

Abstract
The facultative intracellular pathogen Brucella abortus interacts with several organelles of the host cell to reach its replicative niche inside the endoplasmic reticulum. However, little is known about the interplay between the intracellular bacteria and the host cell mitochondria. Here, we showed that B. abortus triggers substantive mitochondrial network fragmentation, accompanied by mitophagy and the formation of mitochondrial Brucella‐containing vacuoles during the late steps of cellular infection. Brucella‐induced expression of the mitophagy receptor BNIP3L is essential for these events and relies on the iron‐dependent stabilisation of the hypoxia‐inducible factor 1α. Functionally, BNIP3L‐mediated mitophagy appears to be advantageous for bacterial exit from the host cell as BNIP3L depletion drastically reduces the number of reinfection events. Altogether, these findings highlight the intricate link between Brucella trafficking and the mitochondria during host cell infection.
Keywords: BNIP3L, Brucella, intracellular trafficking, iron, mitophagy
Subject Categories: Microbiology, Virology & Host Pathogen Interaction
ER‐replicating intracellular pathogen Brucella abortus relocalizes to mitochondria and induces their fragmentation during late stages of cellular infection.
Introduction
Bacteria of the genus Brucella are Gram‐negative, facultative, intracellular pathogens responsible for brucellosis, a worldwide zoonosis affecting various hosts including domestic animals (mainly cattle, sheep and goats) and humans (Atluri et al, 2011; González‐Espinoza et al, 2021). Brucella spp. has evolved several strategies to enter, survive and proliferate inside host cells including professional phagocytes (macrophages and dendritic cells) as well as non‐phagocytic cells (trophoblasts, fibroblasts and epithelial cells; von Bargen et al, 2012). After its internalisation inside the host cell, Brucella is found within a membrane‐bound vacuole, the Brucella‐containing vacuole (BCV), which traffics along the endocytic pathway to reach an acidified compartment, the endocytic BCV (eBCV), around 8 h post‐infection (pi) (Celli et al, 2003). This endocytic stage is necessary for the production of Brucella VirB Type IV secretion system (T4SS) (Boschiroli et al, 2002) which injects effector proteins inside the host cell (Starr et al, 2008). Most of the described Brucella effectors were shown to be required for the establishment of the replicative niche inside of the endoplasmic reticulum (ER) (Ke et al, 2015; Smith et al, 2020). Indeed, Brucella effectors coordinate the interaction between the eBCV and the ER exit sites (ERES) (Celli et al, 2005) as well as the manipulation of the Golgi vesicular trafficking (Miller et al, 2018; Borghesan et al, 2021) to generate a replicative BCV (rBCV), a compartment in continuity with the ER in which bacteria start a massive proliferation around 12 h pi (Celli et al, 2003; Sedzicki et al, 2018). In the third step, around 48 h pi, the rBCVs get engulfed inside LAMP‐1‐positive autophagic membranes, converting them into autophagic BCVs (aBCVs), in which Brucella subverts a part of the autophagy initiation complex machinery, such as ULK1, Beclin1 and ATG14L, to eventually promote bacterial egress and further reinfection of surrounding cells (Starr et al, 2012).
Regarding its intracellular life cycle, Brucella affects the functions and/or morphology of most of the host cell organelles such as lysosomes, the ER and the Golgi apparatus, leading to stress and adaptive responses in the host cell (Smith et al, 2013; Byndloss et al, 2019). However, the putative interplay between Brucella and the mitochondrial population of infected cells is still poorly studied. The ER, lysosomes and mitochondria are able to physically and functionally interact with each other (Hamasaki et al, 2013; Boutry & Kim, 2021). As the central role of mitochondria in innate immunity is now well recognised (Mills et al, 2017; Missiroli et al, 2020), we hypothesised that mitochondria might be affected and play a role during Brucella infection. Our previous work pointed out that BCVs might physically interact with mitochondria of host cells both in cellulo and in vivo (Lobet et al, 2018). In addition, we showed that Brucella abortus triggers the fragmentation of the mitochondrial network in myeloid and non‐myeloid cells at 48 h pi (Lobet et al, 2018). Mitochondrial fission plays a major role in mitochondrial quality control as it allows the segregation of damaged mitochondria upon several stresses such as calcium‐mediated apoptosis (Szabadkai et al, 2004), oxidative stress through reactive oxygen species (ROSs) (Wang et al, 2012) or lipopolysaccharide (LPS)‐induced inflammation (Shi et al, 2019). Eventually, damaged mitochondrial fragments can be cleared through mitophagy, a selective autophagy process targeting mitochondria for degradation by the lysosomal compartments (Onishi & Okamoto, 2021).
During mitophagy, targeted mitochondria interact with the ATG8 family proteins (including LC3, GABARAP and GABE16) which are inserted in autophagic phagophores, and recruited around their cargo, leading to the formation of mitophagosomes (Kabeya et al, 2003; Zachari & Ktistakis, 2020). Besides its physiological roles in basal mitochondrial recycling or establishment of controlled cell differentiation processes (Cairns et al, 2020), mitophagy can be triggered by several exogenous and endogenous stresses which can lead to pathological removal of mitochondria (Sandoval et al, 2008). These stresses may include mitochondrial depolarisation (Narendra et al, 2008; Matsuda et al, 2010), nutrient starvation (Eiyama et al, 2013), hypoxia (Liu et al, 2012; Fu et al, 2020), iron depletion (Allen et al, 2013; Hara et al, 2020) or mitochondrial DNA oxidative damage (Shu et al, 2021). The diversity of mitophagosome formation pathways was recently reviewed (Ganley & Simonsen, 2022). Mitophagy pathways are classified based on their dependency on ubiquitin or not. Canonical ubiquitin‐dependent mitophagy mainly, but not only, relies on the activity of PINK‐1/Parkin. Upon loss of mitochondrial membrane potential (MMP), the PINK1 kinase is stabilised, recruits and activates the Parkin E3‐ubiquitin ligase which ubiquitinates mitochondrial outer membrane proteins leading to the recruitment of receptor proteins (such as p62 or OPTN) that bind to LC3 (Agarwal & Muqit, 2022). On the other hand, ubiquitin‐independent mitophagy is mediated by several mitophagy receptors such as FUNDC1, BNIP3 and BNIP3L (also called NIX), FKBP8 or Bcl2‐L‐13, which directly bind to LC3 (Onishi et al, 2021).
Here, we show, for the first time, that the mitochondrial network fragmentation induced by B. abortus in host cells is accompanied by an induction of mitophagy in the late steps of infection. BNIP3L, a gene for which expression is regulated by HIF‐1α in an iron‐dependent manner during infection, is responsible for B. abortus‐induced mitophagy. Moreover, BNIP3L‐mediated mitophagy is necessary for proper B. abortus egress as BNIP3L depletion reduces aBCV formation and prevents reinfection. Furthermore, we also discovered a new type of BCV, that we called mitochondrial BCV (mBCV), characterised by the presence of B. abortus inside swollen mitochondria during the late steps of infection. Altogether, our discovery of a functional crosstalk taking place between Brucella and host cell mitochondria highlight what could be a crucial step for this bacterial infectious cycle.
Results
B. abortus triggers mitochondrial network fragmentation
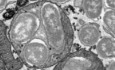
We previously showed that B. abortus induces the mitochondrial network fragmentation of myeloid (RAW264.7 macrophages and bone marrow‐derived macrophages [BMDM]) and non‐myeloid cells (HeLa) at 48 h pi but not at 24 h pi (Lobet et al, 2018). However, the kinetics of mitochondrial fragmentation, especially between 24 and 72 h pi, was not studied in detail. To determine the precise onset of mitochondrial fragmentation induced by B. abortus, we analysed the morphological changes of mitochondria by assessing the aspect ratio (AR) and end‐point/branched‐point ratio (EBR) in infected HeLa cells displaying mitochondria immuno‐stained for TOMM20 (translocase of the outer mitochondrial membrane of 20 kDa) between 8 and 72 h pi (Fig 1A). While the mitochondrial network was significantly fragmented at 48 h pi, this phenotype already appeared progressively from 32 h pi and is maintained until 72 h pi (Fig 1B–D).
Figure 1. B. abortus triggers mitochondrial network fragmentation during the late steps of infection in HeLa cells.
-
ARepresentative confocal micrographs of HeLa cells infected or not with B. abortus 544 for the indicated times, then fixed and immunostained for TOMM20 (Alexa Fluor 488—green) and B. abortus LPS (Alexa Fluor 568—Magenta). DNA was stained with Hoechst 33258 (blue). Scale bars: 20 μm.
-
BSchematic summary of the calculation of the aspect ratio (AR) and the end‐point/branched‐point ratio (EBR) of the mitochondrial network. See the “Quantification of mitochondrial network morphology” paragraph in the Methods section for further information. Created with Biorender.com.
-
C, DQuantification of the mitochondrial population morphology by assessing the AR (C) and EBR (D) of the mitochondria of HeLa cells infected or not (NI) with B. abortus 544 for the indicated times from micrographs shown in (A). Data are presented as means ± SD from n = 5 (biological replicates) independent experiments (the numbers indicated in the columns represent the number of cells analysed per condition). Statistical analyses were performed using a multiple Mann–Whitney test followed by a Holm‐Šidàk's multiple comparisons test; asterisks indicate significant differences compared to the control (NI); *P < 0.05.
Source data are available online for this figure.
B. abortus triggers Parkin‐independent mitophagy
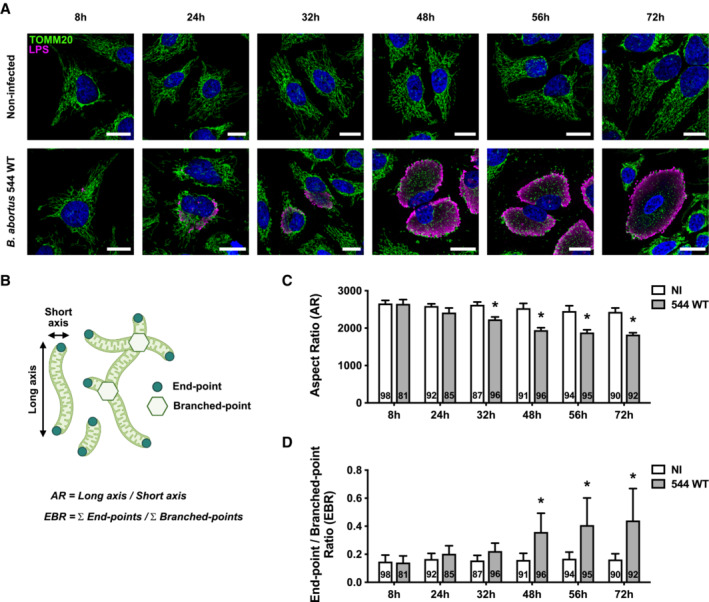
As mitochondrial network fragmentation is usually a major hallmark of subsequent mitophagy, we analysed whether mitophagy was initiated in B. abortus‐infected cells by monitoring the recruitment of phagophores at the mitochondria. To address this question, we quantified the co‐localisation between LC3 and the β‐subunit of the ATP synthase in infected HeLa cells (Fig 2A and B; Appendix Fig S1). A significant increase in the frequency of co‐localisation events between the two proteins was observed at 48 and 72 h pi when compared with non‐infected cells, while no significant difference was found at 24 h pi (Fig 2B). In order to assess the mitophagy at a functional level, we used a FIS1‐GFP‐mCherry reporter construct (also called mito‐QC) consisting of the FIS1 (Fission 1 mitochondrial receptor) gene sequence which is tandem‐tagged with the GFP and mCherry sequences. This tool allows the discrimination of acidified mitochondria (which are FIS1‐mCherry‐positive and GFP‐negative when GFP fluorescence is quenched below pH 6 as inside of lysosomes) from neutral mitochondria (which are FIS1‐GFP and ‐mCherry‐positive; Allen et al, 2013). The number of FIS1‐mCherry‐positive and GFP‐negative structures was significantly higher in B. abortus‐infected cells when compared to non‐infected cells (Fig 2C and D), and was similar when compared with cells treated with deferoxamine (DFO) an iron chelator known to induce mitophagy, used as a positive control (Appendix Fig S2; Allen et al, 2013). These results confirm that B. abortus triggers mitophagy in infected cells.
Figure 2. B. abortus triggers mitophagy in HeLa cells.
-
ARepresentative confocal micrographs of HeLa cells infected or not with B. abortus 544 GFP for 48 h, then fixed and immunostained for the β‐subunit of the ATP synthase (Alexa Fluor 633—magenta) and LC3 (Alexa Fluor 568—green). DNA was stained with Hoechst 33258 (blue). Arrows indicate LC3‐ATP synthase β‐positive punctae. Scale bars: 20 μm. Inset scale bars: 5 μm.
-
BQuantification of the number of LC3‐β‐subunit of the ATP synthase‐positive punctae per HeLa cell infected or not (NI) with B. abortus 544 GFP for 24, 48 and 72 h from micrographs shown in (A) and (Fig EV3A–C). Data are presented as means ± SD from n = 3 (biological replicates independent experiments; the numbers indicated in the columns represent the number of cells analysed per condition). Statistical analyses were performed using a two‐way ANOVA followed by a Šidàk's multiple comparisons test; asterisks indicate significant differences compared to the control (NI); ns: not significant; ***P < 0.001; ****P < 0.0001; hashtags indicate significant differences between infected conditions throughout time; # P < 0.05; ## P < 0.01.
-
CRepresentative confocal micrographs of HeLa cells transfected with a FIS1‐GFP (green)‐mCherry (magenta) expression construct, infected or not with B. abortus 544 for 48 h, then fixed and immunostained for B. abortus LPS (Alexa Fluor 633—Red). DNA was stained with Hoechst 33258 (blue). Arrows indicate FIS1‐mCherry‐positive‐GFP‐negative punctae. Scale bars: 20 μm. Inset scale bars: 5 μm.
-
DQuantification of the number of FIS1‐mCherry‐positive‐GFP‐negative punctae per HeLa cell infected or not (NI) with B. abortus 544 for 48 h from micrographs shown in (C). Data are presented as means ± SD from n = 3 (biological replicates) independent experiments (the numbers indicated in the columns represent the number of cells analysed per condition). Statistical analyses were performed using an unpaired two‐tailed Student's t‐test; ***: P < 0.001 (P = 0.0008).
Source data are available online for this figure.
