

Abstract
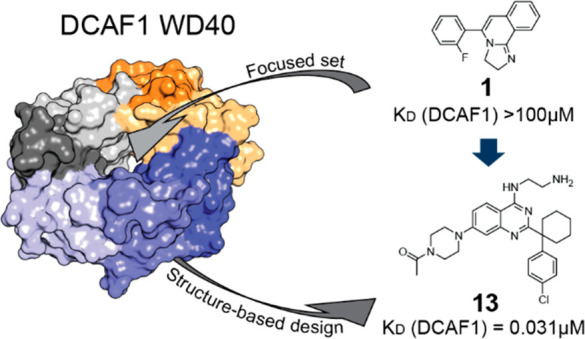
In this study, we describe the rapid identification of potent binders for the WD40 repeat domain (WDR) of DCAF1. This was achieved by two rounds of iterative focused screening of a small set of compounds selected on the basis of internal WDR domain knowledge followed by hit expansion. Subsequent structure-based design led to nanomolar potency binders with a clear exit vector enabling DCAF1-based bifunctional degrader exploration.
Keywords: Virtual screening, Structure-based design, Biophysics, DCAF1, E3 ligase, TPD
DCAF1 (DDB1 and CUL4-associated factor 1) protein is a substrate receptor of the Cullin4-RING E3 ubiquitin ligase complex that was originally discovered as the cellular target of the HIV-1 accessory viral protein R (Vpr) and, hence, was originally named Vpr binding protein (VprBP).1 Since the discovery of its association with CRL4, DCAF1 has attracted the interest of investigators working in the targeted protein degradation (TPD) field.2 The C-terminal domain of DCAF1 has the characteristic of a seven-blade β-propeller with a central cavity (also called donut hole), where each blade comprises a four-stranded antiparallel β-sheet (Figure 1). Strand d of each blade forms the outer surface. Each blade contains approximately 40 amino acids, which often terminates with a tryptophan-aspartate (WD) motif. The WD40 repeat domain (WDR) proteins typically act as a scaffold and have several surfaces for the interactions with multiple binding partners:3 the “top” and “bottom” faces and the circumference. For instance, the WDR protein embryonic ectoderm development (EED) binds to the trimethylated lysine 27 of the histone H3 (H3K27Me3) at the central binding pocket of the “top” face and interacts with the N-terminal helix (residues 38–68) of EZH2 (enhancer of zeste homologue 2) at the “bottom” face of the propeller. Recently, Huang et al. have reported the discovery of potent inhibitors occupying the central pocket of EED.4 In other cases, such as for the cell division cycle 20 (Cdc20) protein, small molecule ligands were found binding at side cavities between the WD40 blades, typically with a weaker affinity.5,6 For an excellent review on WDR proteins, see reference (7). Another important aspect of the WDR protein family is its diversity.8 Even those with very similar fold structures, i.e., with the same number of blades, which can vary from four to a few, display a low conserved primary sequence. Moreover, as in EED, the cavities are dynamic in their nature and can undergo conformational changes upon binding.
Figure 1.
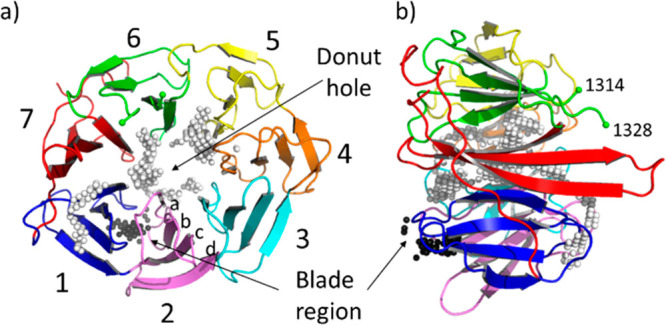
(a) Top view and (b) side view of the crystal structure (PDB ID: 4CC9) of the human E3 ligase substrate adaptor DCAF1 extracted from the ternary complex with the viral accessory protein X (Vpx) and the carboxy-terminal region of human SAMHD1 (not shown). The “top” and “bottom” surfaces are displayed on the right and left of panel (b), respectively. Residues 1315–1327 are not visible in the crystal structure. The Cα atom of the last visible residues, 1314 and 1328 belonging to blade 6, are marked with a green sphere. The white and black spheres indicate the two pocket locations as identified by SiteMap and fpocket. The white spheres correspond to the large ligandable donut hole pocket. The black spheres indicate the location of the blade region, an additional less druggable cavity that lies between blade 1 (colored in blue) and blade 2 (in pink). Each blade (numbered in the picture from 1 to 7) is constituted by a four-stranded antiparallel β-sheet. Strands are labeled from (a) to (d) from the inside toward the outside of the propeller.
