

Abstract
Recent clinical reports have highlighted the need for wild-type (WT) and mutant dual inhibitors of c-MET kinase for the treatment of cancer. We report herein a novel chemical series of ATP competitive type-III inhibitors of WT and D1228V mutant c-MET. Using a combination of structure-based drug design and computational analyses, ligand 2 was optimized to a highly selective chemical series with nanomolar activities in biochemical and cellular settings. Representatives of the series demonstrate excellent pharmacokinetic profiles in rat in vivo studies with promising free-brain exposures, paving the way for the design of brain permeable drugs for the treatment of c-MET driven cancers.
Introduction
Mesenchymal-to-epithelial transition factor (c-MET) is a receptor tyrosine kinase whose only known natural ligand is hepatocyte growth factor (HGF). The HGF-dependent activation of c-MET plays a crucial role in the stimulation of epithelial-mesenchymal transition in tumor cells, which leads to increased tumor cell proliferation, angiogenesis, tumor survival and metastasis.1 c-MET hyperactivity has been linked to numerous cancers including nonsmall cell lung carcinoma (NSCLC), papillary renal cell and hepatocellular carcinoma.1 Targeting the HGF/c-MET signaling pathway is being explored in numerous clinical trials, and a number of small molecule inhibitors including crizotinib,2 cabozantinib3 and savolitinib4,5 have been approved by the FDA.6−8 Notably, c-MET overexpression and/or amplification is also a prevalent resistance mechanism to both first- and second-line treatment with the third-generation epidermal growth factor receptor (EGFR) inhibitor osimertinib,9 in patients with advanced NSCLC.10−12 These findings have resulted in the design of recent clinical trials investigating the efficacy of combination treatments of osimertinib with the c-MET inhibitor savolitinib (1).13,14
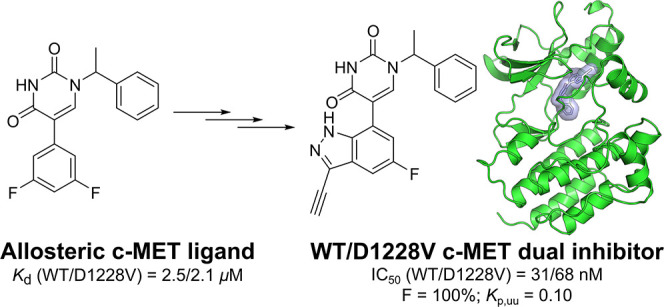
Numerous c-MET inhibitors have been reported, broadly falling into two categories:15 type-I and type-II inhibitors,7 both of which bind to the ATP binding site of the kinase. Type-I c-MET inhibitors such as crizotinib,2,16 PF-0421790317 and savolitinib4 (1) (Figure 1a) are typically exquisitely selective for c-MET over other kinases, with the selectivity being mainly driven by a highly specific π-stacking interaction between the ubiquitous aromatic system of these inhibitors and Y1230's side chain within the activation loop (A-loop) of the kinase (Figure 1b).18 However, recent findings describe the emergence of A-loop mutations as resistance mechanisms to these selective inhibitors, rendering them ineffective. D1228V19 and Y1230H20 represent two examples of clinically relevant single-point mutations of c-MET. It is postulated that the latter leads to a direct disruption of the crucial π-stacking interaction to type-I inhibitors.18 The former leads to an indirect disruption following the misalignment of the Y1230 residue through the loss of the salt bridge between D1228 and the catalytic lysine residue K1110.
Figure 1.
(a) Type I c-MET inhibitor savolitinib (1). (b) Crystal structure of WT c-MET bound by savolitinib (1) demonstrating the characteristic π-stacking interaction with the Y1230 side chain.
Type-II inhibitors of c-MET such as foretinib21 and cabozantinib,3 which bind to the active site of the kinase and further extend into the back-pocket, have been reported to be active against WT and the D1228V mutation.18,19,22 However, these inhibitors commonly suffer from suboptimal physicochemical properties, inferior selectivity for c-MET across the kinome and thus a higher potential risk of off-target toxicity.19 Together, these features of type-II inhibitors significantly limit the feasibility of addressing brain tumors23 related to an overexpression of c-MET24 and brain metastases of c-MET related cancers.25,26 c-MET amplification and overexpression has been associated with 5 and 13% of glioblastomas respectively,24 emphasizing the need for brain penetrant c-MET inhibitors.27
There is therefore an unmet need for brain penetrant c-MET inhibitors, active against WT and the reported resistance mutants, to enable thorough probing of their efficacy in the treatment of c-MET driven cancers. We disclose herein, the discovery of a novel ATP competitive type-III c-MET inhibitor series toward this unique profile.
Results and Discussion
Hit Identification
A lead generation campaign aimed at discovering c-MET inhibitors with novel binding modes and mechanisms of action (MOAs) was carried out. It was postulated that orthosteric inhibitors (type-I and type-II), which do not rely on the aforementioned π-stacking interaction with Y1230, and/or allosteric inhibitors (type-III and type-IV),15,28 could potentially provide the desired activity profile. In order to cover a broad chemical space efficiently29 and to probe for novel binding modes, a fragment-based biophysical screening approach was used. By having a small and efficient starting point, an inhibitor’s polar surface area (PSA), hydrogen-bond donor (HBD) and hydrogen-bond acceptor (HBA) count can be regulated from an early stage in the hit to lead optimization process and thus maximize chances of achieving brain permeability.
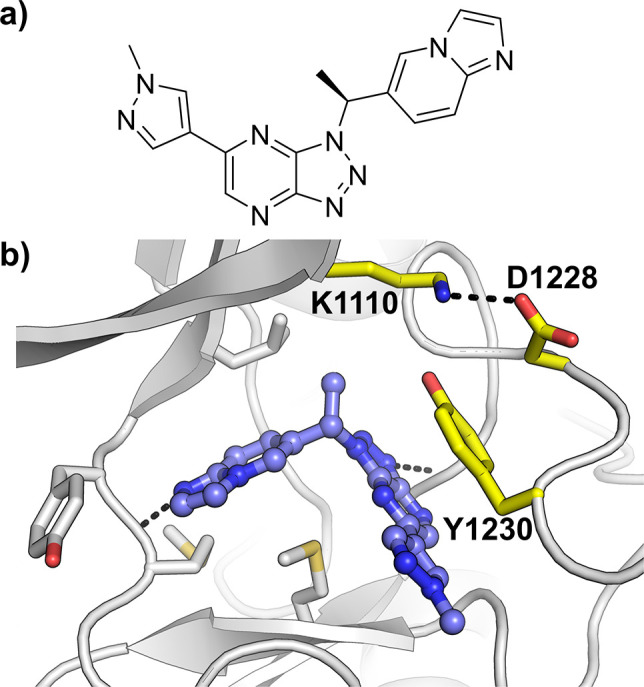
A previously described 1D ligand-observed NMR method was used to identify fragments that bind to WT c-MET.30 The screen was carried out in the presence of adenosine, as a reporter of hinge binding, in order to get an early appreciation of the binding mode of the active fragments. Subsequently, the binding affinities of the hits were assessed in both WT and the clinically relevant D1228V c-MET mutant by SPR. Alongside AstraZeneca’s NMR-subset fragment collection,31,32 a focused set of fragments and low molecular weight lead-like molecules,33 which have shown novel binding modes and/or high ligand efficiency (LE)34,35 for kinases from past internal programs, was also screened. These efforts resulted in the identification of the ligand efficient pyrimidine-2,4-dione 2 (LE = 0.33 kcal mol–1 per heavy atom), which was shown to be equipotent against both WT and D1228V c-MET, with SPR Kd values of 2.5 and 2.1 μM respectively (Figure 2a and Table 1).
Figure 2.
(a) Novel c-MET ligand 2. LE expressed in kcal mol–1 per heavy atom. (b) Crystal structure of D1228V c-MET bound by 2.
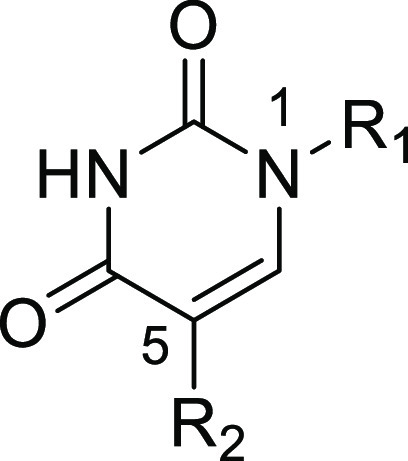
Table 1. Early SAR Exploration of the Pyrimidine-2,4-dione Scaffold.
Affinity data based on n ≥ 2 with SEM within 0.2 log units, unless otherwise stated.
LLE = pKd – log D.
Log D measured via shake-flask method in octanol and water at pH7.4.
Rat hepatocytes.
Human liver microsomes.
n = 1.
SEM = 0.25 log units.
Biophysical and Structural Characterization of 2 for c-MET
1D ligand-observed NMR experiments, where 2 was added to a pre-equilibrated sample containing adenosine in the presence of WT c-MET, demonstrated clear competition for binding to the kinase (Supporting Information, Figure S1). However, co-crystal structures of 2 bound to WT c-MET and the D1228V mutant (Figure 2b and Supporting Information, Figure S2a) revealed the compound to bind exclusively to the back-pocket of the kinase in both instances, with no interactions with the hinge region nor with the key residue Y1230. As shown in Figure 2b, two hydrogen bonds (H-bonds) from the pyrimidine-2,4-dione to the backbone atoms of D1222, anchor the compound in the back-pocket binding site with the unsubstituted phenyl ring residing in the region normally occupied by the F1223 of the conserved aspartate-phenylalanine-glycine (DFG) motif. Interestingly, by displacing the DFG motif, 2 locks F1223 in an orientation that significantly overlaps with the position of ATP as seen in the ATP-bound WT c-MET crystal structure.36 This provides a clear structural rationale for the adenosine competition observed in the aforementioned NMR experiment. By binding exclusively to the kinase back-pocket, the binding mode of 2 falls under the “type-III” category,15,28 and as far as we are aware, this is the first example of this allosteric binding mode for c-MET.
In Silico Binding-Site Analysis
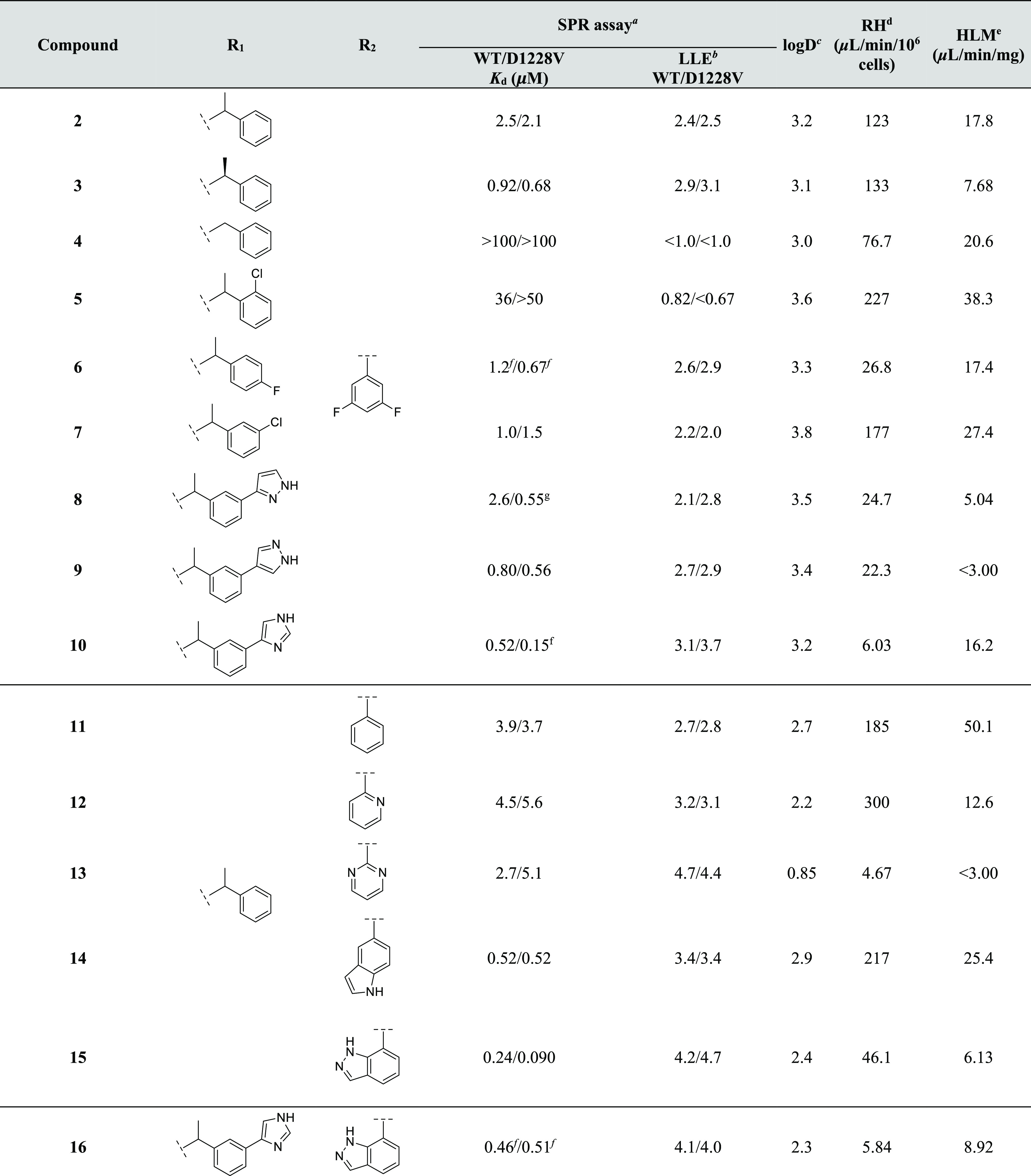
In order to guide structure-based design, the binding pocket of the D1228V c-MET structure was characterized using the program SiteMap (Figure 3a).37 Overall, the site appears to be “druggable”, with reported SiteScore (1.225) and Dscore (1.259) values above the average of 1.0 for sub-μM compatible binding sites.38 The pocket is almost fully enclosed by protein and has an approximate volume of 250 Å3. The two phenyl rings of 2 occupy hydrophobic regions of the pocket as does a large region of the central pyrimidine-2,4-dione ring. Its hydrophilic portion interacts with the D1222 residue, as described above. The ligand’s second oxygen is in the vicinity of K1110 but does not appear to form a strong interaction with the basic nitrogen. There are two HBD interaction sites near F1124 and L1140, and 2 appears to satisfy both through weak aromatic H-bonds to the backbone carbonyl of each residue. There is a cluster of several HBD/HBA interaction sites near H1202, D1222 and R1227. Compound 2 does not reach into this polar subpocket, but a crystallographic water (I in Figure 2b) binds here and is within H-bonding distance to the H1202 carbonyl and D1222 carboxylate oxygens (2.9 and 2.8 Å, respectively).
Figure 3.
(a) SiteMap analysis of the D1228V c-Met backpocket with 2 shown in green ball-and-stick representation. The molecular surface of the binding site is shown in gray. Hydrophobic, HBD and HBA interaction sites are represented by yellow, blue and red surfaces, respectively. H-bonds and aromatic H-bonds are represented by yellow and blue dashed lines, respectively. (b) WaterMap analysis of the co-crystal structure of 2 bound to the D1228V c-MET back-pocket (Chain A). Water sites identified by WaterMap inside the pocket are shown as spheres and are colored by calculated excess free energy ranging from stable (green) to unstable (red).
To further characterize the c-MET back-pocket, WaterMap39,40 was applied to identify putative water sites and to estimate their thermodynamic profiles relative to bulk water. The program relies on molecular dynamics (MD) simulation and clustering of explicit water molecules in a pocket. Furthermore, it applies inhomogeneous solvation theory41 to determine the excess free energy (ΔG), enthalpy (ΔH), and entropy (−TΔS) of identified water sites. Four putative hydration sites were identified around 2 inside the pocket (Figure 3b), all with ΔG > 0 (i.e., “unstable”). Two of the water sites (A and B) look accessible by substitution from either phenyl ring of 2; both sites are roughly along the extended planes of the rings and proximal to the ligand. A, matching the crystallographic water found in chain A of the corresponding co-crystal structure (I in Figure 2b), is at a distance of 4.1 Å from the nearest carbon of the unsubstituted phenyl ring, with ΔGA = +4.7 kcal/mol (ΔHA = −0.13 kcal/mol, –TΔSA = +4.8 kcal/mol). B is 3.3 Å from the nearest carbon in the difluorophenyl ring, with ΔGB = +5.2 kcal/mol (ΔHB = +0.86 kcal/mol, –TΔSB = +4.4 kcal/mol). Thus, both sites A and B interact enthalpically like bulk water, but displacing either water should be entropically highly favorable. A third predicted water site C (ΔGC= +3.3 kcal/mol) in the polar subpocket, proximal to A1226 and R1227, is 5 Å away from the ligand and not easily accessed. Finally, a fourth site D near K1110 was not considered further due to the uncertainty of the side chain’s coordinates.
The evidence of perturbed hydration, taken together with the physicochemical character of the pocket, suggested that the identified c-MET back-pocket would be a promising site to target for inhibition of the kinase.
Preliminary Hit Optimization
The unique binding mode of 2 for WT and D1228V c-MET, coupled with the opportunities revealed by the in silico analysis, encouraged the further investigation of this scaffold. At this stage of the campaign, SPR was used to assess the structural affinity relationship of matched molecular pairs (MMPs) of 2 for both WT and D1228V c-MET (Table 1).
Removal of the methyl from the benzylic position (4) leads to a complete loss of binding affinity for c-MET while removal of the two fluorines (11) is tolerated. Furthermore, chiral separation of 2 helped determine that the binding affinity in both kinases is mainly driven by the S-enantiomer (3), as observed in the co-crystal structures. The R-enantiomer’s binding affinity (not shown) is below the limit of detection of the SPR assay.
Exploration of the periphery of the pyrimidine-2,4-dione scaffold was carried out by modifying the N-1 and C-5 positions independently. Close inspection of the binding pocket around the benzyl ring of the WT and D1228V co-crystal structures of 2 indicated that the meta-position was the most amenable to substitution, with the meta-chloro-substituted benzyl (7) showing an improvement in Kd over 2. Chlorination of the ortho-position (5) leads to a significant drop in binding affinity, whereas para-fluoro substitution (6) appears to be well tolerated.
Encouraged by the meta-position’s amenability to substitution, introduction of more polar and functionalized groups was attempted next. As the water site A, proximal to the meta-position, has a predicted highly unfavorable entropic term (vide supra), MMPs of 2 were designed, with the goal of displacing this water while mimicking its potentially favorable H-bond to the backbone carbonyl of H1202 (Table 1: 8, 9 and 10). Of these compounds, the imidazole 10 provided a significant improvement in both potency and ligand-lipophilicity efficiency (LLE),42−44 albeit with an undesired 3.5-fold selectivity for the D1228V mutant over WT c-MET. Pleasingly, co-crystallization of 10 with the D1228V mutant revealed a binding mode in line with the prediction, with an H-bond from the imidazole group to H1202 clearly visible (Supporting Information, Figure S2b).
In parallel to investigating the N-1 substitution of 2, replacements of the 3,5-difluorophenyl group were also explored. Considering the proximity of the backbone carbonyl of F1124 to the 4-position of the difluorophenyl group (3.5 Å, interaction shown in Figure 3a) designs aimed at increasing the HBD strength of the aromatic hydrogen were explored. A nitrogen scan around the ring’s ortho- and meta-positions established that the 2-pyridyl (12) and 2-pyrimidyl (13) replacements were tolerated for both enzymes, with 13 demonstrating an improvement in LLE compared to the direct MMP 11. A design exploring a formal H-bond through replacing the difluorophenyl with an indole moiety (14) gave an ∼7-fold increase in binding affinity over 11 and also an improvement in LLE for both enzymes. Unfortunately, attempts to co-crystallize this ligand with c-MET to confirm the interaction to the backbone carbonyl of F1124 were unsuccessful.
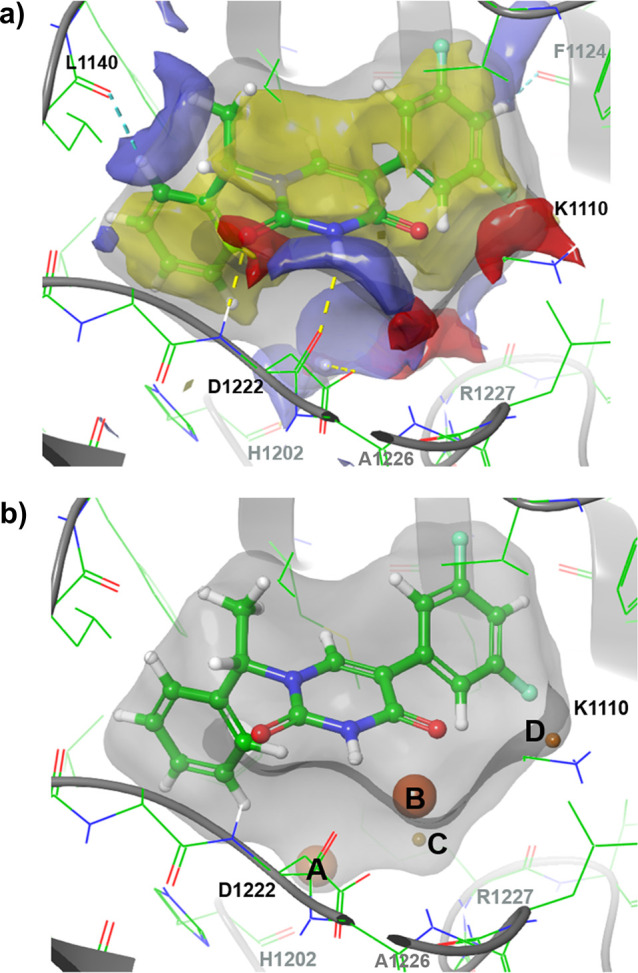
As the approach of targeting the water network proved effective for the N-1 benzyl fragment optimization, probing the region near the highly unstable modeled water site B followed. Analysis of the structure of c-MET bound by 2 suggested that bicyclic replacements of the difluorophenyl may be a suitable means to this end. These could potentially displace, mimic or even interact with the water while simultaneously probing for fruitful interactions in the polar subpocket proximal to residues R1227 and A1226. A design incorporating an indazole moiety (15) gave the first significant breakthrough with respect to potency in the hit-to-lead optimization. 15 offers a sub-μM binding affinity coupled with a significant improvement in LLE over 2 and 11, with some apparent preference for the D1228V mutant over WT c-MET (90 nM vs 240 nM respectively). A co-crystal structure of 15 in complex with D1228V c-MET (Figure 4a) demonstrates that the indazole is positioned in the region of the modeled water B (see Figure 3b) and makes a strong H-bond with the backbone NH of R1227. Furthermore, a new crystallographic water (II) now H-bonds to the indazole’s N-1 position, the L1225 backbone carbonyl and K1110s side chain nitrogen. The lysine’s nitrogen in turn interacts with both the second carbonyl of the ligand and the D1222 backbone carbonyl. Overall, an intricate water-bridged H-bonding network involving three ligand atoms, four protein atoms, one water molecule and six H-bonds is formed in addition to the two H-bonds seen in the initial hit’s co-crystal structure (Figure 2b).
