches for the management of M. catarrhalis epidemics. We have analyzed only complete annotation of GenBank files datasets. In addition, we have searched three antibiotic resistance database, while several databases are available. There is also a lack of systematic antibiotic resistance gene collection predicted by different databases and further evaluate the reliability of wet lab experiment.
Key Points
The fundamental underpinnings of antibiotic susceptibility in the pathogenic bacterial strain Moraxella catarrhalis were discovered using genomic interaction study relying on system biology.
The PPI network uncovered important hub-proteins that might be explored to develop potential pharmacological candidates against antimicrobial-resistant bacteria.
Cluster analysis was used to generate useful and long-lasting groupings of similar factors for biological characterization and assessment that coincided with resistance mechanisms along with corresponding drug classes.
Functional enrichment was carried out to identify several biological processes, cellular functions and molecular processes as well as some multidrug-sensitive mechanisms which were closely associated with AMR genes of M. catarrhalis.
The molecular connections underlying mechanisms of the AMR genes were investigated in this study, which is important in the creation of new and efficient treatments for infectious diseases.
Supplementary Material
Acknowledgements
We would like to thank the team members of the Center for Advanced Bioinformatics Artificial Intelligent Research headed by Dr Md Habibur Rahman who helped us and provided Valuable insight into the research.
Author Biographies
Sadia Afrin Bristy currently works in Bioinformatics and Biomedical Research Network of Bangladesh, Dhaka, Bangladesh. Her research interests include computational biology, system biology, drug design and bioinformatics.
Md. Arju Hossain is a thesis student (Master of Science) of Biotechnology and Genetic Engineering, Mawlana Bhashani Science and Technology University. He focuses on his study innumerous areas, including System Biology, Bioinformatics, Pharmacogenomics, Immunology, Metabolomics and Microbiology.
Dr. Md Habibur Rahman has received his B.Sc in Computer Science and Engineering (CSE) from the Dept. of Computer Science and Engineering, Islamic University, Kushtia-7003, Bangladesh in 2006. Later on, he has finished his M.Sc. in CSE from the same University in 2007. Recently, he has received his PhD in Pattern Recognition and Intelligent Systems from The University of Chinese Academy of Sciences, Beijing, China in 2020. His research interest encompasses Bioinformatics, Computational Biology, Machine Learning, Deep Learning and Drug Discovery. He is currently working as an Associate Professor at the Dept. of CSE, Islamic University, Kushtia-7003, Bangladesh. He is also investigating some projects as principal investigator and co-investigator and supervising several undergraduate and post-graduate students. He has presented several papers in peer-reviewed journals and has published numerous studies in science cited journals. He was an Awardee of the CAS-TWAS Presidential Fellowship, from 2016 to 2020.
Md Imran Hasan is a graduate student of the Department of Computer Science and Engineering, Islamic University. His research priorities include Bioinformatics, Machine learning, Deep learning, Medical Imaging, and Networking Dr. S. M. Hasan Mahmud received a Ph.D. degree in Computer Science and Technology from the Universityof Electronic Science and Technology of China, China. He received his B.Sc.degree in software engineering from the Shenyang University of Chemical Technology, China, and his M.Sc. degree in software engineering from Hohai University, China. He is working as an Assistant professor with the Department of Computer Science, American International University Bangladesh (AIUB). Besides he is also a research scientist at Mahidol University, Thailand. Before joining AIUB, he served as a Lecturer in the Software Engineering department at Daffodil International University, Bangladesh. He has published many research articles in reputed journals and conferences. His research interests include machine learning, deep learning, bioinformatics, drug discovery, and pattern recognition. He received several Best Paper Awards from the IEEE Conferences.
