

Abstract
The rise of multidrug-resistant (MDR) Gram-negative bacteria is a major global health problem necessitating the discovery of new classes of antibiotics. Novel bacterial topoisomerase inhibitors (NBTIs) target the clinically validated bacterial type II topoisomerases with a distinct binding site and mechanism of action to fluoroquinolone antibiotics, thus avoiding cross-resistance to this drug class. Here we report the discovery of a series of NBTIs incorporating a novel indane DNA binding moiety. X-ray cocrystal structures of compounds 2 and 17a bound to Staphylococcus aureus DNA gyrase–DNA were determined, revealing specific interactions with the enzyme binding pocket at the GyrA dimer interface and a long-range electrostatic interaction between the basic amine in the linker and the carboxylate of Asp83. Exploration of the structure–activity relationship within the series led to the identification of lead compound 18c, which showed potent broad-spectrum activity against a panel of MDR Gram-negative bacteria.
Keywords: Antimicrobial, antibacterial, DNA gyrase, topoisomerase, NBTI
The rapid increase in the emergence and spread of drug-resistant pathogens is an urgent global health problem. Of particular concern are life-threatening multidrug-resistant (MDR) bacterial infections caused by the so-called ESKAPE pathogens, i.e., Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species.1 The last four of these are classified as Gram-negative bacteria, which pose particularly formidable challenges for the development of effective new antibiotics due to the presence of an outer membrane comprising tightly packed lipopolysaccharides that preclude passive diffusion of most small molecules. In addition to permeation barriers, bacteria have developed a wide array of adaptive resistance mechanisms such as efflux pumps which further impede accumulation of drugs in the cytosol and in the periplasm of Gram-negative bacteria.2
Bacterial type II topoisomerases DNA gyrase and topoisomerase IV (Topo IV) are essential enzymes involved in regulating the topology of DNA during replication and transcription.3 DNA gyrase introduces negative supercoils into and removes positive supercoils from DNA, while Topo IV decatenates tangles after replication of circular DNA such as bacterial chromosomes and plasmids. Type II topoisomerases are A2B2 heterotetramers of GyrA2GyrB2 (DNA gyrase) and ParC2ParE2 (Topo IV), where the GyrA/ParC subunits contain the catalytic domains and the GyrB/ParE subunits harbor the ATPase domains.
The bacterial type II topoisomerases are the biological target for the well-established fluoroquinolone class of antibiotics, which unfortunately suffer from the increasing emergence of resistance.4,5 The antibiotic novobiocin binds to the ATPase domain of DNA gyrase and Topo IV but has been withdrawn from the market due to safety and efficacy concerns. New structural classes of inhibitors with this mechanism of action have been an area of active research for more than 50 years.5−7
Novel bacterial topoisomerase inhibitors (NBTIs) are a promising new class of antimicrobial compounds acting on DNA gyrase and TopoIV first described by GSK and subsequently investigated by many other groups.8−10,23 The structures of NBTIs are characterized by two heteroaromatic moieties joined by a linker that typically contains a basic amine (Figure 1). The binding mode of GSK299423 to S. aureus DNA gyrase was elucidated by X-ray crystallography.8 One heteroaromatic moiety intercalates into the DNA, and the other binds to a pocket at the C2-symmetric GyrA dimer interface (part of the so-called DNA-gate). No experimental structure of an NBTI bound to Topo IV has been reported, but due to the high homology between the DNA gyrase and Topo IV enzymes in this region, modeling studies predict that NBTIs bind Topo IV in the same manner,11 consistent with the observation that NBTIs are generally dual inhibitors of both enzymes. This is a highly attractive feature of NBTIs since inhibition of two targets is associated with lower potential for resistance development.2,9 Moreover, the binding site and mode of action for NBTIs are distinct from those of the fluoroquinolones, indicating that cross-resistance is unlikely.5,8
Figure 1.

Structure of a representative NBTI.