Figure 4.
(a) Crystal structure of D1228V c-MET bound by 15. (b) WaterMap analysis of the co-crystal structure of 15 bound to the D1228V c-MET back-pocket. Water sites identified by WaterMap inside the pocket are shown as spheres and are colored by calculated excess free energy ranging from stable (green) to unstable (red).
The position of the aforementioned crystallographic water molecule (II) was also closely reproduced in a WaterMap analysis (E in Figure 4b; the distance between experimental water II and the modeled water site E is 0.54 Å) and determined to be highly ordered but overall stable with ΔGE = −2.3 kcal/mol (ΔHE = −7.7 kcal/mol, – TΔSE = +5.4 kcal/mol). Consequently, future designs aimed at maintaining this favorable H-bonding network.
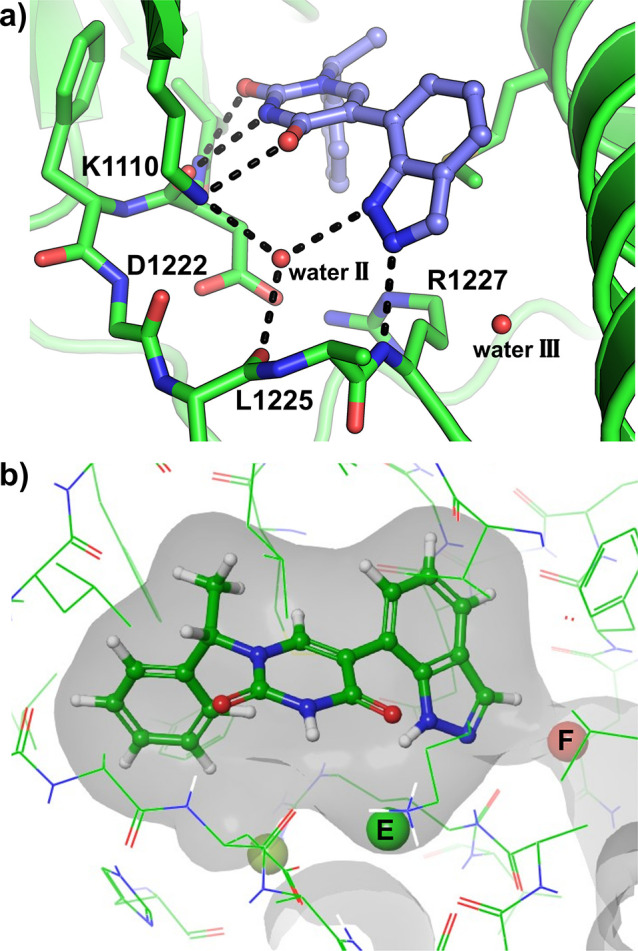
A combination of the most optimal N-1 (ethylphenyl imidazole) and C-5 (indazole) substitutions of the pyrimidine-2,4-dione was attempted in the hybrid 16. Disappointingly, however, the SAR of the two extremities of the core is not additive and a lower than expected binding affinity was observed for 16 in both enzymes (see Supporting Information Figure S2c for the crystal structure of D1228V c-MET bound by compound 16).
FEP Guided Optimization of 15
Despite the lack of additive SAR, the significant improvement in the binding affinity of 15 over 2 was encouraging. Furthermore, as the molecular weight and PSA contributions of the imidazole substitution in 10 may have proven detrimental to permeability, especially across the blood–brain barrier (BBB), a focused exploration of the indazole-containing 15 was prioritized.
In the co-crystal structure of 15 with the D1228V mutant c-MET, a new crystallographic water (III) proximal to the 3-C position of the indazole was noted (Figure 4a). WaterMap reproduced and calculated this water (labeled F in Figure 4b and positioned 0.43 Å from III) to be highly unstable with ΔGF = +6.8 kcal/mol (ΔHF = +3.0 kcal/mol, −TΔSF = +3.8 kcal/mol). Contrary to modeled water E, F was therefore considered a target for displacement. A limited set of free energy perturbation (FEP) calculations45,46 were applied to predict the binding affinities resulting from point changes to the inhibitor, and thus to prioritize designs primarily aimed at further improving binding affinity. The observed interactions and binding-site conformations of 2 and 15 (cf. Figures 2b and 4a, respectively) were considered to be mutually too different to be captured appropriately in the same set of FEP calculations. We therefore proceeded without first attempting to validate an FEP protocol based on the affinity data obtained so far in the campaign, which would normally be done before attempting prospective predictions. The FEP protocol was based on the structure of the mutant D1228V co-crystallized with 15 and the calculations were performed on enantiopure ligands (S-enantiomers). Considering the S-enantiomer was mostly, if not solely responsible for the binding affinity for c-MET (see Table 1: 2 vs 3), racemates were made and tested for synthetic expediency (Table 2).
Table 2. Evaluation of FEP-Guided Designs.

FEP predictions were performed using the corresponding S-enantiomers.
Affinity data based on n ≥ 2 with SEM within 0.2 log units, unless otherwise stated.
Activity data based on n ≥ 3 with SEM within 0.2 log units.
LLE = pIC50 – log D.
Log D measured via shake-flask method in octanol and water at pH7.4.
n = 1.
In agreement with the WaterMap analysis, FEP calculations suggested that displacement of the water proximal to the 3-position of the indazole would be highly favorable as demonstrated by the ΔGs of the 3-F (17), 3-Me (18) and 3-CN (19) indazole substituted ligands. This is particularly reflected in their binding affinities for the mutant and to a lesser extent for WT c-MET. Probing the remaining positions of the indazole was predicted to be detrimental to binding, with the exception of minor substituents on the 5-position as confirmed by 21’s ∼2-fold improvement in binding affinity for both enzymes, compared to 15. The ΔG for the corresponding 4-fluoro-substituted indazole (not shown) was calculated at +1.87 kcal/mol and was thus never prioritized for synthesis. The 6-fluoro analogue (20) was synthesized; however, as predicted (ΔG = +0.750 kcal/mol), it has a weaker binding affinity than the series’ parent, 15. The FEP protocol identified a nitrile and fluoro as optimal substituents for the indazole’s 3- and 5-positions, respectively. As predicted by FEP (ΔG = −3.080 kcal/mol), combination of this SAR in compound 22 leads to a significant improvement in both WT and D1228V mutant Kd values (87 and 11 nM, respectively).
Lead Optimization
The ability of compounds 15, 17–22 to inhibit the enzymatic activity of WT and D1228V c-MET was assessed using ADP-Glo activity assays (Table 2). As anticipated, based on the biophysical and structural studies on the early hit (2), all compounds of this novel series clearly inhibit c-MET, with single digit micromolar to nanomolar IC50 values, coupled with lead-like LLEs for both WT and D1228V c-MET. Interestingly, despite the compounds demonstrating some selectivity for the D1228V mutant in the SPR experiments, they appear to be less selective in the ADP-Glo activity assays.
Despite the overall advances with respect to affinity and inhibitory activity in the series, a suboptimal metabolic profile in rat hepatocytes (RH) and human liver microsomes (HLM), common to most inhibitors (Tables 1 and 2), needed addressing. Initial efforts focused on reducing the log D of 22 by replacing the phenyl ring with a pyridyl (Table 3: 23). Although this modification improves metabolism significantly, it leads to an undesired reduction in potency for both enzymes. Due to the risk of CYP-mediated oxidation at the para-position of the phenyl ring,47−49 we next turned our attention to blocking this potential metabolic soft-spot. Earlier SAR, demonstrating the tolerance of a para-fluoro group (Table 1: 6) encouraged the pursuit of this substitution in 24 (Table 3). This does indeed result in a significant reduction in metabolism, however, it also leads to a slight reduction in potency and LLE. Notably, the corresponding single enantiomer 25, supersedes the inhibitory profile of 22 yet is devoid of its higher metabolism, thus providing a useful tool for further studies (vide infra). Unfortunately, the analogous substitution on 21’s benzyl position does not have the same dramatic effect on metabolism (see 21 vs 26/27).
Table 3. SAR Combination and Lead Optimization on the Indazole-Substituted Pyrimidine-2,4-dione Scaffold.

Activity data based on n ≥ 3 with SEM within 0.2 log units, unless otherwise stated.
LLE = pIC50 – log D.
NCI-H1993 cell lines; activity data based on n ≥ 3 with SEM within 0.2 log units, unless otherwise stated.
Log D measured via shake-flask method in octanol and water at pH7.4.
n = 2.
SEM = 0.22 log units.
During our exploration, it was noted that substitution of the indazole with the polar nitrile group results in an increase in log D (see 15 vs 19 and 21 vs 22 in Table 2). This is presumably due to the opposing dipole moments of the nitrile substitution and the indazole’s nitrogen atoms. Considering the nitrile’s subtle effect on the electronics of the indazole ring, a less polar isosteric replacement, also capable of displacing the crystallographic water (III in Figure 4a), was attempted. Gratifyingly, an acetylene substitution has a dramatic impact on the overall profile of the inhibitors. Comparing the two MMPs of 19 vs 28 and 22 vs 29, the acetylene analogues display significantly improved potency. In fact the enantiopure inhibitor 30, has IC50 values of 13 and 20 nM against WT and D1228V c-MET respectively. Noteworthy is also the improved metabolic profile over their nitrile counterparts, despite their isolipophilic nature. Finally, a pyridyl replacement of the phenyl ring in 29 leads to inhibitor 31, that has similar potency and improved LLE. Co-crystallization studies of the optimized doubly substituted indazole inhibitor 29 with the D1228V mutant c-MET reveal this to maintain the H-bond to the R1227 backbone and successfully displace the crystallographic water (III in Figure 4a), as anticipated (Supporting Information, Figure S2d).
Next, a selection of potent nM inhibitors were assessed for their ability to inhibit WT and D1228V c-MET in a cellular setting using a previously reported homogeneous time-resolved fluorescence (HTRF) assay in parental and D1228V mutant NCI-H1993 cell lines18,30 (Table 3). The cellular AC50 of the inhibitors was determined by incubating them for 4 h in the respective cell lines and subsequently monitoring the HTRF signal of phosphorylated c-MET (residues Y1234/1235). Despite the high concentration of ATP in cells, all compounds show low micromolar to nanomolar activities, with the pyridyl-containing inhibitors 31 and 32 achieving nanomolar AC50s in both the WT and D1228V c-MET cell lines.
Lead Compound Profiles
Kinase Selectivity and Off-Target Pharmacology Assessment of 30 and 32
The single enantiomers 30 and 32 were chosen as potent representative examples of the c-MET allosteric series and screened in a panel of 140 kinases at a single concentration of 100 nM to assess their selectivity profiles (Figure 5 and Supporting Information, Table S2 and Table S3; Table S4 contains the selectivity data for the racemate 31, tested at a 1 μM concentration). Both 30 and 32 show excellent selectivity across the kinome, inhibiting only one kinase, c-MER proto-oncogene kinase (MER), by >50%. MER and c-MET display a high degree of structural homology in the back-pocket (Supporting Information, Figure S2e), which could provide some rationale for this specific off-target activity. The high selectivity may in part be a reflection of the type-III binding mode, as it does not involve binding in the highly conserved ATP pocket of kinases.50
Figure 5.

Kinase selectivity of (a) 30 and (b) 32 tested at 100 nM in a panel of 140 kinases. Inset: The four most inhibited kinases are represented for each compound.
30 and 32 were further profiled for off-target pharmacology in a diverse panel of radioligand binding, enzyme activity and cellular functional assays covering 26 targets, with testing in dose response. Both are >206-fold and >285 fold-selective, respectively, for c-MET (WT and D1228V) over the targets in the panel.
In Vitro and In Vivo ADME Profile of 25, 27, 29, 31
A selection of potent and metabolically stable compounds, with sufficient material availability, were subsequently progressed for permeability assessment prior to performing in vivo experiments (Table 4). These were screened in a human colon carcinoma cell line (Caco2) assay, to determine their intrinsic passive permeability, and an MDR1-transfected Madin-Darby canine kidney cell line (MDCK) assay with overexpressed P-glycoprotein (P-gp) transporters to assess their efflux ratio. The latter assay was used to evaluate whether the compounds could potentially cross the BBB. Encouragingly, the tested compounds are highly permeable, as reflected by the Caco2 assay data, though they demonstrate varying degrees of efflux in the MDR1-MDCK assay.
Table 4. In Vitro and In Vivo ADME Profile of Lead Compounds.
| MDCK
(10–6 cm/s) |
Hepatocytes
(μL/min/106 cells) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Caco2 A to B Papp (10–6 cm/s) | AB/BA | Efflux ratio | Rat | Human | HLM (μL/min/mg) | PSA | Bioavailability (%)/CL (mL/min/kg)/Vss (L/kg)a | Kp,uu | MPO scoreb |
| 27 | 97.1 | 3.44/22.4 | 6.51 | 14.8 | 1.81 | 5.01 | 78 | 57/74/4.3 | 0.11 | 5.0 |
| 25 | 51.6 | 0.740/25.6 | 34.6 | 5.13 | 2.48 | <3.00 | 97 | 96/3.0/1.5 | 0.014 | 4.5 |
| 29 | 64.1 | 3.60/14.2 | 3.94 | 5.99 | 4.27 | 12.2 | 78 | 100/60/4.6 | 0.10 | 4.7 |
| 31 | 73.9 | 2.04/37.9 | 18.6 | 4.81 | 1.29 | <3.00 | 88 | 91/13/1.6 | 0.022 | 5.2 |
Averaged data from 2 male Han Wistar rats per dose group (see Supporting Information for further details).
Score for the CNS MPO algorithm as defined by Wager et al.
The compounds were assessed in human hepatocyte (HH) and HLM stability assays to determine their intrinsic clearance (Clint). Compounds 25 and 31 in particular, show lead-like profiles with low HH and HLM Clint values (Table 4). The pharmacokinetic profile of this selection of compounds was further examined in rat and the data is captured in Table 4. All compounds show excellent bioavailability, despite varying degrees of plasma clearance ranging from low to high. In vivo clearance values matched expectations based on scaling of RH Clint values. Furthermore, relatively low volumes of distribution were noted, reflective of neutral compounds.
The drug likeness central nervous system multiparameter optimization (CNS MPO) score, as defined by Wager et al.,51 for all molecules is >4 thus classing them as promising candidates for permeating the BBB. The MPO score was utilized as an initial metric to support the progression of compounds to rat brain exposure studies. The unbound brain-to-plasma ratio (Kp,uu) across compounds however, mainly correlates with the MDR1-MDCK efflux ratio. Compounds such as 25 and 31, characterized by high efflux ratios, demonstrated limited brain penetration, with a Kp,uu of 0.014 and 0.022, respectively, while 27 and 29, with moderate efflux ratios of <7, gave promising Kp,uu values of 0.11 and 0.10 respectively.
Collectively, the activity data and the aforementioned data from the in vivo studies represent a lead-like profile for these inhibitors, thus stimulating further interest in the series, the advances of which will be communicated in future publications.
Synthetic Chemistry
The majority of the compound syntheses broadly fall in two categories: (a) late stage N-1 alkylation in examples where the C-5 position is substituted with the 3,5-fluorophenyl group and (b) late stage C-5 cross-coupling in examples where the N-1 position is substituted with the phenylethyl group.
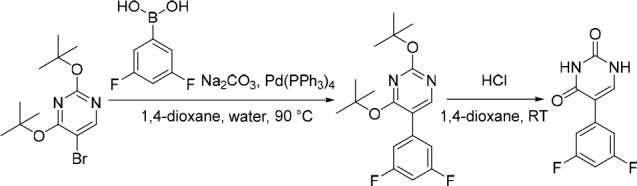
A representative example of the first approach—late stage N-1 alkylation—is captured in Scheme 1. Synthesis of the common intermediate 33 was achieved via a Suzuki cross-coupling of the commercially available 5-bromo-2,4-di-tert-butoxypyrimidine (34) with (3,5-difluorophenyl)boronic acid followed by a deprotection under acidic conditions to afford 33 in 84% yield over two steps. Alkylation of this core using (1-bromoethyl)benzene under basic conditions in DMF/water afforded 2 in 68% yield.
Scheme 1. Synthesis of 2.

Reagents and conditions: (a) (3,5-difluorophenyl)boronic acid, Pd(PPh3)4, Na2CO3, 1,4-dioxane/water, 90 °C; (b) HCl, 1,4-dioxane, rt, 84% over two steps; (c) (1-bromoethyl)benzene, KOH, DMF/water, 60 °C, 68%.
A representative example of the second approach (late stage C-5 cross-coupling) is captured in Scheme 2. Alkylation of 5-bromouracil (35) with 1-(1-bromoethyl)-4-fluorobenzene, under basic conditions, afforded 36 in 81% yield. Cross-coupling of bis(pinacolato)diboron with the commercially available bromo indazole 37 afforded 5-fluoro-1H-indazole-7-boronic acid pinacol ester (38) which was subsequently subjected to Suzuki cross-coupling conditions with 36 to afford inhibitor 26 in 34% yield over two steps. Further functionalization of the indazole substituent could be performed via an iodination on the 3-position and a subsequent late stage palladium catalyzed cyanation to provide inhibitor 24 in 30% yield.
Scheme 2. Synthesis of 26 and 24.

Reagents and conditions: (a) 1-(1-bromoethyl)-4-fluorobenzene, K2CO3, DMSO, 60 °C, 81%; (b) Pd(dppf)Cl2, KOAc, bis(pinacolato)diboron, 1,4-dioxane, 100 °C; (c) 36, Pd(PPh3)4, Na2CO3, 1,4-dioxane/water, 100 °C, 34% over two steps; (d) NaOH, NIS, DMF, rt, 84%; (e) Zn(CN)2, Pd(PPh3)4, DMA, 100 °C, 30%.
Finally, the synthetic route for the lead compound 30 is depicted in Schemes 3 and 4. A high yielding selective iodination on the 3-position of 37 afforded 39 which was subjected to Sonogashira cross-coupling conditions to provide the advanced ethynyl substituted indazole 40 in 44% yield (Scheme 3). A cross-coupling using bis(pinacolato)diboron provided the key boronate intermediate 41 in 76% yield. As an alternative to the alkylation described in Scheme 2, Mitsunobu conditions in the presence of (R)-1-phenylethan-1-ol, were used to alkylate 35 and afford the enantioenriched (S)-5-bromo-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (42 in Scheme 4). This was subsequently coupled to the aforementioned boronate 41 to provide 43 in 26% yield. Deprotection of the trimethylsilyl group using potassium carbonate in methanol at room temperature, was followed by chiral chromatography to ensure a high enantiopurity of the lead compound 30 which was afforded in 43% yield and >99% ee.
Scheme 3. Synthesis of Boronate Intermediate 41.

Reagents and conditions: (a) I2, Ag2SO4, EtOH, rt, 95%; (b) ethynyltrimethylsilane, CuI, Pd(PPh3)4, NEt3, MeCN, 80 °C, 44%; (c) Pd(dppf)Cl2·CH2Cl2, KOAc, bis(pinacolato)diboron, 1,4-dioxane, 80 °C, 76%.
Scheme 4. Synthesis of 30.

Reagents and conditions: (a) DIAD, PPh3, (R)-1-phenylethan-1-ol, THF, rt, 21%; (b) 41, Pd(dppf)Cl2, K3PO4, toluene/water, 80 °C, 26%; (c) K2CO3, MeOH, rt, 43%.
Conclusions
In summary, a novel allosteric binder of c-MET was discovered and optimized into a series of selective inhibitors, with WT and mutant activity, using a combination of structure-based drug design and computational analyses. Members of the series show nanomolar biochemical and cellular activities against both WT and D1228V mutant c-MET, while demonstrating excellent selectivity across the kinome and an extended panel of targets. Furthermore, optimized leads have excellent pharmacokinetic profiles and promising free-brain exposures. The novel compounds reported herein will assist in gaining a deeper understanding into the type-III binding mode and its potential as a fruitful MOA for small molecule therapeutics in the context of c-MET driven cancers.
Experimental Methods
General Procedures
In vitro assays
SPR, ADP-Glo and cell assays for WT and D1228V mutant c-MET were carried out as described previously.18,30
Chemistry General Experimental
Reagents and solvents (all anhydrous HPLC-grade) were obtained from commercial suppliers and used without any further purification unless otherwise stated. All reagents were weighed and handled in air unless otherwise stated. Brine refers to a saturated solution of NaCl. Concentration under reduced pressure refers to the use of a rotary evaporator.
Spectroscopy
1H and 13C NMR spectra were recorded using Bruker Avance III, Avance III HD or Avance III NEO spectrometers at a proton frequency of 300 MHz, or a Bruker Avance Neo spectrometer at a proton frequency of 500 MHz. All 1H and 13C NMR are quoted in ppm for measurement against TMS or residual solvent peaks as internal standards. Unless otherwise stated all experiments were carried out using DMSO-d6 as solvent. 1H NMR chemical shifts (δ) are given in ppm ±0.01, and coupling constants (J) are given in Hz ± 0.1 Hz. The 1H NMR spectra are reported as follows: δ/ppm (multiplicity, coupling constant(s) J/Hz, number of protons). Multiplicity is abbreviated as follows: s = singlet, br s = broad singlet, d = doublet, br d = broad doublet, dd = doublet of doublets, t = triplet, dt = doublet of triplets, q = quartet, dq = doublet of quartets, quint = quintet, m = multiplet. 13C NMR chemical shifts (δ) are given in ppm ±0.1.