Dr Mohammad Ali Moni holds a PhD in Artificial Intelligence & Clinical Informatics in 2015 from the University of Cambridge, UK followed by postdoctoral training at the University of New South Wales, University of Sydney Vice-chancellor fellowship, and Senior Data Scientist at the University of Oxford. Dr Moni then joined as a research senior lecturer University of Queensland in 2021. He is an Artificial Intelligence, Computer Vision & Machine learning, Digital Health Data Science, Health Informatics and Bioinformatics researcher developing interpretable and clinical applicable machine learning and deep learning models to increase the performance and transparency of AI-based automated decision making systems. He has published over 200 journal articles in many top tier journals.
Contributor Information
Sadia Afrin Bristy, Bioinformatics and Biomedical Research Network of Bangladesh, Dhaka, Bangladesh.
Md Arju Hossain, Biotechnology and Genetic Engineering, Mawlana Bhashani Science and Technology University, Tangail, Bangladesh.
Md Imran Hasan, Department of Computer Science and Engineering, Islamic University, Kushtia 7003, Bangladesh.
S M Hasan Mahmud, Department of Computer Science, American International University, Dhaka, Bangladesh.
Mohammad Ali Moni, School of Health and Rehabilitation Sciences, Faculty of Health and Behavioural Sciences, The University of Queensland, St Lucia, QLD 4072, Australia.
Md Habibur Rahman, Department of Computer Science and Engineering, Islamic University, Kushtia 7003, Bangladesh; Center for Advanced Bioinformatics and Artificial Intelligent Research, Islamic University, Kushtia 7003, Bangladesh.
Data Availability Statement
The used datasets are publicly available and the research data can be accessible on request.
Funding
There is no funding of interest.
Author Contribution
Conceptualization: Md Habibur Rahman, Md. Arju Hossain; Investigation: Sadia Afrin Bristy, Md Imran Hasan, Md. Arju Hossain; Writing (Original Draft): Sadia Afrin Bristy; Writing (Review and Editing): Md Habibur Rahman and Md. Arju Hossain. Supervision: Mohammad Ali Moni and Md Habibur Rahman.
References
- 1. Cassir N, Rolain JM, Brouqui P. A new strategy to fight antimicrobial resistance: the revival of old antibiotics. Front Microbiol 2014;5:551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brusselaers N, Vogelaers D, Blot S. The rising problem of antimicrobial resistance in the intensive care unit. Ann Intensive Care 2011;1(1):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Karam G, Chastre J, Wilcox MH, Vincent JL. Antibiotic strategies in the era of multidrug resistance. Crit Care 2016;20(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ren D, Pichichero ME. Vaccine targets against Moraxella catarrhalis. Expert Opin Ther Targets 2016;20(1):19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Verduin CM, Hol C, Fleer A, et al. Moraxella catarrhalis: from emerging to established pathogen. Clin Microbiol Rev 2002;15(1):125–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davie JJ, Earl J, de Vries SP, et al. Comparative analysis and supragenome modeling of twelve Moraxella catarrhalis clinical isolates. BMC Genomics 2011;12(1):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goldstein EJ, Murphy TF, Parameswaran GI. Moraxella catarrhalis, a human respiratory tract pathogen. Clin Infect Dis 2009;49(1):124–31. [DOI] [PubMed] [Google Scholar]
- 8. Aebi C. Moraxella catarrhalis–pathogen or commensal? In: Hot Topics in Infection and Immunity in Children VII, Advances in Experimental Medicine and Biology, New York, NY: Springer, 2011, 697:107–16. [DOI] [PubMed] [Google Scholar]
- 9. Alnahas S, Hagner S, Raifer H, et al. IL-17 and TNF-α are key mediators of Moraxella catarrhalis triggered exacerbation of allergic airway inflammation. Front Immunol 2017;8:1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Berman S. Otitis media in developing countries. Pediatrics 1995;96(1):126–31. [PubMed] [Google Scholar]