The most advanced NBTI, gepotidacin (GSK2140944),12 recently completed a Phase III trial for the treatment of uncomplicated urinary tract infection (UTI) and is currently in Phase III clinical trials for the treatment of infection with Neisseria gonorrheae (gonorrhea). Early NBTIs including gepotidacin show potent activity against Gram-positive bacteria but lack broad-spectrum potency against many of the key Gram-negative ESKAPE pathogens, including P. aeruginosa and A. baumannii, which cause lung infections that are more difficult to treat than UTIs and gonorrhea. The reduced potency against Gram-negatives is attributable to the increased barriers to accumulation in the cell interior afforded by their cell envelope differences and a wide array of efflux pumps.13
More recently, NBTIs showing improved potency on a broad range of Gram-negative bacteria have been described.14−16 One such compound, BWC0977, is in Phase I clinical trials.10
In a program aimed at developing novel classes of antibiotics for curing infections caused by MDR bacteria, we undertook a phenotypic screen to identify novel chemical matter with activity against Gram-negative bacteria.17
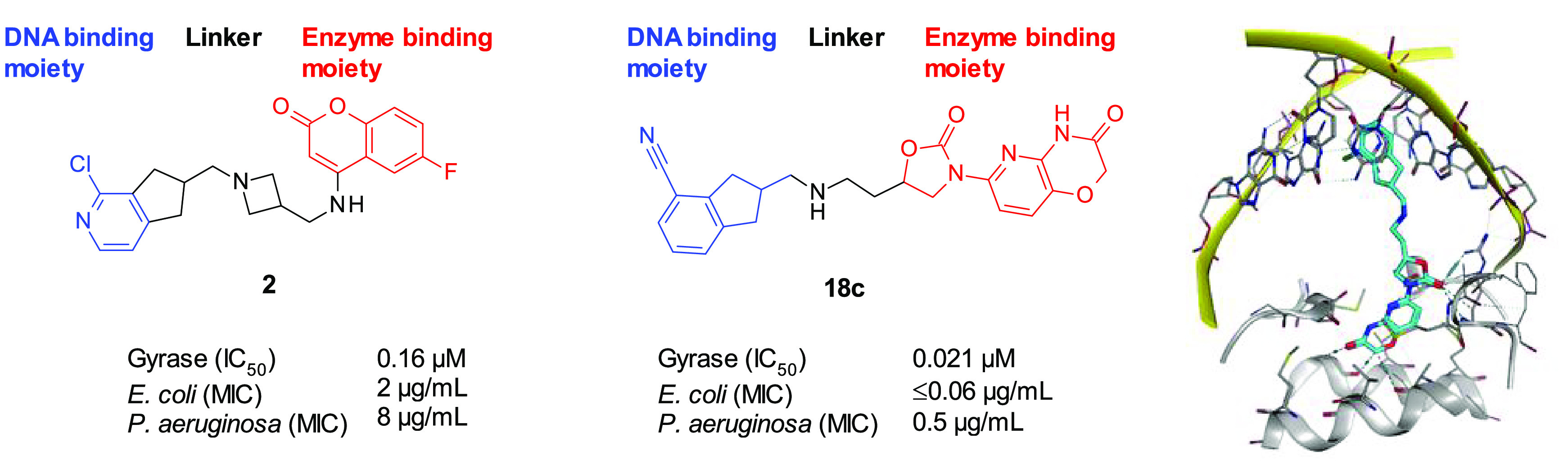
Coumarin–indane compound 1 (Figure 2) was identified as a singleton hit. Further investigation revealed that close analogues of 1 showed specific inhibition of DNA synthesis in Escherichia coli along with potent activity versus E. coli DNA gyrase and topoisomerase IV in topological assays. The compounds displayed effects on DNA synthesis characteristic of the NBTI class of antibiotics.8 The structures follow the general architecture of NBTIs of two aromatic moieties joined by a linker containing a basic amine (Figure 1) but with atypical aromatic groups.9 It was not clear from molecular modeling which end of the molecule intercalates into the DNA and which binds the enzyme pocket. To determine the binding mode, an X-ray cocrystal structure of compound 2 (Figure 2) bound to S. aureus DNA gyrase and DNA was determined at 2.23 Å resolution.18 The structure (Figure 3) shows the aromatic ring of the azaindane moiety intercalating the DNA bases, while the coumarin sits in the enzyme pocket at the GyrA dimer interface. The coumarin ring engages in van der Waals interactions with the side chain of Met121, while the coumarin carbonyl group has a sulfur σ* interaction with Met75. A weak hydrogen-bonding interaction is apparent between the acid carbonyl of Asp83 and the polarized CH2 in the linker close to the coumarin at a distance of 3.1 Å. The basic nitrogen in the linker makes long-range electrostatic interactions with the acids of Asp83 on both sides of the pocket at a distance of 4.8–4.9 Å. An NBTI containing a 4-aminocoumarin motif was modeled into the S. aureus DNA gyrase apo crystal structure (PDB code 2XCS) by co-workers at Angelini.19 The overlay of the 4-aminocoumarins in our experimental structure and the Angelini model is very good, and the space occupied by the linkers also corresponds very well.
Figure 2.

Structures of coumarin–indane NBTIs
Figure 3.
Binding mode of 2 bound to S. aureus DNA gyrase–DNA determined by X-ray crystallography (PDB code 7FVS).