Mass Spectrometry
LC-MS experiments were performed using a Shimadzu LCMS-2020 with electrospray ionization in positive ion detection mode with 20ADXR pump, SIL-20ACXR autosampler, CTO-20AC column oven, M20A PDA Detector and LCMS 2020 MS detector. LC was run in two set ups: (1) Halo C18 column (2.0 μm, 3.0 × 30 mm) in combination with a gradient (5–100% B in 1.2 min) of water and FA (0.1%) (A) and CH3CN and FA (0.1%) (B) at a flow rate of 1.5 mL/min; (2) Poroshell HPH C18 column (2.7 μm, 3.0 × 50 mm) in combination with a gradient (5–95% B in 2 min) of aqueous 46 mM ammonium carbonate/ammonia buffer at pH 10 (A) and MeCN (B) at a flow rate of 1.2 mL/min; (3) Halo C18 column (2.0 μm, 3.0 × 30 mm) in combination with a gradient (5–95% B in 2 min) of water and TFA (0.05%) (A) and CH3CN and TFA (0.05%) at a flow rate of 1.5 mL/min (B). The Column Oven (CTO-20AC) temperature was 40.0 °C. The injection volume was 1 μL. PDA (SPD-M20A) detection was in the range 190–400 nm. The MS detector was configured with electrospray ionization as ionizable source; acquisition mode: scan; nebulizing gas flow: 1.5 L/min; drying gas flow: 15 L/min; detector voltage: tuning voltage ±0.2 kV; DL temperature: 250 °C; heat block temperature: 250 °C; scan range: 90.00–900.00 m/z.
High-Resolution Mass Spectrometry
Accurate mass data of samples were obtained using HRMS system with Waters Acquity I Class UPLC and Xevo G2-XS Q-TOF (Waters Corp., Milford, MA, USA). The samples were separated on reversed phase ACQUITY UPLC BEH C18 column (2.1 × 50 mm, 1.7 μm) using gradient elution with 0.1% FA in H2O as mobile phase A and 0.1% FA in MeCN as mobile phase B. Acquity PDA Detector was in the range 210–400 nm. The injection volume was 0.1 μL. Analytes were separated by a gradient method (Supporting Information, Table S5). The column temperature was set at 40 °C and the flow rate at 0.4 mL/min. Instrument control and accurate mass data were processed using Masslynx software. The MS data were acquired in positive ionization mode with the following conditions: MS equipped with ESI, sensitivity mode, capillary voltage at 2.5 kV, sampling cone voltage at 40.0 V, source temperature at 100 °C, cone gas flow at 50 L/h, desolvation gas flow at 600.0 (L/h), acquisition mass range of 50–1200 Da.
Flash Column Chromatography
Chromatographic purification of products was accomplished with a CHEETAH MP200 system with integrated UV detection, using column chromatography on (A) silica gels (40–60 μm), eluted with petroleum ether and ethyl acetate or dichloromethane and methanol or (B) C18 spherical (20–35 μm), eluted with water and acetonitrile with formic acid (0.1%) or ammonium formate (10 mmol) modifier.
Preparative HPLC
Preparative HPLC was performed with a Waters MassLynx system with integrated MS detection and equipped with Prep C18 OBD 5 μm, 30 × 150 mm columns from XBridge or Xselect CSH. Alternatively Gilson GX-281 with integrated UV detection was used, equipped with either XBridge or Sunfire C18 10 μm, 19 × 150 ID or 19 × 250 mm. As eluent (acidic) gradients of water/MeCN/FA acid (95/5/0.1) or water/0.05% TFA (A) and MeCN/MeOH (B) or 0.05% ammonia in water/10 mmol NH4HCO3 (A) and MeCN/MeOH (B) were applied.
Preparative Chiral-HPLC
Preparative chiral HPLC was performed with a Gilson GX-281 system with integrated UV detection and equipped with one of Chiralpak AS, AD, Chiralcel OD, OJ Chiralpak IA, IB, IC, ID, IE, IF, IG, IH columns (Daicel Chemical Industries, Ltd.) (R,R)-Whelk-O1, (S,S)-Whelk-O1 columns (Regis technologies, Inc.) CHIRAL Cellulose-SB, SC, SA columns (YMC Co., Ltd.) at different column size (250 × 20 mm, 250 × 30 mm) with noted percentage of either ethanol in hexane (%Et/Hex) or isopropanol in hexane (%IPA/Hex) as isocratic solvent systems.
Compound Naming
Compound names are those generated by ChemDraw 19.0.
Compound Purity
Purity for all tested compounds was assessed either via the analytical LC-MS method detailed above or via the HPLC method below. The purity of target compounds was >95% unless otherwise stated.
HPLC Analytical Methods
All HPLC methods were performed on an Agilent 1200 system (Agilent, Santa Clara, CA) comprising 2 G1312B ultrahigh-pressure binary pumps, a G1315C Diode Array detector, a G1316B Column Compartment, a G1379B Micro Degasser, a G1367C Micro Well Plate Autosampler and a 35900E analog-to-digital converter. The eluent from the HPLC was split between an Agilent 6140 single quad mass spectrometer equipped with a multimode source and an ESA (ESA, Chelmsford, MA) Corona charged aerosol detector. HPLC reversed-phase separations were performed on a 50 × 2 mm Kinetex 2.6 μm C18 column (Phenomenex, Torrance, CA) at a flow rate of 700 μL per minute using a gradient comprising (A) HPLC grade water (VWR) with 0.05% formic acid (Sigma-Aldrich) and (B) HPLC grade acetonitrile (Honeywell) with 0.05% formic acid. The conditions for pump 1 were at time 0, A = 90%, with a linear gradient such that after 2 min, B = 100%, which was held for 0.5 min before returning to starting conditions. The gradient conditions for pump 2 were the opposite of pump 1 so as to combine the 2 solvent streams after the column and produce a constant 50:50 volume/volume mix of A and B at the detectors. The Agilent 6140 MS acquired from 100 to 1000 Da in sequential positive and negative ion modes with a total cycle time of 1 s, and the output from the Corona CAD (ESA) detector was acquired at a rate of 5 Hz through the analog-to-digital converter. The diode array detector (DAD) scanned from 220 to 300 nm at a rate of 20 Hz. All data were acquired into Chemstation Version B.03.01 (Agilent). The data from the DAD and CAD detectors were integrated automatically within Chemstation.
Target Compound and Intermediate Synthesis
5-(3,5-Difluorophenyl)pyrimidine-2,4(1H,3H)-dione (33)
Pd(PPh3)4 (6.290 g, 5.443 mmol) was added in one portion to a mixture of 5-bromo-2,4-di-tert-butoxypyrimidine (34) (33.00 g, 108.8 mmol), Na2CO3 (23.07 g, 217.7 mmol) and (3,5-difluorophenyl)boronic acid (18.10 g, 114.6 mmol) in 1,4-dioxane (500 mL) and water (80 mL) under nitrogen at rt. The resulting suspension was stirred at 90 °C for 2 h. The reaction mixture was quenched with saturated brine (100 mL) and then extracted with EtOAc (3 × 500 mL). The combined organic extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to afford a pale yellow solid. The crude product was purified by flash silica chromatography, elution gradient 0–10% MeOH in CH2Cl2. Fractions containing the desired compound were evaporated to dryness. To this solid was added HCl in 1,4-dioxane (4 M, 500 mL, 2000 mmol) and the solution was stirred overnight at rt. The solvent was removed under reduced pressure and the crude product was purified by crystallization from EtOAc/dioxane to afford 5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (20.60 g, 84% over two steps) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 11.38 (br s, 2H), 7.88 (d, J = 6.1 Hz, 1H), 7.44–7.35 (m, 2H), 7.18–7.10 (m, 1H); MS (ESI) m/z calculated for [C10H5F2N2O2]− [M – H]−= 223.0325, found = 223.1.
5-Bromo-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione
To a solution of 5-bromopyrimidine-2,4(1H,3H)-dione (35) (20.00 g, 104.7 mmol) and KOH (11.75 g, 209.4 mmol) in DMF (200 mL) and water (25 mL) under nitrogen at 60 °C, was added (1-bromoethyl)benzene (23.26 g, 125.7 mmol) in one portion. The resulting solution was stirred overnight at 60 °C. The reaction was concentrated under reduced pressure and subsequently quenched with saturated NH4Cl (100 mL) and extracted with EtOAc (3 × 100 mL). The combined organic extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to afford a white solid. The crude product was purified by crystallization from EtOAc/petroleum ether to afford 5-bromo-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (21.45 g, 69%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 11.83 (s, 1H), 8.10 (s, 1H), 7.47–7.26 (m, 5H), 5.74 (q, J = 7.2 Hz, 1H), 1.71 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C12H12BrN2O2]+ [M + H]+ = 295.0077, found = 294.9.
5-(3,5-Difluorophenyl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (2)
(1-Bromoethyl)benzene (198 mg, 1.07 mmol) was added to a solution of 5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (33) (400 mg, 1.78 mmol) and KOH (250 mg, 4.46 mmol) in DMF (15 mL) and water (4.5 mL) under nitrogen at rt. The resulting solution was stirred at 60 °C for 18 h. The reaction mixture was filtered through filter paper and the solvent of the filtrate was removed under reduced pressure. The crude product was purified by preparative HPLC (XBridge Prep OBD C18 Column, 5 μm, 19 × 250 mm) using decreasingly polar mixtures of water (10 mmol/L NH4HCO3) and MeCN (elution gradient 46–47%). Fractions containing the desired compound were evaporated to dryness to afford 5-(3,5-difluorophenyl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (400 mg, 68%) as a white solid. 1H NMR (400 MHz, Methanol-d4) δ 7.67 (s, 1H), 7.51–7.40 (m, 4H), 7.36 (ddd, J = 9.3, 3.9, 1.8 Hz, 1H), 7.18–7.08 (m, 2H), 6.89 (tt, J = 9.1, 2.3 Hz, 1H), 5.96 (q, J = 7.2 Hz, 1H), 1.82 (d, J = 7.2 Hz, 3H); 19F NMR (376 MHz, Methanol-d4) δ −112.22; MS (ESI) m/z calculated for [C18H15F2N2O2]+ [M + H]+ = 329.1096, found = 329.2.
(S)-5-(3,5-Difluorophenyl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (3)
The enantiomeric mixture above (2) was purified by preparative chiral-HPLC (Phenomenex Lux cellulose-4 Axia packed, 5 μm, 21.2 × 250 mm), isocratic 40% MeOH in scCO2. Fractions containing the desired products were evaporated to dryness to afford enantiopure (S)-5-(3,5-difluorophenyl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (RT: 5.82 min, 100 mg, 25%) and enantiopure (R)-5-(3,5-difluorophenyl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (RT: 7.29 min, 97 mg, 24%) as white solids. Data for 3: NMR and MS (ESI) results are identical to 2; ee = 97%. Data for (S)-5-(3,5-difluorophenyl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione: ee = 98%.
1-Benzyl-5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (4)
To solution of 5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (33) (150 mg, 0.669 mmol) and KOH (56.3 mg, 1.00 mmol) in DMF (10 mL) and water (2 mL) under nitrogen at rt, was added (bromomethyl)benzene (114 mg, 0.667 mmol) in one portion. The resulting solution was stirred at 60 °C for 10 h. The reaction mixture was subsequently neutralized with 2 M HCl, the solvent was removed under reduced pressure and the crude product was purified by preparative HPLC (XBridge Prep C18 OBD Column, 5 μm, 19 × 150 mm) using decreasingly polar mixtures of water (10 mmol/L NH4HCO3) and MeCN (elution gradient 43–44%). Fractions containing the desired compound were evaporated to dryness to afford 1-benzyl-5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (98.0 mg, 47%) as a white solid. 1H NMR (400 MHz, Methanol-d4) δ 8.00 (s, 1H), 7.42–7.29 (m, 5H), 7.26–7.19 (m, 2H), 6.88 (tt, J = 9.1, 2.3 Hz, 1H), 5.02 (s, 2H); 19F NMR (376 MHz, Methanol-d4) −112.28; MS (ESI) m/z calculated for [C17H13F2N2O2]+ [M + H]+ = 315.0940, found = 314.9.
1-(1-(2-Chlorophenyl)ethyl)-5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (5)
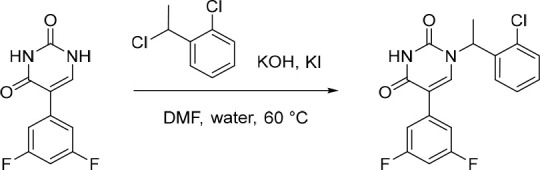
To a solution of 5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (33) (150 mg, 0.669 mmol), KI (111 mg, 0.669 mmol) and 1-chloro-2-(1-chloroethyl)benzene (117 mg, 0.668 mmol) in DMF (4 mL) and water (1 mL) under nitrogen at rt, was added KOH (75.0 mg, 1.34 mmol). The resulting solution was stirred at 60 °C overnight. Subsequently, the reaction was concentrated and purified by flash silica chromatography, elution gradient 0–20% MeOH in CH2Cl2. Fractions containing the desired compound were evaporated to dryness to afford 1-(1-(2-chlorophenyl)ethyl)-5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (89.0 mg, 37%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 11.69 (s, 1H), 7.84 (s, 1H), 7.60 (dd, J = 7.5, 1.4 Hz, 1H), 7.49 (dd, J = 7.5, 1.8 Hz, 1H), 7.48–7.28 (m, 4H), 7.15 (tt, J = 9.3, 2.2 Hz, 1H), 5.91 (q, J = 7.0 Hz, 1H), 1.76 (d, J = 7.1 Hz, 3H); MS (ESI) m/z calculated for [C18H14ClF2N2O2]+ [M + H]+ = 363.0706, found = 363.2.
5-(3,5-Difluorophenyl)-1-(1-(4-fluorophenyl)ethyl)pyrimidine-2,4(1H,3H)-dione (6)
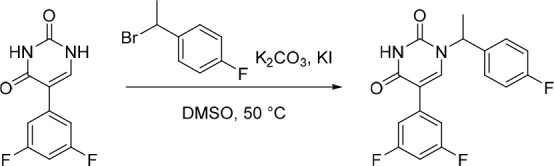
1-(1-Bromoethyl)-4-fluorobenzene (181 mg, 0.891 mmol) was added in one portion to a solution of 5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (33) (200 mg, 0.892 mmol), KI (29.6 mg, 0.178 mmol) and K2CO3 (370 mg, 2.68 mmol) in DMSO (10 mL) under nitrogen at rt. The resulting suspension was stirred at 50 °C for 12 h. The reaction mixture was subsequently filtered through filter paper. The filtrate was purified by preparative HPLC (Xselect CSH OBD column, 5 μm, 30 × 150 mm) using decreasingly polar mixtures of water (containing 0.05% TFA) and MeCN (elution gradient 43–54%). Fractions containing the desired compound were evaporated to dryness to afford 5-(3,5-difluorophenyl)-1-(1-(4-fluorophenyl)ethyl)pyrimidine-2,4(1H,3H)-dione (127 mg, 41%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 11.66 (s, 1H), 8.01 (s, 1H), 7.52–7.43 (m, 2H), 7.42–7.31 (m, 2H), 7.26–7.11 (m, 3H), 5.75 (q, J = 7.1 Hz, 1H), 1.77 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C18H14F3N2O2]+ [M + H]+ = 347.1002, found = 347.2.
1-(1-(3-Chlorophenyl)ethyl)-5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (7)
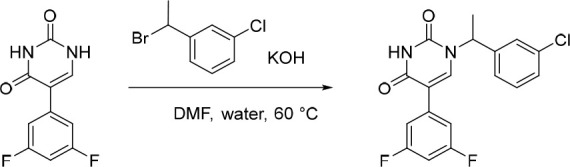
To a solution of 5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (33) (150 mg, 0.669 mmol) and 1-(1-bromoethyl)-3-chlorobenzene (147 mg, 0.670 mmol) in DMF (4 mL) and water (1 mL) under nitrogen at rt, was added KOH (75.0 mg, 1.34 mmol). The resulting solution was stirred at 60 °C overnight. The reaction was concentrated under reduced pressure and the crude product was purified by preparative HPLC (Xselect CSH OBD, 5 μm, 30 × 150 mm) using decreasingly polar mixtures of water (containing 0.1% FA) and MeCN (elution gradient 40–70%). Fractions containing the desired compound were evaporated to dryness to afford 1-(1-(3-chlorophenyl)ethyl)-5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (144 mg, 59%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 11.68 (s, 1H), 8.08 (s, 1H), 7.52 (s, 1H), 7.48–7.32 (m, 5H), 7.16 (tt, J = 9.3, 2.4 Hz, 1H), 5.74 (q, J = 7.2 Hz, 1H), 1.79 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C18H14ClF2N2O2]+ [M + H]+ = 363.0706, found = 363.2.
1-(1-(3-Bromophenyl)ethyl)-5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione
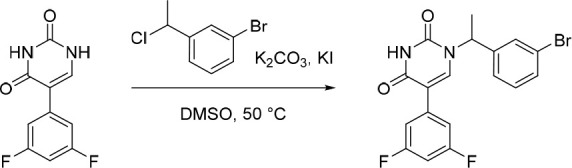
KI (592 mg, 3.57 mmol) was added in one portion to a solution of 5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (33) (800 mg, 3.57 mmol), 1-bromo-3-(1-chloroethyl)benzene (862 mg, 3.93 mmol) and K2CO3 (1.48 g, 10.7 mmol) in DMSO (10 mL) under nitrogen at rt. The resulting suspension was stirred at 50 °C overnight. The suspension was then filtered and the filtrate was directly purified by C18-flash chromatography, elution gradient 0–60% MeCN in water. Fractions containing the desired compound were evaporated to dryness to afford impure 1-(1-(3-bromophenyl)ethyl)-5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (750 mg, 42% pure by LC-MS) as a gray solid. This material was used in the subsequent reactions without further purification. MS (ESI) m/z calculated for [C18H13F2N2O2]+ [M + H]+ = 407.0202 and 409.0181, found = 408.8.
1-(1-(3-(1H-Pyrazol-3-yl)phenyl)ethyl)-5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (8)
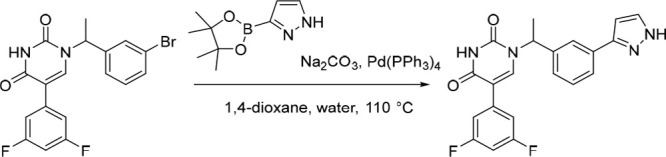
Pd(PPh3)4 (25.5 mg, 0.0221 mmol) was added in one portion to a mixture of 1-(1-(3-bromophenyl)ethyl)-5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (180 mg, 0.442 mmol; material assumed pure for the purposes of reagent equivalent calculation), Na2CO3 (117 mg, 1.10 mmol) and 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (129 mg, 0.665 mmol) in 1,4-dioxane (12 mL) and water (3 mL) under nitrogen at rt. The resulting solution was stirred at 110 °C for 5 h. The reaction was then concentrated under reduced pressure and purified by preparative HPLC (Xselect CSH OBD, 5 μm, 30 × 150 mm) using decreasingly polar mixtures of water (containing 0.1% FA) and MeCN (elution at 35% isocratic). Fractions containing the desired compound were evaporated to dryness to afford 1-(1-(3-(1H-pyrazol-3-yl)phenyl)ethyl)-5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (70.0 mg, 40%) as a white solid. 1H NMR (300 MHz, Methanol-d4) δ 7.86 (br s, 1H), 7.79–7.70 (m, 2H), 7.67 (br s, 1H), 7.52–7.37 (m, 2H), 7.21–7.07 (m, 2H), 6.86 (tt, J = 9.2, 2.4 Hz, 1H), 6.70 (d, J = 2.3 Hz, 1H), 5.98 (q, J = 7.1 Hz, 1H), 1.85 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C21H17F2N4O2]+ [M + H]+ = 395.1314, found = 395.2.
1-(1-(3-(1H-Pyrazol-4-yl)phenyl)ethyl)-5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (9)
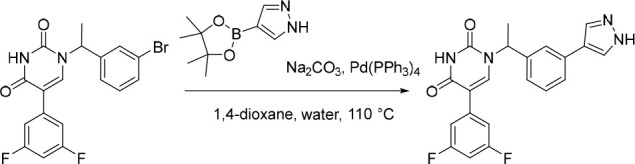
Pd(PPh3)4 (25.5 mg, 0.0221 mmol) was added in one portion to a mixture of 1-(1-(3-bromophenyl)ethyl)-5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (180 mg, 0.442 mmol; material assumed pure for the purposes of reagent equivalent calculation), Na2CO3 (141 mg, 1.33 mmol) and 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (103 mg, 0.531 mmol) in 1,4-dioxane (8 mL) and water (2 mL) under nitrogen at rt. The resulting suspension was stirred at 110 °C for 5 h. The mixture was diluted with EtOAc (10 mL) and washed sequentially with water (10 mL) and saturated brine (2 × 10 mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by preparative HPLC (XBridge Shield RP18 OBD, 5 μm, 19 × 150 mm) using decreasingly polar mixtures of water (10 mmol/L NH4HCO3 + 0.1% NH3·H2O) and MeOH (elution at 58% isocratic). Fractions containing the desired compound were evaporated to dryness to afford 1-(1-(3-(1H-pyrazol-4-yl)phenyl)ethyl)-5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (60.0 mg, 34%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.05 (s, 2H), 7.96 (s, 1H), 7.62 (d, J = 1.9 Hz, 1H), 7.53 (dt, J = 7.8, 1.4 Hz, 1H), 7.36 (t, J = 7.7 Hz, 1H), 7.35–7.26 (m, 2H), 7.22–7.16 (m, 1H), 7.11 (tt, J = 9.4, 2.4 Hz, 1H), 5.76 (q, J = 7.1 Hz, 1H), 1.79 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C21H17F2N4O2]+ [M + H]+ = 395.1314, found = 395.1.
5-(3,5-Difluorophenyl)-1-(1-(3-(1-trityl-1H-imidazol-4-yl)phenyl)ethyl)pyrimidine-2,4(1H,3H)-dione
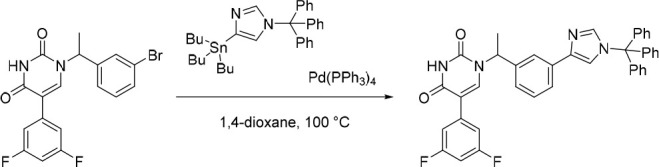
Pd(PPh3)4 (56.8 mg, 0.0492 mmol) was added in one portion to a mixture of 4-(tributylstannyl)-1-trityl-1H-imidazole (294 mg, 0.490 mmol) and 1-(1-(3-bromophenyl)ethyl)-5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (200 mg, 0.491 mmol; material assumed pure for the purposes of reagent equivalent calculation) in 1,4-dioxane (20 mL) under nitrogen at rt. The resulting suspension was stirred at 100 °C for 5 h. The crude product was purified by C18-flash chromatography, elution gradient 30–40% MeCN in water. Fractions containing the desired compound were evaporated to dryness to afford impure 5-(3,5-difluorophenyl)-1-(1-(3-(1-trityl-1H-imidazol-4-yl)phenyl)ethyl)pyrimidine-2,4(1H,3H)-dione (200 mg) as a white solid. The impure material was used in the subsequent reaction without further purification. MS (ESI) m/z calculated for [C40H31F2N4O2]+ [M + H]+ = 637.2410, found = 637.3 (weak detection of desired peak, ESI was dominated by the trityl cation (m/z = 243.2)).