- 11. Shaheen MM, Raquib A, Ahmad SM. Prevalence and associated socio-demographic factors of chronic suppurative otitis media among rural primary school children of Bangladesh. Int J Pediatr Otorhinolaryngol 2012;76(8):1201–4. [DOI] [PubMed] [Google Scholar]
- 12. de Vries SP, Eleveld MJ, Hermans PW, Bootsma HJ. Characterization of the molecular interplay between Moraxella catarrhalis and human respiratory tract epithelial cells. PloS one. 2013;8(8):e72193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ferretti S, Bonneau O, Dubois GR, et al. IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J Immunol 2003;170(4):2106–12. [DOI] [PubMed] [Google Scholar]
- 14. Bakaletz LO. Bacterial biofilms in otitis media: evidence and relevance. Pediatr Infect Dis J 2007;26(10):S17–9. [DOI] [PubMed] [Google Scholar]
- 15. Post JC, Hiller NL, Nistico L, et al. The role of biofilms in otolaryngologic infections: update 2007. Curr Opin Otolaryngol Head Neck Surg 2007;15(5):347–51. [DOI] [PubMed] [Google Scholar]
- 16. Hong W, Mason K, Jurcisek J, et al. Phosphorylcholine decreases early inflammation and promotes the establishment of stable biofilm communities of nontypeable Haemophilus influenzae strain 86-028NP in a chinchilla model of otitis media. Infect Immun 2007;75(2):958–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jurcisek J, Greiner L, Watanabe H, et al. Role of sialic acid and complex carbohydrate biosynthesis in biofilm formation by nontypeable Haemophilus influenzae in the chinchilla middle ear. Infect Immun 2005;73(6):3210–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jefferson KK, Goldmann DA, Pier GB. Use of confocal microscopy to analyze the rate of vancomycin penetration through Staphylococcus aureus biofilms. Antimicrob Agents Chemother 2005;49(6):2467–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ceri H, Olson ME, Stremick C, et al. The Calgary biofilm device: new technology for rapid determination of antibiotic susceptibilities of bacterial biofilms. J Clin Microbiol 1999;37(6):1771–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kyd MJ, McGrath J, Krishnamurthy A. Mechanisms of bacterial resistance to antibiotics in infections of COPD patients. Curr Drug Targets 2011;12(4):521–30. [DOI] [PubMed] [Google Scholar]
- 21. Perez AC, Pang B, King LB, et al. Residence of Streptococcus pneumoniae and Moraxella catarrhalis within polymicrobial biofilm promotes antibiotic resistance and bacterial persistence in vivo. Pathog Dis 2014;70(3):280–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Narinesingh SP, Whitby DJ, Davenport PJ. Moraxella catarrhalis: an unrecognized pathogen of the oral cavity? Cleft Palate Craniofac J 2011;48(4):462–4. [DOI] [PubMed] [Google Scholar]
- 23. Hoban D, Felmingham D. The PROTEKT surveillance study: antimicrobial susceptibility of Haemophilus influenzae and Moraxella catarrhalis from community-acquired respiratory tract infections. J Antimicrob Chemother 2002;50(suppl_2):49–59. [DOI] [PubMed] [Google Scholar]
- 24. Jetter M, Heiniger N, Spaniol V, et al. Outer membrane porin M35 of Moraxella catarrhalis mediates susceptibility to aminopenicillins. BMC Microbiol 2009;9(1):1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Z, Yang Z, Xiang X, et al. Mutation of TonB-dependent receptor encoding gene MCR_0492 potentially associates with macrolides resistance in Moraxella catarrhalis isolates. Infect Drug Resist 2022;15:2419–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen L, Zhang YH, Wang S, et al. Prediction and analysis of essential genes using the enrichments of gene ontology and KEGG pathways. PloS one 2017;12(9):e0184129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Berk SL, Kalbfleisch JH, Chemotherapy APCGJJOA . Antibiotic susceptibility patterns of community-acquired respiratory isolates of Moraxella catarrhalis in Western Europe and in the USA. Journal of Antimicrobial Chemotherapy 1996;38(Suppl_A):85–96. [DOI] [PubMed] [Google Scholar]