The coumarin–indane compounds showed good potency against E. coli and A. baumannii but rather inferior activity against K. pneumoniae and P. aeruginosa (see Table 1 for the MIC values for compound 2). In addition, the compounds were potent inhibitors of the hERG ion channel, which is a common problem in the field of NBTI drug discovery.20,21
Table 1. Activity Data for Compounds 2 and 17–20.
| minimum
inhibitory concentration (μg/mL) |
|||||
|---|---|---|---|---|---|
| compound | E. coli ATCC 25922 | A. baumannii ATCC 19606 | K. pneumoniae ATCC BAA-1705 | P. aeruginosa ATCC 27853 | E. coli DNA gyrase IC50 (μM) |
| 2 | 2 | 4 | 32 | 8 | 0.16 |
| 17a | 0.125 | 0.125 | 2 | 2 | 0.090 |
| 17b | 2 | 4 | 16 | 16 | 0.29 |
| 17c | 0.25 | 0.5 | 2 | 2 | 0.072 |
| 17d | 8 | 4 | >64 | >64 | 0.58 |
| 17e | 0.25 | 0.25 | 4 | 1 | 0.090 |
| 17f | 0.5 | 1 | 8 | 4 | 0.12 |
| 17g | 0.5 | 1 | 32 | 8 | 0.23 |
| 18a | 1 | 2 | 8 | 8 | 0.18 |
| 18b | 0.5 | 0.5 | 4 | 4 | 0.10 |
| 18c | ≤0.06 | 0.125 | 1 | 0.5 | 0.021 |
| 19a | 2 | 8 | 16 | 8 | 0.27 |
| 19b | 0.5 | 1 | 4 | 4 | 0.12 |
| 19c | 2 | 4 | 8 | 16 | 0.25 |
| 19d | 1 | 4 | 8 | 16 | 0.16 |
| 19e | 0.25 | 0.5 | 2 | 2 | 0.057 |
| 20 | 2 | 8 | 16 | 8 | 0.19 |
With the knowledge that the indane moiety binds to the DNA, we decided to explore replacing the coumarin moiety with an oxazolidinone–pyridooxazinone enzyme pocket binding motif.9,16 The novel indane-containing compounds were synthesized as shown in Scheme 1.22 Intermediate 9a containing a one-carbon spacer between the oxazolidinone and the oxygen atom that will later be converted into the amino group in the linker was prepared from commercially available 7, while the two- and three-carbon spacer versions 9b and 9c were synthesized in three or four steps from the TBDMS-protected alcohols 8a and 8b, respectively. Ullmann-type cross-coupling of 9 with 2-bromopyridine 10 gave intermediate 11; subsequent reduction of the nitro group afforded oxazinone 12. The protected alcohol was converted to primary amine 16 in four steps. Finally, reductive amination with ketones 3 and 6 or aldehydes 4 and 5 afforded the final compounds 17–20.
