

Abstract
Bisphenol A is an endocrine disruptor that has been shown to have testicular toxicity in animal models. Its structural analog, including bisphenol S (BPS), bisphenol AF (BPAF), and tetrabromobisphenol A (TBBPA) have been introduced to the market as BPA alternatives. Previously, we developed high-content analysis (HCA) assays and applied machine learning to compare the testicular toxicity of BPA and its analogs in spermatogonial cells and testicular cell co-culture models. There are diverse cell populations in the testis to support spermatogenesis, their cell type-specific toxicities are still not clear. The purpose of this study is to examine the selective toxicity of BPA, BPS), BPAF, and TBBPA on these testicular cells, including Sertoli cell, Leydig cells, and spermatogonia cell. We developed a high-content image-based single-cell analysis and measured a broad spectrum of adverse endpoints related to the development of reproductive toxicology, including cell number, nuclear morphology, DNA synthesis, cell cycle progression, early DNA damage response, cytoskeleton structure, DNA methylation status, and autophagy. We introduced an HCA index and spectrum to reveal multiple HCA parameters and observed distinct toxicity profiling of BPA and its analogs among three testicular types. The HCA spectrum reveals the dynamic, chemical-specific, dose-dependent changes of each HCA parameter. Each chemical displayed a unique dose-dependent profile within each type of cell. All three types of cells showed the highest response to BPAF at 10 μM across all endpoints measured. BPAF targeted spermatogonial cell (C18) more significantly at 5 μM. BPS more likely targeted Sertoli cell (TM4) and Leydig cell (TM3) and less at spermatogonia cells. TBBPA targeted spermatogonia, Sertoli cells, and less at TM3 cells. BPA is mainly targeted at TM4, followed by TM3 cells, and less at spermatogonial cells. Most importantly, we observed BPAFR induced a dose-dependent increase in spermatogonia cell, not in Sertoli cells and Leydig cells. germ In summary, our current HCA assays revealed the cell-type-specific toxicities of BPA and its analogs in different testicular cells. Multinucleation induced by BPAF along with increased DNA damage and synthesis at low doses, could possibly have a profound long-term effect on reproductive systems.
Keywords: Bisphenol A, Analogs, High-content analysis, Cell type-specific toxicity, Spermatogonial cells, Sertoli cells, Leydig cells
Introduction
The incidence of male reproductive disorders has increased in recent decades. Exposure to environmental toxicants and pharmaceuticals has been proposed to impact male reproductive health (Nordkap, Joensen et al. 2012, Knez 2013, Jeng 2014, Jenardhanana, Panneerselvama et al. 2016, Leung, Phuong et al. 2016, Skakkebaek, Rajpert-De Meyts et al. 2016). The decline of human semen quality, such as sperm count and motility, has been reported to be linked to exposure to environmental contaminants (Nordkap, Joensen et al. 2012, Knez 2013, Jeng 2014, Skakkebaek, Rajpert-De Meyts et al. 2016). Bisphenol A (BPA) is a high-production volume chemical used extensively in various consumer products (Rochester 2013, Siracusa, Yin et al. 2018). BPA in urine was detectable in over 90% of the United States population (Calafat, Ye et al. 2008, Lakind and Naiman 2011, CDC 2022). As an endocrine-disrupting chemical, BPA has shown various adverse health effects, such as reproduction and neurotoxicity (Rubin 2011, Peretz, Vrooman et al. 2014, Santoro, Chianese et al. 2019). BPA exposure was found to affect oocyte quality and maturation adversely, decrease sperm production and quality, damage testicular cells, perturb hormone levels, and disrupt ovary function and uterine morphology in animal models (Delclos, Camacho et al. 2014, Qi, Fu et al. 2014, Moore-Ambriz, Acuna-Hernandez et al. 2015, Vigezzi, Bosquiazzo et al. 2015, Barbonetti, Castellini et al. 2016, Berger, Ziv-Gal et al. 2016, Ferris, Mahboubi et al. 2016, Wang, Han et al. 2016). Human exposure to BPA leaches out of consumer products mainly through ingestion, inhalation, and dermal absorption (Kang, Kondo et al. 2006). Although FDA determined the use of BPA at current level is safe as supported by the CLARITY-BPA studieas (Heindel, Newbold et al. 2015, Camacho, Lewis et al. 2019) (FDA. 2012, FDA. 2013, EU. 2016), widespread exposure and potential hazards of BPA in humans drove manufacturers to abandon the use of BPA and introduce analogous chemicals as crosslinking reagents and flame retardants in the plastics industry to produce “BPA-free” products. There is a lack of production data for these analogs, and the usage of these chemicals is rising globally since these are not all strictly used as replacements for BPA, but have a variety of commercial uses. BPA analogs are found in the environment, foods, consumer products, and human urine samples (Geens, Roosens et al. 2009, Liao, Liu et al. 2012, Liao, Liu et al. 2012, Shi, Jiao et al. 2013, Ye, Wong et al. 2015). With high degrees of structural similarities to BPA, these analogs potentially have a similar endocrine-disrupting capacity and exert adverse effects on the reproductive system. Emerging evidence suggests that BPA analogs act with various physiological receptors (Kitamura, Suzuki et al. 2005, Stossi, Bolt et al. 2014). Recently, the National Toxicology Program reported on the biological activity of BPA and several of its analogs, Bisphenol S (BPS), Bisphenol F (BPF), and Bisphenol AF (BPAF), and suggested BPA analogs causing mammalian reproductive toxicity (Pelch, Wignall et al. 2017, Pelch, Wignall et al. 2019). So far, 16 bisphenol analogs have been detected in house dust, drinking water, serum, and human milk (Deceuninck, Bichon et al. 2015, Owczarek, Kubica et al. 2018, Zhang, Zhang et al. 2019, Yang, Shi et al. 2022).
Compared to BPA, little is known about the reproductive toxicity of these analogs. In our previous study, we applied the testicular cell co-culture model and employed a high-content image (HCA) based single-cell analysis to compare the testicular toxicities of BPA and its analogs (Yin, Siracusa et al. 2020). We found dose and time-dependent changes in a broad spectrum of adverse endpoints, including nuclear morphology, DNA synthesis, DNA damage, and cytoskeletal structure in the testicular co-culture model. Furthermore, the co-cultured testicular model was more sensitive than C18 spermatogonial cells in response to BPA and its analogs (Liang, Yin et al. 2017). Sertoli, Leydig, and germ cells have been reported to respond differently compared to BPA, and it is unclear if Sertoli and Leydig cells are more sensitive than germ cells to these BPA analogs. The purpose of this study is to examine the selective toxicity of BPA, bisphenol S (BPS), bisphenol AF (BPAF), and tetrabromobisphenol A (TBBPA) on these testicular cells, including Sertoli cell, Leydig cells, and spermatogonia cell. This study employed a high-content image-based single-cell analysis and measured a broad spectrum of adverse endpoints related to the development of reproductive toxicology, including cell number, nuclear morphology, DNA synthesis, cell cycle progression, early DNA damage response, cytoskeleton structure, DNA methylation status, and autophagy. We introduced an HCA spectrum to reveal multiple HCA parameters and observed distinct toxicity profiling of BPA and its analogs among three testicular types. These findings leveraged the specific-cell target and toxicity mechanism action for each BPA and its analogs, which provided a novel methodology to efficiently identify cell targets and valuable mechanical information in interpreting in vivo reproductive toxicity of BPA and its selected analogs.
Material and Methods:
Chemicals
Dulbecco’s Modified Eagle Medium (DMEM), Modified Eagle’s Medium/Nutrient Mixture F-12 (DMEM/F12), fetal bovine serum (FBS), horse serum, and penicillin-streptomycin were purchased from Thermo Scientific (Waltham, MA). 4,4’-(propane-2,2-diyl) diphenol (BPA, ≥99%), 4,4’-sulfonyldiphenol (BPS, 98%), and 2,2’,6,6’-Tetrabromo-4,4’-isopropylidenediphenol (TBBPA, 97%) were obtained from Sigma-Aldrich (St Louis, MO). 4-[1,1,1,3,3,3-Hexafluoro-2-(4-hydroxyphenyl)propan-2-yl]phenol (BPAF, 98%) was obtained from Alfa Aesar (Ward Hill, MA). Nu-Serum was purchased from BD BioScience (Redford, MA). 5-Bromo-2’-deoxyuridine (BrdU, 99%) was purchased from Thermo Scientific (Waltham, MA). 4% Paraformaldehyde was purchased from Boston Bioproducts (Ashland, MA).
Cell Culture and treatment:
C18-4 mouse spermatogonial cell line was established from germ cells isolated from the testes of 6-day-old Balb/c mouse as reported previously (Hofmann et al., 2005). This cell line exhibits morphological features of type A spermatogonia and expresses testicular germ cell-specific genes such as GFRA1, Dazl, and Ret (Hofmann et al., 2005). Mouse TM3 Leydig and TM4 Sertoli cell lines were purchased from ATCC (Manassas, VA). All the cell culture procedures were conducted as reported previously (Liang, Yin et al. 2017). Briefly, C18-4 spermatogonial cells were maintained in DMEM composed of 5% FBS, 100 U/ml streptomycins/penicillin in a 33°C, 5% CO2 humidified environment in a sub-confluent condition with passaging every 3-4 days. Leydig and Sertoli cells were cultured in DME/F12 composed of 2.5% FBS, 5.0 % horse serum, 100 U/ml streptomycin/penicillin at 37°C, 5% CO2 in a sub-confluent condition with passaging every 2-3 days. When cells reached 70-80% confluence, the spermatogonial cells were inoculated with 1.2 ×104 cells per well, and Leydig and Sertoli cells were inoculated with 1.5 ×104 cells per well into a 96-well plate. Cells were cultured overnight reaching 70-80% confluence, then treated with doses of BPA, BPS (0, 5, 50 and 100 μM), BPAF and TBBPA (0, 1, 5 and 10 μM) at for 24 and 48 h. The experiment was completed with technical triplicates for each concentration, and experiments were repeated 3 or 4 times.
Fluorescence staining and image acquisition
For BrdU incorporation, cells were incubated with BrdU (40 μM) for three hours before fixation with 4% paraformaldehyde for 30 min at room temperature, followed by three times washing with phosphate-buffered saline (PBS) (Liang, Yin et al. 2017). After fixation, cells were permeabilized by 0.1% Triton X-100 in PBS, blocked with PBS/3% BSA, and incubated with a mouse anti-BrdU antibody (Thermo Scientific, MA) in PBS/BSA/0.5% Tween 20 overnight at 4 °C. After washing twice with PBS/BSA, the cells were incubated with goat anti-mouse DyLight 488 and Hoechst 33342 (Thermo Scientific, MA) in PBS/BSA solution for 90 mins at room temperature.