- 28. Sun X, Zhang B, Xu G, et al. In vitro activity of the novel tetracyclines, tigecycline, eravacycline, and omadacycline, against Moraxella catarrhalis. Ann Lab Med 2021;41(3):293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miryala SK, Anbarasu A, Ramaiah S. Discerning molecular interactions: a comprehensive review on biomolecular interaction databases and network analysis tools. Gene 2018;642:84–94. [DOI] [PubMed] [Google Scholar]
- 30. Anitha P, Anbarasu A, Ramaiah S. Gene network analysis reveals the association of important functional partners involved in antibiotic resistance: a report on an important pathogenic bacterium Staphylococcus aureus. Gene 2016;575(2):253–63. [DOI] [PubMed] [Google Scholar]
- 31. Anitha P, Bag S, Anbarasu A, Ramaiah S. Gene and protein network analysis of AmpC β lactamase. Cell Biochem Biophys 2015;71(3):1553–67. [DOI] [PubMed] [Google Scholar]
- 32. Gonzalez MW, Kann MG. Chapter 4: protein interactions and disease. PLoS Comput Biol 2012;8(12):e1002819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bag S, Ramaiah S, Anbarasu A. fabp4 is central to eight obesity associated genes: a functional gene network-based polymorphic study. J Theor Biol 2015;364:344–54. [DOI] [PubMed] [Google Scholar]
- 34. Tatusova T, DiCuccio M, Badretdin A, et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 2016;44(14):6614–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McArthur AG, Waglechner N, Nizam F, et al. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother 2013;57(7):3348–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jia B, Raphenya AR, Alcock B, et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res 2016;45:gkw1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wattam AR, Abraham D, Dalay O, et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res 2014;42(D1):D581–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zankari E, Hasman H, Cosentino S, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 2012;67(11):2640–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sun F, Mu C, Kwok HF, et al. Capivasertib restricts SARS-CoV-2 cellular entry: a potential clinical application for COVID-19. Int J Biol Sci 2021;17(9):2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bardou P, Mariette J, Escudié F, et al. jvenn: an interactive Venn diagram viewer. BMC Bioinformatics 2014;15(1):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kapli P, Yang Z, Telford MJ. Phylogenetic tree building in the genomic age. Nat Rev Genet 2020;21(7):428–44. [DOI] [PubMed] [Google Scholar]
- 42. Kumar S, Stecher G, Li M, et al. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 2018;35(6):1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bristy SA, Islam AH, Andalib KS, et al. Determination of molecular signatures and pathways common to brain tissues of autism spectrum disorder: insights from comprehensive bioinformatics approach. Inform Med Unlocked 2022;29:100871. [Google Scholar]
- 44. Rahman MH, Rana HK, Peng S, et al. Bioinformatics and machine learning methodologies to identify the effects of central nervous system disorders on glioblastoma progression. Brief Bioinform 2021;22(5):bbaa365. [DOI] [PubMed] [Google Scholar]
- 45. Rahman MH, Peng S, Xiyuan H, et al. A network-based bioinformatics approach to identify molecular biomarkers for type 2 diabetes that are linked to the progression of neurological diseases. Int J Environ Res Public Health 2020;17(3):1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rahman MH, Peng S, Xiyuan H, et al. Bioinformatics methodologies to identify interactions between type 2 diabetes and neurological comorbidities. IEEE Access 2019;7:183948–70. [Google Scholar]
- 47. Rahman MR, Islam T, Huq F, et al. Identification of molecular signatures and pathways common to blood cells and brain tissue of amyotrophic lateral sclerosis patients. Inform Med Unlocked 2019;16:100193. [Google Scholar]
- 48. Rahman MH, Rana HK, Seng Peng M, et al. Bioinformatics and system biology approaches to identify pathophysiological impact of COVID-19 to the progression and severity of neurological diseases. Comput Biol Med 2021;138:104859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Van Hoek AH, Mevius D, Guerra B, et al. Acquired antibiotic resistance genes: an overview. Front Microbiol 2011;2:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lima TB, Pinto MF, Ribeiro SM, et al. Bacterial resistance mechanism: what proteomics can elucidate. FASEB J 2013;27(4):1291–303. [DOI] [PubMed] [Google Scholar]