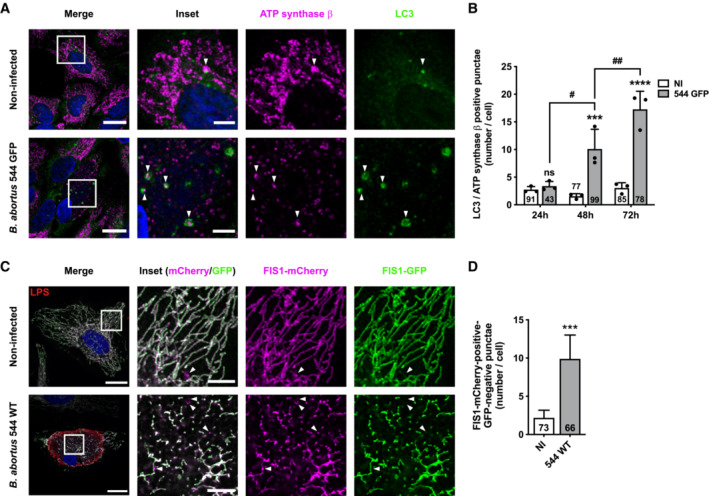
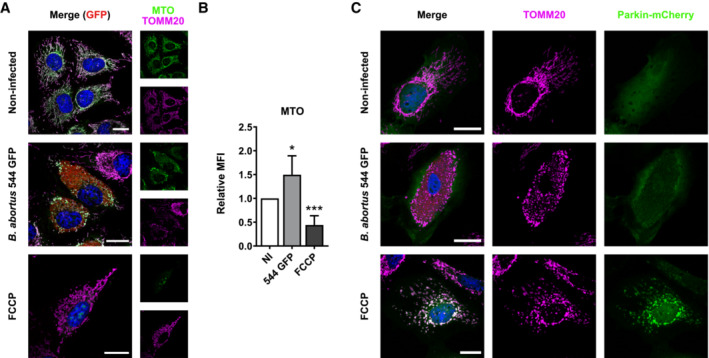
Mechanisms leading to mitophagy pathways can be either ubiquitin‐dependent (such as the PINK1/Parkin pathway in response to a drop in MMP) or receptor‐mediated. To identify whether the PINK1/Parkin pathway is triggered in B. abortus‐infected cells, we analysed the MMP in infected HeLa cells using the MitoTracker™ Orange (MTO) fluorescent probe, as well as the localisation of Parkin expressed in cells transfected with a Parkin‐mCherry construct as HeLa cells do not express the gene encoding Parkin (Denison et al, 2003). B. abortus‐infected cells did not show any loss of MMP nor Parkin co‐localisation with TOMM20, as observed by confocal microscopy and flow cytometry (Fig EV1A–C). The FCCP treatment was used here as a positive control since it is well‐known to induce a drop in MMP, stabilise PINK1 and recruit Parkin at the surface of mitochondria which initiates mitophagy (Narendra et al, 2008; Fig EV1A–C). Altogether, these observations strongly suggest that B. abortus‐mediated mitophagy is Parkin‐independent.
Figure EV1. B. abortus triggers Parkin‐independent mitophagy.
-
ARepresentative confocal micrographs of HeLa cells infected or not with B. abortus 544 GFP (red) for 48 h, stained with 100 nM of MTO fluorescent probe (green) for 30 min before analysis, then fixed and immunostained for TOMM20 (Alexa Fluor 633—Magenta). DNA was stained with Hoechst 33258 (blue). HeLa cells treated with 20 μM FCCP for 30 min were used as a positive control. Scale bars: 20 μm.
-
BRelative median fluorescence intensity (MFI) of the MTO fluorescent probe of HeLa cells infected or not (NI) with B. abortus 544 GFP for 48 h as measured by flow cytometry. HeLa cells treated with 20 μM FCCP for 30 min were used as a positive control. Data are presented as means ± SD from n = 5 (biological replicates) independent experiments (9,718 cells analysed in total per condition). Statistical analyses were performed using a one‐way ANOVA followed by a Tukey's multiple comparisons test; asterisks indicate significant differences compared to the control (NI); *P < 0.05; ***P < 0.001.
-
CRepresentative confocal micrographs of HeLa cells transfected with a Parkin‐mCherry (green) expression construct, infected or not with B. abortus 544 GFP (red) for 48 h, then fixed and immunostained for TOMM20 (Alexa Fluor 647—magenta). DNA was stained with Hoechst 33258 (blue). HeLa cells treated with FCCP (20 μM for 30 min) were used as a positive control. Scale bars: 20 μm.
Source data are available online for this figure.
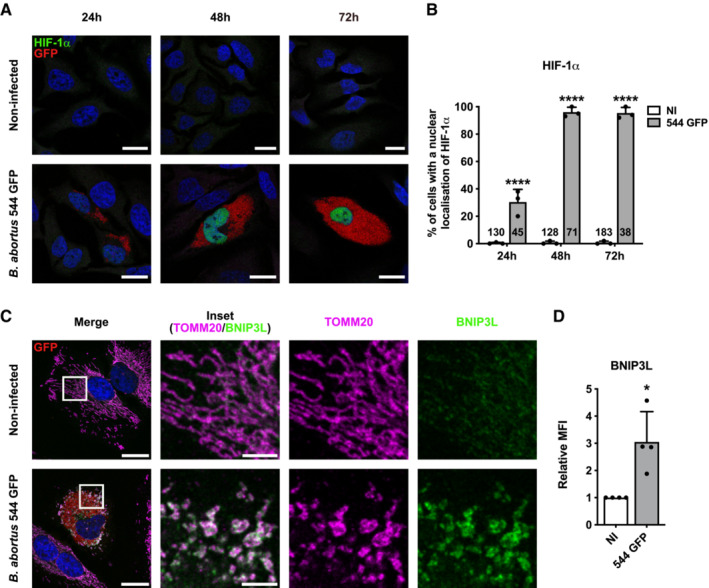
B. abortus induces HIF‐1α stabilisation and BNIP3L expression in a hypoxia‐independent manner
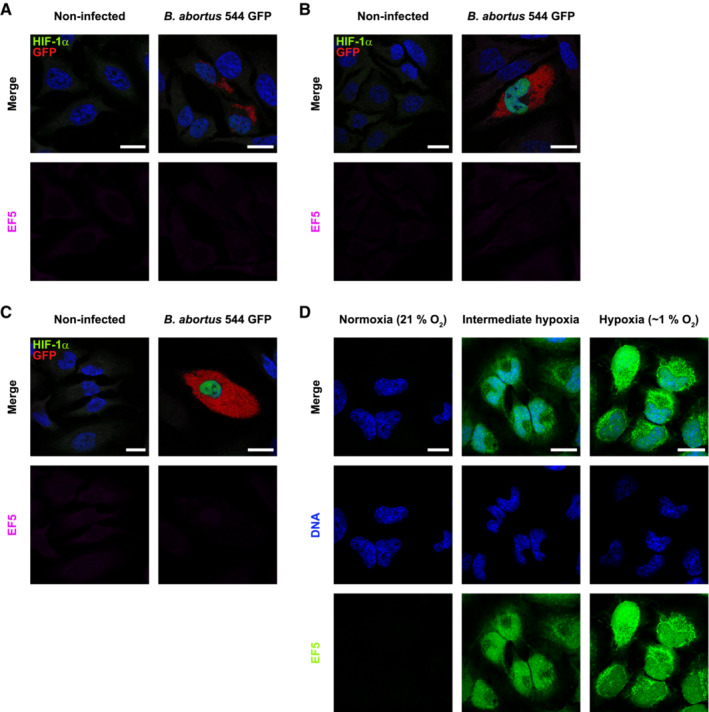
The occurrence of some receptor‐mediated mitophagy was then investigated. Since B. abortus is an aerobic pathogen and regarding the massive load of bacteria observed in host cells at 48 and 72 h pi, we first tested whether a hypoxic environment was generated in infected cells, by monitoring the activation of the HIF‐1α/BNIP3L axis. HIF‐1α is the α subunit of the HIF‐1 (hypoxia inducible factor 1) transcription factor which is the master regulator of the cell response to hypoxia (Lee et al, 2020). BNIP3L (BCL2/adenovirus E1B 19 kDa protein‐interacting protein 3‐like) is a mitophagy receptor found at the OMM and the gene encoding this protein is directly regulated by HIF‐1 (Daskalaki et al, 2018). B. abortus‐infected cells showed a nuclear localisation of HIF‐1α starting at 24 h pi and reaching almost all the infected cell population at 48 and 72 h pi (Fig 3A and B). In parallel, the abundance of BNIP3L was significantly increased in infected cells at 48 h pi when compared with non‐infected cells and the protein was localised at the mitochondria as expected (Fig 3C and D), suggesting that the HIF‐1α/BNIP3L axis is activated by B. abortus. However, using the EF5 compound known to form adducts upon hypoxic stress that can be detected by specific antibodies (Conway et al, 2018), we could not detect the occurrence of a hypoxic environment in infected cells, no matter the time point (Fig EV2A–C), while the EF5 compound could be abundantly detected in cells exposed to mild or severe hypoxia (Fig EV2D). These results suggest that B. abortus triggers HIF‐1α stabilisation by a hypoxia‐independent mechanism.
Figure 3. B. abortus induces HIF‐1α stabilisation and BNIP3L expression in HeLa cells.
-
ARepresentative confocal micrographs of HeLa cells infected or not with B. abortus 544 GFP (red) for 24, 48 and 72 h then fixed and immunostained for HIF‐1α (Alexa 568—green). DNA was stained with Hoechst 33258 (Blue). Scale bars: 20 μm.
-
BQuantification of the percentages of cells positive for a nuclear localisation of HIF‐1α from HeLa cells infected or not (NI) with B. abortus 544 GFP for 24, 48 and 72 h from micrographs shown in (A). Data are presented as means ± SD from n = 3 (biological replicates) independent experiments (the numbers indicated in the columns represent the number of cells analysed per condition). Statistical analyses were performed using a two‐way ANOVA followed by a Šidàk's multiple comparisons test; asterisks indicate significant differences compared to the control (NI); ****P < 0.0001.
-
CRepresentative confocal micrographs of HeLa cells infected or not with B. abortus 544 GFP (red) for 48 h, then fixed and immunostained for BNIP3L (Alexa 568—green) and TOMM20 (Alexa 633—magenta). DNA was stained with Hoechst 33258 (blue). Scale bars: 20 μm.
-
DRelative median fluorescence intensity (MFI) of BNIP3L immunostaining from HeLa cells infected or not (NI) with B. abortus 544 GFP for 48 h as measured by flow cytometry. Data are presented as means ± SD from n = 4 (biological replicates) independent experiments (8,296 cells analysed in total per condition). Statistical analyses were performed using a one sample t‐test; *P < 0.05 (P = 0.0351).
Source data are available online for this figure.
Figure EV2. B. abortus induces HIF‐1α stabilisation in a hypoxia‐independent manner.
-
A–CRepresentative confocal micrographs of HeLa cells infected or not with B. abortus 544 GFP (red) for 24 h (A), 48 h (B) and 72 h (C), treated with 150 μM of the EF5 compound for 3 h before analysis, then fixed and immunostained for EF5 (Anti‐EF5 Cy5 conjugate—magenta) and HIF‐1α (Alexa 568—green). DNA was stained with Hoechst 33258 (blue). Scale bars: 20 μm. Figure panels EV2A–C, reuse the same experiment as described in Fig 3A.
-
DRepresentative confocal micrographs of HeLa cells treated with 150 μM of the EF5 compound, exposed to normoxia (21% O2), hypoxia (1% O2) or an intermediate hypoxia (between 21% and 1% O2) for 3 h, then fixed and immunostained for EF5 (anti‐EF5 Cy5 conjugate—green). DNA was stained with Hoechst 33258 (blue). Scale bars: 20 μm.
Source data are available online for this figure.
Iron prevents B. abortus‐induced HIF‐1α/BNIP3L pathway activation
Regulation of HIF‐1α is mainly controlled by the activity of the prolyl hydroxylase dioxygenases (PHDs) which use molecular oxygen, Fe2+ and alpha‐ketoglutarate (αKG) to catalyse the hydroxylation of HIF‐1α, leading to its proteasome‐mediated degradation in normoxic conditions (Schofield & Ratcliffe, 2004). Hence, in addition to a decrease in oxygen concentration, other factors including ROS (Bell et al, 2007) and iron chelators such as deferoxamine (Guo et al, 2015) are also known to inhibit PHDs and therefore provoke HIF‐1α stabilisation. As mitochondrial‐derived ROS (mtROS) production is often associated with mitochondrial dysfunction, we first tested whether mtROS production is triggered upon B. abortus infection by using the MitoSOX™ fluorescent probe that can be oxidised by the superoxide anion radical () (Mukhopadhyay et al, 2007). In our experimental conditions, no increase in the MitoSOX™ fluorescence intensity was observed in B. abortus‐infected cells when compared to non‐infected cells (Fig EV3A). Moreover, neither MitoTEMPOL (a mitochondrial antioxidant) nor N‐acetyl‐L‐cysteine (NAC, a cytosolic antioxidant) could prevent B. abortus‐induced HIF‐1α stabilisation and nuclear localisation (Fig EV3B–D), suggesting that HIF‐1α stabilisation is not mediated by ROS.
Figure EV3. B. abortus‐induced HIF‐1α stabilisation is not mediated by mitochondrial ROS in HeLa cells.

-
ARepresentative wide‐field micrographs of HeLa cells infected or not (NI) with B. abortus 544 GFP (green) for 48 h, treated or not (ctrl) with 10 μM MTEMPOL for 24 h before analysis, and then stained with 0.5 μM MitoSOX™ fluorescent probe (red) for 30 min. Samples were observed under live‐imaging conditions with the Nikon Eclipse Ti2‐inverted epifluorescence microscope. Scale bars: 20 μm.
-
BRepresentative confocal micrographs of HeLa cells infected or not (NI) with B. abortus 544 GFP (red) for 48 h, treated or not (ctrl) with 10 μM MTEMPOL for 24 h before analysis, and then fixed and immunostained for HIF‐1α (Alexa 568—green). DNA was stained with Hoechst 33258 (blue). Scale bars: 20 μm.
-
CQuantification of the percentages of cells positive for a nuclear localisation of HIF‐1α from HeLa cells infected or not (NI) with B. abortus 544 GFP and treated or not (ctrl) with 10 μM MTEMPOL for 24 h before analysis from micrographs shown in (B). Data are presented as means from n = 1 experiment (the numbers indicated in the columns represent the number of cells analysed per condition).
-
DRepresentative confocal micrographs of HeLa cells infected or not (NI) with B. abortus 544 GFP (red) for 48 h, treated or not (ctrl) with 5 mM NAC for 24 h before analysis, and then fixed and immunostained for HIF‐1α (Alexa 568—green). DNA was stained with Hoechst 33258 (blue). Scale bars: 20 μm.
Source data are available online for this figure.