DNA damage responses was detected by anti-phospho-histone-H2AX (Ser139) (γ-H2AX) and cytoskeleton analysis was performed by F-actin staining with AlexaFluor 488 Phalloidin (Cell Signaling 8878) following the protocol reported previously (Liang, Yin et al. 2016). For epigenetic or autophagy evaluation, antibodies against Methyl CpG Binding Protein 1 (MBD1) (Developmental Studies Hybridoma Bank, http://dshb.biology.uiowa.edu, Iowa) and LC-3B (ThermoFisher Scientific, MA) were used to incubate with the fixed cells and followed by the corresponding secondary antibodies for imaging acquisition.
Multi-channel images were automatically acquired using an Arrayscan™ VTI HCS reader (Thermo Scientific, MA). Forty-nine fields per well were acquired at 20x and 40X magnification using Hamamatsu ROCA-ER digital camera in combination with 0.63x coupler and Carl Zeiss microscope optics in auto-focus mode. Channel one (Ch1) applied the BGRFR 386_23 for Hoechst 33342 auto-focus, object identification, and segmentation. For MBD1 and LC3B staining, channel two (Ch2) was assigned to BGRFR 485_20 for MBD1 detection, and channel three (Ch3) was assigned to BGRFR 650_13 for LC3B detection. Channel one was set to BGRFR 386_23 for Hoechst 33342 for all images and used for auto-focus, object identification and segmentation.” For F-actin and γ-H2AX staining, Ch2 was assigned to BGRFR 485_20 for F-actin, and Ch3 was set to BGRFR 650_13 for γ-H2AX visualization.
High-content images analysis
Multi-channel images were analyzed following the procedure described in the previous study (Yin, Siracusa et al. 2020) using HCS Studio™ 2.0 TargetActivation BioApplication. Multi-parameters of nuclei were characterized in HCA, including the nuclei number, nuclear area, shape, and total DNA intensity. The total intensity of BrdU, γ-H2AX, and F-actin, MBD1, and LC3B of the individual cell ere also quantified from HCA. Nuclear shape measurement included P2A (nuclear perimeter2/4 π * nuclear area) to evaluate nuclear smoothness and LWR (nuclear length/width) to measure nuclear roundness. For a fairly round and smooth object, the values for P2A and LWR are close to 1.0. The total intensity was defined as the total pixel intensities within a cell in the respective channel. With forty-nine fields of each well, at least 1000 cells were analyzed per well, and single-cell-based data were exported for further analysis. The experiments were performed with at least four biological replicates and repeated twice.
HCA-based cell cycle analysis was conducted as previously described (Liang, et al., 2016; Roukos et al., 2015). Briefly, the histogram of total DNA intensity of each replicate in various experimental conditions was constructed in a custom script written in Python 2.7.12 (Python Software Foundation, OR; this script is freely available from the authors upon request). The input data were gated using the nuclear area and total DNA intensity to exclude cell debris and clumps. Cell populations in sub-G1 (apoptotic cells), G0/1, S, and G2/M in the controls and treatments were quantified by the appropriate selection of gating threshold.
Statistical Analysis
Data obtained from the HCS StudioTM 2.0 TargetActivation BioApplication were exported and further analyzed using the JMP statistical analysis package (SAS Institute, NC). A nucleus with an area larger than 1000 μm2 in a 20X image or 2000 μM2 in a 40X image was excluded as cell clumps. For each plate, the vehicle control showed consistent measurements for all endpoints tested. For intra-plate normalization, data were normalized to the overall scaling factors, which was the mean of medians of vehicle controls in each plate. The single cell-based data were averaged for the well-based condition. BrdU-positive cells were set by the total intensity of BrdU in the Control over 25,000 pixels. γ-H2AX positive cells were set by the total intensity of γ-H2AX in the Control over 140,000 pixels. Data were presented as the mean ± standard deviation (SD). Statistical significance was determined using one-way ANOVA followed by Tukey-Kramer all-pairs comparison. A P value less than 0.05 denoted a significant difference compared to the vehicle control (*). To compare the multiplexed HCA parameters among BPA and its analogs between the three cell types, C18-4 spermatogonial, TM3 Leydig, and TM4 Sertoli cells, we introduced the HCA spectrum to reveal the dynamic, chemical-specific, dose-dependent changes of each HCA assay. We further developed an HCA index to integrate these parameters from diverse HCA assays to show dose-dependent dynamic changes in these cellular markers. HCA spectrum reveals the deviation (fold changer from the Control) of each assay, including cell number, nuclear area, the marker for shape measurements (P2A and LWR), DNA synthesis (BrdU positive cells), cell cycle phases (cell population in subG1 phase, G0/G1 phase, S phase, and G2/M phase), DNA damage response (γ-H2AX positive cells), F-actin total intensity, MBD1 total intensity and LC3B total intensity. For each feature parameter, deviation from Control as reflected by the fold change over the mean of the Control was calculated and then log2-transformed. HCA index is the sum of the absolute value of each log2 transformed fold change.
Results:
Cell viability and nuclear morphological alterations
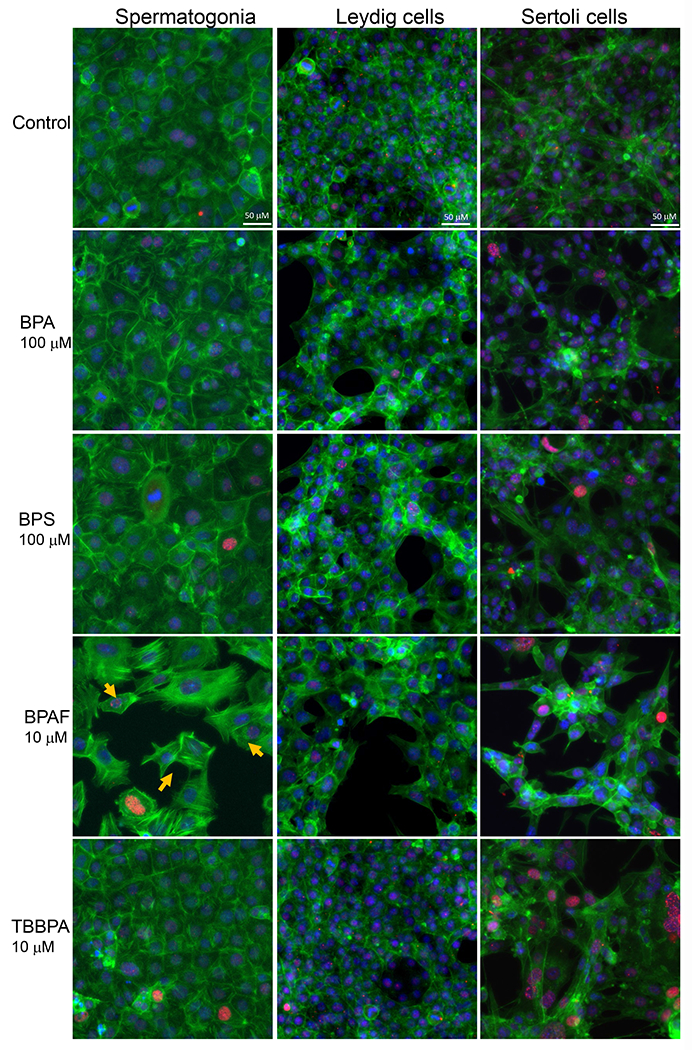
Image-based HCA was used to measure multiple parameters, such as cell number and nuclear morphology changes after 24 and 48h of BPA analogs treatment (O’Brien, Irwin et al. 2006, Yin, Siracusa et al. 2020). We treated three types of testicular cells with concentrations of 0, 5, 50 and 100 μM for BPA and BPS, and 0, 1, 5, and 10 μM for BPAF and TBBBPA based on a previous study (Liang, Yin et al. 2017, Yin, Siracusa et al. 2020). Statistical analysis with doses, cell types, and chemicals was conducted (ANOVA) at each time-point. Dose-dependent changes in cell number were observed in all three types of testicular cells at both 24 and 48 h (Figure 1). BPA induced a dose-dependent decrease in cell number in all testicular cell types. The reduction of cell number is most evident in C18-4 spermatogonial cells at 24h. However, TM4 Sertoli cells lost more cells than C18-4 or TM3 at 48h treatment of BPA. BPS also induced a dose-dependent decrease in cell number at 24h and 48h, and there was no difference among the three types of testicular cells at 24h. Significant reductions in TM3 and TM4 cells were observed compared to spermatogonia cells at 48h. BPAF induced the most considerable cell number decrease among all tested compounds at both time points. The spermatogonial cells decreased more at 5 μmM compared to other types of cells at both 24 and 48h. TBBPA treatment showed distinct responses among the three types of cells, and a dose-dependent decrease of cell number in both spermatogonia cell and TM4 cells was observed but not in TM3 cells. TBBPA induced a more significant reduction in cell number in spermatogonia cells than in TM4 cells. Figure 2A shows the representative nuclear morphology following 24 or 48h treatments. Significant induction of multinucleated cells was only observed in spermatogonial cells treated with BPAF at 5 μM or more. Quantification of nuclear morphology demonstrated exposure to BPA and its analogs induced significant changes in the nuclear area and nuclear shape measurement parameters, P2A, and LWR (Figure 2B–D). As reflected in the nuclear area, the nuclear size decreased at 48h as compared to 24h for the three types of cells (Figure 2B). BPA treatment at 100 μM reduced the size of C18-4 cells at both timepoints, reduced the size of TM3 at 100 μM and TM4 at 50 and 100 μM at 24 h, but increased the size of TM3 and TM4 at both 50 and 100 μM. BPS treatment only increased the nuclear size of C18-4 at 100 μM, TM3 at doses above 5 μM, and TM4 at 50 and 100 μM. BPAF treatment at 100μM increased the nuclear size of C18-4 cells at 48h, increased the size of TM3 at 100 μM and TM4 at 50 and 100 μM at 24 h, and 5, 50 and 100 mM at 48h. TBBPA treatment at 100 μM reduced the size of C18-4 cells at both time points, reduced the size of TM3 at 100 μM and TM4 at 50 and 100 μM at 24 h, increased the size of TM3 and TM4 at both 50, and 100 μM at 48h and increased the size of TM3 and TM4 at both 50, and 100 μM. Nuclear shape measurements included nuclear smoothness P2A and nuclear roundness LWR were quantified (Figure 2C–D). BPA treatment at 100 μM increased the P2A and LWR in C18-4 cells at 48h, reduced P2A and LWR in TM3 cells at 50, 100 μM at 24h and at 5, 50, 100 μM at 48h, and TM4 cell only at 24 h. BPS treatment reduced P2A and LWR in TM3 and TM4 cells at 50, 100 μM at both time points. BPAF treatment significantly increased P2A and LWR in C18-4 cells at 5 μM at 24h and 5 and 10 μM at 48 h, in TM3 at 100 μM at both timepoints, TM4 at 50 and 100 μM at 48h. TBBPA treatment significantly decreased LWR in C18-4 cell at 5 μM at 48 h and decreased P2A and LWR in TM3 at concentration 5 μM or over at both 24h and 48 h, and P2A in TM4 at 100 μM at both time points, TM4 at 50 and 100 μM at 48h. No changes in P2A in BPA, BPS, and TBBPA treatment in C18-4 cells at both time points were observed. Overall, BPA and its analogs have different in vitro adverse effects among these testicular cells and induce distinct changes in nuclear morphology. BPAF was the most cytotoxic compound in all three testicular cells and showed unique multinucleation in spermatogonial cells.