In this work, we describe hit finding approaches used for identifying novel DCAF1 binders that could serve as anchor points for the development of bifunctional degraders (also known as PROTAC) by using the CRL4-based degradation machinery.9
We started by applying two in silico pocket identification tools (SiteMap10 and fpocket11) on the crystal structure of DCAF1 in ternary complex with the viral accessory protein X (Vpx) and the carboxy-terminal region of human SAMHD1 (PDB ID: 4CC9).12 The Vpx protein wraps tightly around the WDR DCAF1 domain and recruits SAMHD1 via its C terminus, thereby making it a competent substrate for proteasomal degradation. Both in silico pocket detection approaches performed on DCAF1 after removal of the other two proteins identified its central region as the major druggable pocket (donut hole of Figure 1; called SiteMap 1 and Fpocket1 in Figure S1). This corresponds to the large cavity in the middle of the WDR domain where the Vpx protein also binds. As discussed in the next sections, the two cavities highlighted in Figure 1 (the donut hole and the blade region), were both confirmed to be ligandable by solving crystal structures of small molecules bound to DCAF1 (prospectively) and EED (retrospectively). Additional cavities, with much lower druggability scores, were also identified (see Figure S1).
A focused set of 21 compounds was assembled by collecting known EED binders from a previous Novartis screening campaign, both in the donut and the blade pockets, with a few additional validated binders of other WDR targets. The small set was screened by 1H NMR spectroscopy (protein observation and ligand T1ρ experiments) against DCAF1, which resulted in two hits of interest that were confirmed as binders by protein observation 2D NMR experiments of DCAF1 with selectively 13Cε-labeled methionines.
For this purpose, the 10 methionine peaks in the 2D [13C,1H]-HMQC spectrum were assigned by site-directed mutagenesis of every methionine residue to leucine (Figure S2). Chemical shift perturbations are observed in the 2D [13C,1H]-HMQC spectrum of DCAF1 in the presence of compound 1, as shown in Figure 2. Compound 1 is a previously disclosed donut pocket EED binder for which the crystal structure in complex with EED and a portion of the histone–lysine N-methyltransferase enzyme EZH2 has been solved (PDB ID: 5H25).4Figure S3 shows the SPR characterization of compound 1 against DCAF1 and EED. Despite the low affinity, we solved the crystal structure of 1 in complex with DCAF1 (Figure 3).
Figure 2.
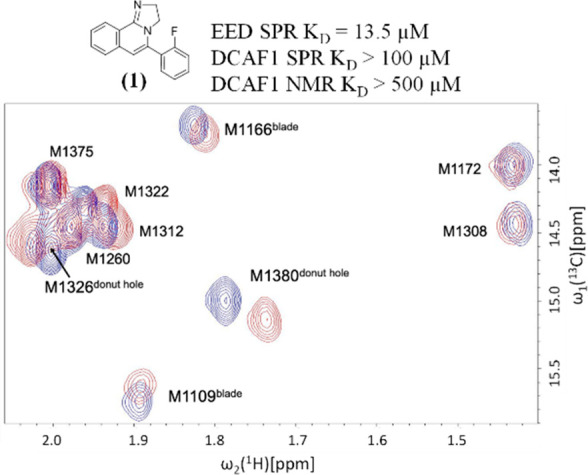
Overlay of the methyl region of 2D [13C,1H]-HMQC spectra of selectively 13Cε-methionine labeled DCAF1 in the absence (blue) and in the presence of the compound 1 (red). The concentrations of the protein and the compound were 15 μM and 480 μM, respectively.
Figure 3.