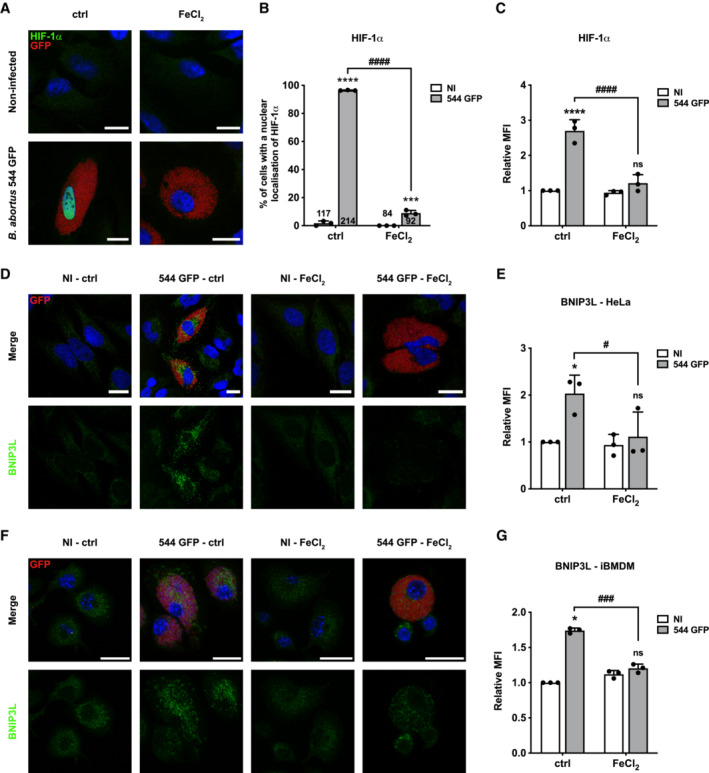
Iron is essential for Brucella virulence (Roop II, 2012). A proteomic study showed that once inside the host cell, B. abortus remodels its iron‐associated proteome through upregulation of different iron import systems, indicating that B. abortus faces iron starvation in the intracellular environment (Roset et al, 2017). We therefore tested the hypothesis that iron depletion could be responsible for B. abortus‐induced HIF‐1α stabilisation. Interestingly, iron supplementation (in the form of FeCl2 in the culture media) totally prevented HIF‐1α nuclear localisation (Fig 4A and B) and suppressed its stabilisation (Fig 4C), as well as the expression of BNIP3L at the mitochondria (Fig 4D and E) in HeLa cells. The inhibitory effect of FeCl2 supplementation on B. abortus‐induced BNIP3L expression was also confirmed in immortalised BMDM (iBMDM) (Fig 4F and G). These results suggest that B. abortus mediates HIF‐1α/BNIP3L pathway activation through an iron starvation response in the host cell.
Figure 4. Iron prevents B. abortus‐induced HIF‐1α/BNIP3L pathway activation in HeLa cells and iBMDM.
-
ARepresentative confocal micrographs of HeLa cells infected or not with B. abortus 544 GFP (red) treated or not (ctrl) with FeCl2 (500 μM) for 48 h, then fixed and immunostained for HIF‐1α (Alexa 568—green). DNA was stained with Hoechst 33258 (blue). Scale bars: 20 μm.
-
BQuantification of the percentages of cells positive for a nuclear localisation of HIF‐1α from HeLa cells infected or not (NI) with B. abortus 544 GFP (red) and treated or not (ctrl) with 500 μM FeCl2 for 48 h from micrographs shown in (A). Data are presented as means ± SD from n = 3 (biological replicates) independent experiments (the numbers indicated in the columns represent the number of cells analysed per condition). Statistical analyses were performed using a two‐way ANOVA followed by a Šidàk's multiple comparisons test; asterisks indicate significant differences compared to the control (NI); ***P < 0.001; ****P < 0.0001; hashtags indicate significant differences compared to the infected condition without FeCl2; #### P < 0.0001.
-
CRelative median fluorescence intensity (MFI) of HIF‐1α immunostaining from HeLa cells infected or not (NI) with B. abortus 544 GFP treated or not (ctrl) with 500 μM FeCl2 for 48 h as measured by flow cytometry. Data are presented as means ± SD from n = 3 independent experiments (10,093 cells analysed in total per condition). Statistical analyses were performed using a two‐way ANOVA followed by a Šidàk's multiple comparisons test; asterisks indicate significant differences compared to the control (NI); ns, not significant; ****P < 0.0001; hashtags indicate significant differences compared to the infected condition without FeCl2; #### P < 0.0001.
-
DRepresentative confocal micrographs of HeLa cells infected or not (NI) with B. abortus 544 GFP (red) treated or not (ctrl) with 500 μM FeCl2 for 48 h, then fixed and immunostained for BNIP3L (Alexa 568—green). DNA was stained with Hoechst 33258 (blue). Scale bars: 20 μm.
-
ERelative median fluorescence intensity (MFI) of BNIP3L immunostaining from HeLa cells infected or not (NI) with B. abortus 544 GFP treated or not (ctrl) with 500 μM FeCl2 for 48 h as measured by flow cytometry. Data are presented as means ± SD from n = 3 (biological replicates) independent experiments (10,199 cells analysed in total per condition). Statistical analyses were performed using a two‐way ANOVA followed by a Šidàk's multiple comparisons test; asterisks indicate significant differences compared to the control (NI); ns, not significant; *P < 0.05; hashtags indicate significant differences compared to the infected condition without FeCl2; # P < 0.05.
-
FRepresentative confocal micrographs of iBMDM infected or not (NI) with B. abortus 544 GFP (red) treated or not (ctrl) with 500 μM FeCl2 for 48 h, then fixed and immunostained for BNIP3L (Alexa 568—green). DNA was stained with Hoechst 33258 (blue). Scale bars: 20 μm.
-
GRelative median fluorescence intensity (MFI) of BNIP3L immunostaining from iBMDM infected or not (NI) with B. abortus 544 GFP treated or not (ctrl) with 500 μM FeCl2 for 48 h as measured by flow cytometry. Data are presented as means ± SD from n = 3 (biological replicates) independent experiments (14,152 cells analysed in total per condition). Statistical analyses were performed using a two‐way ANOVA followed by a Šidàk's multiple comparisons test; asterisks indicate significant differences compared to the control (NI); ns, not significant; *P < 0.05; hashtags indicate significant differences compared to the infected condition without FeCl2; ### P < 0.001.
Source data are available online for this figure.
BNIP3L depletion prevents B. abortus‐mediated mitophagy
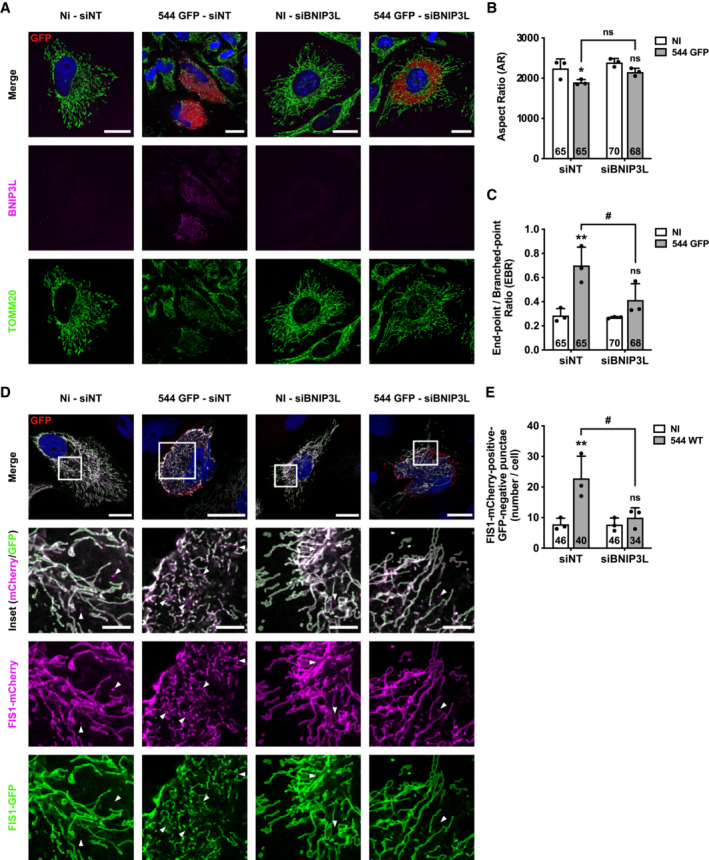
To validate the putative role of BNIP3L as a mitophagy receptor induced by B. abortus, a gene expression silencing approach was used to knock‐down the BNIP3L gene. The efficiency of BNIP3L depletion in cells transfected with a siRNA SMARTpool targeting BNIP3L mRNA on whole cell population, treated or not with CoCl2 known to stabilise and activate HIF‐1α (Dai et al, 2012), was assessed by western blot analysis. The results showed that BNIP3L knock‐down was very efficient in HeLa cells at least until 72 h post‐transfection when compared with cells transfected with a non‐targeting control siRNA (Appendix Fig S3A). Mitochondrial network morphology was first monitored in response to BNIP3L depletion in B. abortus‐infected HeLa cells (Fig 5A). BNIP3L depletion significantly prevented B. abortus‐induced mitochondrial network fragmentation (Fig 5B and C). Furthermore, using the FIS1‐GFP‐mCherry reporter construct, infected cells depleted for BNIP3L had significantly fewer FIS1‐mCherry‐positive and GFP‐negative structures when compared with infected cells transfected with a non‐targeting siRNA, maintaining the number close to that of non‐infected control cells (Fig 5D and E). Altogether, these results demonstrate that BNIP3L receptor is an important actor that controls the activation of mitophagy in B. abortus‐infected HeLa cells.
Figure 5. BNIP3L depletion prevents B. abortus‐induced mitochondrial network fragmentation and mitophagy in HeLa cells.
-
ARepresentative confocal micrographs of HeLa cells transfected with a non‐targeting siRNA pool (siNT—40 nM) or a BNIP3L siRNA SMARTpool (siBNIP3L—40 nM), infected or not (NI) with B. abortus 544 GFP (red) for 48 h, then fixed and immunostained for BNIP3L (Alexa Fluor 568—magenta) and TOMM20 (Alexa Fluor 633—green). DNA was stained with Hoechst 33258 (blue). Scale bars: 20 μm.
-
B, CQuantification of the mitochondrial population morphology by assessing the AR (B) and EBR (C) of the mitochondria of HeLa cells from micrographs shown in (A). Data are presented as means ± SD from n = 3 (biological replicates) independent experiments (the numbers indicated in the columns represent the number of cells analysed per condition). Statistical analyses were performed using a multiple Mann–Whitney test followed by a Holm‐Šidàk's multiple comparisons test; asterisks indicate significant differences compared to the control (NI); *P < 0.05; **P < 0.01; hashtags indicate significant differences compared to the infected condition transfected with a siNT; # P < 0.05.
-
DRepresentative confocal micrographs of HeLa cells transfected with a FIS1‐GFP (green)‐mCherry (magenta) expression construct, infected or not (NI) with B. abortus 544 for 48 h while being transfected with a non‐targeting siRNA pool (siNT—40 nM) or a BNIP3L siRNA SMARTpool (siBNIP3L—40 nM), then fixed and immunostained for B. abortus LPS (Alexa Fluor 633—red). DNA was stained with Hoechst 33258 (blue). Arrows indicate FIS1‐mCherry‐positive‐GFP‐negative punctae. Scale bars: 20 μm. Inset scale bars: 5 μm.
-
EQuantification of the number of FIS1‐mCherry‐positive‐GFP‐negative punctae per cell of HeLa cells from micrographs shown in (D). Data are presented as means ± SD from n = 3 (biological replicates) independent experiments (the numbers indicated in the columns represent the number of cells analysed per condition). Statistical analyses were performed using a two‐way ANOVA followed by a Šidàk's multiple comparisons test; asterisks indicate significant differences compared to the control (NI); ns: not significant; **P < 0.01; hashtags indicate significant differences compared to the infected condition transfected with a siNT; # P < 0.05.
Source data are available online for this figure.
BNIP3L depletion and iron supplementation limit aBCV formation and prevent reinfection events
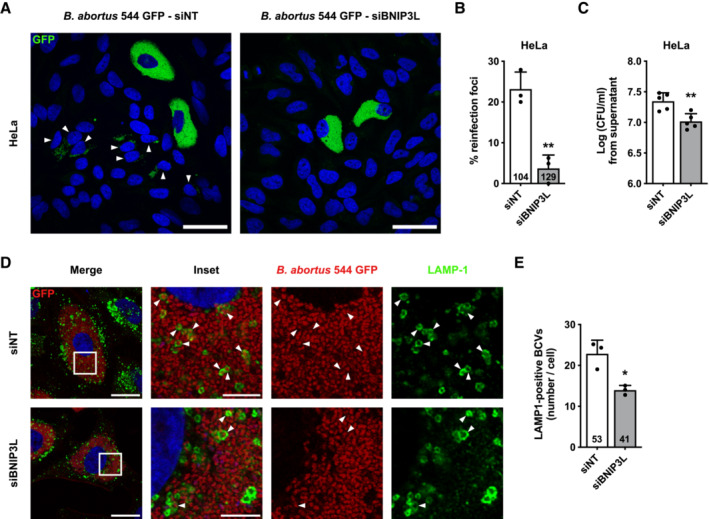
Given the importance of mitochondrial function and dynamics upon bacterial infections (Spier et al, 2019), we next addressed the functional role of the BNIP3L‐mediated mitophagy in B. abortus infectious cycle. Even though BNIP3L depletion does not seem to affect B. abortus intracellular replication (Fig EV4), we hypothesised that B. abortus egress could be controlled by a BNIP3L‐mediated mitophagy. Indeed, in reinfection‐permissive conditions, the formation of reinfection foci was significantly decreased upon BNIP3L depletion in infected HeLa cells at 72 h pi (Fig 6A and B). Consistent with these results, and in the same reinfection‐permissive conditions, the number of bacteria found in the supernatant at 72 h pi was significantly reduced by two‐fold in BNIP3L‐depleted infected HeLa cells (Fig 6C). As bacterial egress was associated with aBCV formation (Starr et al, 2012), we next counted the number of LAMP1‐positive BCVs in infected HeLa cells depleted or not for BNIP3L. We found that the number of LAMP‐1‐positive BCVs was significantly reduced in cells depleted for BNIP3L to a similar extent (Fig 6D and E).
Figure EV4. BNIP3L depletion does not impair B. abortus intracellular replication in HeLa cells.
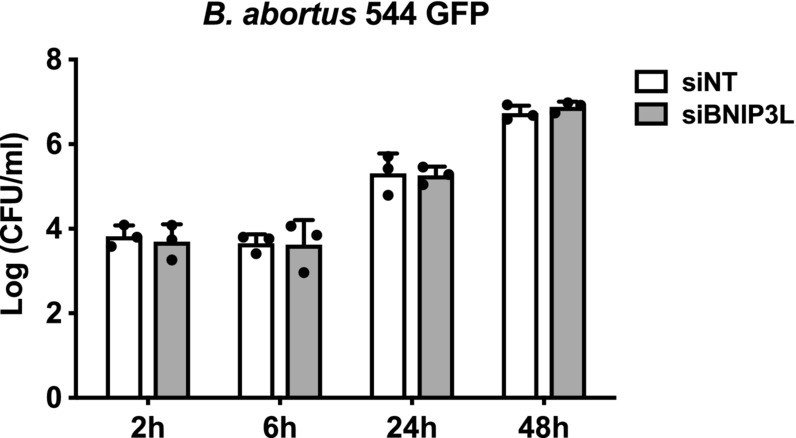
CFU assay expressing Log (CFU/ml) from HeLa cells transfected with a non‐targeting siRNA pool (siNT—40 nM) or a BNIP3L siRNA SMARTpool (siBNIP3L—40 nM), then infected with B. abortus 544 GFP for the indicated times. Data are presented as means ± SD from n = 3 (biological replicates) independent experiments; Statistical analyses were performed using a two‐way ANOVA followed by a Šidàk's multiple comparisons test; no significant differences were found. Source data are available online for this figure.
Figure 6. BNIP3L depletion limits aBCV formation and prevents reinfection events in HeLa cells.