Scheme 1. Synthesis of Compounds 17–20.
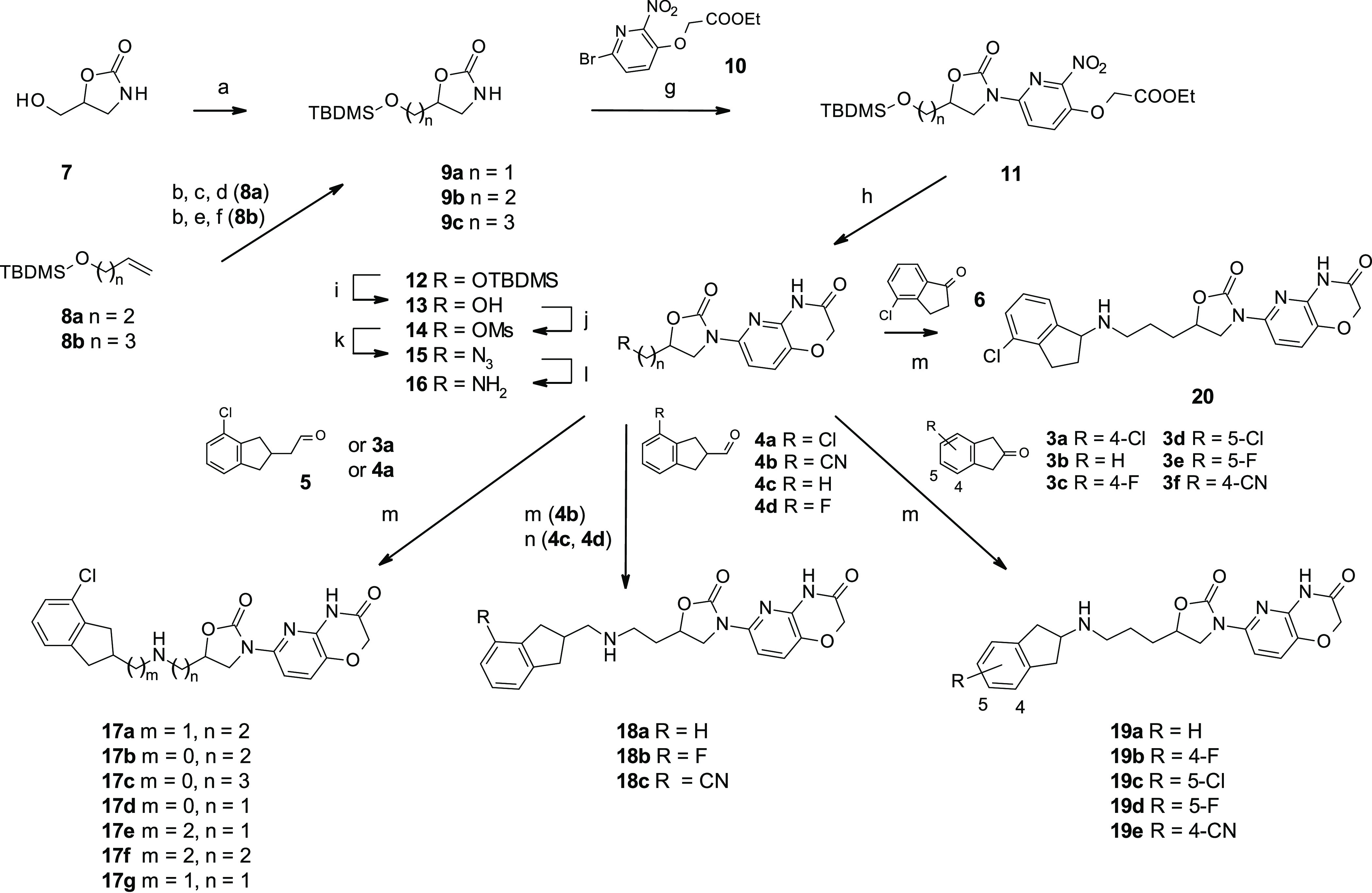
Reagents and conditions: (a) TBDMSCl, imidazole, CH2Cl2, 15 °C; (b) mCPBA, CH2Cl2, 0 °C; (c) NH4OH, MeOH, 25 °C; (d) CO(CCl3)2, NEt3, CH2Cl2, 0 to 25 °C; (e) (i) NaN3, NH4Cl, MeOH, 80 °C, (ii) H2, Pd/C, THF, 25 °C; (f) CDI, THF, 50 °C; (g) 10, CuI (0.2 equiv), Me2NCH2CH2NMe2 (0.4 equiv), K2CO3, 1,4-dioxane, 100 °C; (h) Fe, AcOH, 70 °C; (i) HCl, MeOH, 25 °C; (j) MsCl, NEt3, CH2Cl2, 0 to 25 °C; (k) NaN3, DMF, 60 °C; (l) H2, Pd/C, MeOH, 25 °C; (m) 3a–f or 4a/4b or 5/6, Na(CN)BH3, MeOH, 25 °C; (n) 4c/4d, NaBH(OAc)3, AcOH, NEt3, 25 °C.
Compounds were tested for antimicrobial susceptibility in a panel of multiple strains of Gram-negative (E. coli, A. baumannii, K. pneumoniae, and P. aeruginosa) and Gram-positive (S. aureus) bacteria. Data for four representative Gram-negative strains are given in Table 1 together with IC50 values for the inhibition of the ATPase activity of the E. coli DNA gyrase enzyme. The representative strains were chosen such that the MIC would be predictive of the expected potency in a broader panel of clinical isolates for the particular bacterial species. MIC values for the full panel are given in Table S2. The strain panel comprises drug-sensitive and -resistant reference strains of five key ESKAPE pathogens commonly used in antibiotic drug discovery. E. coli BW25113 and ATCC 25922 are drug-sensitive strains, and ATCC 35218 and ATCC BAA-2340 are resistant strains. A. baumannii ATCC 19606 is considered a sensitive wild-type strain, while ATCC 51432 and ATCC BAA-747 are MDR. ATCC 10031 is a commonly used sensitive K. pneumoniae control, whereas ATCC 700603, ATCC BAA-1705, and ATCC-2146 are model strains for the production of extended-spectrum β-lactamases (ESBLs), K. pneumoniae carbapenemase (KPC), and New Delhi metallostalactamase 1 (NDM-1), respectively. ATCC 27853 is a P. aeruginosa wild-type strain, while NCTC13437 and NCTC11451 are MDR. Lastly, ATCC 29213 and ATCC BAA-1556 (USA300) are commonly used methicillin-susceptible S. aureus (MSSA) and methicillin-resistant S. aureus (MRSA) strains.