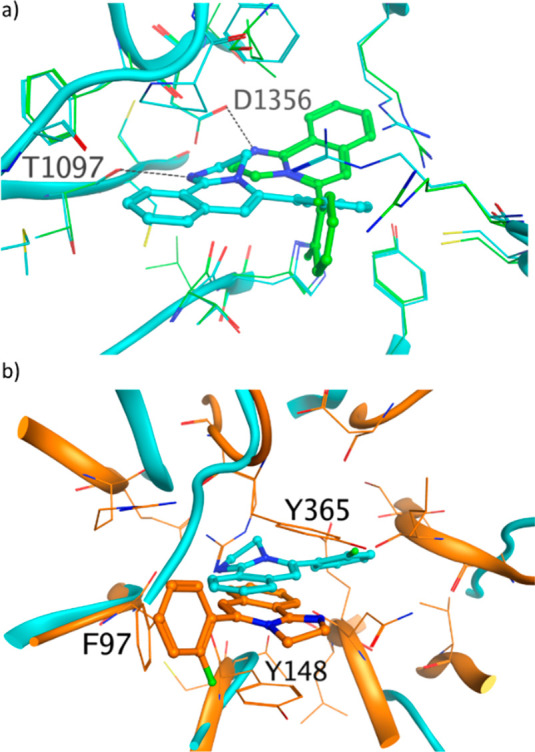
(a) Compound 1 binds to the central donut cavity of DCAF1 in two orientations. In both crystal structures, the protonated imidazoline moiety makes a hydrogen bond, with the carbonyl of T1097 in pose 1 (PDB ID: 8OG6, cyan) and with the carboxyl moiety of D1356 side chain in pose 2 (PDB ID: 8OG5, green). The hydrogen bonds are shown as dashed black lines. (b) Overlay of crystal structure of 1 bound to DCAF1 (PDB ID: 8OG6, cyan) and to EED (PDB ID: 5H25, orange). The three residues forming the aromatic cages in EED are labeled.
The ligand 1 binds to the central donut cavity of DCAF1 in two orientations. In both poses, the protonated imidazoline moiety (experimental pKa of 10.2) makes a hydrogen bond, with the carbonyl of T1097 in pose 1 and the carboxyl moiety of D1356 side chain in pose 2 (Figure 3a).
The protonated nitrogen of the imidazoline moiety nicely mimics a water mediating the interaction between the K84 residue of Vpx and the T1097 carbonyl of DCAF1 in the 4CC9 PDB crystal structure.12 In EED, 1 binds to the same central area as in DCAF1, but the binding mode and the binding location are different (Figure 3b). As previously reported, the binding of compound 1 to EED induces a substantial conformational change of side chains of W364 and Y365, in addition to R367 in the central cavity of EED.4,13 The fused tricyclic core forms interactions in the so-called aromatic cage: cation−π and π–π interactions with Y148 and Y365 and an edge-to-face interaction with the phenyl ring of F97 (Figure 3b).
The crystal structure of the other DCAF1 hit is reported in Figure 4. In this case, the hit 2, which is also an EED binder, binds to DCAF1 and EED in the region, which corresponds to the blade region identified by the SiteMap and fpocket in silico approaches (Figure 1). Compound 2 has a SPR KD = ∼360 μM for EED and a SPR KD > 100 μM for DCAF1 (Figure S4). The electron density is not well-defined for the amidine moiety of the ligand pointing into the solvent. In the EED-1-EZH2 crystal structure, the chlorophenyl moiety of the ligand overlaps with the V68 side chain of EZH2 peptide in its interactions with EED, whereas in the DCAF1-2 crystal structure, the chlorophenyl moiety displaces the side chain of the Y1131 residue of DCAF1.
Figure 4.
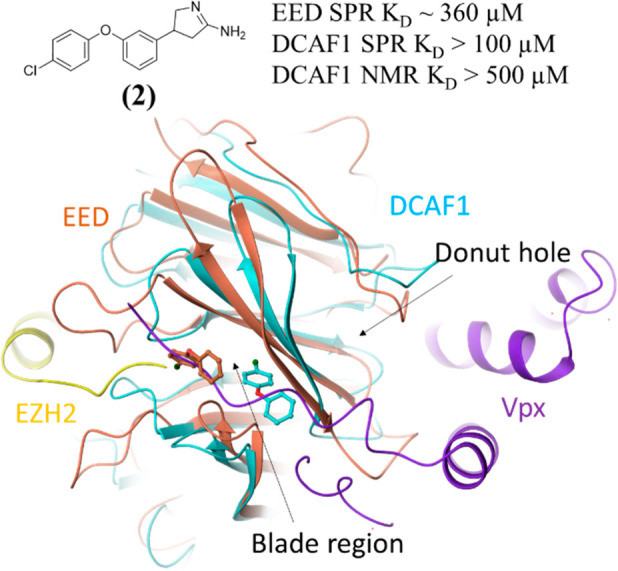
Crystal structures of 2 bound to the blade region of DCAF1 (PDB ID: 8OG7, cyan) and of EED (orange). Only the most buried portion of the ligand is built in the density in both crystal structures. The two crystal structures are overlaid with the DCAF1-Vpx-SAMHD1 crystal structure (PDB ID: 4CC9), from which only the Vpx protein is shown in violet, and with the EED-1-EZH2 crystal structure (PDB ID: 5H25), from which only the EZH2 protein is shown in yellow.