- 51. Zelle JM. Python Programming: An Introduction to Computer Science. Franklin: Beedle & Associates, Inc., 2004. [Google Scholar]
- 52. Hasan MI, Hossain MA, Piplu Bhuiyan M, et al. A system biology approach to determine therapeutic targets by identifying molecular mechanisms and key pathways for type 2 diabetes that are linked to the development of tuberculosis and rheumatoid arthritis. Life Sci 2022;IF(5.037):120483. [DOI] [PubMed] [Google Scholar]
- 53. Holmes AH, Moore LS, Sundsfjord A, et al. Understanding the mechanisms and drivers of antimicrobial resistance. Lancet 2016;387(10014):176–87. [DOI] [PubMed] [Google Scholar]
- 54. Darling AE, Jospin G, Lowe E, et al. PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ 2014;2:e243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tan A, Blakeway LV, Bakaletz LO, et al. Complete genome sequence of Moraxella catarrhalis strain CCRI-195ME, isolated from the middle ear. Genome Announc 2017;5(21):e00384–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Morris DE, Osman KL, Cleary DW, Clarke SC. The characterization of Moraxella catarrhalis carried in the general population. Microbial Genomics 2022;8(5):000820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wilkinson TM, Aris E, Bourne S, et al. A prospective, observational cohort study of the seasonal dynamics of airway pathogens in the aetiology of exacerbations in COPD. Thorax 2017;72(10):919–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen X, Arachchilage GM, Breaker RR. Biochemical validation of a second class of tetrahydrofolate riboswitches in bacteria. RNA 2019;25(9):1091–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bertacine Dias MV, Santos JC, Libreros-Zúñiga GA, et al. Folate biosynthesis pathway: mechanisms and insights into drug design for infectious diseases. Future Med Chem 2018;10(8):935–59. [DOI] [PubMed] [Google Scholar]
- 60. Chung PY. Novel targets of pentacyclic triterpenoids in Staphylococcus aureus: A systematic review. Phytomedicine 2020;73:152933. [DOI] [PubMed] [Google Scholar]
- 61. Waldrop GL. Acetyl-CoA carboxylase as a target for antibacterial development. In: Antimicrobial Drug Discovery: Emerging Strategies, 2012, 208–19.
- 62. Fujita Y, Matsuoka H, Hirooka K. Regulation of fatty acid metabolism in bacteria. Mol Microbiol 2007;66(4):829–39. [DOI] [PubMed] [Google Scholar]
- 63. Schramm T, Lempp M, Beuter D, et al. High-throughput enrichment of temperature-sensitive argininosuccinate synthetase for two-stage citrulline production in E. coli. Metab Eng 2020;60:14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Otsuka T, Kirkham C, Brauer A, et al. The vaccine candidate substrate binding protein SBP2 plays a key role in arginine uptake, which is required for growth of Moraxella catarrhalis. Infect Immun 2016;84(2):432–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zaprasis A, Hoffmann T, Wünsche G, et al. Mutational activation of the RocR activator and of a cryptic rocDEF promoter bypass loss of the initial steps of proline biosynthesis in Bacillus subtilis. Environ Microbiol 2014;16(3):701–17. [DOI] [PubMed] [Google Scholar]
- 66. Gardan R, Rapoport G, Débarbouillé M. Expression of therocDEFOperon involved in arginine catabolism in Bacillus subtilis. J Mol Biol 1995;249(5):843–56. [DOI] [PubMed] [Google Scholar]
- 67. Stokes JM, Lopatkin AJ, Lobritz MA, Collins JJ. Bacterial metabolism and antibiotic efficacy. Cell Metab 2019;30(2):251–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Martínez I, Mohamed ME, Santos VE, et al. Metabolic and process engineering for biodesulfurization in gram-negative bacteria. J Biotechnol 2017;262:47–55. [DOI] [PubMed] [Google Scholar]