Figure 1. High-content image-based analysis (HCA) of single cell numbers changes in C18-4 spermatogonial cells, TM3 Leydig cells, and TM4 Sertoli cells.
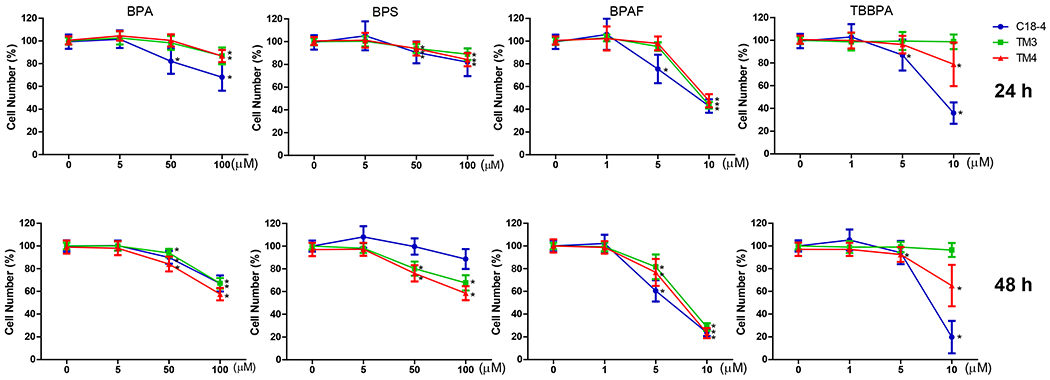
Cells were treated with various concentrations of BPA and BPS (5, 50, and 100 μM), and BPAF and TBBPA (1, 5, and 10 μM) for 24 and 48 h. Data were presented as mean ± SD, n = 10. Five bilogical replicates in two independent experiments were included. One-way ANOVA conducted statistical analysis followed by Tukey-Kramer multiple comparisons (*P < .05).
Figure 2. Effect of BPA and its analogs on nuclear morphology of C18-4 spermatogonial, TM3 Leydig and TM4 Sertoli cells.
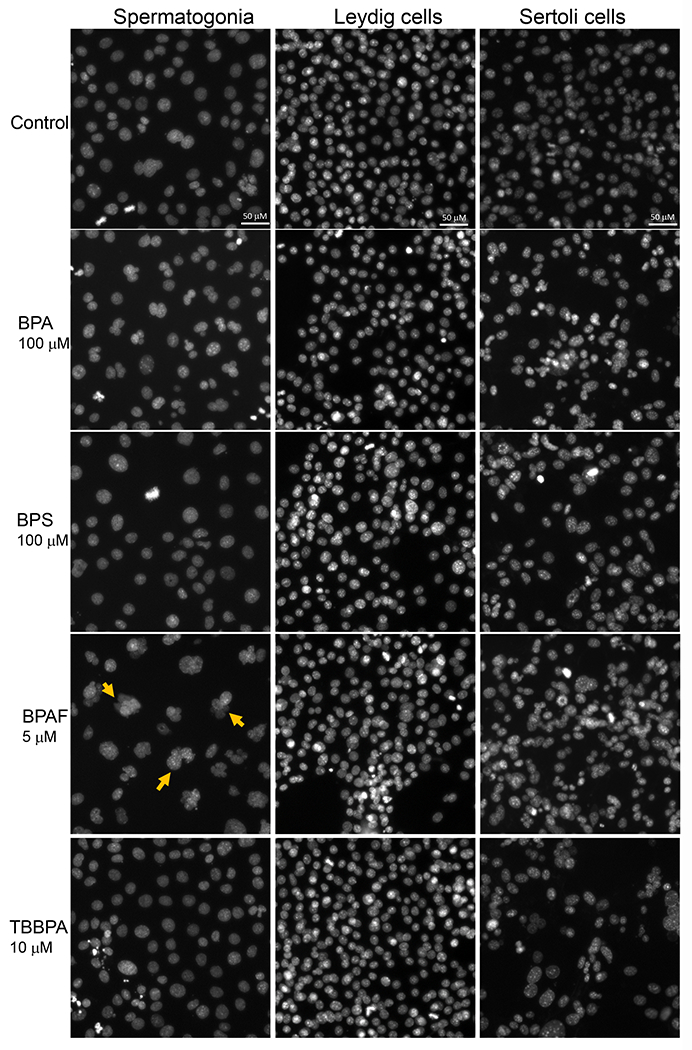
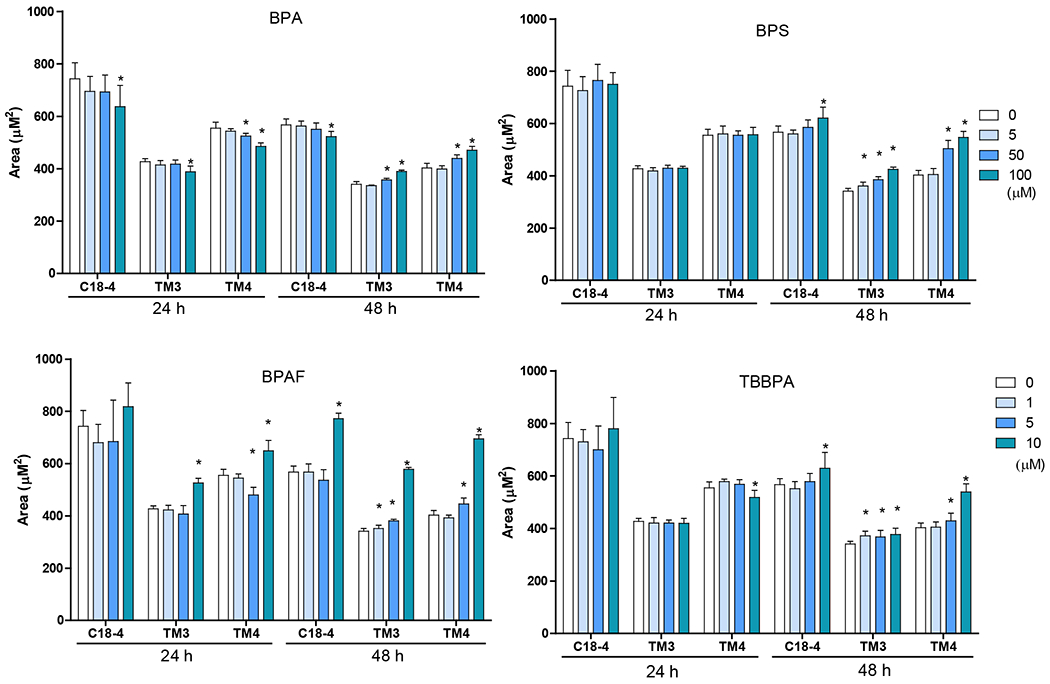
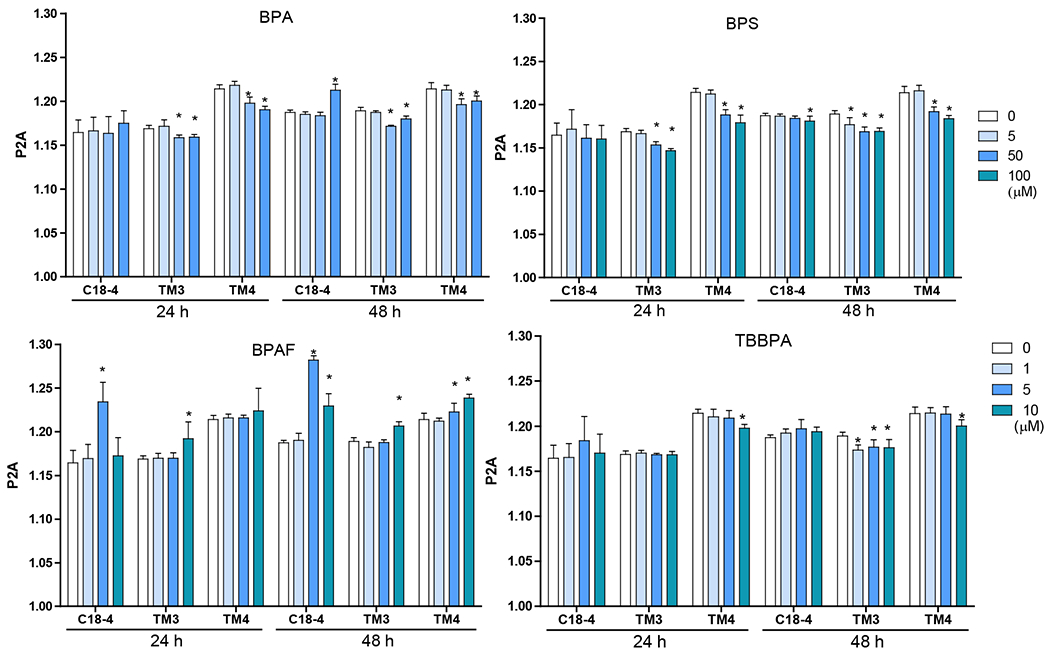
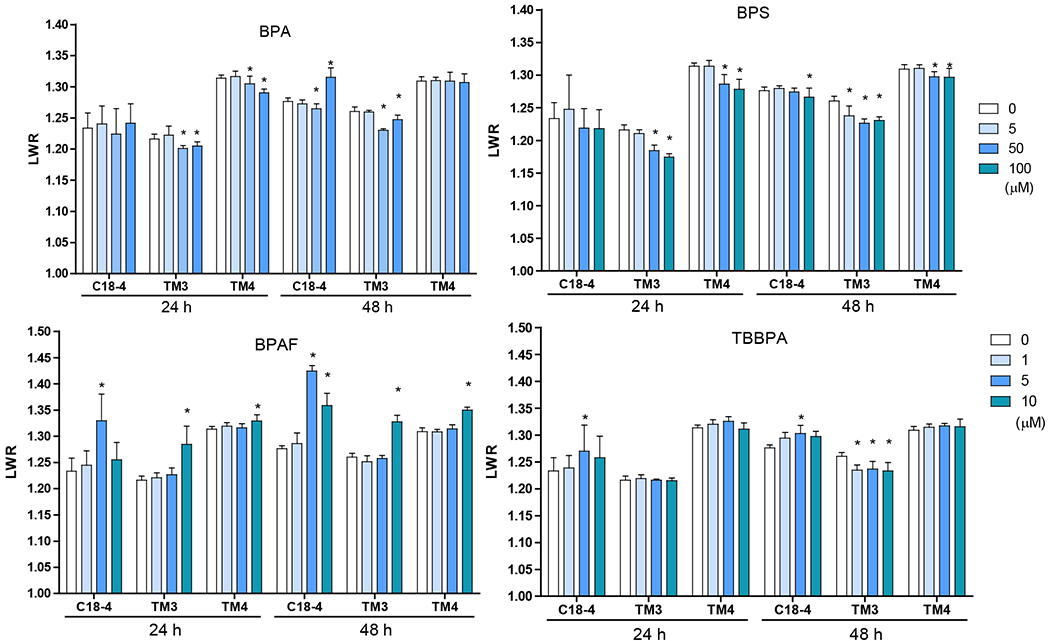
A. Representative images of nuclear morphology in spermatogonial, Leydig, and Sertoli cells treated with BPA, BPS, BPAF, and TBBPA for 24h. The nuclei were stained with Hoechst 33342, and images were automatically obtained with a 40× objective, 49 fields per well. Yellow arrows indicate the multinucleated cells. Scale bar = 50 μm. B–D, Quantification of absolute nuclear area (μm2) (B), nuclear shape parameters, including P2A for smoothness (C) and LWR for nuclear roundness (D) of the 3 testicular cell types treated with BPA and BPS (5, 50, and 100 μM) and BPAF and TBBPA (1, 5, and 10 μM) for 24 and 48 h. Data were presented as mean ± SD, n = 9. Three replicates in 3 separate experiments were included. Statistical analysis was conducted by 1-way ANOVA followed by Tukey-Kramer multiple comparisons (*P < .05).