It is worth noting that, despite the very low sequence similarity between DCAF1 and EED (sequence similarity < 25% and identity < 10%), the focused set approach, selected on the basis of the WDR fold rationale, was very successful in quickly providing low affinity hits. The analysis of the recent new tetrameric structure CUL4A–DDB1–DCAF1 (PDB ID: 7OKQ)14 suggests that there is no evidence that the blade region binders would prevent DDB1-DCAF1 binding. However, the identified donut pocket binder 1 was considered a better anchor point for DCAF1-based bifunctional degrader optimization considering its more attractive pocket ligandability.
To explore the structure–activity relationship (SAR) around compound 1, 30 compounds were selected from the Novartis archive by using in silico approaches, such as 2D similarity followed by docking and 3D similarity (using the field-based Cresset similarity approach15 and the shape-based OpenEye ROCS approach16). Among these 30 compounds, we succeeded in solving the crystal structures for two of them with DCAF1, that is, compounds 3 and 4 (Scheme 1), with 2D NMR-measured KD values of 40 and 13 μM, respectively, against DCAF1 and IC50 values, in a biochemical assay with H3K27Me3 peptide as substrate against EED, of 21 and 25 μM, respectively.
Scheme 1. Analogues of Hit 1.
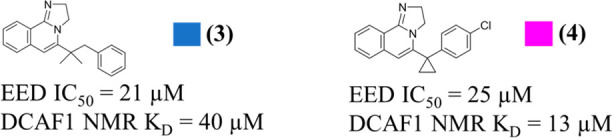
These compounds, in addition of binding to the donut hole, were also found bound to the blade region, as shown in the crystal structures (Figure 5) and by NMR (Figure S5a,b). In the donut cavity (Figure 5a) the binding modes resemble pose 1 of compound 1 (Figure 3a), with the tricyclic core nicely overlapping below the side chain of P1329. However, in the blade region, the two compounds bind differently by pointing either the tricyclic core or the p-chlorophenyl group toward the most buried and hydrophobic part of the blade cavity (Figure 5b).
Figure 5.
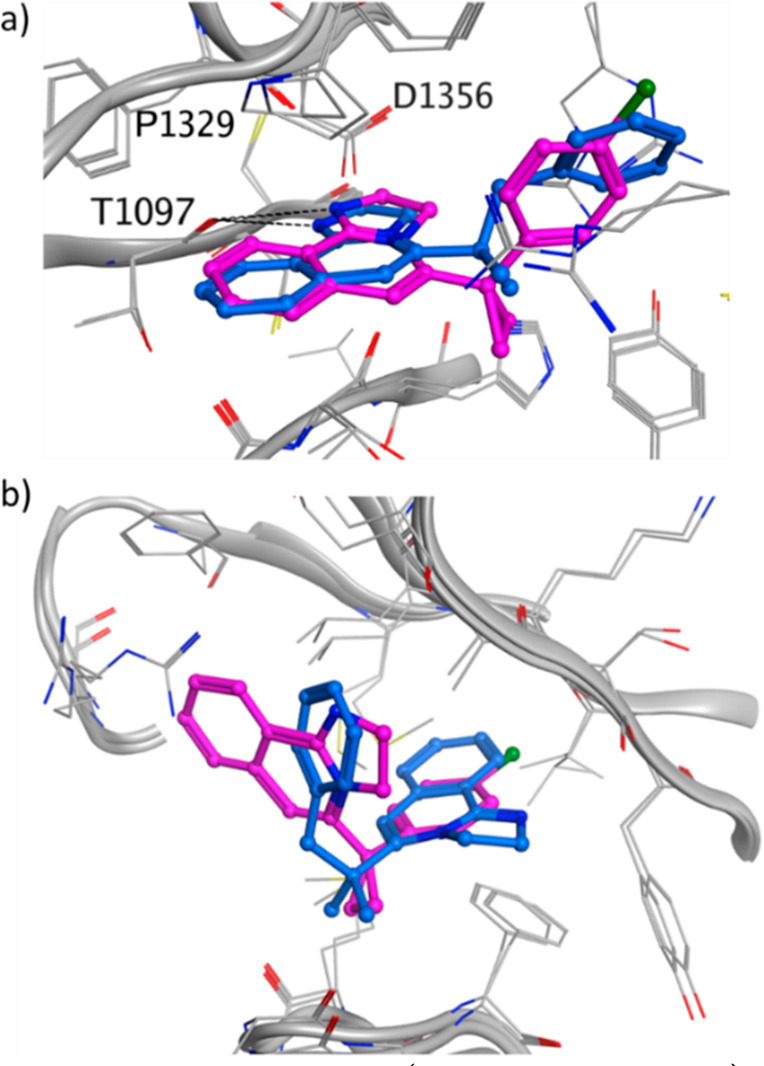
Crystal structure of 3 (PDB ID: 8OG8, blue) and 4 (PDB ID: 8OG9, purple) in (a) the donut hole cavity and in (b) the blade region. For 3 only one of the blade poses is shown. The other pose has the tricyclic core flipped by 180°. The DCAF1 protein is shown in gray, and the hydrogen bonds are shown as dashed black lines.