-
ARepresentative confocal micrographs of HeLa cells infected with B. abortus 544 GFP (green) and transfected with a non‐targeting siRNA pool (siNT—40 nM) or a BNIP3L siRNA SMARTpool (siBNIP3L—40 nM) for 48 h, then incubated under reinfection‐permissive conditions for 24 h before analysis at 72 h pi. Cells were fixed and DNA was stained with Hoechst 33258 (blue). Arrows indicate reinfected cells. Scale bars: 50 μm.
-
BQuantification of the percentages of reinfection foci per infected cell at 72 h pi of HeLa cells from micrographs shown in (A). Data are presented as means ± SD from n = 3 (biological replicates) independent experiments (the numbers indicated in the columns represent the number of cells analysed per condition). Statistical analyses were performed using an unpaired two‐tailed Student's t‐test; **P < 0.01 (P = 0.0032).
-
CCFU assay expressing Log (CFU/ml) from the supernatant of HeLa cells infected with B. abortus 544 GFP (green) that were previously transfected with a non‐targeting siRNA pool (siNT—40 nM) or a BNIP3L siRNA SMARTpool (siBNIP3L—40 nM) for 48 h, then incubated under reinfection‐permissive conditions for 24 h before collecting the supernatant at 72 h pi for analysis. Data are presented as means ± SD from n = 5 (biological replicates) independent experiments; Statistical analyses were performed using an unpaired two‐tailed Student's t‐test; **P < 0.01 (P = 0.0042).
-
DRepresentative confocal micrographs of HeLa cells infected with B. abortus 544 GFP (Red) and transfected with a non‐targeting siRNA pool (siNT—40 nM) or a BNIP3L siRNA SMARTpool (siBNIP3L—40 nM) for 72 h pi, then fixed and immunostained for LAMP‐1 (Alexa Fluor 568—green). DNA was stained with Hoechst 33258 (blue). Arrows indicate LAMP‐1‐positive BCVs (aBCVs). Scale bars: 20 μm. Inset scale bars: 5 μm.
-
EQuantification of the number of LAMP‐1‐positive BCVs (aBCVs) per infected HeLa cells from micrographs shown in (E). Data are presented as means ± SD from n = 3 (biological replicates) independent experiments (the numbers indicated in the columns represent the number of cells analysed per condition). Statistical analyses were performed using an unpaired two‐tailed Student's t‐test; *P < 0.05 (P = 0.0117).
Source data are available online for this figure.
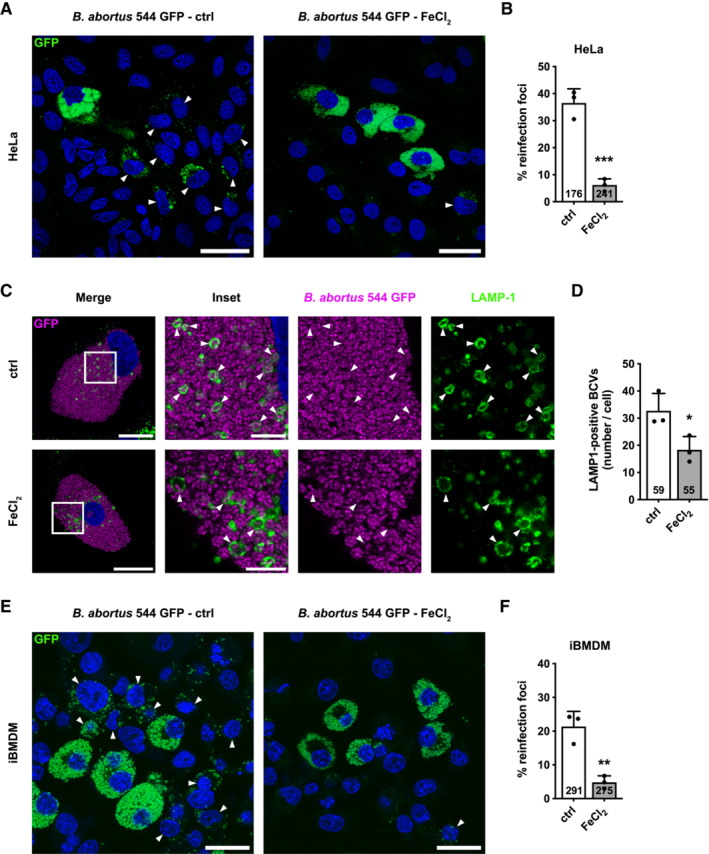
As we showed that B. abortus‐mediated BNIP3L expression is iron‐dependent (Fig 5), we next tested whether iron supplementation could, by itself, mimic the effect of BNIP3L depletion on B. abortus egress. Interestingly, the formation of reinfection foci in reinfection‐permissive conditions (Fig 7A and B), as well as the number of LAMP‐1‐positive BCVs (Fig 7C and D) were significantly decreased upon FeCl2 supplementation in infected HeLa cells at 72 h pi. These results suggest a strong connexion between the iron starvation response induced by B. abortus infection and BNIP3L‐mediated aBCV formation and bacterial egress.
Figure 7. Iron limits aBCV formation and prevents reinfection events in HeLa cells and iBMDM.
-
ARepresentative confocal micrographs of HeLa cells infected with B. abortus 544 GFP (Green) treated or not (ctrl) with FeCl2 (500 μM) for 48 h, then incubated under reinfection‐permissive conditions (with or without FeCl2) for 24 h before analysis at 72 h pi. Cells were fixed and DNA was stained with Hoechst 33258 (blue). Arrows indicate reinfected cells. Scale bars: 50 μm.
-
BQuantification of the percentages of reinfection foci per infected cell at 72 h pi of HeLa cells from micrographs shown in (A). Data are presented as means ± SD from n = 3 (biological replicates) independent experiments (the numbers indicated in the columns represent the number of cells analysed per condition). Statistical analyses were performed using an unpaired two‐tailed Student's t‐test; ***P < 0.001 (P = 0.0008).
-
CRepresentative confocal micrographs of HeLa cells infected or not with B. abortus 544 GFP (Magenta) treated or not (ctrl) with FeCl2 (500 μM) for 72 h, then fixed and immunostained for LAMP‐1 (Alexa Fluor 568—green). DNA was stained with Hoechst 33258 (blue). Arrows indicate LAMP‐1‐positive BCVs (aBCVs). Scale bars: 20 μm. Inset scale bars: 5 μm.
-
DQuantification of the number of LAMP‐1‐positive BCVs (aBCVs) per infected HeLa cells from micrographs shown in (C). Data are presented as means ± SD from n = 3 (biological replicates) independent experiments (the numbers indicated in the columns represent the number of cells analysed per condition). Statistical analyses were performed using an unpaired two‐tailed Student's t‐test; *P < 0.05 (P = 0.0368).
-
ERepresentative confocal micrographs of iBMDM infected with B. abortus 544 GFP (Green) treated or not (ctrl) with FeCl2 (500 μM) for 48 h, then incubated under reinfection‐permissive conditions (with or without FeCl2) for 24 h before analysis at 72 h pi. Cells were fixed and DNA was stained with Hoechst 33258 (blue). Arrows indicate reinfected cells. Scale bars: 50 μm.
-
FQuantification of the percentages of reinfection foci per infected cell at 72 h pi of iBMDM from micrographs shown in (E). Data are presented as means ± SD from n = 3 (biological replicates) independent experiments (the numbers indicated in the columns represent the number of cells analysed per condition). Statistical analyses were performed using an unpaired two‐tailed Student's t‐test; **P < 0.01 (P = 0.0043).
Source data are available online for this figure.
Since the silencing of BNIP3L expression with siRNA was not as efficient in iBMDM than in HeLa cells (Appendix Fig S3B), we directly tested whether iron supplementation could also, by itself, mimic the effect of BNIP3L depletion on B. abortus egress in iBMDM. As expected, in reinfection‐permissive conditions, the formation of reinfection foci was also significantly decreased in infected iBMDM at 72 h pi in response to FeCl2 supplementation (Fig 7E and F). These results confirm the observations obtained in HeLa cells in a more relevant cell infection model. This connexion between iron and bacterial egress reinforces the argument that the iron‐dependent activation of BNIP3L‐mediated mitophagy controls B. abortus exit of the host cell.
B. abortus is observed inside a fraction of swollen mitochondria in infected cells
Alongside the established mitochondrial network fragmentation triggered by B. abortus (Fig 1), another change in mitochondrial morphology was observed between 48 and 72 h pi (Appendix Fig S4A). Indeed, in about 10% of infected HeLa cells, the TOMM20 signal was completely rearranged into large vesicles of a diameter of up to 5 μm. This phenotype was more frequently observed over time reaching 25% of infected cells at 72 h pi (Appendix Fig S4B). These TOMM20‐positive structures were confirmed to be mitochondria since they co‐localise with other mitochondrial markers such as the β‐subunit of the ATP synthase (marker of the inner mitochondrial membrane, IMM) and the MTO fluorescent probe which labels mitochondria in a membrane potential‐dependent manner (Appendix Fig S4C and D).
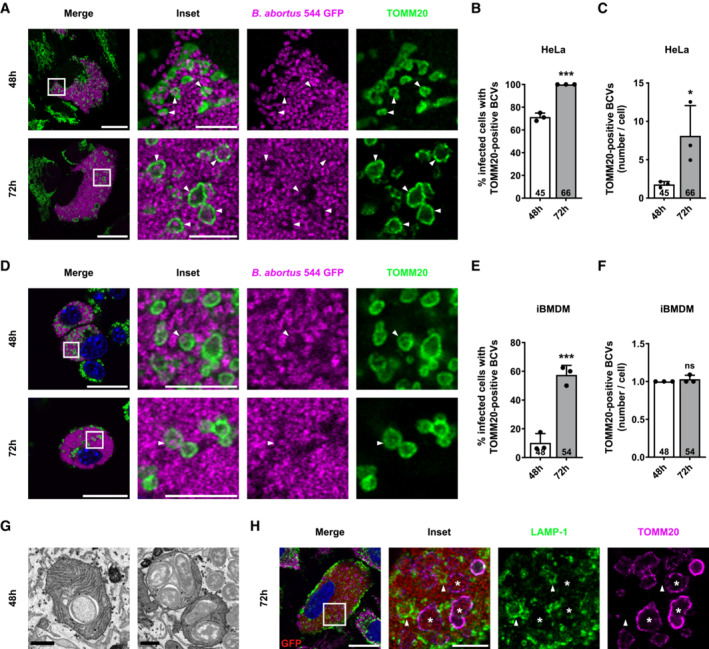
Based on these observations, we wondered whether bacteria could be found inside of these swollen mitochondria. To address this question, we used a high‐resolution Airyscan microscope and were able to detect one or few bacteria in some of these mitochondria at 48 and 72 h pi, in both HeLa cells (Fig 8A–C and Movie EV1) and iBMDM (Fig 8D–F). In rare cases, at 72 h pi, we even observed extreme phenotypes with swollen mitochondria that were full of bacteria, as depicted in stimulated emission depletion (STED) microscopy micrographs (Fig EV5A). Counting of these new Brucella‐containing compartments, that we called mitochondrial BCVs (mBCVs), showed that the percentages of infected cells with mBCVs are higher at 72 h pi in HeLa and iBMDM (Fig 8B and E) and that mBCVs are more abundant at 72 h pi in HeLa cells (Fig 8C). In addition, focused ion beam/scanning electron microscopy (FIB/SEM) confirmed the presence of B. abortus within the mitochondrial intermembrane space (MIS) of infected HeLa cells (Fig 8G; Movie EV2). Considering the role of BNIP3L‐mediated mitophagy in aBCV formation and the observation that the morphologies of mBCVs and aBCVs are very similar, we next explored whether both structures might be the same BCVs. To test this hypothesis, we performed co‐immunostainings of TOMM20 and LAMP‐1 in HeLa infected cells. Strikingly, our observations clearly demonstrated that TOMM20‐positive BCVs were negative for LAMP‐1 (Fig 8H), which confirms that mBCVs and aBCVs are distinct structures.
Figure 8. B. abortus is observed inside a fraction of swollen mitochondria in HeLa cells and iBMDM during the late steps of infection.
-
ARepresentative confocal micrographs of HeLa cells infected with B. abortus 544 GFP (magenta) for 48 and 72 h, then fixed and immunostained for TOMM20 (Alexa Fluor 647—green). DNA was stained with Hoechst 33258 (blue). Arrows indicate when B. abortus was found inside a mitochondrion (mBCVs). Scale bars: 20 μm. Inset scale bars: 5 μm.
-
BQuantification of the percentages of infected HeLa cells displaying TOMM20‐positive BCVs (mBCVs) at the indicated times, from micrographs shown in (A). Data are presented as means ± SD from n = 3 (biological replicates) independent experiments (the numbers indicated in the columns represent the number of cells analysed per condition). Statistical analyses were performed using an unpaired two‐tailed Student's t‐test; ***P < 0.001 (P = 0.0001).
-
CQuantification of the number of TOMM20‐positive BCVs (mBCVs) per infected HeLa cells, at the indicated times, from micrographs shown in (E). Data are presented as means ± SD from n = 3 (biological replicates) independent experiments (the numbers indicated in the columns represent the number of cells analysed per condition). Statistical analyses were performed using an unpaired two‐tailed Student's t‐test; *P = 0.05.
-
DRepresentative confocal micrographs of iBMDM infected with B. abortus 544 GFP (magenta) for 48 and 72 h, then fixed and immunostained for TOMM20 (Alexa Fluor 647—green). DNA was stained with Hoechst 33258 (blue). Arrows indicate when B. abortus was found inside a mitochondrion (mBCVs). Scale bars: 20 μm. Inset scale bars: 5 μm.
-
EQuantification of the percentages of infected iBMDM displaying TOMM20‐positive BCVs (mBCVs) at the indicated times, from micrographs shown in (D). Data are presented as means ± SD from n = 3 (biological replicates) independent experiments (the numbers indicated in the columns represent the number of cells analysed per condition). Statistical analyses were performed using an unpaired two‐tailed Student's t‐test; ***P < 0.001 (P = 0.0009).
-
FQuantification of the number of TOMM20‐positive BCVs (mBCVs) per infected HeLa cells, at the indicated times, from micrographs shown in (E). Data are presented as means ± SD from n = 3 (biological replicates) independent experiments (the numbers indicated in the columns represent the number of cells analysed per condition). Statistical analyses were performed using an unpaired two‐tailed Student's t‐test; ns: not significant (P = 0.3739).
-
GFIB/SEM micrographs of HeLa cells infected with B. abortus 2308 RFP for 48 h showing bacteria within mitochondria. Scale bars: 600 nm.
-
HRepresentative confocal micrographs of HeLa cells infected with B. abortus 544 GFP (red) for 72 h, then fixed and immunostained for LAMP‐1 (Alexa Fluor 568—green) and TOMM20 (Alexa Fluor 633—magenta). DNA was stained with Hoechst 33258 (blue). Arrows indicate LAMP‐1‐positive BCVs (aBCVs). Stars indicate TOMM20‐positive BCVs (mBCVs). Scale bars: 20 μm. Inset scale bars: 5 μm.
Source data are available online for this figure.
Figure EV5. Neither BNIP3L depletion nor iron supplementation alters mBCV occurrence in HeLa cells.