BPA and its analogs altered cell cycle progression
The formation of the multinucleated cell was potentially due to asynchronous nuclear division and dysregulation of the cell cycle control network (Gladfelter, Hungerbuehler et al. 2006). We previously developed multiparametric HCA assays to measure cell cycle progression (Liang, et al., 2017). DNA total intensity histogram was constructed, and the proportion of cells in SubG1, G0/1, S, and G2/M phases of the cell cycle were quantified (Figure 3). Among the three types of testicular cells, alterations in the cell cycle were observed with all four treatments. Among the treated compounds, BPAF induced dose-dependent alterations in all three types of cells at doses of 5 and 10 μM at both time points. In C18-4 cells, both BPA at 100 μM and TBBPA at 10 μM, significantly increased cells in G2/M, and reduced cells in G0/1 and S phases at both 24 and 48 h time points. In TM3 Leydig cells (Figure 3), significant increases in cells in the G0/1 phase and decreases in the G2/M phase were observed in the BPA and BPS treatment at 50 and 100 μM at both 24 and 48 h. In comparison, reductions of cells in the G0/1 phase and increases of cells in the G2/M phase were observed in BPAF treatment at a dose of 10 μM for 24 and 48 h. A significant decrease of cells in the S phase was observed in BPA treatment at 100 μM for 24 h, 50 and 100 μM for 48 h, BPS treatment at 50 and 100 μM for 24 and 48 h, and TBBPA treatment at 10 μM for 24 h. In contrast, accumulation of cells in the S phase was observed in BPAF treatment at a dose of 10 μM for 48 h, accompanied by a significant increase of cells in the sub-G1 phase. In TM4 Sertoli cells (Figure 3), significant decreases in cell proportion in G0/1 phase and increases in the proportion of cells in the G2/M phase were observed in BPA and BPS treatment at 100 μM for 24 h, 50, and 100 μM for 48 h BPAF treatment at 10 μM for 24 h, 5 and 10 μM for 48 h, and TBBPA treatment at 10 μM for 24 and 48 h. Significant decreases in the proportion of cells in S phase were observed in BPA and BPS treatment at 100 μM for 24 h, 50 and 100 μM for 48 h, and TBBPA treatment at 10 μM for 48 h. In contrast, the accumulation of cells in S phase was observed in BPAF treatment at a dose of 10 μM. Increases of cells in the sub-G1 phase were observed in BPAF treatment at 5 μM for 24 and 48 h, and TBBPA treated at 5 and 10 μM for 24 h. Therefore, BPA and its analogs caused differential cell cycle arrest among different types of testicular cells.
Figure 3. Cell cycle analysis of C18-4 spermatogonial cells, TM3 Leydig cells, and TM4 Sertoli cells.
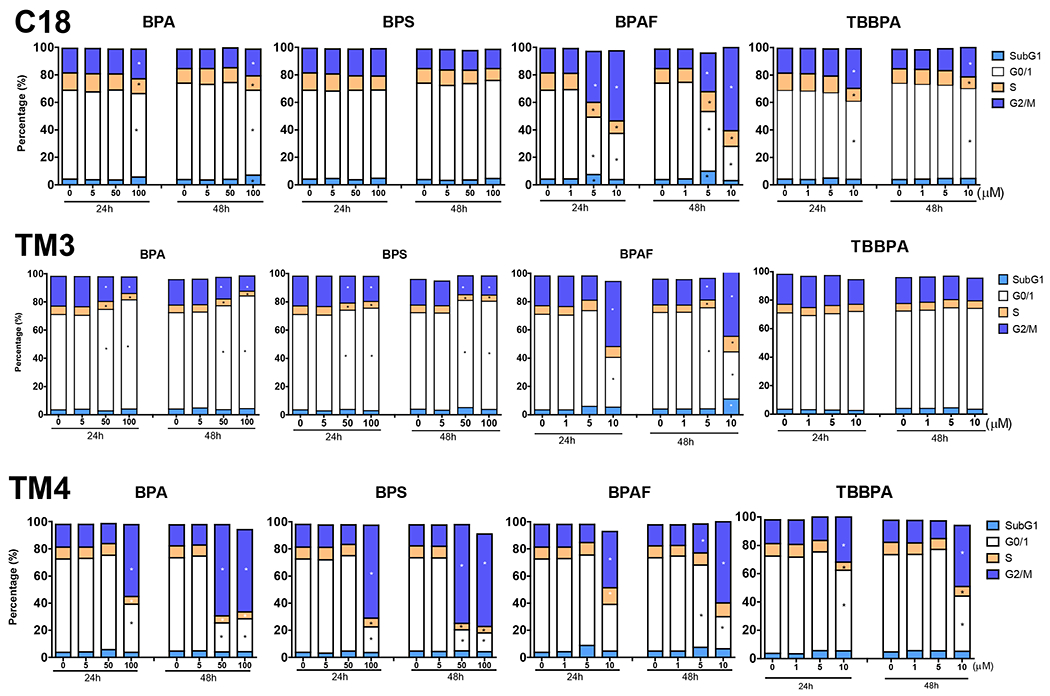
Cells were treated with various concentrations of BPA and BPS (5, 50, and 100 μM), and BPAF and TBBPA (1, 5, and 10 μM) for 24 and 48 h. The percentage of cells in each cell cycle stage was determined by creating gating parameters on histograms of total DNA intensity per cell (by Hoechst 33342 staining) per well at sub-G1, G0/G1, S, and G2M phases using Python script.
Data were presented as mean ± SD, n = 10. Five replicates in two independent experiments were included. One-way ANOVA conducted statistical analysis followed by Tukey-Kramer multiple comparisons (*P < .05).
BPA and its analogs induced alterations in DNA synthesis
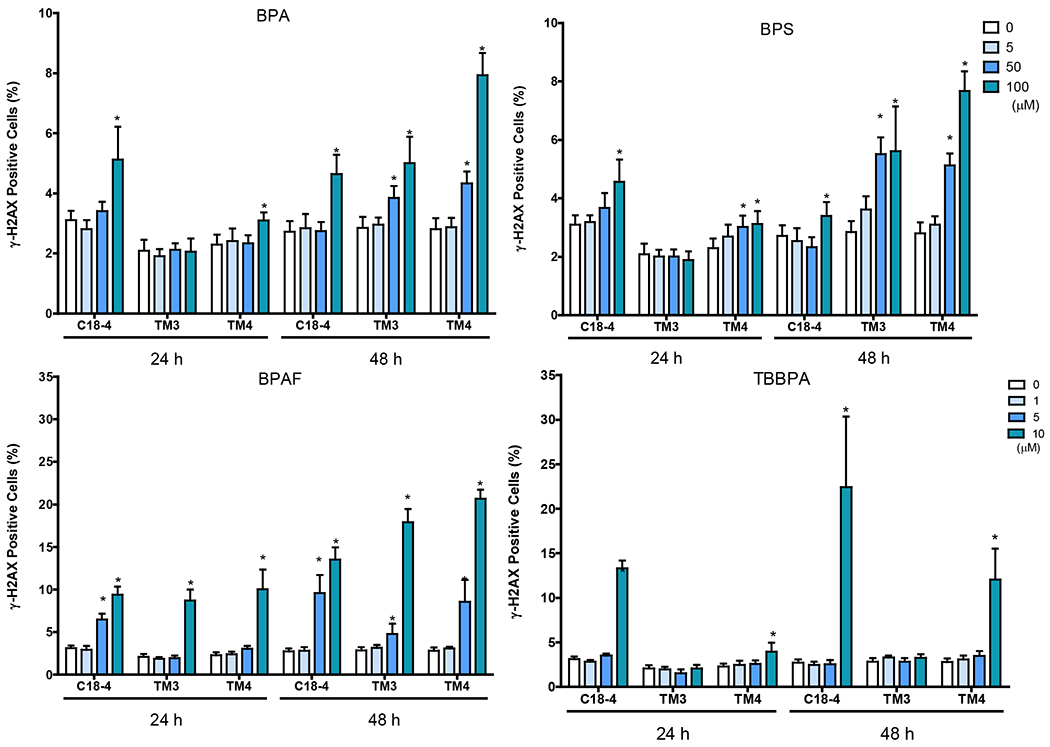
BrdU labeling is a marker of DNA synthesis and not S-phase of the cell cycle or cell division. Representative morphology (Figure 4A) at 24h and HCA quantification (Figure 4B) of BrdU incorporation at both time points revealed differential effects of BPA and its analog in these cells. The percentage of BrdU positive cells was 40%, 32%, 33% in C18-4, TM3 and TM4 cells individually at 24h and 30%, 20%, 15% in C18-4, TM3 and TM4 cells, respectively at 48h. BPA dose-dependently reduced the number of BrdU positive cells in C18-4, TM3 and TM4 at 24h, significantly at 50 and 100 μM. A significant decrease in the number of BrdU positive cells at 50 and 100 μM in C18-4 cells and TM3 at 100 μM at 48h was observed. BPS dose-dependently reduced the number of BrdU positive cells in C18-4, TM3 and TM4 at 24h, significantly at 50 and 100 μM. A significant decrease in the percentage of BrdU positive cells at 50 and 100 μM in C18-4 cells, TM3 at 100 μM, and TM4 at 100 μM at 48h were observed. BPS dose-dependently reduced DNA synthesis in all three types of testicular cells at 24h, significantly at 50 and 100 μM. At 48h, significant reductions were observed in C18-4 and TM3 at 50 and 100 μM and TM4 at 100 μM for 48 h. TBBPA reduced the percentage of BrdU positive cells in spermatogonial C18-4 cells at 5 and 10 μM at 24h and 10 μM at 48h. Interestingly, a decrease in the number of BrdU positive cells was only observed in TM4 Sertoli cells at 10 μM at 24h. In contrast to BPA, BPS and TBBPA, BPAF induced a different change pattern of in BrdU synthesis. In C18-4 spermatogonia cells, a dose-dependent decrease of BrdU positive cells was observed, significantly at 10 μM at 24h. However, a dose-dependent increase of BrdU positive cells was observed at 48, significantly at both 5 and 10 μM. Decreases of BrdU positive cells in TM3 and TM4 at 10 μM were observed at 24h; however, a significant increase of BrdU positive cells at 10 μM TM3 and TM4 cells at 48 were found.