To avoid binding to the blade region, on the basis of the structural information, we decided to remove the chlorine in the para position of the benzo-ring of compound 4 (Figure 5b, purple binding pose) and add a substituent on the phenyl of the tricyclic core. These two changes aimed at disrupting the binding at the blade region. The replacement of chlorine with a methoxy slightly reduced the affinity toward DCAF1, which was recovered by growing on the tricyclic core (compound 6, Scheme 2). The chlorine of 4 appeared to mediate stronger interactions than the methoxy group of 5 in the blade pocket (see 2D [13C,1H]-HMQC spectra of Figure S5b,c). The crystal structure of DCAF1 in complex with compound 6 (Figure S6) indicated that the extension out of the donut hole was tolerated and no binding to the blade region was observed, as also confirmed by NMR (Figure S5d).
Scheme 2. Structure-Based Design Strategies (KD Values from SPR).

We then morphed the core of compound 5 (Scheme 2) by opening the imidazoline moiety (compound 7). This modification was designed to retain the hydrogen bond donor function at the position of the protonated nitrogen and place a basic amine deeper into the donut hole cavity.
The methylpiperazine substitution on the phenyl of the quinazoline scaffold of compound 7 led to compound 8 (Scheme 2) with a SPR-measured KD of 5.6 μM. The piperazine is located at the entrance of the donut hole surrounded by the side chain of F1355 and A1137. This region is underneath the loop connecting the 12 residues (from 1315 to 1327) not visible in the crystal structures (see also Figure 1b). The DCAF1 crystal structure in complex with 8 (Figure 6a) showed that the amino group of the amino-quinazoline is too distant to make an efficient hydrogen bond with the carbonyl of T1097, as observed in the DCAF1-1 crystal structure. Instead, it makes a hydrogen bond with the carbonyl of F1355. In addition, the terminal protonated amine of the ethylamine moiety makes three hydrogen bonds with the side chain of D1356 and H1140 and with the backbone carbonyl of T1139. Replacement of the methyl with an acetyl moiety at the solvent-exposed region of the piperazine (compound 9) further improved the affinity by 3.7-fold with respect to compound 8 (Scheme 2).
Figure 6.
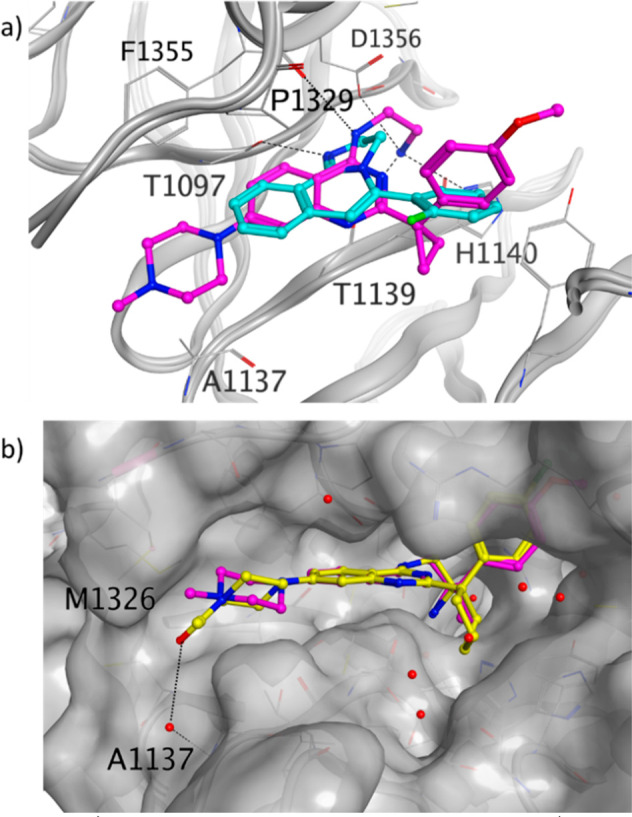
(a) DCAF1 crystal structure in complex with 8 (PDB ID: 8OGB, purple) overlaid with the DCAF1 crystal structure in complex with 1 (PDB ID: 8OG6, cyan). (b) DCAF1 crystal structure in complex with 11 (PDB ID: 8OGC, yellow) overlaid with the DCAF1 crystal structure in complex with 8 (purple), of which only the ligand is shown. The DCAF1 protein surface is shown in gray, and the hydrogen bonds are shown as dashed black lines.