1-(1-(3-(1H-Imidazol-5-yl)phenyl)ethyl)-5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (10)
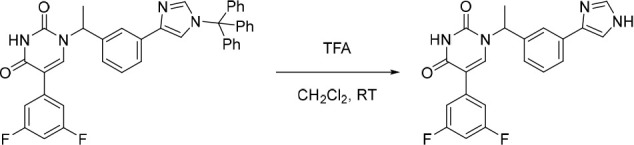
TFA (3.00 mL, 39.2 mmol) was added in one portion to a solution of the above impure 5-(3,5-difluorophenyl)-1-(1-(3-(1-trityl-1H-imidazol-5-yl)phenyl)ethyl)pyrimidine-2,4(1H,3H)-dione in CH2Cl2 (6 mL) under nitrogen at rt. The resulting solution was stirred at rt for 3 h. The solvent was removed under reduced pressure and the crude product was purified by preparative HPLC (XBridge Prep C18 OBD column, 5 μm, 30 × 100 mm) using decreasingly polar mixtures of water (containing 0.5% FA) and MeCN. Fractions containing the desired compound were evaporated to dryness to afford 1-(1-(3-(1H-imidazol-5-yl)phenyl)ethyl)-5-(3,5-difluorophenyl)pyrimidine-2,4(1H,3H)-dione (60.0 mg, 31% over two-steps) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 12.19 (s, 1H), 11.70 (s, 1H), 8.03 (s, 1H), 7.83 (s, 1H), 7.78–7.59 (m, 3H), 7.48–7.31 (m, 3H), 7.23 (d, J = 7.4 Hz, 1H), 7.16 (tt, J = 9.1, 2.1, 1H), 5.82 (q, J = 6.6 Hz, 1H), 1.82 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C21H17F2N4O2]+ [M + H]+ = 395.1314, found = 395.2.
5-Phenyl-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (11)
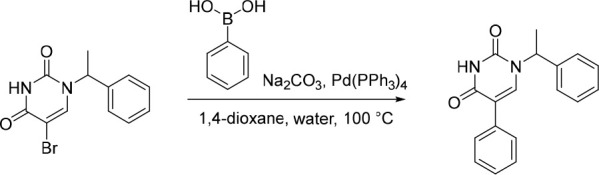
Pd(PPh3)4 (78.0 mg, 0.0675 mmol) was added in one portion to a mixture of 5-bromo-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (200 mg, 0.678 mmol), Na2CO3 (144 mg, 1.36 mmol) and phenylboronic acid (83.0 mg, 0.681 mmol) in 1,4-dioxane (10 mL) and water (2.5 mL) under nitrogen at rt. The resulting solution was stirred at 100 °C for 2 h. The solvent was removed under reduced pressure and the crude product was purified by preparative HPLC (Xselect CSH OBD, 5 μm, 30 × 150 mm) using decreasingly polar mixtures of water (containing 0.1% FA) and MeCN (elution gradient 40–50%). Fractions containing the desired compound were evaporated to dryness to afford 5-phenyl-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (82.9 mg, 42%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 11.56 (s, 1H), 7.74 (s, 1H), 7.53–7.23 (m, 10H), 5.82 (q, J = 7.1 Hz, 1H), 1.77 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C18H17N2O2]+ [M + H]+ = 293.1285, found = 292.9.
1-(1-Phenylethyl)-5-(pyridin-2-yl)pyrimidine-2,4(1H,3H)-dione (12)
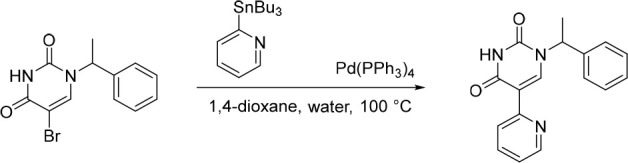
Pd(PPh3)4 (58.7 mg, 0.0508 mmol) was added in one portion to a mixture of 5-bromo-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (150 mg, 0.508 mmol) and 2-(tributylstannyl)pyridine (187 mg, 0.508 mmol) in 1,4-dioxane (10 mL) and water (2.5 mL) under nitrogen at rt. The resulting solution was stirred at 100 °C for 2 h. The solvent was removed under reduced pressure and the crude product was purified by preparative HPLC (XBridge Prep OBD C18, 5 μm, 30 × 150 mm) using decreasingly polar mixtures of water (10 mmol/L NH4HCO3 + 0.1% NH3.H2O) and MeCN (elution gradient 28–48%). Fractions containing the desired compound were evaporated to dryness to afford 1-(1-phenylethyl)-5-(pyridin-2-yl)pyrimidine-2,4(1H,3H)-dione (66.0 mg, 44%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 11.67 (s, 1H), 8.49 (ddd, J = 4.8, 1.7, 0.8 Hz, 1H), 8.35 (s, 1H), 8.23 (d, J = 8.1 Hz, 1H), 7.77 (td, J = 7.8, 1.9 Hz, 1H), 7.46–7.28 (m, 5H), 7.24 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H), 5.88 (q, J = 7.1 Hz, 1H), 1.73 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C17H16N3O2]+ [M + H]+ = 294.1237, found = 294.1.
1′-(1-Phenylethyl)-[2,5′-bipyrimidine]-2′,4′(1′H,3′H)-dione (13)
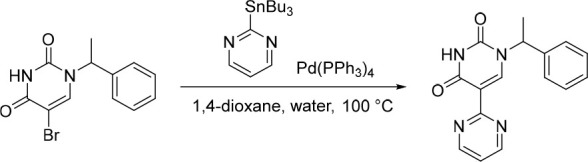
Pd(PPh3)4 (58.7 mg, 0.0508 mmol) was added in one portion to a mixture of 5-bromo-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (150 mg, 0.508 mmol) and 2-(tributylstannyl)pyrimidine (188 mg, 0.509 mmol) in 1,4-dioxane (10 mL) and water (2.5 mL) under nitrogen at rt. The resulting solution was stirred at 100 °C for 2 h. The solvent was removed under reduced pressure and the crude product was purified by preparative HPLC (Xselect CSH OBD, 5 μm, 30 × 150 mm) using decreasingly polar mixtures of water (containing 0.1% FA) and MeCN (elution gradient 25–39%). Fractions containing the desired compound were evaporated to dryness to afford 1′-(1-phenylethyl)-[2,5′-bipyrimidine]-2′,4′(1′H,3′H)-dione (30.0 mg, 20%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 11.63 (s, 1H), 8.81 (d, J = 4.9 Hz, 2H), 8.21 (s, 1H), 7.48–7.27 (m, 6H), 5.86 (q, J = 7.1 Hz, 1H), 1.74 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C16H15N4O2]+ [M + H]+ = 295.3215, found = 295.2.
5-(1H-Indol-5-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (14)
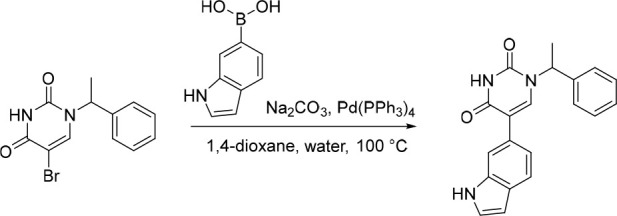
Pd(PPh3)4 (39.2 mg, 0.0339 mmol) was added to a mixture of 5-bromo-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (100 mg, 0.339 mmol), Na2CO3 (71.8 mg, 0.677 mmol) and (1H-indol-5-yl)boronic acid (82.0 mg, 0.509 mmol) in 1,4-dioxane (10 mL) and water (2.5 mL) under nitrogen at rt. The resulting solution was stirred at 100 °C for 2 h. The reaction mixture was diluted with EtOAc (100 mL) and washed with saturated brine (2 × 50 mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by preparative HPLC (XBridge Prep C18 OBD, 5 μm, 19 × 150 mm) using decreasingly polar mixtures of water (10 mmol/L NH4HCO3) and MeOH (elution at 50% isocratic). Fractions containing the desired compound were evaporated to dryness to afford 5-(1H-indol-5-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (20.0 mg, 18%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.46 (s, 1H), 11.08 (s, 1H), 7.62 (d, J = 11.3 Hz, 2H), 7.49–7.26 (m, 7H), 7.16 (dd, J = 8.5, 1.7 Hz, 1H), 6.53–6.32 (m, 1H), 5.83 (q, J = 7.2 Hz, 1H), 1.77 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C20H18N3O2]+ [M + H]+ = 332.1394, found = 332.1.
5-(1H-Indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (15)
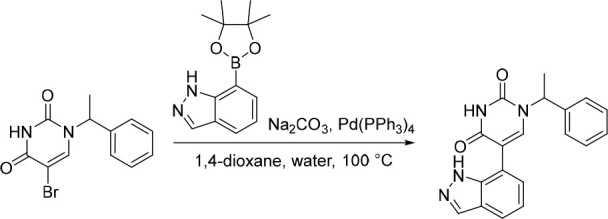
Pd(PPh3)4 (29.4 mg, 0.0254 mmol) was added in one portion to a mixture of 5-bromo-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (150 mg, 0.508 mmol), Na2CO3 (135 mg, 1.27 mmol) and 7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indazole (149 mg, 0.610 mmol) in 1,4-dioxane (12 mL) and water (3 mL) under nitrogen at rt. The resulting solution was stirred at 100 °C for 5 h. The solvent was removed under reduced pressure and the crude product was purified by preparative HPLC (Xselect CSH OBD Column, 5 μm, 30 × 150 mm) using decreasingly polar mixtures of water (containing 0.1% FA) and MeCN (elution gradient 30–40%). Fractions containing the desired compound were evaporated to dryness to afford 5-(1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (120 mg, 71%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 12.78 (s, 1H), 11.60 (s, 1H), 8.07 (d, J = 1.3 Hz, 1H), 7.82 (s, 1H), 7.75 (d, J = 7.6 Hz, 1H), 7.46–7.35 (m, 4H), 7.34–7.27 (m, 1H), 7.18–7.07 (m, 2H), 5.81 (q, J = 7.2 Hz, 1H), 1.77 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C19H17N4O2]+ [M + H]+ = 333.1346, found = 333.0.
1-(3-(1-Trityl-1H-imidazol-4-yl)phenyl)ethan-1-ol
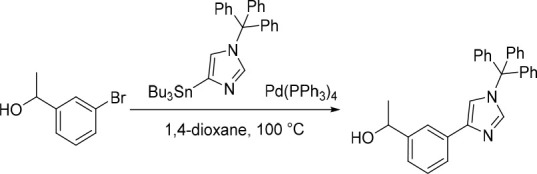
Pd(PPh3)4 (193 mg, 0.167 mmol) was added to a mixture of 4-(tributylstannyl)-1-trityl-1H-imidazole (1.00 g, 1.67 mmol) and 1-(3-bromophenyl)ethan-1-ol (335 mg, 1.67 mmol) in 1,4-dioxane (10 mL) under nitrogen at rt. The resulting solution was stirred at 100 °C for 4 h. The reaction was then concentrated under reduced pressure and purified by flash silica chromatography, elution gradient 0–50% EtOAc in petroleum ether. Fractions containing the desired compound were evaporated to dryness to afford 1-(3-(1-trityl-1H-imidazol-4-yl)phenyl)ethan-1-ol (500 mg, 70%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 7.71 (t, J = 1.7 Hz, 1H), 7.58 (dt, J = 7.8, 1.5 Hz, 1H), 7.47–7.38 (m, 12H), 7.25 (t, J = 7.7 Hz, 1H), 7.21–7.16 (m, 6H), 5.12 (d, J = 4.3 Hz, 1H), 4.76–4.60 (m, 1H), 1.31 (d, J = 6.4 Hz, 3H); MS (ESI) m/z calculated for [C30H26N2NaO]+ [M + Na]+ = 453.1937, found = 453.2.
4-(3-(1-Bromoethyl)phenyl)-1-trityl-1H-imidazole
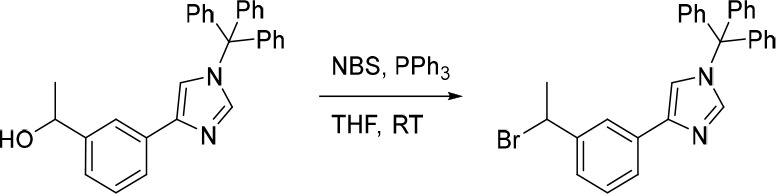
N-Bromosuccinimide (165 mg, 0.927 mmol) was added portion wise to a mixture of PPh3 (244 mg, 0.930 mmol) and 1-(3-(1-trityl-1H-imidazol-4-yl)phenyl)ethan-1-ol (400 mg, 0.929 mmol) in THF (10 mL) under nitrogen at 0 °C. The resulting mixture was stirred at rt for 1 h. The reaction was then concentrated under reduced pressure to afford the crude 4-(3-(1-bromoethyl)phenyl)-1-trityl-1H-imidazole (500 mg). The crude product was used in the subsequent reaction without further purification. MS (ESI) no product mass signal detected. ESI detection was dominated by the trityl cation (m/z = 243.2).
5-Bromo-1-(1-(3-(1-trityl-1H-imidazol-4-yl)phenyl)ethyl)pyrimidine-2,4(1H,3H)-dione
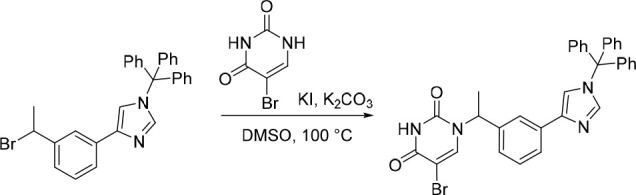
K2CO3 (280 mg, 2.03 mmol) was added to a mixture of 4-(3-(1-bromoethyl)phenyl)-1-trityl-1H-imidazole (500 mg, 1.01 mmol; material assumed pure for the purposes of reagent equivalent calculation), KI (168 mg, 1.01 mmol) and 5-bromopyrimidine-2,4(1H,3H)-dione (35) (194 mg, 1.01 mmol) in DMSO (10 mL) at rt. The resulting mixture was stirred at 100 °C for 12 h. The solvent was removed under reduced pressure and the crude product was purified by preparative TLC (petroleum ether: EtOAc = 1:1), to afford 5-bromo-1-(1-(3-(1-trityl-1H-imidazol-4-yl)phenyl)ethyl)pyrimidine-2,4(1H,3H)-dione (200 mg, 36% over two steps) as a white solid. MS (ESI) m/z calculated for [C34H27BrN4NaO2]+ [M + Na]+ = 625.1210, 627.1190, found = 627.1 (weak detection of desired peak, ESI was dominated by the trityl cation (m/z = 243.2)).
1-(1-(3-(1H-Imidazol-5-yl)phenyl)ethyl)-5-(1H-indazol-7-yl)pyrimidine-2,4(1H,3H)-dione (16)
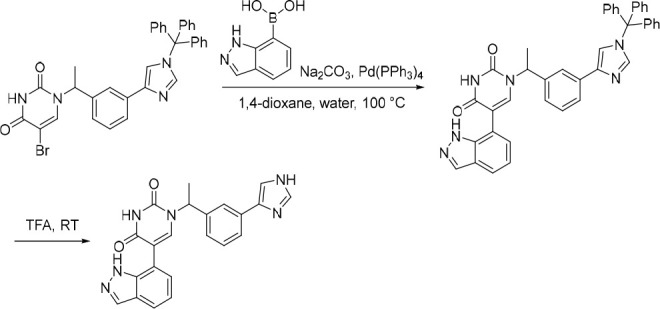
Pd(PPh3)4 (38.3 mg, 0.0331 mmol) was added in one portion to a mixture of 5-bromo-1-(1-(3-(1-trityl-1H-imidazol-4-yl)phenyl)ethyl)pyrimidine-2,4(1H,3H)-dione (200 mg, 0.331 mmol), Na2CO3 (52.7 mg, 0.497 mmol) and (1H-indazol-7-yl)boronic acid (81.0 mg, 0.500 mmol) in 1,4-dioxane (2 mL) and water (1 mL) under nitrogen at rt. The resulting mixture was stirred at 100 °C for 4 h. The reaction mixture was then diluted with EtOAc (25 mL) and washed sequentially with water (25 mL) and saturated brine (25 mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to afford the crude product which was used in the subsequent reaction without further purification. MS (ESI) m/z calculated for [C41H33N6O2]+ [M + H]+ = 641.2660, found = 641.6 (weak detection of desired peak, ESI was dominated by the trityl cation (m/z = 243.2)).
The above crude product was added to TFA (2 mL). The resulting mixture was stirred at rt for 1 h. The solvent was removed under reduced pressure and the crude product was purified by preparative HPLC (XBridge Prep OBD C18 Column, 5 μm, 30 × 150 mm) using decreasingly polar mixtures of water (10 mmol/L NH4HCO3) and MeCN (elution gradient 25–28%). Fractions containing the desired compound were evaporated to dryness to afford 1-(1-(3-(1H-imidazol-5-yl)phenyl)ethyl)-5-(1H-indazol-7-yl)pyrimidine-2,4(1H,3H)-dione (13.0 mg, 10% over two steps) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 12.76 (br s, 1H), 8.07 (s, 1H), 7.85 (s, 1H), 7.80 (s, 1H), 7.76–7.64 (m, 3H), 7.59 (s, 1H), 7.35 (t, J = 7.7 Hz, 1H), 7.24 (d, J = 7.8 Hz, 1H), 7.16 (d, J = 6.9 Hz, 1H), 7.13–7.06 (m, 1H), 6.06 (br s, 1H), 5.83 (q, J = 7.0 Hz, 1H), 1.78 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C22H19N6O2]+ [M + H]+ = 399.1564, found = 399.0.
7-Bromo-3-fluoro-1H-indazole
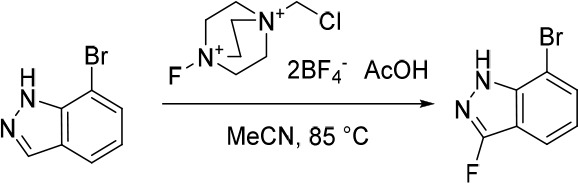
AcOH (3.05 g, 50.8 mmol) was added in one portion to a mixture of 7-bromo-1H-indazole (1.00 g, 5.08 mmol) and 1-(chloromethyl)-4-fluoro-1,4-diazabicyclo[2.2.2]octane-1,4-diium tetrafluoroborate (3.60 g, 10.2 mmol) in MeCN (30 mL) under nitrogen at rt. The resulting solution was stirred at 85 °C for 2 h. The reaction mixture was diluted with EtOAc (200 mL) and washed with saturated Na2CO3 (150 mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash silica chromatography, elution gradient 0–30% EtOAc in petroleum ether. Fractions containing the desired compound were evaporated to dryness to afford 7-bromo-3-fluoro-1H-indazole (600 mg, 55%) as a pale yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.05 (s, 1H), 7.80–7.67 (m, 2H), 7.18–7.06 (m, 1H); MS (ESI) m/z calculated for [C7H5BrFN2]+ [M + H]+ = 214.9615, found = 215.1.
5-(3-Fluoro-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (17)
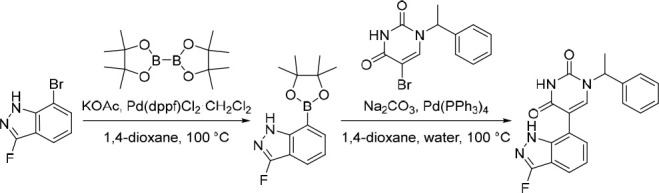
Pd(dppf)Cl2·CH2Cl2 (106 mg, 0.130 mmol) was added in one portion to a mixture of 7-bromo-3-fluoro-1H-indazole (350 mg, 1.63 mmol), KOAc (240 mg, 2.44 mmol) and 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(1,3,2-dioxaborolane) (455 mg, 1.79 mmol) in 1,4-dioxane (20 mL) under nitrogen at rt. The resulting suspension was stirred at 100 °C for 12 h to give a black suspension. To this suspension was added Pd(PPh3)4 (94.0 mg, 0.0813 mmol), Na2CO3 (518 mg, 4.88 mmol), 5-bromo-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (480 mg, 1.63 mmol) and water (5 mL) under nitrogen at rt. The resulting suspension was stirred at 100 °C for 3 h. The reaction mixture was diluted with EtOAc (100 mL) and washed with water (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by preparative HPLC (XSelect CSH Prep C18 OBD column, 5 μm, 19 × 150 mm) using decreasingly polar mixtures of water (containing 0.05% FA) and MeCN (elution gradient 38–48%). Fractions containing the desired compound were evaporated to dryness to afford 5-(3-fluoro-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (150 mg, 26% over two steps) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 12.28 (s, 1H), 11.64 (s, 1H), 7.87 (s, 1H), 7.68 (d, J = 8.0 Hz, 1H), 7.45–7.35 (m, 4H), 7.34–7.22 (m, 2H), 7.22–7.11 (m, 1H), 5.79 (q, J = 7.1 Hz, 1H), 1.75 (d, J = 7.2 Hz, 3H). 19F NMR (282 MHz, DMSO-d6) δ −136.61; MS (ESI) m/z calculated for [C19H16FN4O2]+ [M + H]+ = 351.1252, found = 350.9.