Figure 4. Effect of BPA and its analogs on DNA synthesis of C18-4 spermatogonial, TM3 Leydig and TM4 Sertoli cells.
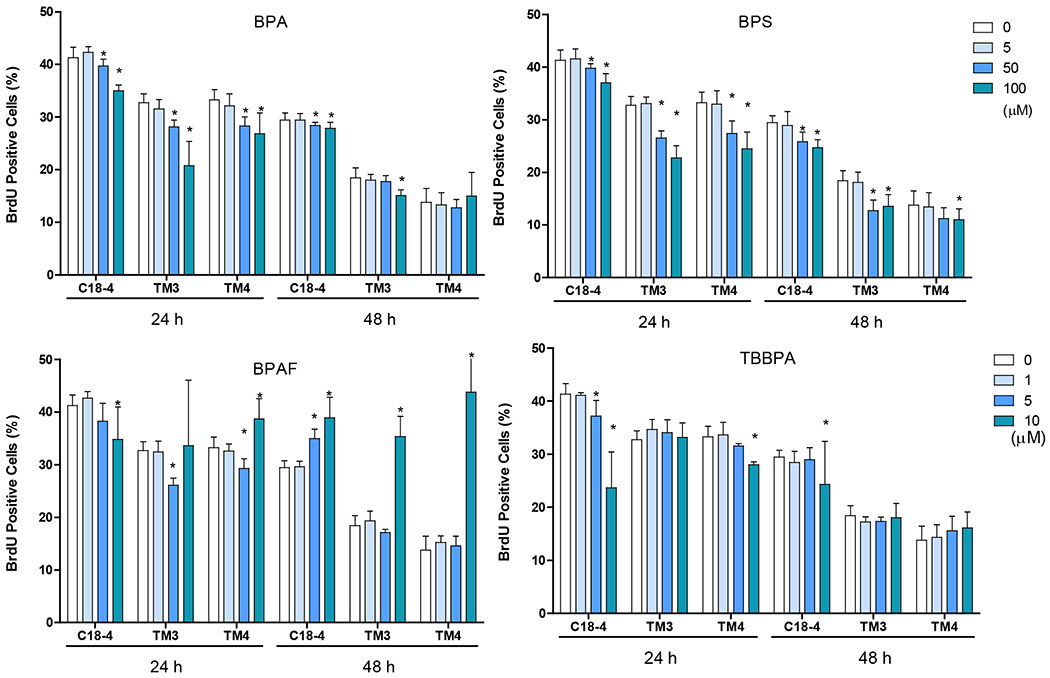
A shows the representative images of BrdU incorporation in spermatogonial (C18-4), Leydig (TM3), and Sertoli (TM4) cells treated with BPA, BPS, BPAF, and TBBPA at various concentrations for 24h. The nuclei were stained with Hoechst 33342, BrdU positive labeling was pseudo-colored in green. Images were automatically obtained by ArrayScan HCA Reader with a 40× objective, 49 fields per well. Scale bar = 50 μm. B Quantification of BrdU incorporation (% BrdU positive cells vs Control) in the 3 testicular cell types treated with BPA and BPS (5, 50, and 100 μM) and BPAF and TBBPA (1, 5, and 10 μM) for 24 and 48 h. Single cell BrdU labeling data were quantified by HCS Studio™ 2.0 TargetActivation BioApplication. Data were presented as mean ± SD, n = 9. Three replicates in 3 separate experiments were included. Statistical analysis was conducted by 1-way ANOVA followed by Tukey-Kramer multiple comparisons (*P < .05).
BPA and its analogs perturbed cytoskeleton and induced early DNA damage responses
Dose-dependent alterations in the cytoskeleton were observed in the treatment with BPA and its analogs (Figure 5). BPA induced dose-dependently increase of F-actin total intensity in all three types of cells, with statistical significance in C18-4 spermatogonial cells at 100 μM at 24h and 50 and 100 μM at 48h, TM3 and TM4 at 50 and 100 μM at both timepoints (Figure 5). BPS significantly increased F-actin total intensity in spermatogonial C18-4 cells at 100 μM at both timepoints, in TM3 Leydig cells at 50 and 100 μM at 24h and at 5 μM or over at 48h, in TM4 Sertoli cells at 50 and 100 μM at both timepoints. BPAF significantly increased F-actin total intensity observed in spermatogonial cells at 10 μM for both timepoints, in TM3 Leydig cells at 5 and 10 μM at 24 h and at 1 or over at 48 h, in TM4 Sertoli cells at 10 μM at 24h and at 5 and 10 μM at 48 h. TBBPA treatment caused F-actin total intensity induction observed in C18-4 spermatogonial and TM4 Sertoli cells at 10 μM for both timepoints, and in TM3 Leydig cells at 1, 5, and 10 μM at 48 h.
Figure 5. BPA and its analogs differentially perturbed cytoskeleton (F-actin) and induced early DNA damage responses (γH2AX) in spermatogonial (C18-4), Leydig (TM3), and Sertoli cells (TM4).
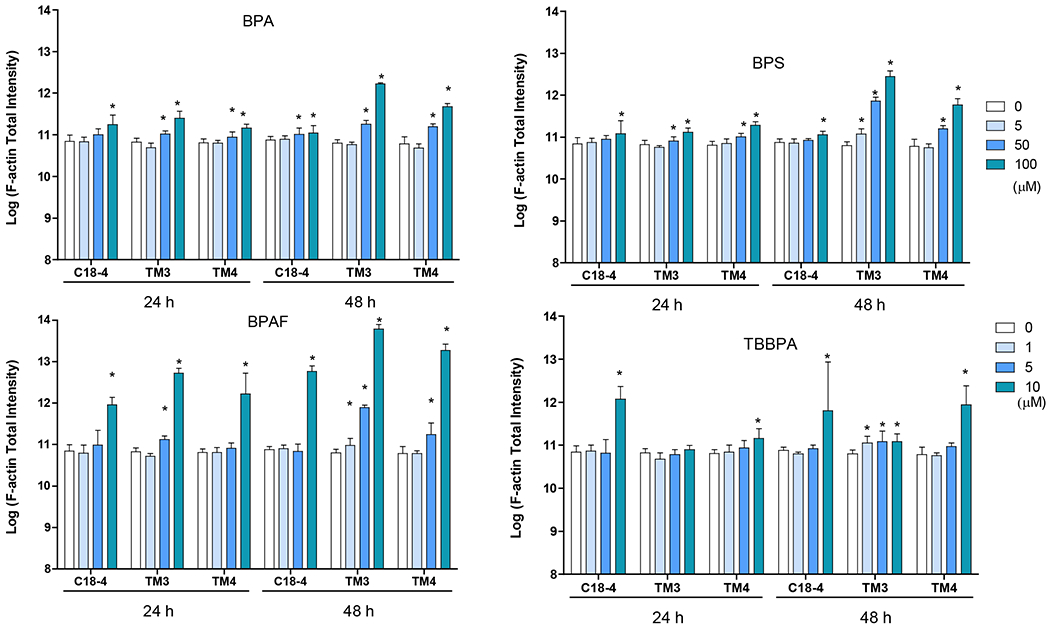
Cells treated with vehicle (0.01% DMSO) were used as negative controls. The nuclei were stained with Hoechst 33342 (blue), F-actin with Phalloidin staining (green), and γ-H2AX with a combination of primary phosphorylated γ–H2AX and secondary Dylight 650 conjugated antibody (red). A shows the representative images of cells treated with BPA and BPS (100 μM), BPAF and TBBPA (10 μM) for 24 h. Arrows indicate dot-like structures. Scale bar = 50 μm. B-C shows the single-cell quantification of the log-transformed total intensity of F-actin (B) and positive γ-H2AX cells (C) of the three testicular cell types treated with various BPA, BPS, BPAF and TBBPA concentrations for 24 and 48h. Data were presented as mean ± SD, n = 6. Three replicates in two separate experiments were included. Statistical analysis was conducted by 1-way ANOVA followed by Tukey-Kramer multiple comparisons (*P < 0.05).
BPA showed significant increases in γ-H2AX positive cells at 100 μM for 24 and 48 h in C18-4 spermatogonial cells, 24 h in TM4 Sertoli cells, 50 and 100 μM for 48 h in TM3 Leydig and TM4 Sertoli cells (Figure 5B). BPS showed accumulation of γ-H2AX positive cells at 100 μM for 24 and 48 h in C18-4 spermatogonial cells, 50 and 100 μM for 24 and 48 h in TM4 Sertoli cells, and 48 h in TM3 Leydig cells. BPAF significantly induced γ-H2AX positive cells at 10 μM for 24 h in TM3 Leydig and TM4 Sertoli cells, 5 and 10 μM for 24 and 48 h in C18-4, and 48 h at TM3 Leydig and TM4 Sertoli cells. TBBPA significantly increased the number of γ-H2AX positive cells at 10 μM for 24 and 48 h in C18-4 spermatogonial and TM4 Sertoli cells.