On the basis of all these findings, we then explored the benzylic position (Scheme 3) using the more potent para-chlorophenyl derivative to better fill the donut cavity. The increased contacts of the cyclopentyl and cyclohexyl with the protein led to potent DCAF1 sub-100 nM binders, which showed a KD > 100 μM against EED, as measured by SPR (see Figure S7).
Scheme 3. Exploration of the Benzylic Position (KD Values from SPR).
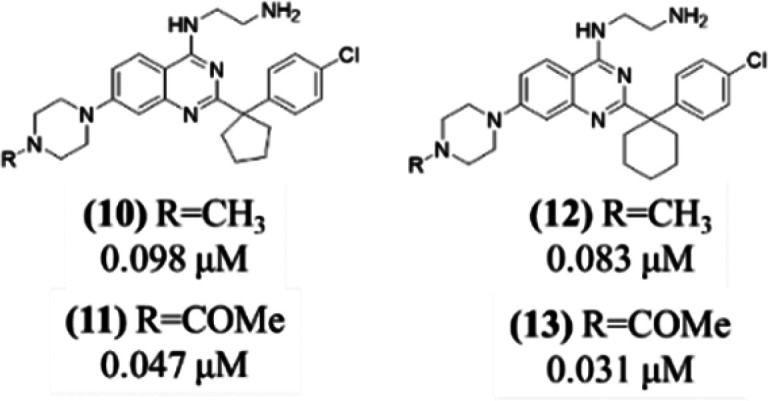
The crystal structure of DCAF1 in complex with 11 (Scheme 3) was also solved, which shows the acetyl moiety making a water-mediated interaction with the NH of the backbone of A1137 at the entrance of the donut hole (Figure 6b). Moreover, the acetyl moiety offers a clear exit vector for DCAF1-based bifunctional degrader exploration, which has been reported in an upcoming paper.17
At the time of the writing of this manuscript, a novel series of small molecule ligands targeting DCAF1 has been reported.18 The ligands of this new chemical class bind in the donut pocket but occupy a different region with respect to the binders reported here (Figure 7a,b) with only a minor overlap (Figure S8). Another crystal structure (PDB ID: 7SSE), not available at the time of our activities, is reported in Figure 7c. Retrospectively, it is interesting to notice that the interactions with the carbonyl of F1355 and the side chain of D1356 involving the ethylamine chain of compound 8 (Figure 7a) are substituted by water molecules in the 7SSE crystal structure, as shown in Figure 7c. The combined information on the bound structures of these chemical classes could be used to guide future DCAF1 drug discovery activities.
Figure 7.

DCAF1 crystal structures in complex with (a) compound 8 (PDB ID: 8OGB) and (b) compound 26e of reference (18) (PDB ID: 8F8E). (c) DCAF1 crystal structure deposited in PDB under the code 7SSE. Hydrogen bonds are shown as dashed black lines, and water molecules are shown as red spheres.
In summary, we have rapidly identified potent DCAF1 binders by combining knowledge-based focused screening with structure-based hit expansion and design. These novel binders enable the exploration of DCAF1 as a ligase for targeted protein degradation.17
Glossary
Abbreviations
- WDR
WD40 repeat domain
- DCAF1
DDB1 and CUL4-associated factor 1
- CRL4
cullin4-RING ubiquitin ligase
- EED
embryonic ectoderm development
- EZH2
enhancer of zeste homologue 2
- Vpr
HIV-1 accessory protein viral protein R
- Vpx
HIV-1 accessory protein viral protein X
- Cdc20
cell division cycle 20
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00104.