-
ASTED micrographs of a HeLa cell infected with B. abortus 544 GFP (magenta) for 72 h, then fixed and immunostained for TOMM20 (Abberior® STAR 635—green). The presented cell displays a rare but spectacular event of enlarged mBCVs with colonisation of the bacteria. Scale bars: 5 μm.
-
BRepresentative confocal micrographs of HeLa cells infected with B. abortus 544 GFP (magenta) and transfected with a non‐targeting siRNA pool (siNT—40 nM) or a BNIP3L siRNA SMARTpool (siBNIP3L—40 nM) for 72 h, then fixed and immunostained for TOMM20 (Alexa Fluor 647—green). DNA was stained with Hoechst 33258 (blue). Arrows indicate when B. abortus was found inside a mitochondrion (mBCVs). Scale bars: 20 μm. Inset scale bars: 5 μm.
-
CQuantification of the number of TOMM20‐positive BCVs (mBCVs) per infected HeLa cells, from micrographs shown in (B). Data are presented as means ± SD from n = 3 (biological replicates) independent experiments (the numbers indicated in the columns represent the number of cells analysed per condition). Statistical analyses were performed using an unpaired two‐tailed Student's t‐test; ns: not significant (P = 0.6742).
-
DRepresentative confocal micrographs of HeLa cells infected with B. abortus 544 GFP (magenta), treated or not (ctrl) with FeCl2 (500 μM) for 72 h, then fixed and immunostained for TOMM20 (Alexa Fluor 647—green). DNA was stained with Hoechst 33258 (blue). Arrows indicate when B. abortus was found inside a mitochondrion (mBCVs). Scale bars: 20 μm. Inset scale bars: 5 μm.
-
EQuantification of the number of TOMM20‐positive BCVs (mBCVs) per infected HeLa cells, from micrographs shown in (D). Data are presented as means ± SD from n = 3 (biological replicates) independent experiments (the numbers indicated in the columns represent the number of cells analysed per condition). Statistical analyses were performed using an unpaired two‐tailed Student's t‐test; ns, not significant (P = 0.4724).
-
FRepresentative confocal micrographs of HeLa cells infected with B. abortus 544 WT for 72 h, then fixed and immunostained for TOMM20 (Alexa Fluor 633—magenta) and LC3 (Alexa Fluor 568—green). DNA (from the HeLa nucleus and B. abortus) was stained with Hoechst 33258 (red). Arrows indicate when B. abortus was found inside a mitochondrion (mBCVs). Scale bars: 20 μm. Inset scale bars: 5 μm.
Source data are available online for this figure.
In an attempt to decipher the cause and/or the origin of mBCV formation in infected cells, we eventually analysed whether mBCV formation could be linked to the BNIP3L‐dependent mitophagy induced by B. abortus or not. We thus tested the putative effects of siRNA‐mediated BNIP3L depletion and iron supplementation on the occurrence of mBCVs in infected cells. We found that neither BNIP3L depletion (Fig EV5B and C) nor FeCl2 supplementation (Fig EV5D and E), altered the number of TOMM20‐positive BCVs in HeLa cells at 72 h pi, suggesting that the BNIP3L‐mediated mitophagy induced by B. abortus is not responsible for mBCV formation. This finding was reinforced by the observation by the exclusion of the LC3 autophagy marker from the membranes of TOMM20‐positive BCVs at 72 h pi (Fig EV5F), suggesting that mBCVs do not result from the biogenesis of mitophagosomes.
Discussion
In this study, we highlighted a new mechanism demonstrating the evidence of an interplay between Brucella and the mitochondrial population of infected cells. We found that B. abortus triggers a BNIP3L‐mediated mitophagy pathway which we showed to be required for the completion of its intracellular cycle up to egress and secondary infection of neighbouring cells (Fig 9A and B). Here, we identified the mitophagy receptor BNIP3L as being required for triggering both B. abortus‐mediated mitochondrial fragmentation and mitophagy since siRNA‐mediated BNIP3L depletion drastically impairs these phenotypes. Interestingly, these results contrast with the general assumption that mitochondrial fission is a prerequisite step for mitophagy (Gomes & Scorrano, 2013). However, it was shown that the formation of the autophagosome around mitochondria actually occurs concurrently with mitochondrial fragmentation in a DRP1‐independent manner in HeLa cells (Yamashita et al, 2016). In addition, apart from their role in mitophagosome formation, several mitophagy receptors, including BNIP3L or FUNDC1, are also able to induce mitochondrial fission by themselves (Chen et al, 2016) (Simpson et al, 2021) (da Silva Rosa et al, 2021). Further research would be required to test whether FUNDC1 could also be involved in B. abortus‐mediated mitochondrial fragmentation and mitophagy since its expression is also regulated by HIF‐1α (Liu et al, 2022). However, regarding the almost complete effect of BNIP3L depletion we observed on mitophagy and mitochondrial morphology readouts, we suggest that BNIP3L is most likely the major mitophagy receptor involved in B. abortus‐mediated mitophagy.
Figure 9. Summary of the interactions between Brucella abortus and the mitochondria of the host cell.
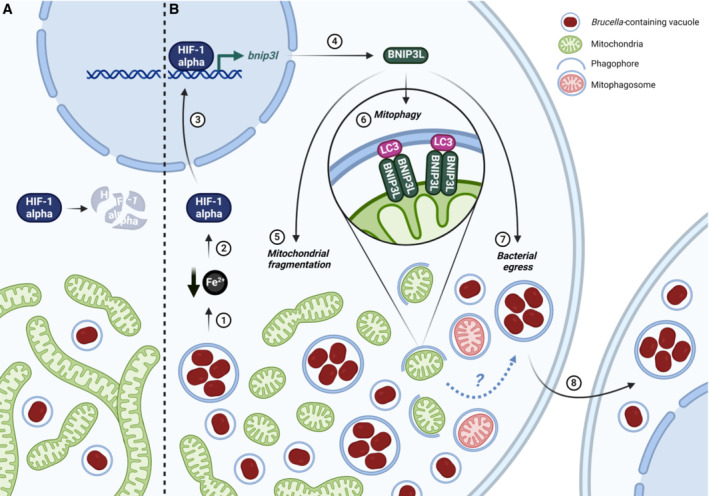
In this study, we showed that until 24 h pi (A), Brucella abortus affects neither the mitochondrial morphology nor the endogenous degradation of HIF‐1α. However, at 48 h pi (B), B. abortus induces an iron starvation response (1) that stabilises and activates HIF‐1α (2) which therefore translocates to the nucleus (3). There, HIF‐1α upregulates the expression of the mitophagy receptor BNIP3L (4) which mediates two phenotypes during the late stages of B. abortus intracellular trafficking. B. abortus‐induced BNIP3L expression mediates mitochondrial network fragmentation (5), one hallmark of mitophagy (6) that is characterised by the engulfment of mitochondrial fragments inside phagophores to specifically degrade and recycle them. Eventually, BNIP3L helps to fulfil B. abortus intracellular cycle as it is necessary for bacterial egress (7) and reinfection of neighbouring cells (8). Created with Biorender.com.
Mechanistically, we showed that the induction of BNIP3L expression by B. abortus is dependent on the stabilisation of HIF‐1α (Fig 9A and B). The HIF‐1α/BNIP3L axis has already been widely described in other mitophagy processes such as mitochondria clearance during erythrocyte maturation (Sandoval et al, 2008). Although mtROS‐dependent HIF‐1α activation has been recently reported in a macrophage model of B. abortus infection to be at the basis of the metabolic reprogramming induced in host cells (Gomes et al, 2021), no mitochondria‐related HIF‐1α downstream signalling was analysed. Our results strongly support that HIF‐1α stabilisation and BNIP3L expression are not mediated by mtROS nor by an intracellular hypoxic environment but are dependent on iron availability. Indeed, we showed that iron supplementation is sufficient to prevent HIF‐1α stabilisation and BNIP3L expression induced by B. abortus. These results are in line with previously reported data suggesting that a drop in iron concentrations upon B. abortus infection is responsible for a major rewiring of the gene expression program leading to the upregulation of different iron uptake processes, such as Irr (iron‐responsive regulator protein, BAB1_2175), a sensor of iron concentration variations, as well as entC (isochorismate synthase, BAB2_0015), one of the enzymes required for siderophore biosynthesis (Roset et al, 2017). Indeed, the host cell represents an iron‐deprived environment for invading pathogens such as Brucella (Roop II et al, 2017), which led to the evolution complex pathogen iron acquisition systems as survival and virulence determinants. Three different iron acquisition systems have been identified in B. abortus (Roop II, 2012), which are (i) the expression of high‐affinity Fe2+ transporters (FtrABCD; Elhassanny et al, 2013), (ii) the use of a heme import system (BhuA heme transporter; Paulley et al, 2007), (iii) the use of two siderophores: 2,3‐DHBA and brucebactin (Anderson et al, 2008). Further research will be necessary to confirm the origin of iron starvation induced by B. abortus and whether it is responsible for the initiation of mitophagy in the host cell. Interestingly, siderophores from Pseudomonas aeruginosa have been described to induce the stabilisation of HIF‐1α, mitochondrial stress and mitophagy in C. elegans (Kang et al, 2018). Our hypothesis is that Brucella could consume the host cell iron, possibly through the subversion of the mitochondrial siderophore 2,5‐DHBA (2,5‐dihydroxybenzoic acid), a molecule with similar structure as the bacterial siderophore 2,3‐DHBA (Liu et al, 2014), a precursor for siderophore synthesis in Brucella spp. (Parent et al, 2002). Since mitochondria are an important reservoir of intracellular iron (Ward & Cloonan, 2019), one could imagine that Brucella depletes the mitochondrial iron, which would lead to mitochondrial stress and subsequent mitophagy.
What could be the advantage for an intracellular pathogen to induce mitochondrial fragmentation and mitophagy? Given their function in energy production and metabolism, as well as in the regulation of inflammatory processes and programmed cell death, mitochondria are a target of choice for invading pathogens, whether they are bacteria (Lobet et al, 2015; Spier et al, 2019), viruses (Li et al, 2021) or protozoa (Medeiros et al, 2021). Even if bacteria such as Chlamydiae spp. inhibit mitochondrial fission to preserve the host cell ATP production providing energy for survival and replication (Chowdhury et al, 2017), other pathogens such as Listeria monocytogenes (Carvalho et al, 2020), Shigella flexneri, (Sirianni et al, 2016), Legionella pneumophila (Escoll et al, 2017) and Mycobacterium tuberculosis (Fine‐Coulson et al, 2015) trigger the disruption of the mitochondrial network to either induce a beneficial glycolytic shift or to control the molecular mechanisms of host cell death (through apoptosis) and/or inflammasome activation that could contribute to clear the pathogen. As we have shown during B. abortus infection, mitophagy is also triggered by some of these pathogens to manipulate the host cell environment to their advantage. On the one hand, for example, upon L. monocytogenes infection, a newly discovered mitophagy receptor, the NOD‐like receptor NLRX1, induces mitophagy to limit mtROS production and therefore bacteria avoid to be killed by oxidant molecules (Zhang et al, 2019). On the other hand, in the case of M. tuberculosis and M. bovis, a transcriptomic study pointed an upregulation of BNIP3L expression in response to infection, leading to mitophagy, necessary for macrophage inflammatory function and resolution of the infection (Mahla et al, 2021). These two recent studies demonstrate that mitophagy can be either beneficial for the pathogen or deleterious as part of the host cell defence. In an evolutionary point of view, the manipulation of mitochondria by intracellular pathogens is thus clearly crucial in a competition for survival between the host and the pathogen.
In the case of B. abortus, the induced BNIP3L‐mediated mitophagy seems beneficial for the bacteria. Indeed, as BNIP3L depletion reduced the number of aBCVs in host cells, the number of bacteria in the supernatant, as well as the number of reinfected cells at 72 h pi, we demonstrated that BNIP3L is required for proper aBCV formation and bacterial egress. Our findings bring novel insights in the importance of autophagy mechanisms during the late steps of Brucella intracellular cycle. BNIP3L is thus a new and key actor in the list of autophagy markers such as ULK1, Beclin1 and ATG14L that were already described for aBCV formation and bacterial egress (Starr et al, 2012). As several organelles, including mitochondria (Hailey et al, 2010), have been proposed to provide membranes for autophagosome formation (Dikic & Elazar, 2018), one hypothesis could be that mitochondrial turnover through mitophagy could supply membranes for aBCV formation. Although we showed that aBCVs are negative for TOMM20, it does not exclude the presence of other OMM proteins, or even the putative loss of some OMM during membrane turnover. A more exhaustive screening of OMM proteins should be performed in order to better characterise the aBCVs membrane markers, which are, to date, quite limited (Starr et al, 2012). Although the exact molecular mechanisms and link between auto−/mito‐phagy and aBCV still remain to be elucidated, we clearly showed that the iron/HIF‐1α/BNIP3L axis is necessary for B. abortus egress as FeCl2 supplementation also limits aBCV formation and reinfection events in both HeLa and iBMDM. These observations suggest that when iron is available, Brucella would rather proliferate and avoid the exit of the host cell. This hypothesis is in line with the fact that Brucella displays a greater proliferation in iron‐rich cell types such as trophoblasts (Salcedo et al, 2013). Interestingly, from an epidemiological point of view, these results are reminiscent from those from several case reports that present patients suffering from acute brucellosis and subsequent autoimmune hemolytic anaemia (Meena et al, 2018). Anaemia is therefore considered as an additional risk factor for developing severe brucellosis (Mak et al, 2019).
Finally, by investigating the changes in mitochondrial morphology caused by B. abortus infection, we discovered a new type of BCV, the mitochondrial BCV (mBCV). Indeed, high‐resolution confocal and electron microscopy approaches revealed the existence of mitochondrial structures and ultra‐structures—positive for several mitochondrial markers (such as TOMM20)—which harbour few bacteria inside the MIS between 48 and 72 h pi in HeLa as well as in iBMDM. The intriguing presence of bacteria inside mitochondria of host cells was also reported for Midichloria mitochondrii, which is able to invade and colonise the MIS of oocytes in the Ixodes ricinus tick, by a still unknown process (Sassera et al, 2006; Stavru et al, 2020). In an effort to better understand the mechanisms of mBCV biogenesis, a tempting hypothesis could be that mBCVs would be a consequence of the mitophagy induced by Brucella and would also play a role in bacterial egress. However, we showed that, unlike aBCV formation, neither iron supplementation nor BNIP3L depletion altered the number of mBCV in infected cells, suggesting that mBCVs are unrelated to the iron/BNIP3L‐mediated mitophagy required for B. abortus egress. The fact that LC3 does not co‐localise with mBCVs is an additional argument to suggest that B. abortus‐mediated mitophagy is not correlated to the formation of mBCVs. Other hypotheses could be considered in the search of the origin of mBCVs. For example, considering mitophagy‐independent mechanisms of mitochondrial vesicle formation, we propose that mitochondria‐derived vesicles (MDVs) are interesting candidates for further study. Indeed, TOMM20, which is the marker of reference we used to detect mBCVs at 48 and 72 h pi, is a marker that is also found in the membrane of MDVs (Soubannier et al, 2012; Soto‐Heredero et al, 2017). Mitofusins (MFNs) could also be candidates of interest in the biogenesis of mBCV as MFN1 and 2 have been reported to be depleted from the mitochondria of Toxoplasma gondii‐infected cells, and relocalised to enlarged structures emerging from mitochondria (Li et al, 2022). However, these hypotheses would not explain how the bacteria could cross the mitochondrial membranes and enter the mitochondria, either by perforating the OMM or by direct fusion of the OMM with the BCV, as it is speculated for Midichloria mitochondrii (Sassera et al, 2006; Stavru et al, 2020). A comprehensive and super‐resolution analysis would be required for the visualisation of interactions, integrity and/or trafficking of the BCV and mitochondrial membranes. The discovery of this new BCV also highlights the diversity of potential fates of intracellular bacteria at late stages of a cellular infection.