Compounds 17a–g were designed to explore the effect of varying the linker between the oxazolidinone and the indane. A linker of length four atoms (17a, 17c, 17e) showed the best potencies, with activity decreasing with linker length for three-atom linkers (17b, 17g) and two-atom linkers (17d). Increasing the linker length to five atoms (17f) resulted in a slight drop in potency, suggesting that four atoms is the optimal length. Comparing the results for different positions of the NH in the four-atom linker compounds showed that 17a had the best activity versus E. coli and A. baumannii, while 17e was the most potent versus P. aeruginosa but was 2-fold less active versus K. pneumoniae than 17a and 17c. The enzyme activities of 17a, 17c, and 17e were very similar, suggesting that the position of the NH is not critical for binding to the target enzyme.
The synthesis route to 17e required a four-step homologation sequence to convert a 2-hydroxymethylindane (synthetic precursor to 4) to a 2-(2-hydroxyethyl)indane (synthetic precursor to 5), and this linker was therefore less attractive for rapid SAR exploration. To optimize the indane substituent, compounds having the NH adjacent to the indane (19a–e) or one atom position closer to the oxazolidinone (18a–c) were synthesized.
The results showed that removing the chloro substituent led to a reduction in potency (compare 18a and 19a with 17a and 17c), replacing chloro with fluoro gave a small drop in potency (18b and 19b), and moving the chloro or fluoro to the indane 5-position also resulted in lower potency (19c and 19d). Replacing the chloro with cyano (18c and 19e) maintained or slightly improved the potency, with compound 18c showing the lowest MICs across all four species. Finally, moving the attachment point of the linker from the indane 2-position to the 1-position resulted in significantly lower potency (compare 20 with 17c).
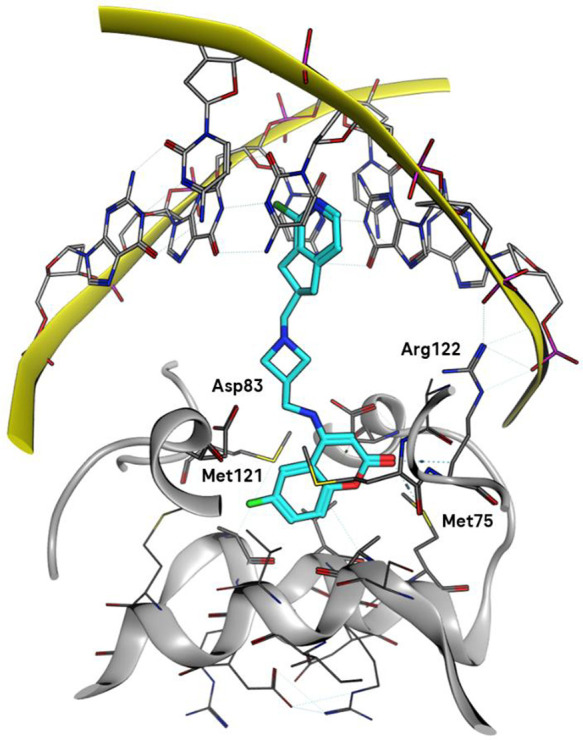
An X-ray cocrystal structure of compound 17a bound to S. aureus DNA gyrase and DNA was determined at 2.16 Å resolution.18 The structure (Figure 4) confirms that the aromatic part of the indane moiety intercalates with DNA while the pyridooxazinone binds the enzyme pocket at the GyrA dimer interface in the same space as the coumarin in compound 2, making mostly van der Waals interactions with the protein. A similar interaction between the aromatic ring and Met121 as was observed in the cocrystal structure of coumarin 2 is apparent. The carbonyl of the oxazinone in 17a fills the same space as the fluorine atom in 2. The oxazolidinone carbonyl of 17a is embedded in a weak hydrogen-bonding network, closest to the backbone NH of Arg122 in GyrA with a distance of 2.9 Å.
Figure 4.