BPA and its analogs differentially induced DNA methylation and activated autophagy
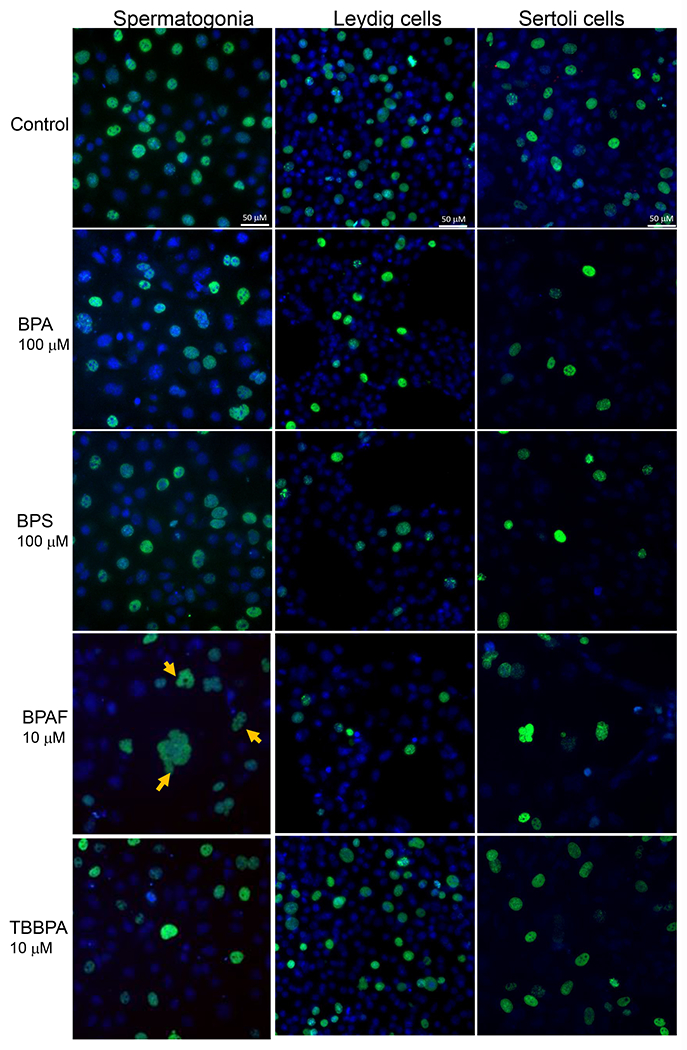
We used Methyl-CpG binding domain 1 (MBD1), a transcriptional repressor that binds to single methylated CpGs as a methylation marker, to quantify the methylation status of cells treated with BPA and its analogs. Figure 6A shows the representative images for MBD1 and LC3B co-staining in three cell types with various treatment conditions after 24h exposure. As shown in Figure 6B, BPA significantly increased MBD1 total intensity at 50 and 100 μM for 24 h in C18-4 spermatogonial, 48 h in TM4 Sertoli cells, 100 μM for 24 h in TM3 Leydig and TM4 Sertoli cells, and 48 h in C18-4 spermatogonial and TM3 Leydig cells.BPS significantly induced MBD1 total intensity at 100 μM in spermatogonial cells, 50 and 100 μM in Leydig and Sertoli cells for 24 and 48 h. BPAF significantly induced MBD1 total intensity at a dose of 10 μM for 24 h, 5 and 10 μM for 48 h in all three cell types. TBBPA increased MBD1 total intensity at 10 μM for 24 h and 48 h in spermatogonial cells and 48 h in Sertoli cells.
Figure 6. BPA and its analogs differentially induced DNA methylation and activated autophagy in spermatogonial (C18-4), Leydig (TM3), and Sertoli cells (TM4).
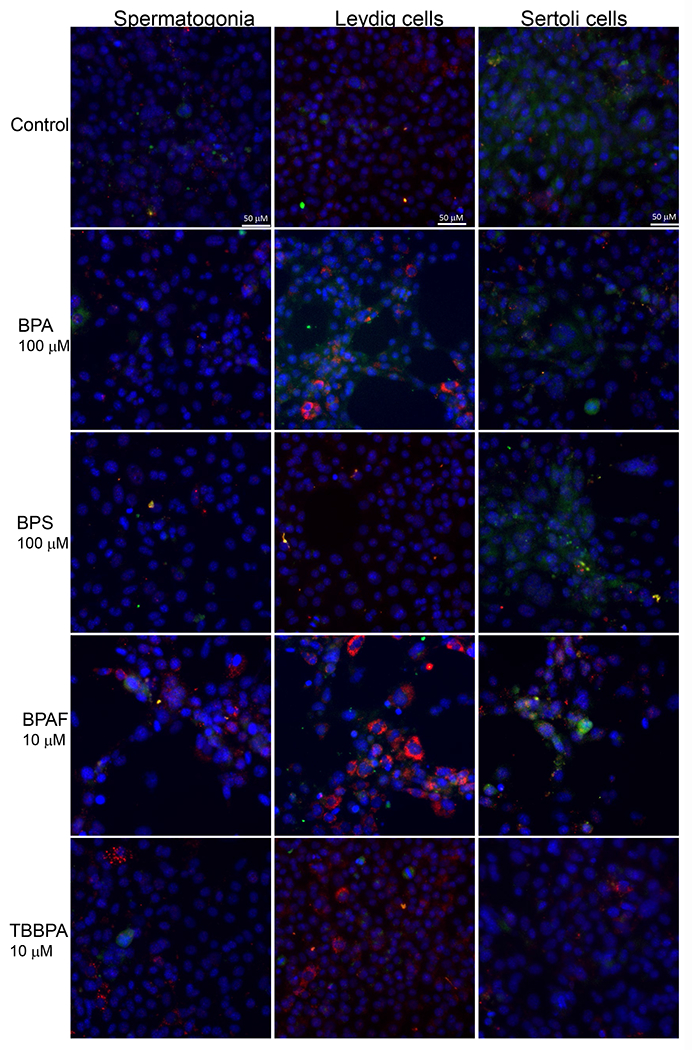
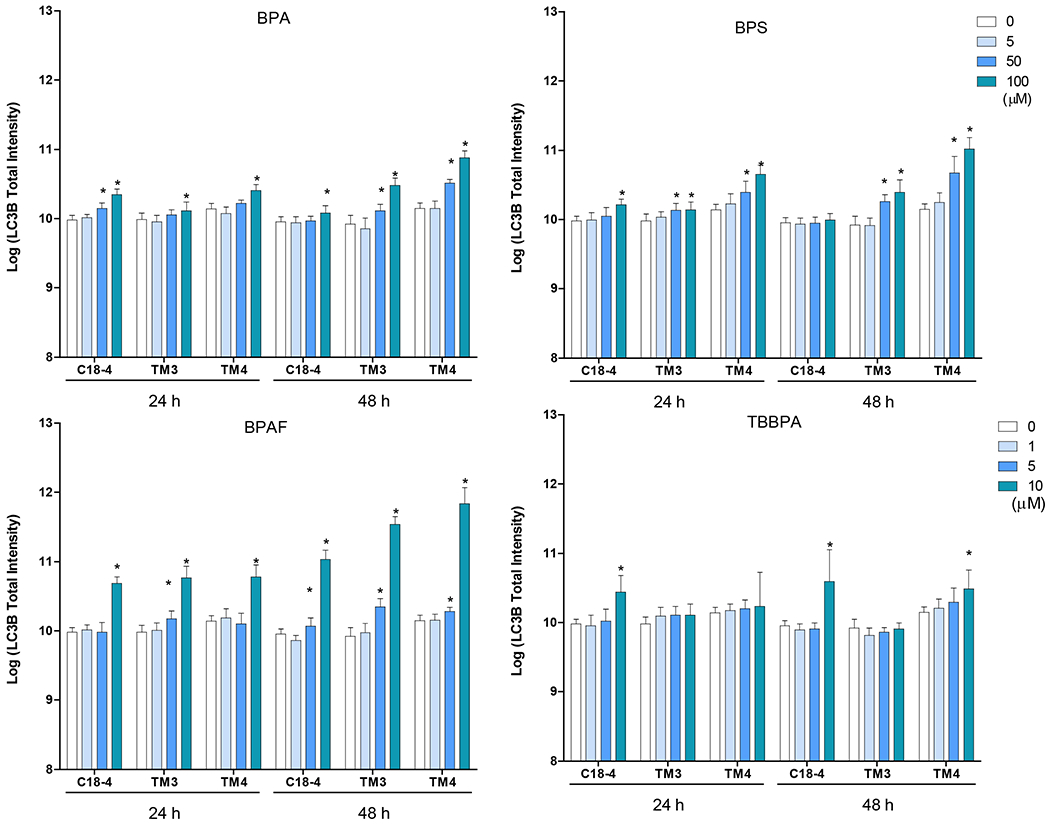
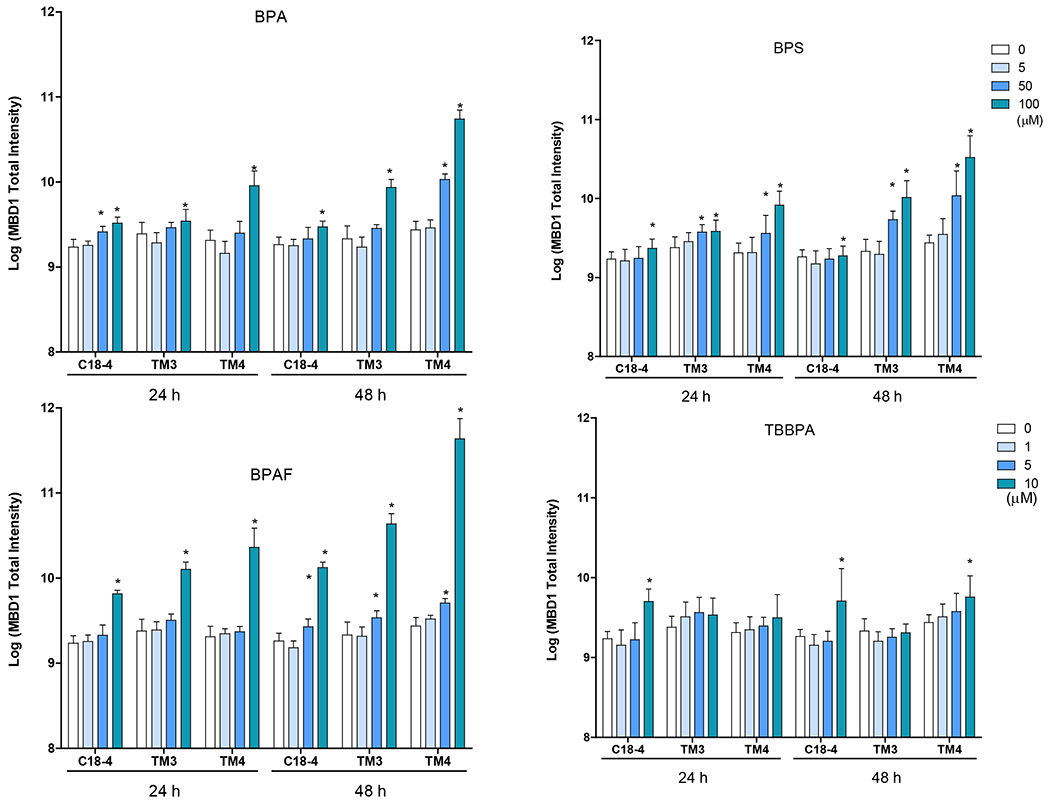
Cells treated with vehicle (0.01% DMSO) were used as negative controls. A shows the representative images of cells treated with BPA and BPS (100 μM), BPAF and TBBPA (10 μM) for 24 h. The nuclei were stained with Hoechst 33342 (blue). Double immunofluorescence in the cell lines using the LC-3B (red) and MBD-1 (green) antibodies. Noted a clearly distinct expression of LC3A in red vacuoles with perinuclear/nuclear localization and of MBD-1 green that has a diffuse, throughout the cytoplasm, localization. Scale bar = 50 μm. B-C shows the single-cell quantification of the log-transformed total intensity of MBD1 (B) and LC3B (C) immuno-labeling of the three testicular cell types treated with various BPA, BPS, BPAF and TBBPA concentrations for 24 and 48h. Data were presented as mean ± SD, n = 6. Three replicates in two separate experiments were included. Statistical analysis was conducted by 1-way ANOVA followed by Tukey-Kramer multiple comparisons (*P < 0.05).