5-(3-Methyl-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (18)
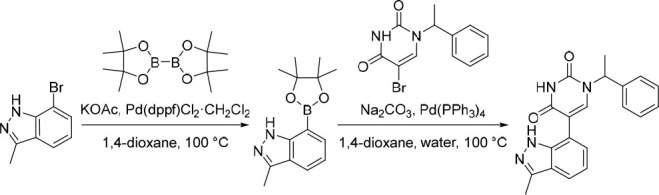
Pd(dppf)Cl2·CH2Cl2 (77.0 mg, 0.0943 mmol) was added in one portion to a mixture of 7-bromo-3-methyl-1H-indazole (250 mg, 1.18 mmol), KOAc (174 mg, 1.77 mmol) and 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(1,3,2-dioxaborolane) (331 mg, 1.30 mmol) in 1,4-dioxane (2 mL) under nitrogen at rt. The resulting suspension was stirred at 100 °C for 12 h to give a black suspension. To this suspension was added Pd(PPh3)4 (68.4 mg, 0.0592 mmol), Na2CO3 (377 mg, 3.56 mmol), 5-bromo-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (350 mg, 1.19 mmol) and water (0.5 mL) under nitrogen at rt. The resulting suspension was stirred at 100 °C for 3 h. The reaction mixture was diluted with EtOAc (100 mL) and washed with water (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by preparative HPLC (XBridge Prep C18 OBD, 5 μm, 30 × 150 mm) using decreasingly polar mixtures of water (containing 10 mmol/L NH4HCO3 + 0.1% NH3.H2O) and MeCN (elution at 34% isocratic). Fractions containing the desired compound were evaporated to dryness to afford 5-(3-methyl-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (133 mg, 33% over two-steps) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 12.32 (s, 1H), 9.96 (br s, 1H), 7.76 (br s, 1H), 7.65 (dd, J = 8.0, 0.9 Hz, 1H), 7.45–7.22 (m, 5H), 7.14–6.99 (m, 2H), 5.78 (q, J = 7.2 Hz, 1H), 2.47 (s, 3H), 1.74 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C20H19N4O2]+ [M + H]+ = 347.1503, found = 347.2.
Methyl 7-Bromo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole-3-carboxylate
NaH (60% dispersion in mineral oil, 306 mg, 7.65 mmol) was added portionwise to a solution of methyl 7-bromo-1H-indazole-3-carboxylate (1.50 g, 5.88 mmol) in THF (100 mL) under nitrogen at 0 °C, over a period of 5 min. The resulting solution was stirred for 30 min at 0 °C and subsequently (2-(chloromethoxy)ethyl)trimethylsilane (1.08 g, 6.48 mmol) was added dropwise. The reaction was allowed to reach rt and stirred for 1 h. The reaction mixture was quenched with saturated NH4Cl (100 mL) and extracted with EtOAc (3 × 100 mL). The combined organic extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure to afford crude methyl 7-bromo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole-3-carboxylate (2.20 g, 97%) as a pale yellow oil. The crude product was used in the subsequent reaction without further purification. MS (ESI) m/z calculated for [C15H22BrN2O3Si]+ [M + H]+ = 385.0578 and 387.0558, found = 387.1.
7-Bromo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole-3-carboxamide
A solution of methyl 7-bromo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole-3-carboxylate (2.20 g, 5.71 mmol) and ammonia in MeOH (8 M, 36.0 mL, 288 mmol) in a 100 mL sealed tube was stirred at 100 °C for 12 h. The solvent was removed under reduced pressure to afford the crude 7-bromo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole-3-carboxamide (2.10 g, 99%) as a pale yellow solid. The crude product was used in the subsequent reaction without further purification. MS (ESI) m/z calculated for [C14H20BrN3NaO2Si]+ [M + Na]+ = 392.0400 and 394.0380, found = 392.2.
7-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole-3-carboxamide
Pd(dppf)Cl2·CH2Cl2 (212 mg, 0.260 mmol) was added in one portion to a mixture of 7-bromo-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole-3-carboxamide (1.20 g, 3.24 mmol), KOAc (477 mg, 4.86 mmol) and 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(1,3,2-dioxaborolane) (905 mg, 3.56 mmol) in 1,4-dioxane (30 mL) under nitrogen at rt. The resulting suspension was stirred at 100 °C for 12 h. The solvent was removed under reduced pressure and the crude product was purified by flash silica chromatography, elution gradient 0–50% EtOAc in petroleum ether. Fractions containing the desired compound were evaporated to dryness to afford 7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole-3-carboxamide (660 mg, 49%) as a pale yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 8.37 (dd, J = 8.1, 1.3 Hz, 1H), 7.82–7.72 (m, 2H), 7.47 (s, 1H), 7.31 (dd, J = 8.2, 6.9 Hz, 1H), 6.09 (s, 2H), 3.27 (t, J = 8.0 Hz, 2H), 1.36 (s, 12H), 0.70 (t, J = 8.0 Hz, 2H), −0.18 (s, 9H); MS (ESI) m/z calculated for [C20H32BN3NaO4Si]+ [M + Na]+ = 440.2147, found = 440.4.
7-(2,4-Dioxo-1-(1-phenylethyl)-1,2,3,4-tetrahydropyrimidin-5-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole-3-carboxamide
Pd(PPh3)4 (120 mg, 0.104 mmol) was added in one portion to a mixture of 5-bromo-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (382 mg, 1.29 mmol), Na2CO3 (343 mg, 3.24 mmol) and 7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole-3-carboxamide (648 mg, 1.55 mmol) in 1,4-dioxane (20 mL) and water (5 mL) under nitrogen at rt. The resulting solution was stirred at 100 °C for 5 h. The solvent was removed under reduced pressure and the crude product was purified by C18-flash chromatography, elution gradient 30–40% MeCN in water. Fractions containing the desired compound were evaporated to dryness to afford 7-(2,4-dioxo-1-(1-phenylethyl)-1,2,3,4-tetrahydropyrimidin-5-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole-3-carboxamide (250 mg, 38%) as a white solid. MS (ESI) m/z calculated for [C26H31N5NaO4Si]+ [M + Na]+ = 528.2038, found = 528.4.
7-(2,4-Dioxo-1-(1-phenylethyl)-1,2,3,4-tetrahydropyrimidin-5-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole-3-carbonitrile
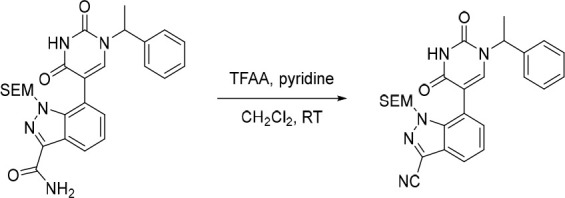
2,2,2-Trifluoroacetic anhydride (110 μL, 0.791 mmol) was added dropwise to a solution of 7-(2,4-dioxo-1-(1-phenylethyl)-1,2,3,4-tetrahydropyrimidin-5-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole-3-carboxamide (200 mg, 0.396 mmol) and pyridine (160 μL, 1.98 mmol) in CH2Cl2 (5 mL) under nitrogen at 0 °C, over a period of 5 min. The resulting solution was stirred at rt for 1 h. The solvent was removed under reduced pressure to afford the crude 7-(2,4-dioxo-1-(1-phenylethyl)-1,2,3,4-tetrahydropyrimidin-5-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole-3-carbonitrile (200 mg) as a pale yellow gum. The crude product was used in the subsequent reaction without further purification. MS (ESI) m/z calculated for [C26H29N5NaO3Si]+ [M + H]+ = 510.1932, found = 510.0.
7-(2,4-Dioxo-1-(1-phenylethyl)-1,2,3,4-tetrahydropyrimidin-5-yl)-1H-indazole-3-carbonitrile (19)
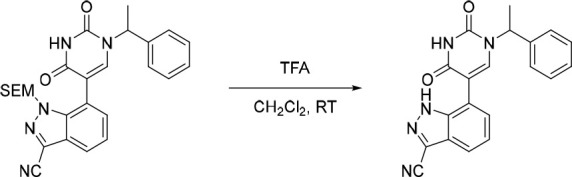
The above crude 7-(2,4-dioxo-1-(1-phenylethyl)-1,2,3,4-tetrahydropyrimidin-5-yl)-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-indazole-3-carbonitrile was diluted in CH2Cl2 (6 mL) and to this solution was added dropwise 2,2,2-trifluoroacetic acid (2.00 mL, 26.1 mmol) under nitrogen at rt. The resulting solution was stirred at rt for 4 h. The solvent was removed under reduced pressure and the crude product was purified by preparative HPLC (Xselect CSH OBD, 5 μm, 30 × 150 mm) using decreasingly polar mixtures of water (containing 0.1% FA) and MeCN (elution gradient 38–48%). Fractions containing the desired compound were evaporated to dryness to afford 7-(2,4-dioxo-1-(1-phenylethyl)-1,2,3,4-tetrahydropyrimidin-5-yl)-1H-indazole-3-carbonitrile (50.0 mg, 35% over two steps) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 14.06 (s, 1H), 11.71 (s, 1H), 7.97 (s, 1H), 7.86 (dd, J = 7.6, 1.4 Hz, 1H), 7.46–7.24 (m, 7H), 5.80 (q, J = 7.3 Hz, 1H), 1.76 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C20H16N5O2]+ [M + H]+ = 358.1299, found = 358.3.
5-(6-Fluoro-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (20)
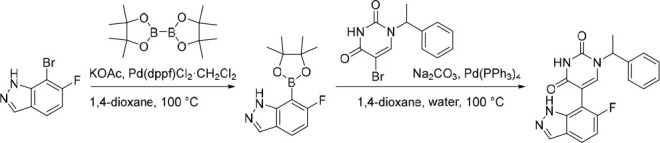
Pd(dppf)Cl2·CH2Cl2 (47.5 mg, 0.0582 mmol) was added in one portion to a mixture of 7-bromo-6-fluoro-1H-indazole (250 mg, 1.16 mmol), KOAc (285 mg, 2.91 mmol) and 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(1,3,2-dioxaborolane) (886 mg, 3.49 mmol) in 1,4-dioxane (20 mL) under nitrogen at rt. The resulting suspension was stirred at 100 °C for 18 h to give a black suspension. The crude product was used as a suspension in the subsequent reaction without further purification. MS (ESI) m/z calculated for [C13H17BFN2O2]+ [M + H]+ = 263.1362, found = 263.1.
Water (5 mL), 5-bromo-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (282 mg, 0.955 mmol), Na2CO3 (303 mg, 2.86 mmol) and Pd(PPh3)4 (55.1 mg, 0.0477 mmol) were added sequentially to the above suspension under nitrogen at rt. The resulting suspension was stirred at 100 °C for 5 h. The reaction mixture was diluted with EtOAc (150 mL) and washed with water (50 mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by preparative HPLC (Xselect CSH OBD, 5 μm, 30 × 150 mm), using decreasingly polar mixtures of water (containing 0.1% FA) and MeOH (elution gradient 50–51%). Fractions containing the desired compound were evaporated to dryness to afford 5-(6-fluoro-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (25.0 mg, 6% over two steps) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 12.98 (s, 1H), 11.66 (s, 1H), 8.08 (s, 1H), 7.97 (s, 1H), 7.77 (dd, J = 8.8, 4.9 Hz, 1H), 7.49–7.28 (m, 5H), 7.00 (t, J = 9.3 Hz, 1H), 5.81 (q, J = 7.1 Hz, 1H), 1.73 (d, J = 7.2 Hz, 3H); 19F NMR (282 MHz, DMSO-d6) δ −119.94; MS (ESI) m/z calculated for [C19H16FN4O2]+ [M + H]+ = 351.1252, found = 351.2.
5-(5-Fluoro-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (21)
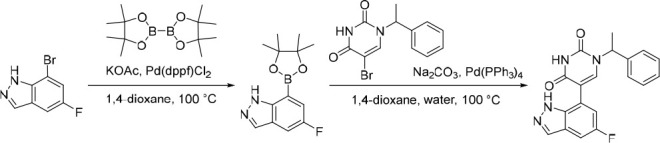
Pd(dppf)Cl2 (197 mg, 0.241 mmol) was added in one portion to a mixture of KOAc (445 mg, 4.53 mmol), 7-bromo-5-fluoro-1H-indazole (37) (650 mg, 3.02 mmol) and 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(1,3,2-dioxaborolane) (844 mg, 3.33 mmol) in 1,4-dioxane (20 mL) under nitrogen at rt. The resulting suspension was stirred at 100 °C for 12 h to give a black suspension. The crude product was used as a suspension in the subsequent reaction without further purification. MS (ESI) m/z calculated for [C13H17BFN2O2]+ [M + H]+ = 263.1362, found = 263.0.
Water (5 mL), 5-bromo-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (803 mg, 2.72 mmol), Na2CO3 (961 mg, 9.07 mmol) and Pd(PPh3)4 (175 mg, 0.151 mmol) were added sequentially to the above suspension under nitrogen at rt. The resulting suspension was stirred at 100 °C for 3 h. The reaction mixture was diluted with EtOAc (150 mL) and washed with water (50 mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by C18-flash chromatography, elution gradient 30–60% MeCN in water. Fractions containing the desired compound were evaporated to dryness to afford 5-(5-fluoro-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (540 mg, 51% over two steps) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 12.93 (s, 1H), 11.59 (s, 1H), 8.07 (s, 1H), 7.96 (s, 1H), 7.56–7.48 (m, 1H), 7.47–7.33 (m, 4H), 7.35–7.25 (m, 1H), 7.15 (s, 1H), 5.78 (q, J = 7.2 Hz, 1H), 1.77 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C19H16FN4O2]+ [M + H]+ = 351.1252, found = 351.0.
5-(3-Bromo-5-fluoro-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione
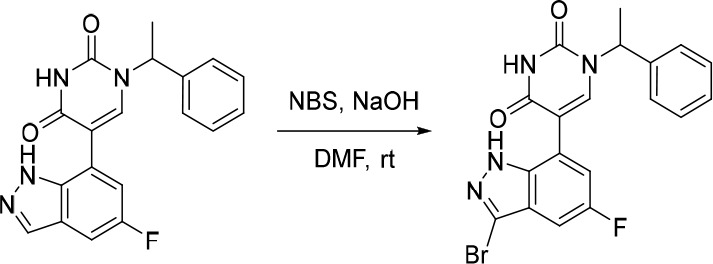
N-Bromosuccinimide (274 mg, 1.54 mmol) was added to a mixture of 5-(5-fluoro-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (21) (540 mg, 1.54 mmol) and NaOH (123 mg, 3.08 mmol) in DMF (6 mL) under nitrogen at rt. The resulting solution was stirred at rt for 30 min to give a black suspension. The crude suspension was purified by C18-flash chromatography, elution gradient 30–70% MeCN in water. Fractions containing the desired compound were evaporated to dryness to afford 5-(3-bromo-5-fluoro-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (400 mg, 61%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 13.28 (br s, 1H), 11.71 (br s, 1H), 8.05 (s, 1H), 7.48–7.24 (m, 7H), 5.78 (q, J = 7.1 Hz, 1H), 1.76 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C19H15BrFN4O2]+ [M + H]+ = 429.0357 and 431.0337, found = 430.9.
7-(2,4-Dioxo-1-(1-phenylethyl)-1,2,3,4-tetrahydropyrimidin-5-yl)-5-fluoro-1H-indazole-3-carbonitrile (22)
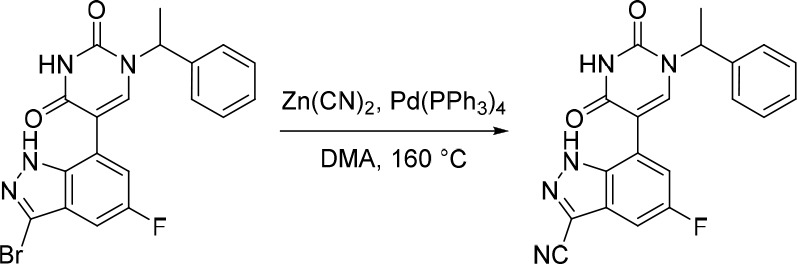
Pd(PPh3)4 (808 mg, 0.699 mmol), 5-(3-bromo-5-fluoro-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (300 mg, 0.699 mmol) and dicyanozinc (164 mg, 1.40 mmol) were suspended in DMA (6 mL) under nitrogen at rt and sealed into a microwave tube. The reaction was heated to 160 °C for 30 min in a microwave reactor and subsequently cooled to rt. The solvent was removed under reduced pressure and the crude product was purified by preparative HPLC (Xselect CSH OBD, 5 μm, 30 × 150 mm) using decreasingly polar mixtures of water (containing 0.1% FA) and MeCN (elution gradient 40–60%). Fractions containing the desired compound were evaporated to dryness to afford to afford 7-(2,4-dioxo-1-(1-phenylethyl)-1,2,3,4-tetrahydropyrimidin-5-yl)-5-fluoro-1H-indazole-3-carbonitrile (50.0 mg, 19%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 14.16 (s, 1H), 11.74 (s, 1H), 8.07 (s, 1H), 7.71 (dd, J = 8.4, 2.3 Hz, 1H), 7.45–7.22 (m, 6H), 5.77 (q, J = 7.2 Hz, 1H), 1.75 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C20H15FN5O2]+ [M + H]+ = 376.1204, found = 376.3.
5-Bromo-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione
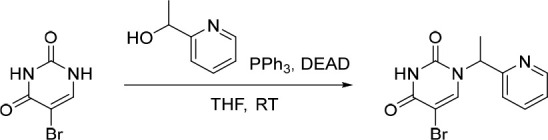
Diethyl azodicarboxylate (1.29 mL, 8.15 mmol) was added dropwise to a mixture of 5-bromopyrimidine-2,4(1H,3H)-dione (35) (1.20 g, 6.28 mmol), 1-(pyridin-2-yl)ethan-1-ol (696 mg, 5.65 mmol) and PPh3 (2.14 g, 8.16 mmol) in THF (50 mL) under nitrogen at rt, over a period of 1 min. The resulting solution was stirred at rt for 2 h. The mixture was filtered through a Celite pad. The solvent of the filtrate was removed under reduced pressure and the crude product was purified by C18-flash chromatography, elution gradient 0–60% MeCN in water. Fractions containing the desired compound were evaporated to dryness to afford 5-bromo-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione (1.10 g, 59%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 11.79 (s, 1H), 8.54 (ddd, J = 4.9, 1.8, 0.9 Hz, 1H), 8.18 (s, 1H), 7.80 (td, J = 7.7, 1.8 Hz, 1H), 7.41 (dd, J = 7.9, 1.3 Hz, 1H), 7.31 (ddd, J = 7.6, 4.8, 1.1 Hz, 1H), 5.72 (q, J = 7.2 Hz, 1H), 1.69 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C11H11BrN3O2]+ [M + H]+ = 296.0030 and 298.0009, found = 296.0.
5-(5-Fluoro-1H-indazol-7-yl)-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione
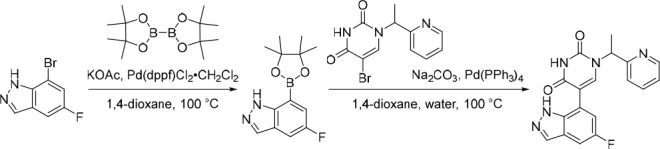
Pd(dppf)Cl2·CH2Cl2 (152 mg, 0.186 mmol) was added in one portion to 7-bromo-5-fluoro-1H-indazole (37) (500 mg, 2.33 mmol), KOAc (571 mg, 5.81 mmol) and 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(1,3,2-dioxaborolane) (709 mg, 2.79 mmol) in 1,4-dioxane (20 mL) under nitrogen at rt. The resulting suspension was stirred at 100 °C for 12 h. The crude product was used as a suspension in the subsequent reaction without further purification. MS (ESI) m/z calculated for [C13H17BFN2O2]+ [M + H]+ = 263.1362, found = 263.2.
Water (5 mL), 5-bromo-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione (600 mg, 2.03 mmol), Na2CO3 (644 mg, 6.08 mmol) and Pd(PPh3)4 (117 mg, 0.101 mmol) were added sequentially to the above suspension under nitrogen at rt. The resulting suspension was stirred at 100 °C for 5 h. The reaction mixture was diluted with EtOAc (150 mL) and washed with water (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by C18-flash chromatography, elution gradient 0–50% MeCN in water. Fractions containing the desired compound were evaporated to dryness to afford 5-(5-fluoro-1H-indazol-7-yl)-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione (330 mg, 40% over two steps) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 12.90 (s, 1H), 11.65 (s, 1H), 8.55 (d, J = 4.1 Hz, 1H), 8.07 (d, J = 1.3 Hz, 1H), 8.03 (s, 1H), 7.82 (td, J = 7.7, 1.8 Hz, 1H), 7.53 (dd, J = 8.9, 2.2 Hz, 1H), 7.45 (d, J = 7.9 Hz, 1H), 7.32 (dd, J = 6.8, 4.9 Hz, 1H), 7.17 (dd, J = 9.8, 2.3 Hz, 1H), 5.82 (q, J = 7.1 Hz, 1H), 1.77 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C18H15FN5O2]+ [M + H]+ = 352.1204, found = 352.2.
5-(3-Bromo-5-fluoro-1H-indazol-7-yl)-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione
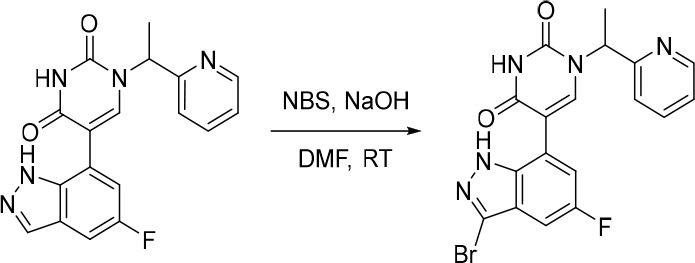
N-Bromosuccinimide (152 mg, 0.854 mmol) was added in one portion to a mixture of 5-(5-fluoro-1H-indazol-7-yl)-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione (300 mg, 0.854 mmol) and NaOH (68.3 mg, 1.71 mmol) in DMF (5 mL) under nitrogen at rt. The resulting solution was stirred at rt for 1 h. The crude product was purified directly by C18-flash chromatography, elution gradient 0–40% MeCN in water. Fractions containing the desired compound were evaporated to dryness to afford 5-(3-bromo-5-fluoro-1H-indazol-7-yl)-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione (250 mg, 68%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 12.65 (br s, 1H), 8.54 (d, J = 4.1 Hz, 1H), 8.37 (br s, 1H), 7.81 (td, J = 7.7, 1.8 Hz, 1H), 7.49–7.23 (m, 4H), 5.82 (q, J = 7.1 Hz, 1H), 1.78 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C18H14BrFN5O2]+ [M + H]+ = 430.0310 and 432.0289, found = 430.2.