- 69. Manskaya SM, Drozdova TV. Geochemistry of Organic Substances: International Series of Monographs in Earth Sciences. V.I Vernedskii Institute of Geochemistry and Analytical Chemistry, USSR Academy of Science, Elsevier, 2013. [Google Scholar]
- 70. Carpinelli EC, Telatin A, Vitulo N, et al. Chromosome scale genome assembly and transcriptome profiling of Nannochloropsis gaditana in nitrogen depletion. Mol Plant 2014;7(2):323–35. [DOI] [PubMed] [Google Scholar]
- 71. Makabenta JM, Nabawy A, Li CH, et al. Nanomaterial-based therapeutics for antibiotic-resistant bacterial infections. Nat Rev Microbiol 2021;19(1):23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cascioferro S, Parrino B, Carbone D, et al. Thiazoles, their benzofused systems, and thiazolidinone derivatives: versatile and promising tools to combat antibiotic resistance. J Med Chem 2020;63(15):7923–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Arraiano CM, Andrade JM, Domingues S, et al. The critical role of RNA processing and degradation in the control of gene expression. FEMS Microbiol Rev 2010;34(5):883–923. [DOI] [PubMed] [Google Scholar]
- 74. Muñoz-Elías EJ, McKinney JD. Carbon metabolism of intracellular bacteria. Cell Microbiol 2006;8(1):10–22. [DOI] [PubMed] [Google Scholar]
- 75. Eisenreich W, Dandekar T, Heesemann J, Goebel W. Carbon metabolism of intracellular bacterial pathogens and possible links to virulence. Nat Rev Microbiol 2010;8(6):401–12. [DOI] [PubMed] [Google Scholar]
- 76. Ishii SI, Suzuki S, Norden-Krichmar TM, et al. Microbial population and functional dynamics associated with surface potential and carbon metabolism. ISME J 2014;8(5):963–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kumar P, Dubey KK. Citric acid cycle regulation: back bone for secondary metabolite production. InNew and future developments in microbial biotechnology and bioengineering. Elsevier 2019;165–81. [Google Scholar]
- 78. Ghannoum MA, Rice LB. Antifungal agents: mode of action, mechanisms of resistance, and correlation of these mechanisms with bacterial resistance. Clin Microbiol Rev 1999;12(4):501–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Aslam B, Wang W, Arshad MI, et al. Antibiotic resistance: a rundown of a global crisis. Infect Drug Resist 2018;11:1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Liu B, Pop M. ARDB—antibiotic resistance genes database. Nucleic Acids Res 2009;37(suppl_1):D443–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Saito R, Nonaka S, Fujinami Y, et al. The frequency of BRO β-lactamase and its relationship to antimicrobial susceptibility and serum resistance in Moraxella catarrhalis. J Infect Chemother 2014;20(1):6–8. [DOI] [PubMed] [Google Scholar]
- 82. Khan MA, Northwood JB, Levy F, et al. Bro β-lactamase and antibiotic resistances in a global cross-sectional study of Moraxella catarrhalis from children and adults. J Antimicrob Chemother 2010;65(1):91–7. [DOI] [PubMed] [Google Scholar]
- 83. Hernando-Amado S, Blanco P, Alcalde-Rico M, et al. Multidrug efflux pumps as main players in intrinsic and acquired resistance to antimicrobials. Drug Resist Updat 2016;28:13–27. [DOI] [PubMed] [Google Scholar]
- 84. Normark BH, Normark S. Evolution and spread of antibiotic resistance. J Intern Med 2002;252(2):91–106. [DOI] [PubMed] [Google Scholar]
- 85. Singpanomchai N, Akeda Y, Tomono K, et al. Rapid detection of multidrug-resistant tuberculosis based on allele-specific recombinase polymerase amplification and colorimetric detection. PloS one 2021;16(6):e0253235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Guitor AK, Wright GD. Antimicrobial resistance and respiratory infections. Chest 2018;154(5):1202–12. [DOI] [PubMed] [Google Scholar]
- 87. Mujawar S, El-Aal A, Ahmed AA, Lahiri C. Variant analysis from bacterial isolates affirms DnaK crucial for multidrug resistance. In: International Work-Conference on Bioinformatics and Biomedical Engineering, 2020. pp. 237–48. Springer, Cham. [Google Scholar]
- 88. Vanghele LM, Ionescu M, Coste H, Ganea E. Association of DnaK and GroEL with antimicrobial resistance in Salmonella Abortusovis. International Journal of Veterinary Medicine: Research & Reports 2016 (2016), 1–11.