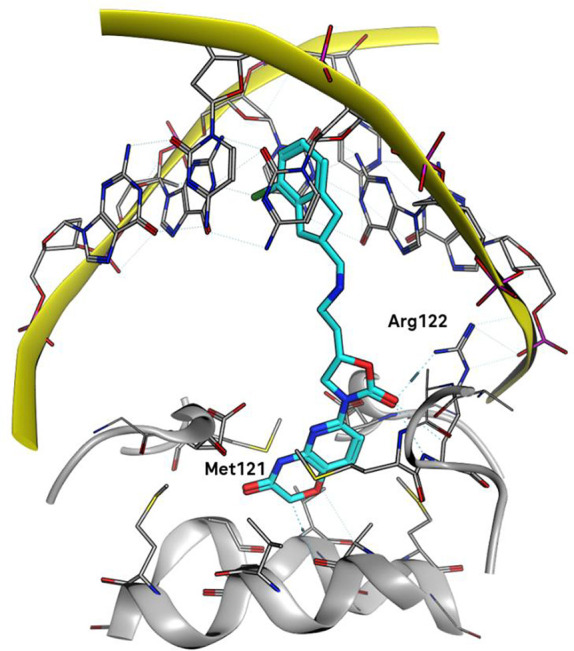
Binding mode of 17a bound to S. aureus DNA gyrase–DNA determined by X-ray crystallography (PDB code 7FVT). The linker region is disordered, chiral centers were assigned as (S,S) based on best geometry, and the rest of the linker was modeled with CSD-compatible torsion angles.
The binding mode is consistent with the observed SAR since the indane moiety in analogues with shorter linkers than in 17a would be expected to be unable to interact with the DNA as effectively, particularly 17d, which indeed has the highest enzyme IC50. Interestingly, the indane in coumarin 2 does not intercalate as deeply into DNA as the indane in 17a. Also, the position of the basic amine in the linker does not affect the enzyme activity significantly, consistent with the observation that it does not make any specific interactions with the protein beyond long-range electrostatic ones with Asp83.
Compounds 17–20 were prepared as 1:1:1:1 mixtures of four stereoisomers. From the observed binding mode, it was anticipated that the individual stereoisomers would have equal potency. This was confirmed for 17a, as the four isomers were prepared individually (see p S12 in the Supporting Information) and were found to have the same MIC values against each strain across the panel (data not shown). It is noteworthy that despite these compounds being racemic 1:1 mixtures of two diastereomers, they appear as single diastereomers in high-field 1H and 13C NMR (see Figures S3–S6) and the diastereomeric mixtures of 17a were separable only by chiral chromatography. From inspection of the compound structures we observe that the two chiral centers are quite distant from each other; moreover, the indane chiral center has some pseudo-C2 symmetry.
The most potent compound, 18c, was further studied in topological assays, measuring its inhibition of DNA supercoiling activity in bacterial DNA gyrase and of decatenation activity in bacterial topoisomerase IV and human topoisomerase IIa. The data (Table 2) show that 18c is a potent and balanced dual inhibitor of both bacterial enzymes, with no observed activity on the human enzyme.
Table 2. Topological Enzyme Activity Data for Compound 18c.
| enzyme | IC50 (μM) |
|---|---|
| E. coli DNA gyrase | 0.10 |
| E. coli topoisomerase IV | 0.16 |
| human topoisomerase IIa | >100 |
Generally for the compounds in Table 1 the enzyme inhibition activity as measured in the DNA gyrase ATPase assay correlated well with the MIC values against individual strains in the panel (Table 1) and also with the IC50 values in the topoisomerase IV ATPase assay (see Table S3 and Figure S8). The biochemical ATPase assays were used in parallel to the MIC panel for routine compound screening because of their higher throughput and reduced compound requirement compared with the gel-based topological enzyme assays. The exact difference in IC50 values between the two targets in the biochemical assays for a given compound may not be significant, considering that these assays measure target inhibition indirectly. In contrast, a comparison of the IC50 values for compound 18c against the two targets in the topological assays (Table 2) is a more reliable indication of the balanced nature of this inhibitor.
DNA gyrase and topoisomerase IV are highly conserved across bacterial species, and it is therefore expected that a compound will have very similar intrinsic potencies on the target in each species. Differences in MIC values between different species and strains are presumed to arise instead from differences in the ability of the compounds to penetrate the outer and inner membranes and overcome efflux pumps to reach the target in the cytosol.
The compounds in Table 1 showed undesirable inhibition of the hERG ion channel; for example, compound 18c had an IC50 of 0.90 μM in an electrophysiological patch clamp assay at 37 °C, showing that the cardiac safety margin needs to be improved for an eventual drug candidate. Otherwise 18c represents a promising starting point for optimization toward a potential antibiotic demonstrating broad-spectrum activity against Gram-negative bacteria.
In summary, we have identified a novel series of NBTIs incorporating a previously unreported DNA binding group: a 4-halo- or 4-cyanoindane. The compounds show potent broad-spectrum activity in vitro against Gram-negative bacteria, including highly resistant strains. The indane moiety appears broadly to confer comparable potencies against Gram-negative bacteria compared with previously reported NBTIs containing the same oxazolidinone–pyridooxazinone enzyme pocket binding group together with different DNA intercalator moieties.16 The discovery of the indane-containing series exemplified by compound 18c provides new options for the identification of an NBTI with the overall profile needed for curing infections caused by MDR Gram-negative bacteria. Further SAR exploration and optimization of this series will be reported in due course.