7-(2,4-Dioxo-1-(1-(pyridin-2-yl)ethyl)-1,2,3,4-tetrahydropyrimidin-5-yl)-5-fluoro-1H-indazole-3-carboxamide
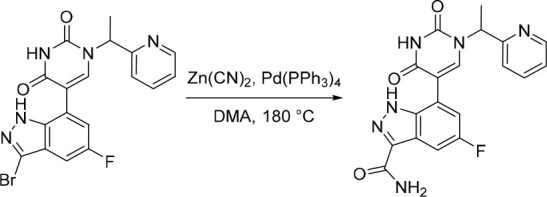
Pd(PPh3)4 (33.6 mg, 0.0291 mmol), 5-(3-bromo-5-fluoro-1H-indazol-7-yl)-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione (250 mg, 0.581 mmol) and dicyanozinc (341 mg, 2.90 mmol) were suspended in DMA (10 mL) under nitrogen at rt and sealed into a microwave tube. The reaction was heated to 180 °C for 2 h in a microwave reactor and subsequently cooled to rt. The solvent was removed under reduced pressure and the crude product was purified by C18-flash chromatography, elution gradient 0–100% MeCN in water. Fractions containing the desired compound were evaporated to dryness to afford 7-(2,4-dioxo-1-(1-(pyridin-2-yl)ethyl)-1,2,3,4-tetrahydropyrimidin-5-yl)-5-fluoro-1H-indazole-3-carboxamide (130 mg, 57%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 13.33 (s, 1H), 11.70 (s, 1H), 8.60–8.51 (m, 1H), 8.08 (s, 1H), 7.88–7.70 (m, 3H), 7.46 (d, J = 7.9 Hz, 1H), 7.38 (br s, 1H), 7.32 (ddd, J = 7.5, 4.8, 0.8 Hz, 1H), 7.23 (dd, J = 9.7, 2.3 Hz, 1H), 5.83 (q, J = 7.1 Hz, 1H), 1.77 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C19H16FN6O3]+ [M + H]+ = 395.1262, found = 395.3.
7-(2,4-Dioxo-1-(1-(pyridin-2-yl)ethyl)-1,2,3,4-tetrahydropyrimidin-5-yl)-5-fluoro-1H-indazole-3-carbonitrile (23)
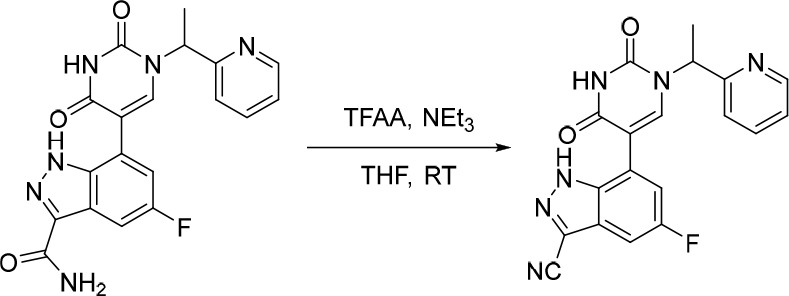
NEt3 (138 μL, 0.990 mmol) was added to a solution of 7-(2,4-dioxo-1-(1-(pyridin-2-yl)ethyl)-1,2,3,4-tetrahydropyrimidin-5-yl)-5-fluoro-1H-indazole-3-carboxamide (130 mg, 0.330 mmol) and 2,2,2-trifluoroacetic anhydride (138 μL, 0.993 mmol) in THF (8 mL) under nitrogen at rt. The resulting suspension was stirred at rt for 2 h. The reaction was concentrated under reduced pressure and purified by C18-flash chromatography, elution gradient 0–100% MeCN in water. Fractions containing the desired compound were evaporated to dryness to afford a white solid as the crude product. The crude product was repurified by preparative HPLC (Xselect CSH OBD, 5 μm, 30 × 150 mm) using decreasingly polar mixtures of water (containing 0.05% TFA) and MeCN (elution gradient 30–60%). Fractions containing the desired compound were evaporated to dryness to afford 7-(2,4-dioxo-1-(1-(pyridin-2-yl)ethyl)-1,2,3,4-tetrahydropyrimidin-5-yl)-5-fluoro-1H-indazole-3-carbonitrile (20.0 mg, 16%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 14.20 (s, 1H), 11.76 (s, 1H), 8.59–8.50 (m, 1H), 8.14 (s, 1H), 7.83 (t, J = 7.5 Hz, 1H), 7.74 (d, J = 7.7 Hz, 1H), 7.51–7.27 (m, 3H), 5.89–5.75 (m, 1H), 1.77 (d, J = 7.4 Hz, 3H); MS (ESI) m/z calculated for [C19H14FN6O2]+ [M + H]+ = 377.1157, found = 377.2.
5-Bromo-1-(1-(4-fluorophenyl)ethyl)pyrimidine-2,4(1H,3H)-dione (36)
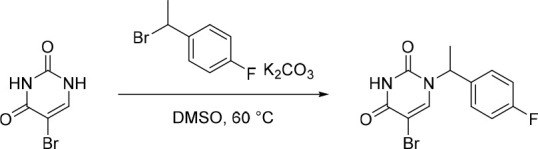
K2CO3 (4.08 g, 29.5 mmol) was added to a solution of 5-bromopyrimidine-2,4(1H,3H)-dione (35) (3.39 g, 17.8 mmol) and 1-(1-bromoethyl)-4-fluorobenzene (2.00 g, 9.85 mmol) in DMSO (18 mL) under nitrogen at rt. The resulting suspension was stirred at 60 °C for 3 h. The reaction was concentrated under reduced pressure and the crude product was purified by C18-flash chromatography, elution gradient 0–100% MeCN in water. Fractions containing the desired compound were evaporated to dryness to afford 5-bromo-1-(1-(4-fluorophenyl)ethyl)pyrimidine-2,4(1H,3H)-dione (2.50 g, 81%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 7.56 (s, 1H), 7.33 (dd, J = 8.6, 5.6 Hz, 2H), 7.15 (t, J = 8.9 Hz, 2H), 5.77 (q, J = 7.2 Hz, 1H), 1.56 (d, J = 7.3 Hz, 3H); MS (ESI) m/z calculated for [C12H11BrFN2O2]+ [M + H]+ = 312.9982, found = 313.1.
5-Fluoro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indazole (38)
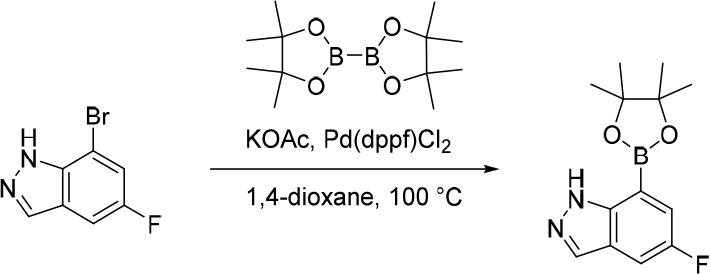
Pd(dppf)Cl2 (408 mg, 0.558 mmol) was added in one portion to a mixture of KOAc (1.64 g, 16.7 mmol), 7-bromo-5-fluoro-1H-indazole (37) (1.20 g, 5.58 mmol) and 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(1,3,2-dioxaborolane) (1.42 g, 5.59 mmol) in 1,4-dioxane (30 mL) under nitrogen at rt. The resulting mixture was stirred at 100 °C for 12 h. The solvent was removed under reduced pressure and the crude product was purified by flash silica chromatography, elution gradient 40–50% EtOAc in petroleum ether. Fractions containing the desired compound were evaporated to dryness to afford 5-fluoro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indazole (1.50 g, 53% pure by LC-MS) as a brown solid. The impure material was used in the next step without further purification. 1H NMR (300 MHz, DMSO-d6) δ 12.68 (s, 1H), 8.09 (s, 1H), 7.69 (dd, J = 9.0, 2.6 Hz, 1H), 7.39 (dd, J = 9.1, 2.5 Hz, 1H), 1.34 (s, 12H); MS (ESI) m/z calculated for [C13H17BFN2O2]+ [M + H]+ = 263.1362, found = 263.2.
5-(5-Fluoro-1H-indazol-7-yl)-1-(1-(4-fluorophenyl)ethyl)pyrimidine-2,4(1H,3H)-dione (26)
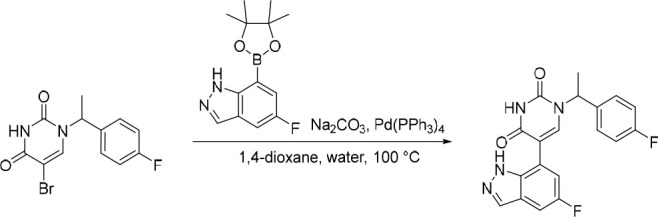
Pd(PPh3)4 (661 mg, 0.572 mmol) was added in one portion to a mixture of 5-bromo-1-(1-(4-fluorophenyl)ethyl)pyrimidine-2,4(1H,3H)-dione (36) (1.79 g, 5.72 mmol), 5-fluoro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indazole (38) (1.50 g, 5.72 mmol; material assumed pure for the purposes of reagent equivalent calculation) and Na2CO3 (1.82 g, 17.2 mmol) in 1,4-dioxane (20 mL) and water (20 mL) under nitrogen at rt. The resulting suspension was stirred at 100 °C for 12 h. The reaction was concentrated under reduced pressure and purified by C18-flash chromatography, elution gradient 50–60% MeCN in water. Fractions containing the desired compound were evaporated to dryness to afford 5-(5-fluoro-1H-indazol-7-yl)-1-(1-(4-fluorophenyl)ethyl)pyrimidine-2,4(1H,3H)-dione (700 mg, 34% over two steps) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 12.61 (br s, 1H), 7.98 (s, 1H), 7.67 (s, 1H), 7.45–7.27 (m, 3H), 7.13 (t, J = 8.9 Hz, 3H), 5.87 (q, J = 7.1 Hz, 1H), 1.67 (d, J = 7.3 Hz, 3H); MS (ESI) m/z calculated for [C19H15F2N4O2]+ [M + H]+ = 369.1158, found = 369.2.
(S)-5-(5-Fluoro-1H-indazol-7-yl)-1-(1-(4-fluorophenyl)ethyl)pyrimidine-2,4(1H,3H)-dione (27)
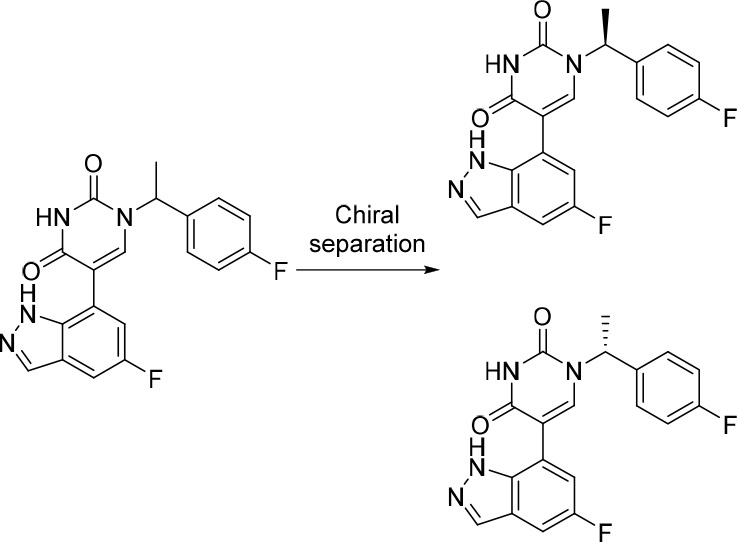
300 mg of an enantiomeric mixture of 5-(5-fluoro-1H-indazol-7-yl)-1-(1-(4-fluorophenyl)ethyl)pyrimidine-2,4(1H,3H)-dione, were purified by preparative chiral-HPLC (CHIRALPAK IG, 5 μm, 20 × 250 mm), isocratic 50% EtOH in hexane (containing 8 mmol/L NH3.MeOH). Fractions containing the desired products were evaporated to dryness to afford enantiopure (S)-5-(5-fluoro-1H-indazol-7-yl)-1-(1-(4-fluorophenyl)ethyl)pyrimidine-2,4(1H,3H)-dione (Rt: 8.65, 30.0 mg, 10%) (27) and (R)-5-(5-fluoro-1H-indazol-7-yl)-1-(1-(4-fluorophenyl)ethyl)pyrimidine-2,4(1H,3H)-dione (Rt: 11.82, 50.0 mg, 17%) as white solids. Data for 27: NMR and MS (ESI) results are equivalent to 26; purity 93–94%; ee >99%. Data for (R)-5-(5-fluoro-1H-indazol-7-yl)-1-(1-(4-fluorophenyl)ethyl)pyrimidine-2,4(1H,3H)-dione: ee = 98%.
5-(5-Fluoro-3-iodo-1H-indazol-7-yl)-1-(1-(4-fluorophenyl)ethyl)pyrimidine-2,4(1H,3H)-dione
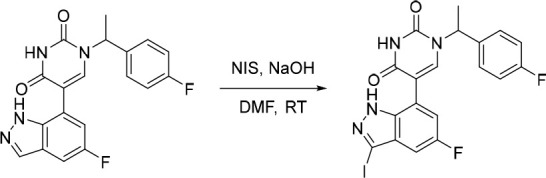
N-Iodosuccinimide (366 mg, 1.63 mmol) was added to a solution of 5-(5-fluoro-1H-indazol-7-yl)-1-(1-(4-fluorophenyl)ethyl)pyrimidine-2,4(1H,3H)-dione (26) (400 mg, 1.09 mmol) and NaOH (87.0 mg, 2.17 mmol) in DMF (20 mL) at rt to give a brown solution. The resulting mixture was stirred at rt for 5 h. The crude product was purified directly by C18-flash chromatography, elution gradient 40–50% MeCN in water. Fractions containing the desired compound were evaporated to dryness to afford 5-(5-fluoro-3-iodo-1H-indazol-7-yl)-1-(1-(4-fluorophenyl)ethyl)pyrimidine-2,4(1H,3H)-dione (450 mg, 84%) as a white solid. 1H NMR (300 MHz, Methanol-d4) δ 7.86 (s, 1H), 7.54–7.41 (m, 3H), 7.22 (dd, J = 9.6, 2.3 Hz, 1H), 7.17–7.04 (m, 2H), 5.93 (q, J = 7.2 Hz, 1H), 1.81 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C19H14F2IN4O2]+ [M + H]+ = 495.0124, found = 495.1.
5-Fluoro-7-(1-(1-(4-fluorophenyl)ethyl)-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)-1H-indazole-3-carbonitrile (24)
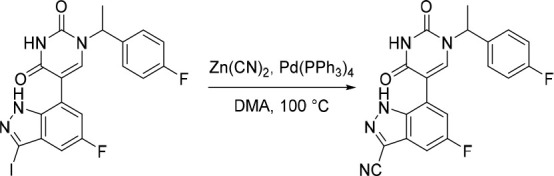
Pd(PPh3)4 (105 mg, 0.0909 mmol) was added to a mixture of 5-(5-fluoro-3-iodo-1H-indazol-7-yl)-1-(1-(4-fluorophenyl)ethyl)pyrimidine-2,4(1H,3H)-dione (450 mg, 0.910 mmol) and dicyanozinc (321 mg, 2.73 mmol) in DMA (20 mL) at rt to give a brown solution. The solution was stirred at 100 °C for 12 h. The reaction was filtered and directly purified by C18-flash chromatography, elution gradient 5–95% MeCN in water. Fractions containing the desired compound were evaporated to dryness to afford the 5-fluoro-7-(1-(1-(4-fluorophenyl)ethyl)-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)-1H-indazole-3-carbonitrile (106 mg, 30%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 14.19 (s, 1H), 11.76 (s, 1H), 8.11 (s, 1H), 7.73 (dd, J = 8.3, 2.1 Hz, 1H), 7.47 (dd, J = 8.6, 5.5 Hz, 2H), 7.39 (dd, J = 9.7, 2.0 Hz, 1H), 7.20 (t, J = 8.8 Hz, 2H), 5.76 (q, J = 6.9 Hz, 1H), 1.76 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C20H14F2N5O2]+ [M + H]+ = 394.1110, found = 394.3.
(S)-5-Fluoro-7-(1-(1-(4-fluorophenyl)ethyl)-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)-1H-indazole-3-carbonitrile (25)
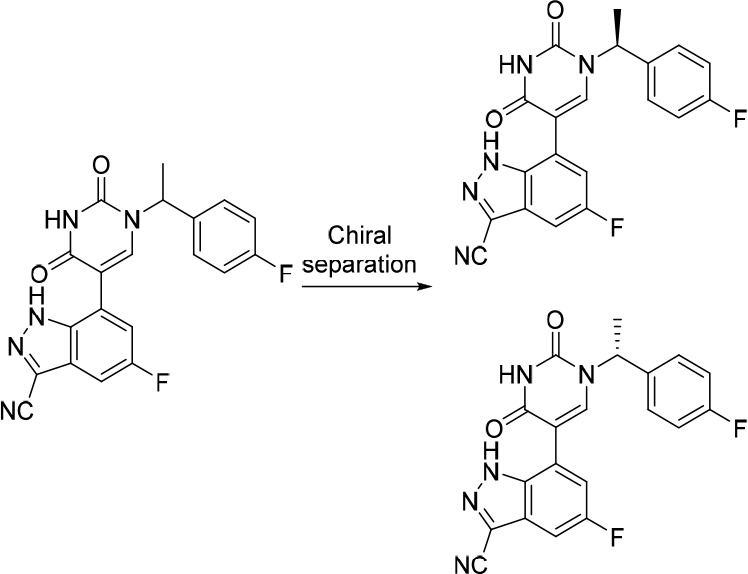
The enantiomeric mixture above (24) was purified by preparative chiral-HPLC (CHIRALPAK IG-03, 5 μm, 20 × 250 mm), isocratic 40% EtOH in hexane (8 mmol/L NH3.MeOH). Fractions containing the desired products were evaporated to dryness to afford enantiopure (S)-5-fluoro-7-(1-(1-(4-fluorophenyl)ethyl)-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)-1H-indazole-3-carbonitrile (Rt: 8.05 min, 30.0 mg, 28%) (25) and enantiopure (R)-5-fluoro-7-(1-(1-(4-fluorophenyl)ethyl)-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)-1H-indazole-3-carbonitrile (Rt: 10.08 min, 30.0 mg, 28%) as white solids. Data for 25: 1H NMR (300 MHz, Methanol-d4) δ 7.87 (s, 1H), 7.55–7.40 (m, 3H), 7.23 (dd, J = 9.6, 2.3 Hz, 1H), 7.17–7.04 (m, 2H), 5.93 (q, J = 7.2 Hz, 1H), 1.81 (d, J = 7.2 Hz, 3H); 13C NMR (75 MHz, Methanol-d4) δ 163.83 (d, JCF = 256.6 Hz), 163.80, 160.6 (d, JCF = 253.2 Hz), 152.4, 143.3, 137.6, 137.1, 130.3 (d, JCF = 8.3 Hz), 126.0 (d, JCF = 11.4 Hz), 121.4 (d, JCF = 9.8 Hz), 118.8 (d, JCF = 28.4 Hz), 116.6 (d, JCF = 21.8 Hz), 114.4, 112.06, 112.04, 103.7 (d, JCF = 25.1 Hz), 55.7, 18.9; 19F NMR (282 MHz, Methanol-d4) δ −115.97, −120.35; MS (ESI) m/z calculated for [C20H14F2N5O2]+ [M + H]+ = 394.1110, found = 394.2; ee = 99%. Data for (R)-5-fluoro-7-(1-(1-(4-fluorophenyl)ethyl)-2,4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)-1H-indazole-3-carbonitrile: ee = 97%
5-(3-Bromo-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione
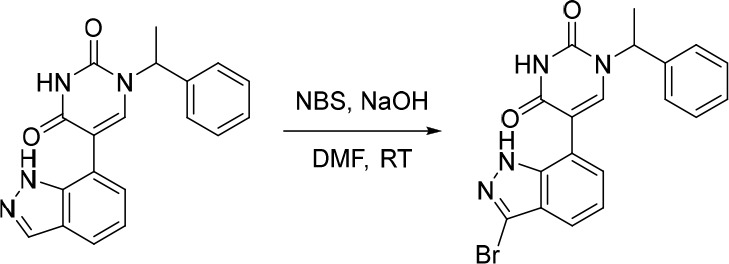
N-Bromosuccinimide (517 mg, 2.91 mmol) was added portionwise to a mixture of 5-(1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (15) (920 mg, 2.77 mmol) and NaOH (221 mg, 5.54 mmol) in DMF (10 mL) under nitrogen at 0 °C. The resulting suspension was stirred at rt for 1 h. The reaction was concentrated under reduced pressure and the crude product purified by C18-flash chromatography, elution gradient 0–40% MeCN in water. Fractions containing the desired compound were evaporated to dryness to afford 5-(3-bromo-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (950 mg, 83%) as a pale yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 12.36 (br s, 1H), 8.08 (s, 1H), 7.51 (dd, J = 8.1, 0.9 Hz, 1H), 7.47–7.26 (m, 6H), 7.17 (dd, J = 8.0, 7.2 Hz, 1H), 5.82 (q, J = 7.1 Hz, 1H), 1.76 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C19H16BrN4O2]+ [M + H]+ = 411.0452 and 413.0431, found = 413.1.
1-(1-Phenylethyl)-5-(3-((trimethylsilyl)ethynyl)-1H-indazol-7-yl)pyrimidine-2,4(1H,3H)-dione
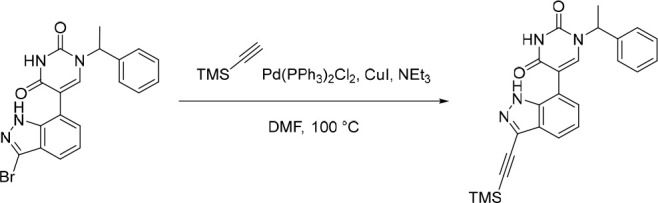
Pd(PPh3)2Cl2 (42.7 mg, 0.0608 mmol) and CuI (11.6 mg, 0.0609 mmol) was added in one portion to a mixture of 5-(3-bromo-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (250 mg, 0.608 mmol), NEt3 (615 mg, 6.08 mmol) and ethynyltrimethylsilane (299 mg, 3.04 mmol) in DMF (5 mL) under nitrogen at rt. The resulting suspension was stirred at 100 °C for 12 h. The reaction was concentrated under reduced pressure and the crude product purified by C18-flash chromatography, elution gradient 0–40% MeCN in water. Fractions containing the desired compound were evaporated to dryness to afford 1-(1-phenylethyl)-5-(3-((trimethylsilyl)ethynyl)-1H-indazol-7-yl)pyrimidine-2,4(1H,3H)-dione (130 mg, 50%) as a pale yellow solid. MS (ESI) m/z calculated for [C24H25N4O2Si]+ [M + H]+ = 429.1741, found = 429.2.