All the experimental details relevant to the protein production, structural characterization, compounds synthesis, and compound testing (PDF)
Accession Codes
Atomic coordinates for the X-ray structures of compounds 1 (PDB: 8OG5 and 8OG6), 2 (PDB: 8OG7), 3 (PDB: 8OG8), 4 (PDB: 8OG9), 6 (PDB: 8OGA), 8 (PDB: 8OGB), and 11 (PDB: 8OGC) in DCAF1 are available from the RCSB Protein Data Bank (www.rscb.org). Authors will release the atomic coordinates upon article publication.
Author Present Address
∥ Ridgeline Discovery GmbH, Aeschenvorstadt 36, Basel 4051, Switzerland
Author Present Address
⊥ Institute of Biochemistry, ETH, Otto-Stern-Weg 3, Zurich 8093, Switzerland
The authors declare no competing financial interest.
Supplementary Material
References
- Xu C.; Bian C.; Yang W.; Galka M.; Ouyang H.; Chen C.; Qiu W.; Liu H.; Jones A. E.; MacKenzie F.; Pan P.; Li S. S.-C.; Wang H.; Min J. Binding of different histone marks differentially regulates the activity and specificity of polycomb repressive complex 2 (PRC2). Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 19266–19271. 10.1073/pnas.1008937107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y.; Remillard D.; Vinogradova E. V.; Yokoyama M.; Banchenko S.; Schwefel D.; Melillo B.; Schreiber S. L.; Zhang X.; Cravatt B. F. Targeted Protein Degradation by Electrophilic PROTACs that Stereoselectively and Site-Specifically Engage DCAF1. J. Am. Chem. Soc. 2022, 144, 18688–18699. 10.1021/jacs.2c08964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Feng Y.; Narayan O.; Zhao L.-J. Cytoplasmic retention of HIV-1 regulatory protein Vpr by protein-protein interaction with a novel human cytoplasmic protein VprBP. Gene 2001, 263, 131–140. 10.1016/S0378-1119(00)00583-7. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Zhang J.; Yu Z.; Zhang H.; Wang Y.; Lingel A.; Qi W.; Gu J.; Zhao K.; Shultz M. D.; Wang L.; Fu X.; Sun Y.; Zhang Q.; Jiang X.; Zhang J.; Zhang C.; Li L.; Zeng J.; Feng L.; Zhang C.; Liu Y.; Zhang M.; Zhang L.; Zhao M.; Gao Z.; Liu X.; Fang D.; Guo H.; Mi Y.; Gabriel T.; Dillon M. P.; Atadja P.; Oyang C. Discovery of First-in-Class, Potent, and Orally Bioavailable Embryonic Ectoderm Development (EED) Inhibitor with Robust Anticancer Efficacy. J. Med. Chem. 2017, 60, 2215–2226. 10.1021/acs.jmedchem.6b01576. [DOI] [PubMed] [Google Scholar]
- Sackton K. L.; Dimova N.; Zeng X.; Tian W.; Zhang M.; Sackton T. B.; Meaders J.; Pfaff K. L.; Sigoillot F.; Yu H.; Luo X.; King R. W. Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature 2014, 514, 646–649. 10.1038/nature13660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W.; Li B.; Warrington R.; Tomchick D. R.; Yu H.; Luo X. Structural analysis of human Cdc20 supports multisite degron recognition by APC/C. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 18419–18424. 10.1073/pnas.1213438109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song R.; Wang Z.-D.; Schapira M. Disease Association and Druggability of WD40 Repeat Proteins. J. Proteome Res. 2017, 16, 3766–3773. 10.1021/acs.jproteome.7b00451. [DOI] [PubMed] [Google Scholar]
- Smith T. F.Diversity of WD-Repeat proteins. In The Coronin Family of Proteins: Subcellular Biochemistry; Clemen C. S., Eichinger L., Rybakin V., Eds.; Springer New York: New York, 2008; pp 20–30. [Google Scholar]
- Ishida T.; Ciulli A. E3 Ligase Ligands for PROTACs: How They Were Found and How to Discover New Ones. SLAS DISCOVERY: Advancing the Science of Drug Discovery 2021, 26, 484–502. 10.1177/2472555220965528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren T. A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49, 377–389. 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