Acknowledgments
The authors thank Eva Anna Krafft, Melodie Zehnder, Linus Oesch, Weizhen Zhang, Chenwei Wu, Futang Lian, Liang Dai, Fajun Miao, and Shoubao Cui for the synthesis of the compounds described. We thank Luisa Vaccaro, Xiangyu Yao, Yi Mao, Guanglei Zhai, Waikwong Wu, and Evgenia Gissinger for biological testing. The research described herein was funded by F. Hoffmann-La Roche AG.
Glossary
Abbreviations
- NBTI
novel bacterial topoisomerase inhibitor
- MDR
multidrug-resistant
- Topo IV
topoisomerase IV
- MIC
minimum inhibitory concentration
- hERG
human ether-à-go-go-related gene
- TBDMS
tert-butyldimethylsilyl
- SAR
structure–activity relationship
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00187.
Synthesis of compounds 2, 17a, and 18c and their analytical data, assay procedures for antimicrobial susceptibility testing, ATPase enzyme assays, topological enzyme assays and hERG assay, protein purification, crystallization and X-ray crystallography procedures for compounds 2 and 17a, MIC values for all panel strains, ATPase enzyme activities, topological enzyme activities for compound 18c and positive controls, X-ray crystallographic data collection and refinement statistics, and electron density images (PDF)
Author Contributions
All of the authors approved the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Pendleton J. N.; Gorman S. P.; Gilmore B. F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti-Infect. Ther. 2013, 11, 297–308. 10.1586/eri.13.12. [DOI] [PubMed] [Google Scholar]
- Silver L. L. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. C. Cellular roles of DNA topoisomerases: a molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- Tse-Dinh Y. C. Targeting bacterial topoisomerases: how to counter mechanisms of resistance. Future Med. Chem. 2016, 8, 1085–1100. 10.4155/fmc-2016-0042. [DOI] [PubMed] [Google Scholar]
- Tomašić T.; Mašič L. P. Prospects for Developing New Antibacterials Targeting Bacterial Type IIA Topoisomerases. Curr. Top. Med. Chem. 2013, 14, 130–151. 10.2174/1568026613666131113153251. [DOI] [PubMed] [Google Scholar]
- Bisacchi G. S.; Manchester J. I. A New-Class Antibacterial—Almost. Lessons in Drug Discovery and Development: A Critical Analysis of More than 50 Years of Effort toward ATPase Inhibitors of DNA Gyrase and Topoisomerase IV. ACS Infect. Dis. 2015, 1, 4–41. 10.1021/id500013t. [DOI] [PubMed] [Google Scholar]
- Hu Y.; Shi H.; Zhou M.; Ren Q.; Zhu W.; Zhang W.; Zhang Z.; Zhou C.; Liu Y.; Ding X.; Shen H. C.; Yan S. F.; Dey F.; Wu W.; Zhai G.; Zhou Z.; Xu Z.; Ji Y.; Lv H.; Jiang T.; Wang W.; Xu Y.; Vercruysse M.; Yao X.; Mao Y.; Yu X.; Bradley K.; Tan X. Discovery of Pyrido[2,3-b]indole Derivatives with Gram-Negative Activity Targeting Both DNA Gyrase and Topoisomerase IV. J. Med. Chem. 2020, 63, 9623–9649. 10.1021/acs.jmedchem.0c00768. [DOI] [PubMed] [Google Scholar]
- Bax B. D.; Chan P. F.; Eggleston D. S.; Fosberry A.; Gentry D. R.; Gorrec F.; Giordano I.; Hann M. M.; Hennessy A.; Hibbs M.; Huang J.; Jones E.; Jones J.; Brown K. K.; Lewis C. J.; May E. W.; Saunders M. R.; Singh O.; Spitzfaden C. E.; Shen C.; Shillings A.; Theobald A. J.; Wohlkonig A.; Pearson N. D.; Gwynn M. N. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 2010, 466, 935–940. 10.1038/nature09197. [DOI] [PubMed] [Google Scholar]
- Kolarič A.; Anderluh M.; Minovski N. Two Decades of Successful SAR-Grounded Stories of the Novel Bacterial Topoisomerase Inhibitors (NBTIs). J. Med. Chem. 2020, 63, 5664–5674. 10.1021/acs.jmedchem.9b01738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai J.; S S.; Kumar S.; Sharma R. Novel Bacterial Topoisomerase inhibitors (NBTIs) - A comprehensive review. Eur. J. Med. Chem. Rep. 2021, 3, 100017. 10.1016/j.ejmcr.2021.100017. [DOI] [Google Scholar]