In conclusion, our findings pave the way for a better understanding of Brucella infection mechanisms, strengthening the intricate links existing between mitochondria of host cells and Brucella intracellular trafficking. Our study also emphasises that mitochondria are a central hub for invading pathogens that need to survive, proliferate and disseminate throughout the host.
Materials and Methods
Reagents and antibodies
When necessary and for the indicated times, culture media were supplemented with: 150 μM deferoxamine mesylate (DFO, Sigma‐Aldrich, D9533), 20 μM carbonyl cyanide‐p‐trifluoromethoxyphenylhydrazone (FCCP; Sigma‐Aldrich, C2920), 100 nM MitoTracker™ Orange CMTMRos fluorescent probe (MTO; Invitrogen, M7510), 150 μM (2‐(2‐nitro‐1H‐imidazol‐1‐yl)‐N‐(2,2,3,3,3‐pentafluoropropyl)) acetamide (EF5 compound; Sigma‐Aldrich, EF5014), 0.5 μM MitoSOX™ fluorescent probe (Invitrogen, M36008), 10 μM MitoTEMPOL (MTEMPOL; Abcam ab144644), 5 mM N‐acetyl‐L‐cysteine (NAC; Sigma‐Aldrich, A9165), 500 μM iron (II) chloride tetrahydrate (FeCl2, Sigma‐Aldrich, 44939), 100 μM cobalt chloride (CoCl2; Sigma‐Aldrich, 15862), according to the manufacturer's instructions.
Antibodies used for immunofluorescence and flow cytometry include: mouse anti‐β subunit of the ATP synthase (1:500 dilution; Invitrogen, A21‐351); rabbit anti‐BNIP3L/NIX (1:100 dilution; Cell Signalling Technology, 12396S); mouse anti‐Brucella smooth LPS (O‐antigen) (A76/12G12 antibody produced as described previously in Cloeckaert et al (1990)); mouse anti‐EF5‐Cyanine 5 conjugate, clone ELK3‐51 (1:100 dilution; Sigma‐Aldrich, EF5012); rabbit anti‐FAM92A (1:100 dilution; Sigma‐Aldrich, HPA034760); rabbit anti‐HIF‐1α (1:100 dilution; Abcam, ab179483); rabbit anti‐LC3B (1:100 dilution; Sigma‐Aldrich, L7543); mouse anti‐TOMM20 (1:100 dilution; Abcam, ab56783); rabbit anti‐TOMM20 (1:200 dilution; Abcam, ab186735); Alexa Fluor 488/568/633/647 goat anti‐rabbit and anti‐mouse IgG conjugates (1:1,000 dilution; Invitrogen); Abberior® STAR 635 goat anti‐rabbit IgG conjugates (1:1,000 dilution; Sigma‐Aldrich). DNA was stained with Hoechst 33258 (1:5,000 dilution; Invitrogen, H3569).
Antibodies used for western blot analysis include: rabbit anti‐BNIP3L/NIX (1:1,000 dilution; Cell Signalling Technology, 12396S); mouse anti‐β actin (1:10,000 dilution; Sigma‐Aldrich, A5441); Ir‐Dye 680/800 goat anti‐rabbit and anti‐mouse IgG conjugates (1:10,000 dilution; Licor Biosciences).
Mammalian cell culture
Human HeLa cells (HeLa CCL‐2; ATCC) were cultured in Minimum Essential Medium (MEM‐Glutamax; Gibco) supplemented with 1% non‐essential amino acids (100 X) (MEM NEAA; Gibco), 1 mM sodium pyruvate (Gibco) and 10% foetal bovine serum (FBS; Gibco) at 37°C in 5% CO2. Immortalised BMDMs (a generous gift from Prof. Suzana Salcedo; Blasi et al, 1987) were cultured in Dulbecco's Modified Eagle Medium (DMEM—4.5 g/l glucose, Gibco) supplemented with 10% heat‐inactivated foetal bovine serum (FBS; Gibco) and 10% conditioned media obtained from cultured L929 fibroblasts providing macrophage colony‐stimulating factor (M‐CSF), at 37°C in 5% CO2. All cell lines were routinely tested for mycoplasma but were not authenticated by DNA profiling.
For hypoxia experiments, HeLa cells were incubated in serum‐free CO2‐independent MEM (Gibco) supplemented with 500 μM L‐glutamine (Merck), and then incubated at 37°C under either normal atmosphere (normoxia, 21% O2), hypoxia (1% O2), or intermediate hypoxia (between 21 and 1% O2) for 3 h. To expose cells to hypoxia, a homemade pressurised incubator was used wherein gaseous N2 was injected to blow out the air contained in the incubator, thereby creating a progressive hypoxic environment. Hypoxia (1% O2) and intermediate hypoxia (between 21 and 1% O2) were triggered by letting air flowing out of the device for 3 min and 90 s respectively.
Cell transfection (plasmids and siRNA)
For Parkin localisation and mitophagy analyses, respectively, a plasmid expressing Parkin‐mCherry (mCherry‐parkin; Addgene plasmid #23956 from R. Youle; Narendra et al, 2008) and a plasmid expressing FIS1‐GFP‐mCherry (mCherry‐GFP‐FIS1mt101‐152, commonly known as mito‐QC; a generous gift from Dr. Pla‐Martín; Allen et al, 2013) were used to transfect HeLa cells 24 h before infection using X‐tremeGENE™ HP DNA Transfection Reagent (Roche, 6366244001), according to the manufacturer's instructions.
For siRNA‐mediated silencing of BNIP3L, HeLa cells were transfected for 24 h before infection using DharmaFECT1 Transfection Reagent (Dharmacon, T‐2001‐01) with 40 nM of the ON‐TARGETplus human BNIP3L siRNA SMARTpool (siBNIP3L; Dharmacon, L‐011815‐00‐0005), according to the manufacturer's instructions. ON‐TARGETplus Non‐targeting pool (siNT; Dharmacon, D‐001810‐10‐20) was used as a control for non‐specific effects. For infections with 72 h pi time points, cell transfections with siRNAs were performed 6 h after infection for 24 h to maintain proper BNIP3L silencing which lasts up to 72 h post‐transfection (pt) in HeLa cells (Appendix Fig S3A).
Bacterial strains and culture
The strains used in this study were the smooth virulent B. abortus strain 544 NalR (spontaneous nalidixic acid resistant mutant strain) which is a B. abortus biovar 1 strain from J.‐M. Verger (INRA, Tours, France) and a variant constitutively expressing the monomeric green fluorescent protein (mGFP) caused by the integration of a plasmid containing the mGFP coding sequence under the control of the sojA promoter (plasmid pSK oriT kan PsojA‐mgfp, where mGFP replaces DsRed in pSK oriT kan PsojA‐dsRed; Copin et al, 2012). All B. abortus strains were grown in Tryptic Soy Broth‐rich medium (3% Bacto™ TSB, BD Biosciences) at 37°C under constant agitation.
All B. abortus strains were handled under Biosafety level 3 (BSL‐3) containment according to the Council Directive 98/81/EC of 26 October 1998, adopted by the Walloon government (4 July 2002).
Cell infection
For infections, HeLa cells and iBMDM were seeded either onto 13 mm glass coverslips (VWR) in 24‐well plates (Corning) (for microscopy: 2 × 104 cells/well for HeLa cells and 7 × 103 cells/well for iBMDM) or in 6‐well plates (Corning) (for flow cytometry: 1 × 105 cells/well for HeLa cells and 5 × 104 cells/well for iBMDM) 16 h before infection allowing cell adhesion and growth. In parallel, B. abortus cultures were incubated for 16 h in TSB medium from isolated colonies. The next day, bacterial cultures were washed twice in phosphate buffer saline (PBS, Lonza) and bacterial exponential growth was controlled by measuring culture optical density at 600 nm. B. abortus infection doses were prepared in complete HeLa culture medium at a multiplicity of infection (MOI) of 2,000 and incubated with the HeLa cells. Infected cells were centrifuged at 400 g for 10 min to favour bacteria‐to‐host cell contact and incubated for 1 h at 37°C in 5% CO2. Cells were then incubated with 50 μg/ml of gentamycin (Gibco) for 1 h to kill the remaining extracellular bacteria and thereafter washed and incubated with 10 μg/ml of gentamycin for the desired time points.
For reinfection experiments, infected cells were incubated in so‐called reinfection‐permissive conditions at 48 h pi in culture media without gentamycin to allow the survival of released bacteria and reinfection of new host cells. Cells were washed once in PBS and incubated with gentamycin‐free HeLa culture media until 72 h pi for further analysis.
CFU assay
To assess B. abortus intracellular replication at several time points (2, 6, 24 and 48 h pi), infected HeLa cells were washed once in PBS then lysed with 0.1% Triton‐X‐100 (Sigma‐Aldrich) in PBS for 10 min at room temperature. Serial supernatant dilutions were then plated onto TSB agar plates, incubated at 37°C for 5 days and colony‐forming units (CFUs) were counted for each condition from three technical replicates (Roba et al, 2022).
To assess B. abortus egress at 72 h pi, serial dilutions of supernatant from infected cells previously incubated in reinfection‐permissive conditions were plated onto TSB agar plates, incubated at 37°C for 5 days and colony‐forming units (CFUs) were counted for each condition from three technical replicates.
Immunofluorescence
At the different time points indicated in figure legends, cells seeded on coverslips were washed once with PBS, fixed with 4% paraformaldehyde (PFA; VWR) for 30 min at 37°C, then washed three times with PBS and permeabilised with 1% Triton‐X‐100 (Carl Roth) in PBS for 10 min. For LC3 immunostainings, fixation and permeabilisation was performed using pure ice‐cold methanol (−20°C) for 15 min at 4°C. Cells were then incubated with a blocking solution (2% bovine serum albumin [BSA; VWR] diluted in PBS) for 30 min at room temperature, and then incubated with the primary antibodies in blocking solution for 2 h at room temperature. Afterwards, cells were washed three times with the blocking solution and incubated with the secondary antibodies in blocking solution for 1 h a room temperature. For LAMP‐1 immunostainings, permeabilisation was performed using 0.2% saponin (Sigma‐Aldrich) diluted in PBS for 30 min, and the blocking solution was composed of 0.2% saponin, 2% BSA, 10% horse serum (HS, Gibco) diluted in PBS. DNA staining using Hoechst 33258 was performed during the incubation with the secondary antibody. Finally, cells were washed twice with blocking buffer and twice with distilled water, then mounted on glass slides in Mowiol (Sigma‐Aldrich) or Fluoromount‐G (Invitrogen™) as appropriate.
Light microscopy
Fixed and immunostained cells were visualised with a Leica TCS SP5 II confocal laser‐scanning microscope equipped with a HCX Plan Apo CS 40× numerical aperture (NA) 1.3 oil immersion objective. For observations requiring a super resolution, a Zeiss LSM 900 confocal laser‐scanning microscope equipped with an Airyscan 2 multiplex system and a Plan Apo 63× NA 1.4 oil immersion objective was used, in particular for the analysis of mitophagy (Figs 2C and 5D), reinfection foci (Figs 6A and 7A and E), aBCVs (Figs 6D, 7C and 8H) and mBCVs (Figs 8 and EV5B–F). For experiments assessing reinfection foci, the same microscope with a Plan Apo 40× NA 1.3 oil immersion objective was used instead. In addition, stimulated emission depletion (STED) microscopy analyses using an Abberior STEDycon attached to a Zeiss Axio Imager Z2 microscope were performed to better observe B. abortus localisation inside of mitochondria (Fig EV5A).
For live cell imaging, observations were made at 37°C with a Nikon Eclipse Ti2 inverted epifluorescence microscope equipped with a Plan Apo λ DM 100 X K 1.45/0.13 PH3 phase‐contrast objective to visualise the MitoSOX™ probe staining. Look‐up tables (LUT) were adjusted to the best signal–background ratio.
Quantitative analyses of fluorescence micrographs
All fluorescence images were analysed using FIJI v.2.1.0, a distribution of ImageJ.
Quantification of mitochondrial network morphology
The length and branching status of mitochondrial network were determined by calculating the aspect ratio (AR) and the end‐point/branched‐point ratio (EBR) of mitochondrial particles in entire cell sections as previously described (De Vos & Sheetz, 2007; Lobet et al, 2018). The AR represents the mean of the ratio between the long axis and the short axis of each mitochondrial fragments of one cell. The AR is therefore proportional to the length of mitochondrial fragments. The EBR represents the mean of the ratio between the end‐points and branched‐points of each mitochondrial fragments of one cell. The EBR is therefore proportional to the disconnected status of the mitochondrial network (Fig 1B).
Quantification of LC3 recruitment at the mitochondria proximity
The quantification of LC3 recruitment at the proximity of mitochondria was determined by analysing the co‐localisation between Alexa Fluor 568 (LC3) and Alexa Fluor 633 (β‐subunit of ATP synthase)‐related fluorescence signals. A signal threshold was thus applied on Alexa Fluor 568 micrographs to dissociate LC3‐type II punctae structures from LC3‐type I diffuse signal, as well as on Alexa Fluor 633 micrographs to delineate the mitochondrial network. The number of co‐localisation events between LC3 punctae and mitochondria was performed by generating on overlay of both processed images and counting the number of LC3‐β‐subunit of ATP synthase‐positive co‐localised events in entire cell sections.
Quantification of the number of mitochondrial fragments in acidified compartments
The number of mitochondrial fragments in acidified structures was determined by counting the number of FIS1‐mCherry(magenta)‐positive but GFP(green)‐negative structures in entire cell sections as previously described (Allen et al, 2013).
Quantification of the percentages of cells with a HIF‐1α nuclear pattern
The percentages of cells with a HIF‐1α nuclear pattern were determined by calculating the ratio between the number of cells that display a nuclear localisation of HIF‐1α and the total number of cells observed from one experiment.
Quantification of the percentages of reinfection events
The percentages of reinfection events were determined by calculating the percentages of reinfection foci as described previously (Starr et al, 2012). Reinfection foci were defined as a primary infected cell (containing a massive number of bacteria—72 h pi) surrounded by at least 4 adjacent cells containing a small number of bacteria (secondary infection—24 h pi).
Quantification of the number of aBCVs and mBCVs
The number of aBCVs was determined by counting the number of LAMP‐1‐positive vesicles harbouring 1 or more GFP‐positive B. abortus 544 bacteria in entire cell sections. The same procedure was applied to mBCVs by counting TOMM20‐positive BCVs.