- Le Guilloux V.; Schmidtke P.; Tuffery P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinformatics 2009, 10, 168. 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwefel D.; Groom H. C. T.; Boucherit V. C.; Christodoulou E.; Walker P. A.; Stoye J. P.; Bishop K. N.; Taylor I. A. Structural basis of lentiviral subversion of a cellular protein degradation pathway. Nature 2014, 505, 234–238. 10.1038/nature12815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Zhang H.; Zhang M.; Zhao M.; Feng L.; Luo X.; Gao Z.; Huang Y.; Ardayfio O.; Zhang J.-H.; Lin Y.; Fan H.; Mi Y.; Li G.; Liu L.; Feng L.; Luo F.; Teng L.; Qi W.; Ottl J.; Lingel A.; Bussiere D. E.; Yu Z.; Atadja P.; Lu C.; Li E.; Gu J.; Zhao K. Discovery and Molecular Basis of a Diverse Set of Polycomb Repressive Complex 2 Inhibitors Recognition by EED. PLoS One 2017, 12, e0169855 10.1371/journal.pone.0169855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed W. I.; Schenk A. D.; Kempf G.; Cavadini S.; Basters A.; Potenza A.; Abdul Rahman W.; Rabl J.; Reichermeier K.; Thomä N. H. The CRL4DCAF1 cullin-RING ubiquitin ligase is activated following a switch in oligomerization state. EMBO Journal 2021, 40, e108008 10.15252/embj.2021108008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseright T. J.; Mackey M. D.; Melville J. L.; Vinter J. G. FieldScreen: Virtual Screening Using Molecular Fields. Application to the DUD Data Set. J. Chem. Inf. Model. 2008, 48, 2108–2117. 10.1021/ci800110p. [DOI] [PubMed] [Google Scholar]
- Hawkins P. C. D.; Skillman A. G.; Nicholls A. Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem. 2007, 50, 74–82. 10.1021/jm0603365. [DOI] [PubMed] [Google Scholar]
- Schroeder M.; Renatus M.; Liang X.; Meili F.; Ferrand S.; Gauter F.; Li X.; Sigoillot F.; Gleim S.; Stachyra M.-T.; Thomas J.; Schirle M.; Zoller T.; Begue D.; Lefeuvre P.; Chung B.; Ma R.; Carbonneau S.; Pinch B.; Schmiedeberg N.; Imbach P.; Hofmann A.; Rey R.; Gorses D.; Calkins K.; Bauer-Probst B.; Maschlej M.; Niederst M.; Maher R.; Henault M.; Alford J.; Ahrne E.; Hollingworth G.; Thomae N. H.; Vulpetti A.; Radimerski T.; Holzer P.; Thoma C. R.. Reinstating targeted protein degradation with DCAF1 PROTACs in CRBN PROTAC resistant settings. bioRxiv, April 9, 2023, 536153. 10.1101/2023.04.09.536153. [DOI]
- Li A. S. M.; Kimani S.; Wilson B.; Noureldin M.; González-Álvarez H.; Mamai A.; Hoffer L.; Guilinger J. P.; Zhang Y.; von Rechenberg M.; Disch J. S.; Mulhern C. J.; Slakman B. L.; Cuozzo J. W.; Dong A.; Poda G.; Mohammed M.; Saraon P.; Mittal M.; Modh P.; Rathod V.; Patel B.; Ackloo S.; Santhakumar V.; Szewczyk M. M.; Barsyte-Lovejoy D.; Arrowsmith C. H.; Marcellus R.; Guié M.-A.; Keefe A. D.; Brown P. J.; Halabelian L.; Al-awar R.; Vedadi M. Discovery of Nanomolar DCAF1 Small Molecule Ligands. J. Med. Chem. 2023, 66, 5041–5060. 10.1021/acs.jmedchem.2c02132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Schroeder M.; Renatus M.; Liang X.; Meili F.; Ferrand S.; Gauter F.; Li X.; Sigoillot F.; Gleim S.; Stachyra M.-T.; Thomas J.; Schirle M.; Zoller T.; Begue D.; Lefeuvre P.; Chung B.; Ma R.; Carbonneau S.; Pinch B.; Schmiedeberg N.; Imbach P.; Hofmann A.; Rey R.; Gorses D.; Calkins K.; Bauer-Probst B.; Maschlej M.; Niederst M.; Maher R.; Henault M.; Alford J.; Ahrne E.; Hollingworth G.; Thomae N. H.; Vulpetti A.; Radimerski T.; Holzer P.; Thoma C. R.. Reinstating targeted protein degradation with DCAF1 PROTACs in CRBN PROTAC resistant settings. bioRxiv, April 9, 2023, 536153. 10.1101/2023.04.09.536153. [DOI]