Autophagy plays an essential role in regulating cell survival by degrading and recycling cytoplasmic components to maintain cellular homeostasis (Baehrecke 2005, Gallagher, Williamson et al. 2016, Hargarten and Williamson 2018). Here we employed an autophagosome membrane protein LC3B to monitor autophagy activity, as shown in Figures 6A and 6C. BPA significantly increased LC3B total intensity at 50 and 100 μM for 24 h in spermatogonial cells, 48 h for Leydig and Sertoli cells, 100 μM for 24 h in Leydig and Sertoli cells, and 48 h in spermatogonial cells. BPS significantly induced LC3B total intensity at 100 μM for 24 h in spermatogonial cells, and 50 and 100 μM for 24 and 48 h in Leydig and Sertoli cells. BPAF induced LC3B expression at 10 μM for 24 h in spermatogonial and Sertoli cells, 5 and 10 μM for 24 h in Leydig cell, and 48 h for all cell types. TBBPA significantly increased LC3B level at 10 μM for 24 h in spermatogonial cells, and 48 h in spermatogonial and Sertoli cells.
BPA and its analogs demonstrated differential toxicity responses
Figure 7A illustrates the differential toxicity spectrum profile of tested compounds in spermatogonial, Sertoli, and Leydig cells (cell types with the indicated concentration of tested chemicals on the left). The HCA spectrum reveals the dynamic, chemical-specific, dose-dependent changes of each HCA parameter. Figure 7A compares multiplexed HCA parameters among BPA and its analogs from three types of testicular cells. Each chemical displayed a unique dose-dependent profile within each type of cell. The aptitudes of these spectra were more prominent at the 48h than at the 24h. Combined with the HCA index (Figure 7B), we found all three types of cells showed the highest response to BPAF at 10 μM across all variables measured. BPAF targeted spermatogonial cell more significantly (C18) at 5 μM. BPS more likely targeted Sertoli cell (TM4) and Leydig cell (TM3) and less at spermatogonia cells. TBBPA targeted spermatogonia, Sertoli cells, and less at TM3 cells. BPA is mainly targeted at TM4, followed by TM3 cells, and less at spermatogonial cells. Finally, we examined the statistical correlations among these HCA parameters, as shown in Table 1. These correlation-coefficient matrices indicate cell type-specific and chemical-specific. For example, cell viability was positively correlated with BrdU and negatively correlated in MBD1, LC3B, and H2AX in BPA, BPS, TBBPA treated C18 cells except for BPAF treatment in which cell viability was negatively associated with BrdU. We also observed that BPAF was negatively associated with BrdU in TM3 and TM4 cells, different from other treatments. Also, we observed cell viability negatively associated with the cell cycle G2M phase in all treatments in C18-4 and TM4 cells but positively associated with G2M in BPA, BPS, and TBBPA in TM3 cells, not BPAF. Further studies would be needed to clarify the mechanisms involved in differential sensitivity among three cell types in response to BPA and its analogs, especially BPAF.
Figure 7. Differential toxic responses of BPA and its analogs on C18 spermatogonial, TM3 Leydig, and TM4 Sertoli cells.
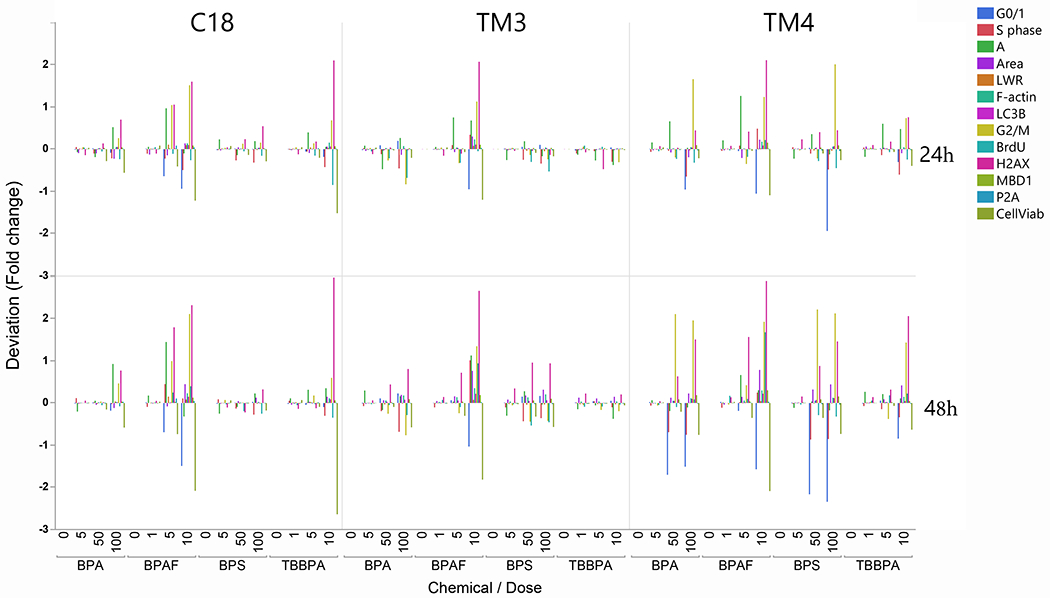
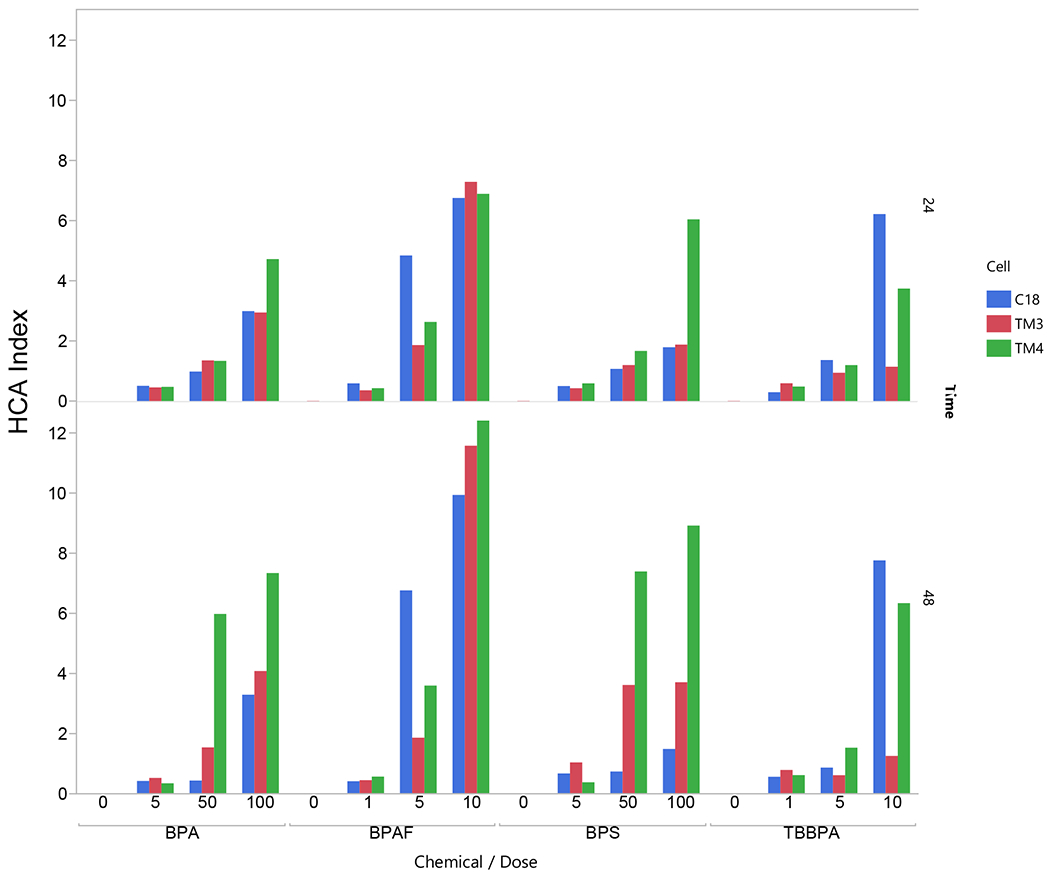
A. HCA toxicity spectrums of BPA and its analogs. Multiplexed HCA parameters among BPA and its analogs among three types of testicular cells. The deviation (fold changer from the Control) of each assay, including cell number, nuclear area, nuclear shape measurements (P2A and LWR), DNA synthesis (BrdU positive cells), cell cycle phases (cell population in subG1 phase, G0/G1 phase, S phase, and G2/M phase), DNA damage response (γ-H2AX positive cells), F-actin total intensity, MBD1 total intensity and LC3B total intensity were calculated, log2-transformed and represented as color-spectrums (by cellular functions). Each chemical displayed a unique dose-dependent profile within each type of cell. The aptitudes of these spectra were more prominent at the 48h time point than at the 24h time point. B. HCA index of BPA and its analogs. HCA index is calculated as the sum of the absolute value of each log2 transformed fold change, representing each chemical’s accumulative toxicity.
Table 1.
Corelation matrix of 13 parameters from the high conetent analsysis in spermatogonial cells, TM4 Setoli cells and TM3Leydig cells. The correlations are estimated by Row-wise method.