Quantification of the percentages of TOMM20‐vesicles‐positive infected cells
The percentages of TOMM20‐vesicles‐positive infected cells was determined by calculating the ratio between the number of infected cells displaying large TOMM20‐positive vesicles and the total number of infected cells from one experiment.
Electron microscopy sample preparation
HeLa cells were seeded onto 32 mm gridded glass coverslips (Ibidi, Martinsried, Germany) in a 6‐well plate at 150,000 cells per well and incubated for 16 h before infection. In parallel, B. abortus 2308 were grown overnight in TSB medium at 37°C to an OD of 0.8–1.0. Bacteria were then diluted in DMEM/10% FCS and added to HeLa cells at a final MOI of 2,000. Plates were centrifuged at 400 g for 20 min at 4°C to synchronise bacterial entry. After 2 h of incubation at 37°C and 5% CO2, extracellular bacteria were killed by replacing the infection medium by DMEM/10% FCS supplemented with 100 μg/ml gentamicin. After the total infection time, cells were fixed using PHEM fixation buffer (4% formaldehyde, 0.2% glutaraldehyde, 60 mM PIPES, 25 mM HEPES, 10 mM EGTA, 4 mM MgCl2) for 90 min at room temperature. Following fixation, the coverslips were washed in PHEM buffer (60 mM PIPES, 25 mM HEPES, 10 mM EGTA, 4 mM MgCl2) and fixed in cacodylate fixation buffer (2.5% glutaraldehyde, 150 mM sodium cacodylate, 2 mM MgCl2) at 4°C overnight. Following overnight fixation, samples were washed 3 times with cacodylate buffer (150 mM sodium cacodylate, 2 mM MgCl2) at 4°C; and then immersed in freshly prepared reduced osmium buffer (2% osmium tetroxide, 150 mM sodium cacodylate, 2 mM MgCl2, 40 mM potassium ferrocyanide) for 1 h at 4°C. After this initial staining/fixation step, the samples were washed with deionised water at room temperature and immersed in 100 mM thiocarbohydrazide solution for 20 min at room temperature, and then washed with deionised water and incubated in 2% osmium tetroxide for 30 min at room temperature. This was followed by overnight incubation in 1% uranyl acetate at 4°C. The following morning, the samples were washed in deionised water and incubated in freshly prepared 20 mM lead aspartate solution for 30 min at 60°C. Samples were then dehydrated with ethanol and immersed in 50% solution of durcupan in ethanol for 1 h. Afterwards, the samples were incubated 2 times in fresh durcupan and placed at 60°C for 48 h for polymerisation.
Focused ion beam/scanning electron microscopy (FIB/SEM)
The cells of interest were located in the polymerised resin block, trimmed and attached to pre‐tilt 45° SEM stubs (Agar Scientific, Stansted, UK) using colloidal silver paint (Ted Pella, Redding, CA), sputter‐coated with platinum and subjected to FIB/SEM tomography. The images were acquired with a Helios NanoLab 650 Dual Beam FIB/SEM using the Slice and View software (FEI, Hillsboro, OR). They had 3072 × 2048 or 2048 × 1780 pixel and were collected using an Elstar in‐lens BSE detector at 1.5 kV with a horizontal field width of 15 μm at a working distance of 4.01 mm. The milling was performed with an FIB operating at 30 kV and 0.78 nA beam current. The thickness of the slices was between 10 and 20 nm. Image stacks were aligned using the TrackEM2 plugin for ImageJ (Cardona et al, 2010).
Flow cytometry
At the different time points indicated in figure legends, cells were washed once with PBS, detached with Trypsin 0.05%—EDTA (Gibco) for 3 min at 37°C and centrifuged at 400 g for 5 min at 4°C. Collected cells were washed twice with an ice‐cold Flow Cytometry buffer (0.5% BSA, 2 mM EDTA diluted in PBS) and then fixed with the IC Intracellular Fixation Buffer (eBioscience™ Invitrogen, 00‐8222‐49) for 30 min at room temperature. Fixed cells were permeabilised with the Permeabilisation Buffer 1 X (eBioscience™ Invitrogen, 00‐8333‐56) for 5 min at room temperature, then incubated with the primary antibodies in Permeabilisation Buffer for 1 h at room temperature. Cells were next washed once with Permeabilisation Buffer and incubated with the secondary antibodies in Permeabilisation Buffer for 1 h a room temperature. Finally, cells were washed once with Permeabilisation Buffer and transferred into glass tubes with Flow Cytometry buffer before analysis with the BD Biosciences FACSVerse™. Data analyses were performed using the FlowJo™ software (BD Biosciences).
Western blot analysis
At the different time points indicated in figure legend, cells were washed once with ice‐cold PBS and lysed in radioimmunoprecipitated assay (RIPA) buffer (150 mM NaCl, 1% NP40, 0.1% SDS, 1% sodium deoxycholate, 25 mM Tris–HCL; pH 7.6) supplemented with complete Protease Inhibitor Cocktail (Roche, 11697498001) and 4% Phosphatase Inhibitor Buffer (25 mM Na3VO4, 250 mM 4‐nitrophenylphosphate, 250 mM β‐glycerophosphate, 125 mM NaF) for 10 min on ice. Cell lysates were centrifuged at 13,000 g for 10 min at 4°C. Clear cell lysates were then incubated for 1 h at 80°C to kill remaining bacteria. Protein concentration was assessed with the Pierce 660 nm Protein Assay Reagent (Thermo Scientific, 22660). Samples of 10 μg of cell lysate proteins were resolved by gel electrophoresis using a 10% acrylamide gel. Proteins were then electro‐transferred onto a polyvinylidene fluoride (PVDF) membrane (0.45 μm; Merck by ice‐cold liquid transfer for 2 h at 70 V). Membranes were then incubated with Licor Intercept (PBS) Blocking buffer (LICOR Biosciences, 927‐70003) diluted twice in PBS for 1 h at room temperature, and then incubated with the primary antibody diluted in Intercept Blocking buffer supplemented with 0.1% Tween20 (Carl Roth, 9127.1) and 0.2% sodium dodecyl sulfate (SDS; VWR, A3942) for 16 h at 4°C. Membranes were then washed three times with 0.1% Tween20 diluted in PBS, and then incubated with the secondary antibody diluted in Intercept Blocking buffer supplemented with 0.1% Tween20 and 0.2% SDS for 1 h at room temperature. Membranes were then washed twice with 0.1% Tween20 in PBS, once in PBS, and the fluorescence intensity of the bands corresponding to the proteins of interest were detected using the Odyssey ODY‐1869 scanner (LICOR). The immunodetection of the β‐actin was used as a loading control.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 9 software. All data are presented as the means ± standard deviation (SD) of results from at least three biological independent experiments (n = 3). Normality of the distribution was assessed using the Shapiro–Wilk tests. Comparisons between two conditions were assessed using an unpaired two‐tailed Student's t‐test, or a one sample t‐test when normality failed. Comparisons between more than two groups involving one single factor were assessed using a one‐way ANOVA followed by a Tukey's multiple comparisons test. Comparisons between more than two groups involving two simultaneous factors were assessed using a two‐way ANOVA followed by a Šidàk's multiple comparisons test, or a multiple Mann–Whitney test followed by a Holm‐Šidàk's multiple comparisons test when normality failed. A P‐value <0.05 was considered as statistically significant.
Author contributions
Jérémy Verbeke: Conceptualization; formal analysis; funding acquisition; investigation; writing – original draft; writing – review and editing. Youri Fayt: Formal analysis; investigation. Lisa Martin: Formal analysis; investigation. Oya Yilmaz: Formal analysis; investigation. Jaroslaw Sedzicki: Resources; formal analysis; investigation; writing – review and editing. Angéline Reboul: Conceptualization; writing – review and editing. Michel Jadot: Conceptualization; writing – review and editing. Patricia Renard: Conceptualization; writing – review and editing. Christoph Dehio: Resources; funding acquisition; writing – review and editing. Henri‐François Renard: Conceptualization; funding acquisition; writing – review and editing. Jean‐Jacques Letesson: Conceptualization; writing – review and editing. Xavier De Bolle: Conceptualization; supervision; funding acquisition; writing – original draft; writing – review and editing. Thierry Arnould: Conceptualization; supervision; funding acquisition; writing – original draft; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
PDF+
Source Data for Expanded View and Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
The authors would like to thank Prof. Suzana Salcedo (University of Lyon, France) for her helpful discussions and for the generous gift of iBMDM, as well as Prof. Carles Canto (Ecole Polytechnique Fédérale de Lausanne, Switzerland) for his helpful discussions. The authors also thank the “Morphology and Imaging” (Morph‐Im) technological platform (University of Namur) and especially Catherine Demazy and Noëlle Ninane for the advice and trainings for the initial use of the Leica TCS SP5 II confocal microscope. We also thank Dr. David Pla‐Martín for generously providing the FIS1‐GFP‐mCherry reporter construct. Jérémy Verbeke is a Research Fellow (2018‐2022) of the F.R.S‐FNRS (Fonds de la Recherche Scientifique, Belgium). This work was also supported by two “Crédit de Recherche” grants (CDR 2019‐2021: “MITOCHOBRU” grant J.0003.20‐AID 35252856 and CDR 2022‐2023: “Brucella and BNIP3L‐mediated mitophagy” grant J.0003.22 AID 40007965) obtained from the F.R.S‐FNRS, as well as by a Start‐Up grant obtained from the Fondation Francqui (to Henri‐François Renard) and the grant 310030B_201273 obtained from the Swiss National Science Foundation (to Christoph Dehio).
The EMBO Journal (2023) 42: e112817
Data availability
All data are available in the main text or the expanded view section. This study includes no data deposited in external repositories.
References
- Agarwal S, Muqit MMK (2022) PTEN‐induced kinase 1 (PINK1) and Parkin: unlocking a mitochondrial quality control pathway linked to Parkinson's disease. Curr Opin Neurobiol 72: 111–119 [DOI] [PubMed] [Google Scholar]
- Allen GFG, Toth R, James J, Ganley IG (2013) Loss of iron triggers PINK1/Parkin‐independent mitophagy. EMBO Rep 14: 1127–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson ES, Paulley JT, Roop RM II (2008) The AraC‐like transcriptional regulator DhbR is required for maximum expression of the 2,3‐dihydroxybenzoic acid biosynthesis genes in Brucella abortus 2308 in response to iron deprivation. J Bacteriol 190: 1838–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atluri VL, Xavier MN, De Jong MF, Den Hartigh AB, Tsolis RM (2011) Interactions of the human pathogenic Brucella species with their hosts. Annu Rev Microbiol 65: 523–541 [DOI] [PubMed] [Google Scholar]
- von Bargen K, Gorvel JP, Salcedo SP (2012) Internal affairs: investigating the Brucella intracellular lifestyle. FEMS Microbiol Rev 36: 533–562 [DOI] [PubMed] [Google Scholar]
- Bell EL, Klimova TA, Eisenbart J, Schumacker PT, Chandel NS (2007) Mitochondrial reactive oxygen species trigger hypoxia‐inducible factor‐dependent extension of the replicative life span during hypoxia. Mol Cell Biol 27: 5737–5745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasi E, Radzioch D, Durum SK, Varesio L (1987) A murine macrophage cell line, immortalized by v‐raf and v‐myc oncogenes, exhibits normal macrophage functions. Eur J Immunol 17: 1491–1498 [DOI] [PubMed] [Google Scholar]
- Borghesan E, Smith EP, Myeni S, Binder K, Knodler LA, Celli J (2021) A Brucella effector modulates the Arf6‐Rab8a GTPase cascade to promote intravacuolar replication. EMBO J 40: 1–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boschiroli ML, Ouahrani‐Bettache S, Foulongne V, Michaux‐Charachon S, Bourg G, Allardet‐Servent A, Cazevieille C, Liautard JP, Ramuz M, O'Callaghan D (2002) The Brucella suis virB operon is induced intracellularly in macrophages. Proc Natl Acad Sci USA 99: 1544–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutry M, Kim PK (2021) ORP1L mediated PI(4)P signaling at ER‐lysosome‐mitochondrion three‐way contact contributes to mitochondrial division. Nat Commun 12: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byndloss MX, Tsai AY, Walker GT, Miller CN, Young BM, English BC, Seyffert N, Kerrinnes T, de Jong MF, Atluri VL et al (2019) Brucella abortus infection of placental trophoblasts triggers endoplasmic reticulum stress‐mediated cell death and fetal loss via type IV secretion system‐dependent activation of CHOP. MBio 10: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns G, Thumiah‐Mootoo M, Burelle Y, Khacho M (2020) Mitophagy: a new player in stem cell biology. Biology (Basel) 9: 1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona A, Saalfeld S, Preibisch S, Schmid B, Cheng A, Pulokas J, Tomancak P, Hartenstein V (2010) An integrated micro- and macroarchitectural analysis of the Drosophila brain by computer-assisted serial section electron microscopy. PLoS Biol 8: e1000502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho F, Spier A, Chaze T, Matondo M, Cossart P (2020) Listeria monocytogenes exploits mitochondrial contact site and cristae organizing system complex subunit Mic10 to. MBio 11: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli J, de Chastellier C, Franchini D‐M, Pizarro‐Cerda J, Moreno E, Gorvel J‐P (2003) Brucella evades macrophage killing via VirB‐dependent sustained interactions with the endoplasmic reticulum. J Exp Med 198: 545–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli J, Salcedo SP, Gorvel J‐P (2005) Brucella coopts the small GTPase Sar1 for intracellular replication. Proc Natl Acad Sci USA 102: 1673–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li Y, Han Z, Chen L, Gao R, Liu L et al (2016) Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 12: 689–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury SR, Reimer A, Sharan M, Kozjak‐Pavlovic V, Eulalio A, Prusty BK, Fraunholz M, Karunakaran K, Rudel T (2017) Chlamydia preserves the mitochondrial network necessary for replication via microRNA‐dependent inhibition of fission. J Cell Biol 216: 1071–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloeckaert A, De Wergifosse P, Dubray G, Limet JN (1990) Identification of seven surface‐exposed Brucella outer membrane proteins by use of monoclonal antibodies: immunogold labeling for electron microscopy and enzyme‐linked immunosorbent assay. Infect Immun 58: 3980–3987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway JRW, Warren SC, Herrmann D, Murphy KJ, Cazet AS, Vennin C, Shearer RF, Killen MJ, Magenau A, Mélénec P et al (2018) Intravital imaging to monitor therapeutic response in moving hypoxic regions resistant to PI3K pathway targeting in pancreatic cancer. Cell Rep 23: 3312–3326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copin R, Vitry MA, Hanot Mambres D, Machelart A, de Trez C, Vanderwinden JM, Magez S, Akira S, Ryffel B, Carlier Y et al (2012) In situ microscopy analysis reveals local innate immune response developed around Brucella infected cells in resistant and susceptible mice. PLoS Pathog 8: e1002575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai ZJ, Gao J, Bin MX, Yan K, Liu XX, Kang HF, Ji ZZ, Guan HT, Wang XJ (2012) Up‐regulation of hypoxia inducible factor‐1α by cobalt chloride correlates with proliferation and apoptosis in PC‐2 cells. J Exp Clin Cancer Res 31: 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daskalaki I, Gkikas I, Tavernarakis N (2018) Hypoxia and selective autophagy in cancer development and therapy. Front Cell Dev Biol 6: 1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Sheetz MP (2007) Visualization and quantification of mitochondrial dynamics in living animal cells. Methods Cell Biol 80: 627–682 [DOI] [PubMed] [Google Scholar]