5-(3-Ethynyl-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (28)
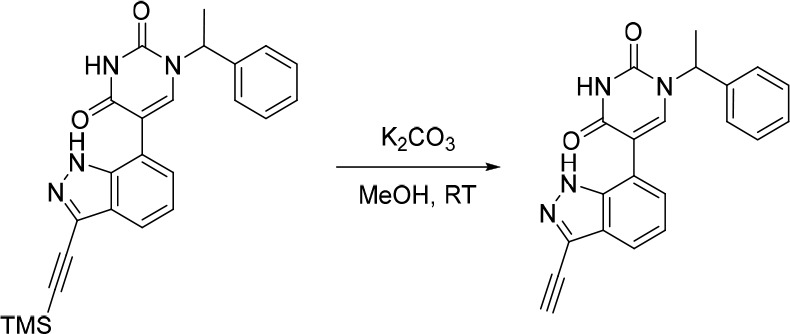
K2CO3 (74.2 mg, 0.537 mmol) was added in one portion to a solution of 1-(1-phenylethyl)-5-(3-((trimethylsilyl)ethynyl)-1H-indazol-7-yl)pyrimidine-2,4(1H,3H)-dione (115 mg, 0.268 mmol) in MeOH (5 mL) under nitrogen at rt. The resulting suspension was stirred at rt for 1 h. The reaction was concentrated under reduced pressure and the crude product purified by preparative HPLC (Xselect CSH OBD, 5 μm, 30 × 150 mm) using decreasingly polar mixtures of water (containing 0.1% FA) and MeCN (elution gradient 38–48%). Fractions containing the desired compound were evaporated to dryness to afford 5-(3-ethynyl-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (39.4 mg, 41%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 13.14 (s, 1H), 11.62 (s, 1H), 7.88 (s, 1H), 7.73–7.64 (m, 1H), 7.47–7.26 (m, 5H), 7.26–7.16 (m, 2H), 5.80 (q, J = 7.1 Hz, 1H), 4.49 (s, 1H), 1.76 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C21H17N4O2]+ [M + H]+ = 357.1346, found = 357.2.
7-Bromo-5-fluoro-3-iodo-1H-indazole (39)
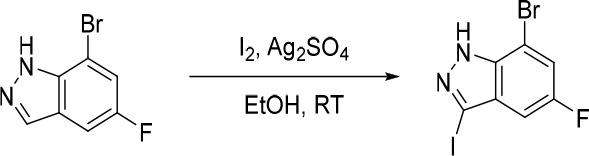
Silver sulfate (5.80 g, 18.6 mmol) was added to a mixture of I2 (4.72 g, 18.6 mmol) and 7-bromo-5-fluoro-1H-indazole (37) (4.00 g, 18.6 mmol) in EtOH (500 mL) under air at rt. The resulting solution was stirred at rt for 17 h. The reaction mixture was filtered through glass fiber paper and the filtrate was concentrated under reduced pressure to afford 7-bromo-5-fluoro-3-iodo-1H-indazole (6.00 g, 95%) as a pale yellow solid. The crude product was used in the subsequent reaction without further purification. 1H NMR (300 MHz, DMSO-d6) δ 8.33 (br s, 1H), 7.71 (dd, J = 8.7, 2.2 Hz, 1H), 7.24 (dd, J = 8.4, 2.2 Hz, 1H); MS (ESI) m/z calculated for [C7H4BrFN2]+ [M + H]+ = 340.8582 and 342.8561, found = 342.9.
7-Bromo-5-fluoro-3-((trimethylsilyl)ethynyl)-1H-indazole (40)

Ethynyltrimethylsilane (1.73 g, 17.6 mmol) was added to a degassed suspension of 7-bromo-5-fluoro-3-iodo-1H-indazole (39) (6.00 g, 17.6 mmol), CuI (335 mg, 1.76 mmol) and Pd(PPh3)4 (2.03 g, 1.76 mmol) in NEt3 (50 mL) and MeCN (50 mL) under nitrogen at rt. The reaction was stirred at 80 °C for 17 h. The solvent was removed under reduced pressure and the crude product was purified by flash silica chromatography, elution gradient 0–20% EtOAc in petroleum ether. Fractions containing the desired compound were evaporated to dryness to afford 7-bromo-5-fluoro-3-((trimethylsilyl)ethynyl)-1H-indazole (2.40 g, 44%) as a pale yellow solid. 1H NMR (500 MHz, Chloroform-d) δ 10.12 (s, 1H), 7.43–7.41 (m, 2H), 0.31 (s, 9H); MS (ESI) m/z calculated for [C12H13BrFN2Si]+ [M + H]+ = 311.0010 and 312.9990, found = 311.0.
5-Fluoro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3-((trimethylsilyl)ethynyl)-1H-indazole (41)

Pd(dppf)Cl2·CH2Cl2 (630 mg, 0.771 mmol) was added to a suspension of 7-bromo-5-fluoro-3-((trimethylsilyl)ethynyl)-1H-indazole (40) (2.40 g, 7.71 mmol), KOAc (2.27 g, 23.1 mmol) and 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(1,3,2-dioxaborolane) (2.94 g, 11.6 mmol) in 1,4-dioxane (100 mL) under nitrogen at rt. The resulting suspension was stirred at 80 °C for 17 h. The solvent was removed under reduced pressure and the crude product was purified by flash silica chromatography, elution gradient 0–5% EtOAc in petroleum ether. Fractions containing the desired compound were evaporated to dryness to afford 5-fluoro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3-((trimethylsilyl)ethynyl)-1H-indazole (2.10 g, 76%) as a pale yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.08 (s, 1H, s), 7.55 (dd, J = 8.4, 2.5 Hz, 1H), 7.46 (dd, J = 9.0, 2.6 Hz, 1H), 1.35 (s, 12H), 0.28 (s, 9H); MS (ESI) m/z calculated for [C18H25BFN2O2Si]+ [M + H]+ = 359.1757, found = 359.2.
5-(3-Ethynyl-5-fluoro-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (29)
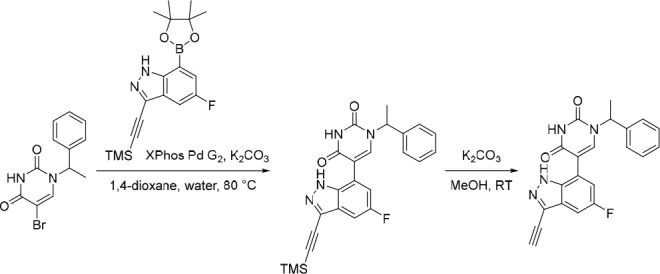
XPhos Pd G2 (42.3 mg, 0.0538 mmol) was added to a solution of 5-fluoro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3-((trimethylsilyl)ethynyl)-1H-indazole (41) (269 mg, 0.751 mmol), 5-bromo-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (148 mg, 0.501 mmol) and K2CO3 (173 mg, 1.25 mmol) in 1,4-dioxane (2.0 mL) and water (0.5 mL) under nitrogen at rt. The reaction was degassed and then stirred at 80 °C for 3 h. After cooling, the reaction was diluted with EtOAc and washed with water. The aqueous layer was then extracted with EtOAc. The combined organic extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash silica chromatography, elution gradient 0–100% EtOAc in heptane. Fractions containing the desired compound were evaporated to dryness.
The above residue was dissolved in MeOH (2.5 mL) before the addition of K2CO3 (500 mg, 3.62 mmol) to the reaction mixture. The reaction was stirred at rt for 1 h and upon completion, the reaction was filtered and the solids washed with CH2Cl2. The filtrate was concentrated under reduced pressure and purified by preparative HPLC (Xselect CSH OBD, 5 μm, 30 × 100 mm) using decreasingly polar mixtures of water (containing 1% NH3) and MeCN. Fractions containing the desired compound were evaporated to dryness to afford 5-(3-ethynyl-5-fluoro-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (46.0 mg, 25% over two steps) as a colorless solid. 1H NMR (500 MHz, DMSO-d6) δ 13.29 (br s, 1H), 11.66 (br s, 1H), 8.07 (s, 1H), 7.44–7.40 (m, 3H), 7.39–7.34 (m, 2H), 7.33–7.22 (m, 2H), 5.78 (q, J = 7.4 Hz, 1H), 4.52 (s, 1H), 1.77 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C21H16FN4O2]+ [M + H]+ = 375.3829, found = 375.4.
(S)-5-Bromo-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (42)
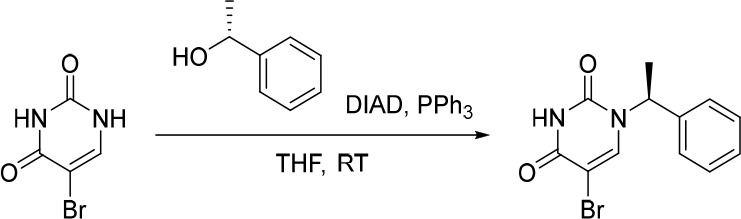
Diisopropyl azodicarboxylate (2.39 mL, 12.3 mmol) was added dropwise to a mixture of PPh3 (3.22 g, 12.3 mmol), (R)-1-phenylethan-1-ol (1.00 g, 8.19 mmol) and 5-bromopyrimidine-2,4(1H,3H)-dione (35) (1.56 g, 8.17 mmol) in THF (40 mL) under nitrogen at rt. The resulting solution was stirred at rt for 17 h. The solvent was removed under reduced pressure and the crude product was purified by flash silica chromatography, elution gradient 0–40% EtOAc in petroleum ether. Fractions containing the desired compound were evaporated to dryness to afford an impure mixture which was repurified by C18-flash chromatography, elution gradient 0–60% MeCN in water (containing 0.1% FA). Fractions containing the desired compound were evaporated to dryness to afford (S)-5-bromo-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (500 mg, 21%) as a pale yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 11.80 (s, 1H), 8.08 (s, 1H), 7.42–7.26 (m, 5H), 5.71 (q, J = 7.2 Hz, 1H), 1.68 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C12H12BrN2O2]+ [M + H]+ = 295.0077 and 292.0057, found = 294.9; ee = 91%.
(S)-5-(5-Fluoro-3-((trimethylsilyl)ethynyl)-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (43)
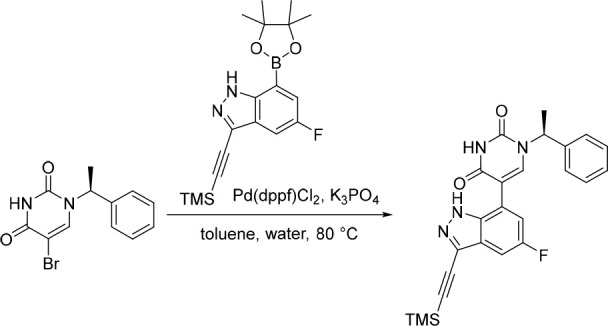
Pd(dppf)Cl2 (199 mg, 0.305 mmol) was added to a suspension of K3PO4 (432 mg, 2.04 mmol), (S)-5-bromo-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (42) (300 mg, 1.02 mmol) and 5-fluoro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3-((trimethylsilyl)ethynyl)-1H-indazole (41) (364 mg, 1.02 mmol) in toluene (24 mL) and water (0.40 mL) under nitrogen at rt. The resulting suspension was stirred at 80 °C for 17 h. The solvent was removed under reduced pressure and the crude product was purified by C18-flash chromatography, elution gradient 0–60% MeCN in water (containing 0.1% FA). Fractions containing the desired compound were evaporated to dryness to afford (S)-5-(5-fluoro-3-((trimethylsilyl)ethynyl)-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (120 mg, 26%) as a pale yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 13.37 (s, 1H), 11.70 (s, 1H), 8.06 (s, 1H), 7.43–7.36 (m, 5H), 7.31–7.23 (m, 2H), 5.78 (q, J = 7.1 Hz, 1H), 1.77 (d, J = 7.3 Hz, 3H), 0.30 (s, 9H); MS (ESI) m/z calculated for [C24H24FN4O2Si]+ [M + H]+ = 447.1647, found = 447.1.
(S)-5-(3-Ethynyl-5-fluoro-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (30)
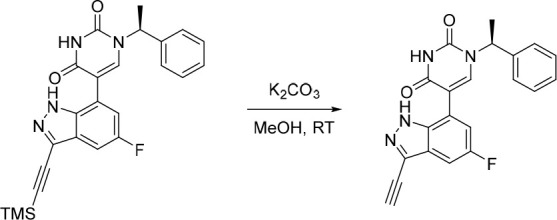
K2CO3 (204 mg, 1.48 mmol) was added to a solution of (S)-5-(5-fluoro-3-((trimethylsilyl)ethynyl)-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (43) (110 mg, 0.246 mmol) in MeOH (10 mL) at rt. The resulting mixture was stirred at rt for 2 h. The solvent was removed under reduced pressure and the crude product was purified by C18-flash chromatography, elution gradient 0–50% MeCN in water (containing 0.1% FA). Fractions containing the desired compound were evaporated to dryness and subsequently repurified by preparative chiral-HPLC (CHIRALPAK IG, 5 μm, 20 × 250 mm), isocratic 30% EtOH in hexane (containing 0.5% 2 M NH3·MeOH). Fractions containing the desired product were evaporated to dryness to afford enantiopure (S)-5-(3-ethynyl-5-fluoro-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione (Rt: 13.70 min, 40.0 mg, 43%) as a pale yellow solid. The impurity (R)-5-(3-ethynyl-5-fluoro-1H-indazol-7-yl)-1-(1-phenylethyl)pyrimidine-2,4(1H,3H)-dione eluted at 18.43 min. 1H NMR (400 MHz, DMSO-d6) δ 13.32 (s, 1H), 11.71 (s, 1H), 8.05 (s, 1H), 7.48–7.39 (m, 3H), 7.37 (t, J = 7.5 Hz, 2H), 7.34–7.21 (m, 2H), 5.78 (q, J = 7.1 Hz, 1H), 4.55 (s, 1H), 1.77 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 161.6, 157.5 (d, JCF = 237.9 Hz), 150.6, 141.8, 140.6, 136.4 (d, JCF = 2.9 Hz), 128.6, 127.7, 126.9, 126.8, 124.2 (d, JCF = 10.9 Hz), 119.6 (d, JCF = 9.7 Hz), 116.9 (d, JCF = 27.9 Hz), 110.1, 102.7 (d, JCF = 23.6 Hz), 84.6, 75.5, 54.3, 18.6; 19F NMR (376 MHz, DMSO-d6) −121.67; MS (ESI) m/z calculated for [C21H16FN4O2]+ [M + H]+ = 375.1252, found = 375.1; HRMS (ESI) m/z calculated for [C21H16FN4O2]+ [M + H]+ = 375.1252, found = 375.1270; ee > 99%.
5-(3-Ethynyl-5-fluoro-1H-indazol-7-yl)-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione (31)
XPhos Pd G2 (42.3 mg, 0.0538 mmol) was added to a solution of 5-fluoro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3-((trimethylsilyl)ethynyl)-1H-indazole (41) (269 mg, 0.751 mmol), 5-bromo-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione (148 mg, 0.500 mmol) and K2CO3 (173 mg, 1.25 mmol) in 1,4-dioxane (2.0 mL) and water (0.50 mL) under nitrogen at rt. The reaction was degassed and then stirred at 80 °C for 3 h. After cooling, the reaction was diluted with EtOAc and washed with water. The aqueous layer was then extracted with EtOAc. The combined organic extracts were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash silica chromatography, elution gradient 0–100% EtOAc in heptane. Fractions containing the desired compound were evaporated to dryness.
The above residue was dissolved in MeOH (2.5 mL) before the addition of K2CO3 (500 mg, 3.62 mmol) to the reaction mixture. The reaction was stirred at rt for 1 h and upon completion, the reaction was filtered and the solids washed with CH2Cl2. The filtrate was concentrated under reduced pressure and purified by preparative HPLC (Xselect CSH OBD, 5 μm, 30 × 100 mm) using decreasingly polar mixtures of water (containing 1% NH3) and MeCN. Fractions containing the desired compound were evaporated to dryness to afford 5-(3-ethynyl-5-fluoro-1H-indazol-7-yl)-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione (24.0 mg, 13% over two steps) as a colorless solid. 1H NMR (500 MHz, DMSO-d6) δ 13.21 (br s, 1H), 11.71 (br s, 1H), 8.56–8.52 (m, 1H), 8.23 (br s, 1H), 7.82 (td, J = 7.7, 1.8 Hz, 1H), 7.45 (d, J = 7.9 Hz, 1H), 7.41 (dd, J = 8.2, 2.0 Hz, 1H), 7.32 (ddd, J = 7.3, 4.7, 0.7 Hz, 2H), 5.82 (q, J = 7.1 Hz, 1H), 4.51 (s, 1H), 1.77 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C20H15FN5O2]+ [M + H]+ = 376.1204, found = 376.4.
(S)-5-Bromo-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione
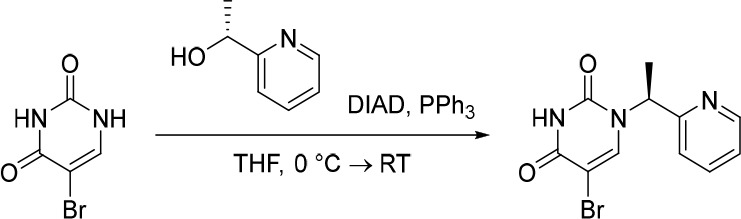
Diisopropyl azodicarboxylate (1.58 mL, 8.13 mmol) was added dropwise to a mixture of PPh3 (2.13 g, 8.12 mmol), (R)-1-(pyridin-2-yl)ethan-1-ol (1.00 g, 8.12 mmol) and 5-bromopyrimidine-2,4(1H,3H)-dione (35) (2.33 g, 12.2 mmol) in THF (20 mL) under nitrogen at 0 °C. The resulting solution was stirred at rt for 20 h. The solvent was removed under reduced pressure and the crude product was purified by flash silica chromatography, elution gradient 0–10% MeOH in CH2Cl2. Fractions containing the desired compound were evaporated to dryness to afford an impure mixture which was repurified by C18-flash chromatography, elution gradient 0–20% MeCN in water (containing 0.1% FA). Fractions containing the desired compound were evaporated to dryness to afford (S)-5-bromo-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione (1.00 g, 42%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.81 (s, 1H), 8.58–8.52 (m, 1H), 8.19 (s, 1H), 7.81 (td, J = 7.7, 1.8 Hz, 1H), 7.42 (d, J = 7.9 Hz, 1H), 7.37–7.29 (m, 1H), 5.74 (q, J = 7.2 Hz, 1H), 1.70 (d, J = 7.2 Hz, 3H); MS (ESI) m/z calculated for [C11H11BrN3O2]+ [M + H]+ = 296.0029 and 298.0009, found = 298.1; ee = 99% (the Rt of the minor peak was determined as the enantiomer following the synthesis of (R)-5-bromo-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione).
(S)-5-(5-Fluoro-3-((trimethylsilyl)ethynyl)-1H-indazol-7-yl)-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione

Pd(dppf)Cl2 (182 mg, 0.279 mmol) was added to a suspension of K3PO4 (741 mg, 3.49 mmol), (S)-5-bromo-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione (413 mg, 1.39 mmol) and 5-fluoro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3-((trimethylsilyl)ethynyl)-1H-indazole (41) (500 mg, 1.40 mmol) in toluene (20 mL) and water (0.33 mL) under nitrogen at rt. The resulting suspension was stirred at 80 °C for 16 h. The solvent was removed under reduced pressure and the crude product was purified by C18-flash chromatography, elution gradient 0–50% MeCN in water (containing 0.1% FA). Fractions containing the desired compound were evaporated to dryness to afford (S)-5-(5-fluoro-3-((trimethylsilyl)ethynyl)-1H-indazol-7-yl)-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione (83.0 mg, 13%) as a brown solid. 1H NMR (300 MHz, DMSO-d6) δ 13.33 (s, 1H), 11.70 (s, 1H), 8.59–8.51 (m, 1H), 8.09 (s, 1H), 7.83 (td, J = 7.7, 1.8 Hz, 1H), 7.46 (d, J = 7.9 Hz, 1H), 7.40 (dd, J = 8.3, 2.0 Hz, 1H), 7.33 (ddd, J = 7.5, 4.8, 1.1 Hz, 1H), 7.27 (d, J = 9.7 Hz, 1H), 5.81 (q, J = 7.2 Hz, 1H), 1.78 (d, J = 7.2 Hz, 3H), 0.30 (s, 9H); MS (ESI) m/z calculated for [C23H23FN5O2Si]+ [M + H]+ = 448.1600, found = 448.2.
(S)-5-(3-Ethynyl-5-fluoro-1H-indazol-7-yl)-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione (32)
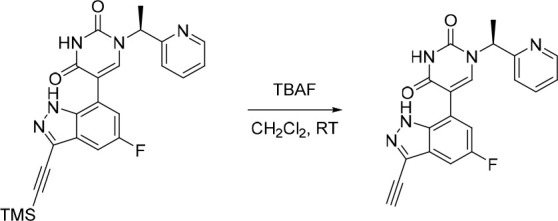
Tetrabutylammonium fluoride in THF (1 M, 174 μL, 0.174 mmol) was added to a solution of (S)-5-(5-fluoro-3-((trimethylsilyl)ethynyl)-1H-indazol-7-yl)-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione (78.0 mg, 0.174 mmol) in CH2Cl2 (5 mL). The resulting solution was stirred at rt for 30 min. The solvent was removed under reduced pressure and the crude product was purified by C18-flash chromatography, elution gradient 0–20% MeCN in water (containing 0.1% NH4HCO3). Fractions containing the desired compound were evaporated to dryness and subsequently repurified by preparative chiral-HPLC (CHIRALPAK ID, 5 μm, 20 × 250 mm), isocratic 50% iPrOH in hexane (containing 0.5% 2 M NH3.MeOH). Fractions containing the desired products were evaporated to dryness to afford enantiopure (S)-5-(3-ethynyl-5-fluoro-1H-indazol-7-yl)-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione (Rt: 14.9 min, 43.0 mg, 66%) as a white solid. The impurity (R)-5-(3-ethynyl-5-fluoro-1H-indazol-7-yl)-1-(1-(pyridin-2-yl)ethyl)pyrimidine-2,4(1H,3H)-dione eluted at 5.9 min. 1H NMR (400 MHz, DMSO-d6) δ 13.30 (s, 1H), 11.72 (s, 1H), 8.55 (d, J = 4.2 Hz, 1H), 8.14 (br s, 1H), 7.82 (td, J = 1.6, 7.7 Hz, 1H), 7.50–7.40 (m, 2H), 7.36–7.23 (m, 2H), 5.82 (q, J = 7.1 Hz, 1H), 4.55 (s, 1H), 1.77 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 161.6, 158.8, 157.5 (d, JCF = 237.9 Hz), 150.6, 149.2, 142.7, 137.2, 136.3, 127.0, 124.3 (d, JCF = 11.0 Hz), 122.9, 121.6, 119.8, 116.6 (d, JCF = 28.0 Hz), 109.4, 102.7 (d, JCF = 23.5 Hz), 84.6, 75.5, 55.6, 18.5; 19F NMR (376 MHz, DMSO-d6) −121.61; MS (ESI) m/z calculated for [C20H15FN5O2]+ [M + H]+ = 376.1204, found = 376.2; HRMS (ESI) m/z calculated for [C20H15FN5O2]+ [M + H]+ = 376.1204, found = 376.1214; ee > 99% (the Rt of the enantiomer was identified following the analytical chiral separation of the racemate).