- Kokot M.; Anderluh M.; Hrast M.; Minovski N. The Structural Features of Novel Bacterial Topoisomerase Inhibitors That Define Their Activity on Topoisomerase IV. J. Med. Chem. 2022, 65, 6431–6440. 10.1021/acs.jmedchem.2c00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson E. G.; Bax B.; Chan P. F.; Osheroff N. Mechanistic and Structural Basis for the Actions of the Antibacterial Gepotidacin against Staphylococcus aureus Gyrase. ACS Infect. Dis. 2019, 5, 570–581. 10.1021/acsinfecdis.8b00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M. F.; Hergenrother P. J. The challenge of converting Gram-positive-only compounds into broad-spectrum antibiotics. Ann. N. Y. Acad. Sci. 2019, 1435, 18–38. 10.1111/nyas.13598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty T. J.; Nayar A.; Newman J. V.; Hopkins S.; Stone G. G.; Johnstone M.; Shapiro A. B.; Cronin M.; Reck F.; Ehmann D. E. NBTI 5463 Is a Novel Bacterial Type II Topoisomerase Inhibitor with Activity against Gram-Negative Bacteria and In Vivo Efficacy. Antimicrob. Agents Chemother. 2014, 58, 2657–2664. 10.1128/AAC.02778-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surivet J.-P.; Zumbrunn C.; Bruyère T.; Bur D.; Kohl C.; Locher H. H.; Seiler P.; Ertel E. A.; Hess P.; Enderlin-Paput M.; Enderlin-Paput S.; Gauvin J.-C.; Mirre A.; Hubschwerlen C.; Ritz D.; Rueedi G. Synthesis and Characterization of Tetrahydropyran-Based Bacterial Topoisomerase Inhibitors with Antibacterial Activity against Gram-Negative Bacteria. J. Med. Chem. 2017, 60, 3776–3794. 10.1021/acs.jmedchem.6b01831. [DOI] [PubMed] [Google Scholar]
- Zumbrunn C. A Short History of Topoisomerase Inhibitors at Actelion Pharmaceuticals. Chimia 2022, 76, 647–655. 10.2533/chimia.2022.647. [DOI] [PubMed] [Google Scholar]
- Zoffmann S.; Vercruysse M.; Benmansour F.; Maunz A.; Wolf L.; Blum Marti R.; Heckel T.; Ding H.; Truong H. H.; Prummer M.; Schmucki R.; Mason C. S.; Bradley K.; Jacob A. I.; Lerner C.; Araujo del Rosario A.; Burcin M.; Amrein K. E.; Prunotto M. Machine learning-powered antibiotics phenotypic drug discovery. Sci. Rep. 2019, 9, 5013. 10.1038/s41598-019-39387-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The coordinates have been deposited in the PDB with accession codes 7FVS for compound 2 and 7FVT for compound 17a.
- Magarò G.; Prati F.; Garofalo B.; Corso G.; Furlotti G.; Apicella C.; Mangano G.; D’Atanasio N.; Robinson D.; Di Giorgio F. P.; Ombrato R. Virtual Screening Approach and Investigation of Structure-Activity Relationships To Discover Novel Bacterial Topoisomerase Inhibitors Targeting Gram-Positive and Gram-Negative Pathogens. J. Med. Chem. 2019, 62, 7445–7472. 10.1021/acs.jmedchem.9b00394. [DOI] [PubMed] [Google Scholar]
- Black M. T.; Coleman K. New Inhibitors of Bacterial Topoisomerase Gyra/Parc Subunits. Curr. Opin. Invest. Drugs 2009, 10, 804–810. [PubMed] [Google Scholar]
- Kolarič A.; Minovski N. Novel Bacterial Topoisomerase Inhibitors: Challenges and Perspectives in Reducing hERG Toxicity. Future Med. Chem. 2018, 10, 2241–2244. 10.4155/fmc-2018-0272. [DOI] [PubMed] [Google Scholar]
- Cumming J. G.; Kramer C; Kreis L; Kuehne H; Schnider P.. Preparation of heterocyclic compounds and their use in the treatment of bacterial infection. WO 2021190727 A1, 2021.
- Black M. T.; Stachyra T.; Platel D.; Girard A.-M.; Claudon M.; Bruneau J.-M.; Miossec C. Mechanism of action of the antibiotic NXL101, a novel non-fluoroquinolone inhibitor of bacterial type II topoisomerases. Antimicrob. Agents Chemother. 2008, 52, 3339–3349. 10.1128/AAC.00496-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.