- Denison SR, Wang F, Becker NA, Schüle B, Kock N, Phillips LA, Klein C, Smith DI (2003) Alterations in the common fragile site gene Parkin in ovarian and other cancers. Oncogene 22: 8370–8378 [DOI] [PubMed] [Google Scholar]
- Dikic I, Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19: 349–364 [DOI] [PubMed] [Google Scholar]
- Eiyama A, Kondo‐Okamoto N, Okamoto K (2013) Mitochondrial degradation during starvation is selective and temporally distinct from bulk autophagy in yeast. FEBS Lett 587: 1787–1792 [DOI] [PubMed] [Google Scholar]
- Elhassanny AEM, Anderson ES, Menscher EA, Roop RM II (2013) The ferrous iron transporter FtrABCD is required for the virulence of Brucella abortus 2308 in mice. Mol Microbiol 88: 1070–1082 [DOI] [PubMed] [Google Scholar]
- Escoll P, Song OR, Viana F, Steiner B, Lagache T, Olivo‐Marin JC, Impens F, Brodin P, Hilbi H, Buchrieser C (2017) Legionella pneumophila modulates mitochondrial dynamics to trigger metabolic repurposing of infected macrophages. Cell Host Microbe 22: 302–316.e7 [DOI] [PubMed] [Google Scholar]
- Fine‐Coulson K, Giguère S, Quinn FD, Reaves BJ (2015) Infection of A549 human type II epithelial cells with Mycobacterium tuberculosis induces changes in mitochondrial morphology, distribution and mass that are dependent on the early secreted antigen, ESAT‐6. Microbes Infect 17: 689–697 [DOI] [PubMed] [Google Scholar]
- Fu ZJ, Wang ZY, Xu L, Chen XH, Li XX, Liao WT, Ma HK, Jiang MD, Xu TT, Xu J et al (2020) HIF‐1α‐BNIP3‐mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol 36: 101671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley IG, Simonsen A (2022) Diversity of mitophagy pathways at a glance. J Cell Sci 135: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Scorrano L (2013) Mitochondrial morphology in mitophagy and macroautophagy. Biochim Biophys Acta 1833: 205–212 [DOI] [PubMed] [Google Scholar]
- Gomes MTR, Guimarães ES, Marinho FV, Macedo I, Aguiar ERGR, Barber GN, Moraes‐Vieira PMM, AlvesFilho JC, Oliveira SC (2021) STING regulates metabolic reprogramming in macrophages via HIF‐1α during Brucella infection. PLoS Pathog 17: 1–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González‐Espinoza G, Arce‐Gorvel V, Mémet S, Gorvel JP (2021) Brucella: reservoirs and niches in animals and humans. Pathogens 10: 1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Zhang YX, Wang T, Zhong ML, Yang ZH, Hao LJ, Chai R, Zhang S (2015) Intranasal deferoxamine attenuates synapse loss via up‐regulating the P38/HIF‐1α pathway on the brain of APP/PS1 transgenic mice. Front Aging Neurosci 7: 101671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailey DW, Rambold AS, Satpute‐Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott‐Schwartz J (2010) Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141: 656–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y et al (2013) Autophagosomes form at ER‐mitochondria contact sites. Nature 495: 389–393 [DOI] [PubMed] [Google Scholar]
- Hara Y, Yanatori I, Tanaka A, Kishi F, Lemasters JJ, Nishina S, Sasaki K, Hino K (2020) Iron loss triggers mitophagy through induction of mitochondrial ferritin. EMBO Rep 21: 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2003) LC3, a mammalian homolog of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 22: 4577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D, Kirienkoa DR, Webster P, Fisher AL, Kirienko NV (2018) Pyoverdine, a siderophore from Pseudomonas aeruginosa, translocates into C. elegans, removes iron, and activates a distinct host response. Virulence 9: 804–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y, Wang Y, Li W, Chen Z (2015) Type IV secretion system of Brucella spp. and its effectors. Front Cell Infect Microbiol 5: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P, Chandel NS, Simon MC (2020) Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol 21: 268–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wu K, Zeng S, Zhao F, Fan J, Li Z, Yi L, Ding H, Zhao M, Fan S et al (2021) Viral infection modulates mitochondrial function. Int J Mol Sci 22: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Straub J, Medeiros TC, Mehra C, Den Brave F, Peker E, Atanassov I, Stillger K, Michaelis JB, Brubridge E et al (2022) Mitochondria shed their outer membrane in response to infection‐induced stress. Science 375: eabi4343 [DOI] [PubMed] [Google Scholar]
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W et al (2012) Mitochondrial outer‐membrane protein FUNDC1 mediates hypoxia‐induced mitophagy in mammalian cells. Nat Cell Biol 14: 177–185 [DOI] [PubMed] [Google Scholar]
- Liu Z, Reba S, Chen WD, Porwal SK, Boom WH, Petersen RB, Rojas R, Viswanathan R, Devireddy L (2014) Regulation of mammalian siderophore 2,5‐DHBA in the innate immune response to infection. J Exp Med 211: 1197–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Xu C, Zhang W, Cao Y, Ye J, Li B, Jia S, Weng L, Liu Y, Liu L et al (2022) FUNDC1‐mediated mitophagy and HIF1α activation drives pulmonary hypertension during hypoxia. Cell Death Dis 13: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobet E, Letesson JJ, Arnould T (2015) Mitochondria: a target for bacteria. Biochem Pharmacol 94: 173–185 [DOI] [PubMed] [Google Scholar]
- Lobet E, Willemart K, Ninane N, Demazy C, Sedzicki J, Lelubre C, De Bolle X, Renard P, Raes M, Dehio C et al (2018) Mitochondrial fragmentation affects neither the sensitivity to TNFα‐induced apoptosis of Brucella‐infected cells nor the intracellular replication of the bacteria. Sci Rep 8: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahla RS, Kumar A, Tutill HJ, Krishnaji ST, Sathyamoorthy B, Noursadeghi M, Breuer J, Pandey AK, Kumar H (2021) NIX‐mediated mitophagy regulate metabolic reprogramming in phagocytic cells during mycobacterial infection. Tuberculosis 126: 102046 [DOI] [PubMed] [Google Scholar]
- Mak WW, Masnammany AM, Kori AN (2019) Brucellosis‐induced autoimmune haemolytic anaemia (AIHA). Med J Malaysia 74: 443–444 [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F et al (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189: 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros TC, Mehra C, Pernas L (2021) Contact and competition between mitochondria and microbes. Curr Opin Microbiol 63: 189–194 [DOI] [PubMed] [Google Scholar]
- Meena DS, Sonwal VS, Rohila AK, Meena V (2018) Acute brucellosis presenting as an autoimmune hemolytic anemia. Case Rep Infect Dis 2018: 1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CN, Smith EP, Cundiff JA, Knodler LA, Bailey J, Lupashin V, Celli J (2018) A Brucella type IV effector targets the COG tethering complex to remodel host secretory traffic and promote intracellular replication. Cell Host Microbe 22: 317–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, Kelly B, O'Neill LAJ (2017) Mitochondria are the powerhouses of immunity. Nat Immunol 18: 488–498 [DOI] [PubMed] [Google Scholar]
- Missiroli S, Genovese I, Perrone M, Vezzani B, Vitto VAM, Giorgi C (2020) The role of mitochondria in inflammation: from cancer to neurodegenerative disorders. J Clin Med 9: 740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Rajesh M, Yoshihiro K, Haskó G, Pacher P (2007) Simple quantitative detection of mitochondrial superoxide production in live cells. Biochem Biophys Res Commun 358: 203–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi M, Okamoto K (2021) Mitochondrial clearance: mechanisms and roles in cellular fitness. FEBS Lett 595: 1239–1263 [DOI] [PubMed] [Google Scholar]
- Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K (2021) Molecular mechanisms and physiological functions of mitophagy. EMBO J 40: 1–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent MA, Bellaire BH, Murphy EA, Roop RM, Elzer PH, Baldwin CL (2002) Brucella abortus siderophore 2,3‐dihydroxybenzoic acid (DHBA) facilitates intracellular survival of the bacteria. Microb Pathog 32: 239–248 [DOI] [PubMed] [Google Scholar]
- Paulley JT, Anderson ES, Roop RM II (2007) Brucella abortus requires the heme transporter BhuA for maintenance of chronic infection in BALB/c mice. Infect Immun 75: 5248–5254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roba A, Carlier E, Godessart P, Naili C, De Bolle X (2022) Histidine auxotroph mutant is defective for cell separation and allows the identification of crucial factors for cell division in Brucella abortus . Mol Microbiol 118: 145–154 [DOI] [PubMed] [Google Scholar]
- Roop RM II (2012) Metal acquisition and virulence in Brucella. Anim Health Res Rev 13: 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roop RM II, Elhassanny AE, Almirón MA, Anderson ES, Atkinson XJ (2017) Chapter 2 iron. In Metals and the biology and virulence of Brucella, Roop RM II, Caswell CC (eds), pp 9–39. Cham: Springer International Publishing; [Google Scholar]
- Roset MS, Alefantis TG, DelVecchio VG, Briones G (2017) Iron‐dependent reconfiguration of the proteome underlies the intracellular lifestyle of Brucella abortus . Sci Rep 7: 10637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo SP, Chevrier N, Lacerda TLS, Ben Amara A, Gerart S, Gorvel VA, De Chastellier C, Blasco JM, Mege JL, Gorvel JP (2013) Pathogenic Brucellae replicate in human trophoblasts. J Infect Dis 207: 1075–1083 [DOI] [PubMed] [Google Scholar]
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal T, Chen M, Wang J (2008) Essential role for nix in autopagic maturation of erythroid cells. Nature 454: 232–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassera D, Beninati T, Bandi C, Bouman EAP, Sacchi L, Fabbi M, Lo N (2006) ‘Candidatus Midichloria mitochondrii’, an endosymbiont of the Ixodes ricinus with a unique intramitochondrial lifestyle. Int J Syst Evol Microbiol 56: 2535–2540 [DOI] [PubMed] [Google Scholar]
- Schofield CJ, Ratcliffe PJ (2004) Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol 5: 343–354 [DOI] [PubMed] [Google Scholar]
- Sedzicki J, Tschon T, Low SH, Willemart K, Goldie KN, Letesson JJ, Stahlberg H, Dehio C (2018) 3D correlative electron microscopy reveals continuity of Brucella‐containing vacuoles with the endoplasmic reticulum. J Cell Sci 131: jcs210799 [DOI] [PubMed] [Google Scholar]
- Shi J, Yu J, Zhang Y, Wu L, Dong S, Wu L, Wu L, Du S, Zhang Y, Ma D (2019) PI3K/Akt pathway‐mediated HO‐1 induction regulates mitochondrial quality control and attenuates endotoxin‐induced acute lung injury. Lab Invest 99: 1795–1809 [DOI] [PubMed] [Google Scholar]
- Shu L, Hu C, Xu M, Yu J, He H, Lin J, Sha H, Lu B, Engelender S, Guan M et al (2021) ATAD3B is a mitophagy receptor mediating clearance of oxidative stress‐induced damaged mitochondrial DNA. EMBO J 40: 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva Rosa SC, Martens MD, Field JT, Nguyen L, Kereliuk SM, Hai Y, Chapman D, Diehl‐Jones W, Aliani M, West AR et al (2021) BNIP3L/nix‐induced mitochondrial fission, mitophagy, and impaired myocyte glucose uptake are abrogated by PRKA/PKA phosphorylation. Autophagy 17: 2257–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson CL, Tokito MK, Uppala R, Sarkar MK, Gudjonsson JE, Holzbaur ELF (2021) NIX initiates mitochondrial fragmentation via DRP1 to drive epidermal differentiation. Cell Rep 34: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirianni A, Krokowski S, Lobato‐Márquez D, Buranyi S, Pfanzelter J, Galea D, Willis A, Culley S, Henriques R, Larrouy‐Maumus G et al (2016) Mitochondria mediate septin cage assembly to promote autophagy of Shigella. EMBO Rep 17: 1029–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JA, Khan M, Magnani DD, Harms JS, Durward M, Radhakrishnan GK, Liu YP, Splitter GA (2013) Brucella induces an unfolded protein response via TcpB that supports intracellular replication in macrophages. PLoS Pathog 9: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EP, Cotto‐Rosario A, Borghesan E, Held K, Miller CN, Celli J (2020) Epistatic interplay between type IV secretion effectors engages the small GTPase Rab2 in the Brucella intracellular cycle. MBio 11: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto‐Heredero G, Baixauli F, Mittelbrunn M (2017) Interorganelle communication between mitochondria and the endolysosomal system. Front Cell Dev Biol 5: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, McBride HM (2012) A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol 22: 135–141 [DOI] [PubMed] [Google Scholar]
- Spier A, Stavru F, Cossart P (2019) Interaction between intracellular bacterial pathogens and host cell mitochondria. Microbiol Spectr 7: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T, Ng TW, Wehrly TD, Knodler LA, Celli J (2008) Brucella intracellular replication requires trafficking through the late endosomal/lysosomal compartment. Traffic 9: 678–694 [DOI] [PubMed] [Google Scholar]
- Starr T, Child R, Wehrly TD, Hansen B, Hwang S, López‐Otin C, Virgin HW, Celli J (2012) Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe 11: 33–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavru F, Riemer J, Jex A, Sassera D (2020) When bacteria meet mitochondria: the strange case of the tick symbiont Midichloria mitochondrii†. Cell Microbiol 22: 1–9 [DOI] [PubMed] [Google Scholar]
- Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ, Rizzuto R (2004) Drp‐1‐dependent division of the mitochondrial network blocks intraorganellar calcium waves and protects against calcium‐mediated apoptosis. Mol Cell 16: 59–68 [DOI] [PubMed] [Google Scholar]
- Wang Y, Nartiss Y, Steipe B, McQuibban GA, Kim PK (2012) ROS‐induced mitochondrial depolarization initiates PARK2/PARKIN‐dependent mitochondrial degradation by autophagy. Autophagy 8: 1462–1476 [DOI] [PubMed] [Google Scholar]
- Ward DM, Cloonan SM (2019) Mitochondrial Iron in human health and disease. Annu Rev Physiol 10: 453–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita SI, Jin X, Furukawa K, Hamasaki M, Nezu A, Otera H, Saigusa T, Yoshimori T, Sakai Y, Mihara K et al (2016) Mitochondrial division occurs concurrently with autophagosome formation but independently of Drp1 during mitophagy. J Cell Biol 215: 649–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachari M, Ktistakis NT (2020) Mammalian mitophagosome formation: a focus on the early signals and steps. Front Cell Dev Biol 8: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yao Y, Qiu X, Wang G, Hu Z, Chen S, Wu Z, Yuan N, Gao H, Wang J et al (2019) Listeria hijacks host mitophagy through an novel mitophagy receptor to evade killing. Nat Immunol 20: 433–446 [DOI] [PubMed] [Google Scholar]