Acknowledgments
We would like to acknowledge the scientific contributions from many of our colleagues at AstraZeneca including Bernard Barlaam, Kun Song and Kristin Goldberg.
Glossary
Abbreviations Used
- BBB
blood–brain barrier
- Clint
intrinsic clearance
- c-MET
mesenchymal-to-epithelial transition factor
- CNS MPO
central nervous system multiparameter optimization
- FEP
free energy perturbation
- H-bonds
hydrogen bonds
- HGF
hepatocyte growth factor
- HH
human hepatocyte
- HLM
human liver microsomes
- HTRF
homogeneous time-resolved fluorescence
- Kp,uu
unbound brain-to-plasma ratio
- LLE
ligand-lipophilicity efficiency
- MDCK
Madin-Darby canine kidney cell line
- MER
c-MER proto-oncogene kinase
- MMPs
matched molecular pairs
- NSCLC
nonsmall cell lung carcinoma
- RH
rat hepatocyte
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c00401.
Additional details are provided for: NMR-, crystallography- and DMPK-experiments; kinase selectivity; computational chemistry; analytical chemistry; synthetic chemistry (including reaction schemes for multistep syntheses of compounds, HPLC traces and NMR spectra for key compounds) (PDF)
Molecular formula strings with associated data and statistical analyses (CSV)
Accession Codes
Crystal structures reported here have been deposited in the Protein Data Bank with the following accession codes: 8OW3 (2, wild-type c-MET), 8OWG (2), 8OV7 (10), 8OUV (15), 8OVZ (16), 8OUU (29). All structures are for D1228V c-MET except where stated otherwise. See Supporting Information, Table S1 for further details.
The authors declare the following competing financial interest(s): At the time of writing, some of the authors were employees of AstraZeneca PLC (see affiliations).
Supplementary Material
References
- Zhang Y.; Xia M.; Jin K.; Wang S.; Wei H.; Fan C.; Wu Y.; Li X.; Li X.; Li G.; Zeng Z.; Xiong W. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 2018, 17 (1), 45. 10.1186/s12943-018-0796-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J. J.; Tran-Dube M.; Shen H.; Nambu M.; Kung P. P.; Pairish M.; Jia L.; Meng J.; Funk L.; Botrous I.; McTigue M.; Grodsky N.; Ryan K.; Padrique E.; Alton G.; Timofeevski S.; Yamazaki S.; Li Q.; Zou H.; Christensen J.; Mroczkowski B.; Bender S.; Kania R. S.; Edwards M. P. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54 (18), 6342–6363. 10.1021/jm2007613. [DOI] [PubMed] [Google Scholar]
- Yakes F. M.; Chen J.; Tan J.; Yamaguchi K.; Shi Y.; Yu P.; Qian F.; Chu F.; Bentzien F.; Cancilla B.; Orf J.; You A.; Laird A. D.; Engst S.; Lee L.; Lesch J.; Chou Y. C.; Joly A. H. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10 (12), 2298–2308. 10.1158/1535-7163.MCT-11-0264. [DOI] [PubMed] [Google Scholar]
- Jia H.; Dai G.; Weng J.; Zhang Z.; Wang Q.; Zhou F.; Jiao L.; Cui Y.; Ren Y.; Fan S.; Zhou J.; Qing W.; Gu Y.; Wang J.; Sai Y.; Su W. Discovery of (S)-1-(1-(Imidazo[1,2-a]pyridin-6-yl)ethyl)-6-(1-methyl-1H-pyrazol-4-yl)-1H-[1,2,3]triazolo[4,5-b]pyrazine (volitinib) as a highly potent and selective mesenchymal-epithelial transition factor (c-Met) inhibitor in clinical development for treatment of cancer. J. Med. Chem. 2014, 57 (18), 7577–7589. 10.1021/jm500510f. [DOI] [PubMed] [Google Scholar]
- Markham A. Savolitinib: First Approval. Drugs 2021, 81 (14), 1665–1670. 10.1007/s40265-021-01584-0. [DOI] [PubMed] [Google Scholar]
- Mo H. N.; Liu P. Targeting MET in cancer therapy. Chronic Dis. Transl. Med. 2017, 3 (3), 148–153. 10.1016/j.cdtm.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh P. K.; Ghate M. D. Recent advances in the discovery of small molecule c-Met Kinase inhibitors. Eur. J. Med. Chem. 2018, 143, 1103–1138. 10.1016/j.ejmech.2017.08.044. [DOI] [PubMed] [Google Scholar]
- Puccini A.; Marin-Ramos N. I.; Bergamo F.; Schirripa M.; Lonardi S.; Lenz H. J.; Loupakis F.; Battaglin F. Safety and Tolerability of c-MET Inhibitors in Cancer. Drug Saf 2019, 42 (2), 211–233. 10.1007/s40264-018-0780-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross D. A.; Ashton S. E.; Ghiorghiu S.; Eberlein C.; Nebhan C. A.; Spitzler P. J.; Orme J. P.; Finlay M. R.; Ward R. A.; Mellor M. J.; Hughes G.; Rahi A.; Jacobs V. N.; Red Brewer M.; Ichihara E.; Sun J.; Jin H.; Ballard P.; Al-Kadhimi K.; Rowlinson R.; Klinowska T.; Richmond G. H.; Cantarini M.; Kim D. W.; Ranson M. R.; Pao W. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discovery 2014, 4 (9), 1046–1061. 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlman C.; Cadranel J.; Rousseau-Bussac G.; Lacave R.; Pujals A.; Girard N.; Callens C.; Gounant V.; Theou-Anton N.; Friard S.; Tredaniel J.; Blons H.; Dujon C.; Duchemann B.; Schischmanoff P. O.; Chinet T.; Giroux Leprieur E. Resistance mechanisms to osimertinib in EGFR-mutated advanced non-small-cell lung cancer: A multicentric retrospective French study. Lung Cancer 2019, 137, 149–156. 10.1016/j.lungcan.2019.09.019. [DOI] [PubMed] [Google Scholar]
- Papadimitrakopoulou V. A.; Wu Y.-L.; Han J.-Y.; Ahn M.-J.; Ramalingam S. S.; John T.; Okamoto I.; Yang J. C.-H.; Bulusu K. C.; Laus G.; Collins B.; Barrett J. C.; Chmielecki J.; Mok T. S. K. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Annals of Oncology 2018, 29, viii741. 10.1093/annonc/mdy424.064. [DOI] [Google Scholar]
- Ramalingam S. S.; Cheng Y.; Zhou C.; Ohe Y.; Imamura F.; Cho B. C.; Lin M.-C.; Majem M.; Shah R.; Rukazenkov Y.; Todd A.; Markovets A.; Barrett J. C.; Chmielecki J.; Gray J. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Annals of Oncology 2018, 29, viii740. 10.1093/annonc/mdy424.063. [DOI] [Google Scholar]
- Sequist L. V.; Lee J. S.; Han J.-Y.; Su W.-C.; Yang J. C.-H.; Yu H.; Ottesen L. H.; Verheijen R. B.; Mellemgaard A.; Wessen J.; Oxnard G.; Cho B. C. Abstract CT033: TATTON Phase Ib expansion cohort: Osimertinib plus savolitinib for patients (pts) with EGFR-mutant, MET-amplified NSCLC after progression on prior third-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI). Cancer Res. 2019, 79, CT033. 10.1158/1538-7445.AM2019-CT033. [DOI] [Google Scholar]
- Yu H.; Ahn M.-J.; Kim S.-W.; Cho B. C.; Sequist L.; Orlov S.; Ottesen L. H.; Verheijen R. B.; Mellemgaard A.; Wessen J.; Han J.-Y. Abstract CT032: TATTON Phase Ib expansion cohort: Osimertinib plus savolitinib for patients (pts) with EGFR-mutant, MET-amplified NSCLC after progression on prior first/second-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI). Cancer Res. 2019, 79, CT032. 10.1158/1538-7445.AM2019-CT032. [DOI] [Google Scholar]
- Roskoski R. Jr. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. 10.1016/j.phrs.2015.10.021. [DOI] [PubMed] [Google Scholar]
- As reported by Cui, J. J. et al. (see ref (17)), crizotinib is a potent inhibitor of c-MET (cell IC50 = 8.0 nM); however, it also inhibits ALK (cell IC50 = 20 nM) and RON (cell IC50 = 80 nM). Cui et al. speculate that crizotinib is less selective for c-MET compared to PF-04217903, due to a weaker interaction with Y1230 and also a lack of interaction with D1222.
- Cui J. J.; McTigue M.; Nambu M.; Tran-Dube M.; Pairish M.; Shen H.; Jia L.; Cheng H.; Hoffman J.; Le P.; Jalaie M.; Goetz G. H.; Ryan K.; Grodsky N.; Deng Y. L.; Parker M.; Timofeevski S.; Murray B. W.; Yamazaki S.; Aguirre S.; Li Q.; Zou H.; Christensen J. Discovery of a novel class of exquisitely selective mesenchymal-epithelial transition factor (c-MET) protein kinase inhibitors and identification of the clinical candidate 2-(4-(1-(quinolin-6-ylmethyl)-1H-[1,2,3]triazolo[4,5-b]pyrazin-6-yl)-1H-pyrazol-1 -yl)ethanol (PF-04217903) for the treatment of cancer. J. Med. Chem. 2012, 55 (18), 8091–8109. 10.1021/jm300967g. [DOI] [PubMed] [Google Scholar]
- Collie G. W.; Koh C. M.; O’Neill D. J.; Stubbs C. J.; Khurana P.; Eddershaw A.; Snijder A.; Mauritzson F.; Barlind L.; Dale I. L.; Shaw J.; Phillips C.; Hennessy E. J.; Cheung T.; Narvaez A. J. Structural and Molecular Insight into Resistance Mechanisms of First Generation cMET Inhibitors. ACS Med. Chem. Lett. 2019, 10 (9), 1322–1327. 10.1021/acsmedchemlett.9b00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahcall M.; Sim T.; Paweletz C. P.; Patel J. D.; Alden R. S.; Kuang Y.; Sacher A. G.; Kim N. D.; Lydon C. A.; Awad M. M.; Jaklitsch M. T.; Sholl L. M.; Janne P. A.; Oxnard G. R. Acquired METD1228V Mutation and Resistance to MET Inhibition in Lung Cancer. Cancer Discovery 2016, 6 (12), 1334–1341. 10.1158/2159-8290.CD-16-0686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrock A. B.; Lai A.; Ali S. M.; Miller V. A.; Raez L. E. Mutation of MET Y1230 as an Acquired Mechanism of Crizotinib Resistance in NSCLC with MET Exon 14 Skipping. J. Thorac. Oncol. 2017, 12 (7), e89–e90. 10.1016/j.jtho.2017.02.017. [DOI] [PubMed] [Google Scholar]
- Qian F.; Engst S.; Yamaguchi K.; Yu P.; Won K. A.; Mock L.; Lou T.; Tan J.; Li C.; Tam D.; Lougheed J.; Yakes F. M.; Bentzien F.; Xu W.; Zaks T.; Wooster R.; Greshock J.; Joly A. H. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 2009, 69 (20), 8009–8016. 10.1158/0008-5472.CAN-08-4889. [DOI] [PubMed] [Google Scholar]
- Reungwetwattana T.; Liang Y.; Zhu V.; Ou S. I. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The Why, the How, the Who, the Unknown, and the Inevitable. Lung Cancer 2017, 103, 27–37. 10.1016/j.lungcan.2016.11.011. [DOI] [PubMed] [Google Scholar]
- Skerratt S. E.; Storer R. I.. Chapter 6: The Design of Brain Penetrant Kinase Inhibitors. In Kinase Drug Discovery: Modern Approaches; The Royal Society of Chemistry, 2019; pp 128–180. [Google Scholar]
- Cruickshanks N.; Zhang Y.; Yuan F.; Pahuski M.; Gibert M.; Abounader R. Role and Therapeutic Targeting of the HGF/MET Pathway in Glioblastoma. Cancers 2017, 9 (7), 87. 10.3390/cancers9070087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedettini E.; Sholl L. M.; Peyton M.; Reilly J.; Ware C.; Davis L.; Vena N.; Bailey D.; Yeap B. Y.; Fiorentino M.; Ligon A. H.; Pan B.-S.; Richon V.; Minna J. D.; Gazdar A. F.; Draetta G.; Bosari S.; Chirieac L. R.; Lutterbach B.; Loda M. Met Activation in Non-Small Cell Lung Cancer Is Associated with de Novo Resistance to EGFR Inhibitors and the Development of Brain Metastasis. Am. J. Pathol. 2010, 177 (1), 415–423. 10.2353/ajpath.2010.090863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella G. M.; Corino A.; Berzero G.; Kolling S.; Filippi A. R.; Benvenuti S. Brain Metastases from Lung Cancer: Is MET an Actionable Target?. Cancers 2019, 11 (3), 271. 10.3390/cancers11030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recent studies describing the successful use of cabozantinib to treat patients with brain tumors have been reported. For some relevant publications see:; a Ciccarese C.; Iacovelli R.; Mosillo C.; Tortora G. Exceptional Response to Cabozantinib of Rapidly Evolving Brain Metastases of Renal Cell Carcinoma: A Case Report and Review of the Literature. Clinical genitourinary cancer 2018, 16 (5), e1069–e1071. 10.1016/j.clgc.2018.06.005. [DOI] [PubMed] [Google Scholar]; b Klempner S. J.; Borghei A.; Hakimian B.; Ali S. M.; Ou S. I. Intracranial Activity of Cabozantinib in MET Exon 14-Positive NSCLC with Brain Metastases. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer 2017, 12 (1), 152–156. 10.1016/j.jtho.2016.09.127. [DOI] [PubMed] [Google Scholar]; c Uche A.; Sila C.; Tanoura T.; Yeh J.; Bhowmick N.; Posadas E.; Figlin R.; Gong J. Brain Complete Response to Cabozantinib prior to Radiation Therapy in Metastatic Renal Cell Carcinoma. Case reports in urology 2019, 2019, 6769017. 10.1155/2019/6769017. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wen P. Y.; Drappatz J.; de Groot J.; Prados M. D.; Reardon D. A.; Schiff D.; Chamberlain M.; Mikkelsen T.; Desjardins A.; Holland J.; Ping J.; Weitzman R.; Cloughesy T. F. Phase II study of cabozantinib in patients with progressive glioblastoma: subset analysis of patients naive to antiangiogenic therapy. Neuro-oncology 2018, 20 (2), 249–258. 10.1093/neuonc/nox154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufkötter O.; Hu H.; Miljković F.; Bajorath J. Structure- and Similarity-Based Survey of Allosteric Kinase Inhibitors, Activators, and Closely Related Compounds. J. Med. Chem. 2022, 65 (2), 922–934. 10.1021/acs.jmedchem.0c02076. [DOI] [PubMed] [Google Scholar]
- Price A. J.; Howard S.; Cons B. D. Fragment-based drug discovery and its application to challenging drug targets. Essays Biochem. 2017, 61 (5), 475–484. 10.1042/EBC20170029. [DOI] [PubMed] [Google Scholar]
- Collie G. W.; Michaelides I. N.; Embrey K.; Stubbs C. J.; Börjesson U.; Dale I. L.; Snijder A.; Barlind L.; Song K.; Khurana P.; Phillips C.; Storer R. I. Structural Basis for Targeting the Folded P-Loop Conformation of c-MET. ACS Med. Chem. Lett. 2021, 12 (1), 162–167. 10.1021/acsmedchemlett.0c00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller N.; Spadola L.; Cowen S.; Patel J.; Schonherr H.; Cao Q.; McKenzie A.; Edfeldt F.; Rabow A.; Goodnow R. An improved model for fragment-based lead generation at AstraZeneca. Drug Discovery Today 2016, 21 (8), 1272–1283. 10.1016/j.drudis.2016.04.023. [DOI] [PubMed] [Google Scholar]
- Lucas S. C. C.; Börjesson U.; Bostock M. J.; Cuff J.; Edfeldt F.; Embrey K. J.; Eriksson P.-O.; Gohlke A.; Gunnarson A.; Lainchbury M.; Milbradt A. G.; Moore R.; Rawlins P. B.; Sinclair I.; Stubbs C.; Storer R. I. Fragment screening at AstraZeneca: developing the next generation biophysics fragment set. RSC Med. Chem. 2022, 13, 1052–1057. 10.1039/D2MD00154C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprea T. I.; Davis A. M.; Teague S. J.; Leeson P. D. Is There a Difference between Leads and Drugs? A Historical Perspective. J. Chem. Inf. Comput. Sci. 2001, 41 (5), 1308–1315. 10.1021/ci010366a. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9 (10), 430–431. 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- Kuntz I. D.; Chen K.; Sharp K. A.; Kollman P. A. The maximal affinity of ligands. Proc. Natl. Acad. Sci. U.S.A. 1999, 96 (18), 9997–10002. 10.1073/pnas.96.18.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan S. G.; Hendle J.; Lee P. S.; Smith C. R.; Bounaud P.-Y.; Jessen K. A.; Tang C. M.; Huser N. H.; Felce J. D.; Froning K. J.; Peterman M. C.; Aubol B. E.; Gessert S. F.; Sauder J. M.; Schwinn K. D.; Russell M.; Rooney I. A.; Adams J.; Leon B. C.; Do T. H.; Blaney J. M.; Sprengeler P. A.; Thompson D. A.; Smyth L.; Pelletier L. A.; Atwell S.; Holme K.; Wasserman S. R.; Emtage S.; Burley S. K.; Reich S. H. SGX523 is an exquisitely selective, ATP-competitive inhibitor of the MET receptor tyrosine kinase with antitumor activity in vivo. Mol. Cancer Ther. 2009, 8 (12), 3181–3190. 10.1158/1535-7163.MCT-09-0477. [DOI] [PubMed] [Google Scholar]
- Halgren T. New Method for Fast and Accurate Binding-site Identification and Analysis. Chem. Biol. Drug Des. 2007, 69 (2), 146–148. 10.1111/j.1747-0285.2007.00483.x. [DOI] [PubMed] [Google Scholar]
- Halgren T. A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49 (2), 377–389. 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
- Abel R.; Young T.; Farid R.; Berne B. J.; Friesner R. A. Role of the active-site solvent in the thermodynamics of factor Xa ligand binding. J. Am. Chem. Soc. 2008, 130 (9), 2817–2831. 10.1021/ja0771033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young T.; Abel R.; Kim B.; Berne B. J.; Friesner R. A. Motifs for molecular recognition exploiting hydrophobic enclosure in protein–ligand binding. Proc. Natl. Acad. Sci. U.S.A. 2007, 104 (3), 808–813. 10.1073/pnas.0610202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis T. Inhomogeneous Fluid Approach to Solvation Thermodynamics. 1. Theory. J. Phys. Chem. B 1998, 102 (18), 3531–3541. 10.1021/jp9723574. [DOI] [Google Scholar]
- Ryckmans T.; Edwards M. P.; Horne V. A.; Correia A. M.; Owen D. R.; Thompson L. R.; Tran I.; Tutt M. F.; Young T. Rapid assessment of a novel series of selective CB(2) agonists using parallel synthesis protocols: A Lipophilic Efficiency (LipE) analysis. Bioorg. Med. Chem. Lett. 2009, 19 (15), 4406–4409. 10.1016/j.bmcl.2009.05.062. [DOI] [PubMed] [Google Scholar]
- Freeman-Cook K. D.; Hoffman R. L.; Johnson T. W. Lipophilic efficiency: the most important efficiency metric in medicinal chemistry. Future Med. Chem. 2013, 5 (2), 113–115. 10.4155/fmc.12.208. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6 (11), 881–890. 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- Cournia Z.; Allen B.; Sherman W. Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J. Chem. Inf. Model. 2017, 57 (12), 2911–2937. 10.1021/acs.jcim.7b00564. [DOI] [PubMed] [Google Scholar]
- Zwanzig R. W. High-Temperature Equation of State by a Perturbation Method. I. Nonpolar Gases. J. Chem. Phys. 1954, 22 (8), 1420–1426. 10.1063/1.1740409. [DOI] [Google Scholar]
- El-Haj B. M.; Ahmed S. B. M.; Garawi M. A.; Ali H. S. Linking Aromatic Hydroxy Metabolic Functionalization of Drug Molecules to Structure and Pharmacologic Activity. Molecules 2018, 23 (9), 2119. 10.3390/molecules23092119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purser S.; Moore P. R.; Swallow S.; Gouverneur V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37 (2), 320–330. 10.1039/B610213C. [DOI] [PubMed] [Google Scholar]
- Shah P.; Westwell A. D. The role of fluorine in medicinal chemistry. J. Enzyme Inhib. Med. Chem. 2007, 22 (5), 527–540. 10.1080/14756360701425014. [DOI] [PubMed] [Google Scholar]
- Wu P.; Clausen M. H.; Nielsen T. E. Allosteric small-molecule kinase inhibitors. Pharmacol. Ther. 2015, 156, 59–68. 10.1016/j.pharmthera.2015.10.002. [DOI] [PubMed] [Google Scholar]
- Wager T. T.; Hou X.; Verhoest P. R.; Villalobos A. Moving beyond Rules: The Development of a Central Nervous System Multiparameter Optimization (CNS MPO) Approach To Enable Alignment of Druglike Properties. ACS Chem. Neurosci. 2010, 1 (6), 435–449. 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.