- 89. Yamada K, Saito R. Molecular analysis of low-level fluoroquinolone resistance in clinical isolates of Moraxella catarrhalis. J Med Microbiol 2014;63(8):1066–70. [DOI] [PubMed] [Google Scholar]
- 90. Attia AS, Lafontaine ER, Latimer JL, et al. The UspA2 protein of Moraxella catarrhalis is directly involved in the expression of serum resistance. Infect Immun 2005;73(4):2400–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Thomy D, Culp E, Adamek M, et al. The ADEP biosynthetic gene cluster in Streptomyces hawaiiensis NRRL 15010 reveals an accessory clpP gene as a novel antibiotic resistance factor. Appl Environ Microbiol 2019;85(20):e01292–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Culp EJ, Sychantha D, Hobson C, et al. ClpP inhibitors are produced by a widespread family of bacterial gene clusters. Nat Microbiol 2022;7(3):451–62. [DOI] [PubMed] [Google Scholar]
- 93. Sekyere JO, Asante J. Emerging mechanisms of antimicrobial resistance in bacteria and fungi: advances in the era of genomics. Future Microbiol 2018;13(2):241–62. [DOI] [PubMed] [Google Scholar]
- 94. Kaur P, Agarwal S, Datta S. Delineating bacteriostatic and bactericidal targets in mycobacteria using IPTG inducible antisense expression. PloS one. 2009;4(6):e5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Martinez JL, Baquero F. Mutation frequencies and antibiotic resistance. Antimicrob Agents Chemother 2000;44(7):1771–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Jacobs MR. Worldwide trends in antimicrobial resistance among common respiratory tract pathogens in children. Pediatr Infect Dis J 2003;22(8):S109–19. [DOI] [PubMed] [Google Scholar]
- 97. Zhang Z, Yang Z, Xiang X, et al. Mutation of TonB-dependent receptor encoding gene MCR_0492 potentially associates with macrolides resistance in Moraxella catarrhalis isolates. Infect Drug Resist 2022;15:2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kasai A, Ohta A, Maeda Y, et al. Novel mechanism responsible for high-level macrolide resistance in Moraxella catarrhalis. Infect Drug Resist 2018;11:2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Liu Y, Xu H, Xu Z, et al. High-level macrolide-resistant Moraxella catarrhalis and development of an allele-specific PCR assay for detection of 23S rRNA gene A2330T mutation: a three-year study at a Chinese tertiary hospital. Microb Drug Resist 2015;21(5):507–11. [DOI] [PubMed] [Google Scholar]
- 100. Fàbrega A, Madurga S, Giralt E, Vila J. Mechanism of action of and resistance to quinolones. J Microbial Biotechnol 2009;2(1):40–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Warner DM, Shafer WM, Jerse AE. Clinically relevant mutations that cause derepression of the Neisseria gonorrhoeae MtrC-MtrD-MtrE efflux pump system confer different levels of antimicrobial resistance and in vivo fitness. Mol Microbiol 2008;70(2):462–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Gygli SM, Borrell S, Trauner A, Gagneux S. Antimicrobial resistance in Mycobacterium tuberculosis: mechanistic and evolutionary perspectives. FEMS Microbiol Rev 2017;41(3):354–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The used datasets are publicly available and the research data can be accessible on request.