| C18 | TM3 | TM4 | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CellViab | G0/1 | S phase | G2/M | A | Area | P2A | LWR | F-actin | BrdU | MBD1 | LC3B | H2AX | CellViab | G0/1 | S phase | G2/M | A | Area | P2A | LWR | F-actin | BrdU | MBD1 | LC3B | H2AX | CellViab | G0/1 | S phase | G2/M | A | Area | P2A | LWR | F-actin | BrdU | MBD1 | LC3B | H2AX | ||||||
| BPA | CellViab | 1 | 0.79 | 0.73 | −0.9 | −0.8 | 0.82 | −0.8 | −0.6 | −0.9 | 0.84 | −1 | −0.83 | −0.95 | CellViab | 1 | −0.9 | 0.96 | 0.83 | −0.3 | −0.6 | 0.44 | 0.37 | −1 | 0.51 | −0.97 | −0.96 | −0.84 | CellViab | 1 | 0.79 | 0.82 | −0.8 | 0.42 | −0.6 | 0.5 | 0.17 | −1 | −0.3 | −1 | −0.97 | −0.99 | ||
| G0/1 | 0.79 | 1 | 0.3 | −1 | −1 | 0.56 | −1 | −0.9 | −0.5 | 0.5 | −0.6 | −0.47 | −0.89 | G0/1 | −0.86 | 1 | −1 | −1 | 0.06 | 0.32 | −0.7 | −0.7 | 0.92 | −0.8 | 0.87 | 0.85 | 0.67 | G0/1 | 0.79 | 1 | 0.97 | −1 | 0.58 | −0.5 | 0.69 | 0.22 | −0.9 | −0 | −0.9 | −0.9 | −0.85 | |||
| S phase | 0.73 | 0.3 | 1 | −0.4 | −0.4 | 0.8 | −0.4 | −0.2 | −0.9 | 0.87 | −0.9 | −0.93 | −0.63 | S phase | 0.96 | −1 | 1 | 0.95 | −0.3 | −0.4 | 0.57 | 0.47 | −1 | 0.71 | −0.92 | −0.89 | −0.74 | S phase | 0.82 | 0.97 | 1 | −1 | 0.52 | −0.4 | 0.75 | 0.41 | −0.9 | 0.12 | −0.9 | −0.9 | −0.86 | |||
| G2/M | −0.9 | −1 | −0.4 | 1 | 0.97 | −0.7 | 0.96 | 0.87 | 0.61 | −0.6 | 0.72 | 0.55 | 0.92 | G2/M | 0.83 | −1 | 0.95 | 1 | −0.2 | −0.2 | 0.62 | 0.5 | −0.9 | 0.89 | −0.8 | −0.76 | −0.53 | G2/M | −0.8 | −1 | −1 | 1 | −0.6 | 0.42 | −0.71 | −0.3 | 0.86 | −0.1 | 0.89 | 0.88 | 0.83 | |||
| A | −0.8 | −1 | −0.4 | 0.97 | 1 | −0.6 | 0.93 | 0.85 | 0.6 | −0.6 | 0.68 | 0.52 | 0.9 | A | −0.28 | 0.06 | −0.3 | −0.2 | 1 | −0.1 | 0.34 | 0.43 | 0.15 | −0.3 | 0.07 | 0.05 | 0.03 | A | 0.42 | 0.58 | 0.52 | −0.6 | 1 | −0.5 | −0.05 | −0.2 | −0.4 | −0.5 | −0.4 | −0.42 | −0.46 | |||
| Area | 0.82 | 0.56 | 0.8 | −0.7 | −0.6 | 1 | −0.6 | −0.5 | −0.9 | 0.84 | −0.9 | −0.94 | −0.73 | Area | −0.64 | 0.32 | −0.4 | −0.2 | −0.1 | 1 | −0.3 | −0.3 | 0.63 | 0.31 | 0.7 | 0.75 | 0.9 | Area | −0.6 | −0.5 | −0.4 | 0.42 | −0.5 | 1 | 0.09 | 0.62 | 0.56 | 0.79 | 0.51 | 0.61 | 0.68 | |||
| P2A | −0.8 | −1 | −0.4 | 0.96 | 0.93 | −0.6 | 1 | 0.95 | 0.49 | −0.5 | 0.66 | 0.53 | 0.87 | P2A | 0.44 | −0.7 | 0.57 | 0.62 | 0.34 | −0.3 | 1 | 0.99 | −0.6 | 0.43 | −0.54 | −0.57 | −0.58 | P2A | 0.5 | 0.69 | 0.75 | −0.7 | −0.1 | 0.09 | 1 | 0.77 | −0.7 | 0.63 | −0.7 | −0.65 | −0.5 | |||
| LWR | −0.6 | −0.9 | −0.2 | 0.87 | 0.85 | −0.5 | 0.95 | 1 | 0.25 | −0.3 | 0.43 | 0.34 | 0.69 | LWR | 0.37 | −0.7 | 0.47 | 0.5 | 0.43 | −0.3 | 0.99 | 1 | −0.5 | 0.28 | −0.49 | −0.54 | −0.59 | LWR | 0.17 | 0.22 | 0.41 | −0.3 | −0.2 | 0.62 | 0.77 | 1 | −0.3 | 0.82 | −0.4 | −0.24 | −0.12 | |||
| F-actin | −0.9 | −0.5 | −0.9 | 0.61 | 0.6 | −0.9 | 0.49 | 0.25 | 1 | −1 | 0.93 | 0.93 | 0.81 | F-actin | −0.98 | 0.92 | −1 | −0.9 | 0.15 | 0.63 | −0.6 | −0.5 | 1 | −0.5 | 0.98 | 0.98 | 0.87 | F-actin | −1 | −0.9 | −0.9 | 0.86 | −0.4 | 0.56 | −0.66 | −0.3 | 1 | 0.1 | 0.97 | 0.99 | 0.97 | |||
| BrdU | 0.84 | 0.5 | 0.87 | −0.6 | −0.6 | 0.84 | −0.5 | −0.3 | −1 | 1 | −0.9 | −0.92 | −0.81 | BrdU | 0.51 | −0.8 | 0.71 | 0.89 | −0.3 | 0.31 | 0.43 | 0.28 | −0.5 | 1 | −0.45 | −0.39 | −0.08 | BrdU | −0.3 | −0 | 0.12 | −0.1 | −0.5 | 0.79 | 0.63 | 0.82 | 0.1 | 1 | 0.07 | 0.15 | 0.28 | |||
| MBD1 | −1 | −0.6 | −0.9 | 0.72 | 0.68 | −0.9 | 0.66 | 0.43 | 0.93 | −0.9 | 1 | 0.93 | 0.86 | MBD1 | −0.97 | 0.87 | −0.9 | −0.8 | 0.07 | 0.7 | −0.5 | −0.5 | 0.98 | −0.5 | 1 | 0.99 | 0.88 | MBD1 | −1 | −0.9 | −0.9 | 0.89 | −0.4 | 0.51 | −0.69 | −0.4 | 0.97 | 0.07 | 1 | 0.99 | 0.96 | |||
| LC3B | −0.8 | −0.5 | −0.9 | 0.55 | 0.52 | −0.9 | 0.53 | 0.34 | 0.93 | −0.9 | 0.93 | 1 | 0.75 | LC3B | −0.96 | 0.85 | −0.9 | −0.8 | 0.05 | 0.75 | −0.6 | −0.5 | 0.98 | −0.4 | 0.99 | 1 | 0.92 | LC3B | −1 | −0.9 | −0.9 | 0.88 | −0.4 | 0.61 | −0.65 | −0.2 | 0.99 | 0.15 | 0.99 | 1 | 0.98 | |||
| H2AX | −1 | −0.9 | −0.6 | 0.92 | 0.9 | −0.7 | 0.87 | 0.69 | 0.81 | −0.8 | 0.86 | 0.75 | 1 | H2AX | −0.84 | 0.67 | −0.7 | −0.5 | 0.03 | 0.9 | −0.6 | −0.6 | 0.87 | −0.1 | 0.88 | 0.92 | 1 | H2AX | −1 | −0.9 | −0.9 | 0.83 | −0.5 | 0.68 | −0.5 | −0.1 | 0.97 | 0.28 | 0.96 | 0.98 | 1 | |||
| CellViab | G0/1 | S phase | G2/M | A | Area | P2A | LWR | F-actin | BrdU | MBD1 | LC3B | H2AX | CellViab | G0/1 | S phase | G2/M | A | Area | P2A | LWR | F-actin | BrdU | MBD1 | LC3B | H2AX | CellViab | G0/1 | S phase | G2/M | A | Area | P2A | LWR | F-actin | BrdU | MBD1 | LC3B | H2AX | ||||||
| BPAF | CellViab | 1 | 0.98 | −0.1 | −1 | 0.17 | −0.8 | −0.5 | −0.6 | −0.9 | −0.4 | −1 | −0.95 | −0.93 | CellViab | 1 | 0.96 | −0.9 | −0.9 | −0.8 | −1 | −0.9 | −0.9 | −1 | −0.8 | −0.99 | −0.98 | −0.99 | CellViab | 1 | 0.99 | −0.7 | −1 | 0.07 | −1 | −0.97 | −1 | −1 | −0.9 | −1 | −0.99 | −0.94 | ||
| G0/1 | 0.98 | 1 | −0 | −1 | 0 | −0.7 | −0.6 | −0.7 | −0.9 | −0.4 | −0.9 | −0.88 | −0.97 | G0/1 | 0.96 | 1 | −0.9 | −1 | −0.8 | −0.9 | −1 | −1 | −0.9 | −0.7 | −0.94 | −0.89 | −0.95 | G0/1 | 0.99 | 1 | −0.8 | −1 | 0.11 | −0.9 | −0.94 | −1 | −1 | −0.9 | −1 | −0.97 | −0.93 | |||
| S phase | −0.1 | −0 | 1 | −0 | 0.46 | 0.01 | 0.32 | 0.22 | −0.2 | 0.83 | −0.1 | −0.11 | 0.18 | S phase | −0.94 | −0.9 | 1 | 0.86 | 0.87 | 0.94 | 0.77 | 0.85 | 0.9 | 0.85 | 0.97 | 0.96 | 0.88 | S phase | −0.7 | −0.8 | 1 | 0.75 | −0 | 0.54 | 0.64 | 0.64 | 0.77 | 0.4 | 0.71 | 0.64 | 0.77 | |||
| G2/M | −1 | −1 | −0 | 1 | 0.01 | 0.68 | 0.63 | 0.76 | 0.86 | 0.29 | 0.93 | 0.87 | 0.96 | G2/M | −0.93 | −1 | 0.86 | 1 | 0.7 | 0.88 | 0.91 | 0.94 | 0.85 | 0.74 | 0.91 | 0.85 | 0.93 | G2/M | −1 | −1 | 0.75 | 1 | −0.2 | 0.96 | 0.94 | 0.93 | 0.98 | 0.87 | 0.97 | 0.96 | 0.94 | |||
| A | 0.17 | 0 | 0.46 | 0.01 | 1 | −0.6 | 0.78 | 0.64 | −0.5 | 0.07 | −0.3 | −0.43 | 0.21 | A | −0.82 | −0.8 | 0.87 | 0.7 | 1 | 0.72 | 0.75 | 0.84 | 0.79 | 0.51 | 0.85 | 0.83 | 0.75 | A | 0.07 | 0.11 | −0 | −0.2 | 1 | −0.2 | 0.07 | −0 | −0.1 | −0.1 | −0.1 | −0.1 | 0.07 | |||
| Area | −0.8 | −0.7 | 0.01 | 0.68 | −0.6 | 1 | −0.1 | 0.1 | 0.93 | 0.45 | 0.86 | 0.9 | 0.6 | Area | −0.96 | −0.9 | 0.94 | 0.88 | 0.72 | 1 | 0.73 | 0.8 | 0.95 | 0.88 | 0.97 | 0.98 | 0.95 | Area | −1 | −0.9 | 0.54 | 0.96 | −0.2 | 1 | 0.95 | 0.94 | 0.94 | 0.96 | 0.95 | 0.97 | 0.87 | |||
| P2A | −0.5 | −0.6 | 0.32 | 0.63 | 0.78 | −0.1 | 1 | 0.97 | 0.16 | 0.22 | 0.34 | 0.22 | 0.76 | P2A | −0.88 | −1 | 0.77 | 0.91 | 0.75 | 0.73 | 1 | 0.98 | 0.82 | 0.44 | 0.85 | 0.79 | 0.88 | P2A | −1 | −0.9 | 0.64 | 0.94 | 0.07 | 0.95 | 1 | 0.97 | 0.97 | 0.9 | 0.96 | 0.96 | 0.95 | |||
| LWR | −0.6 | −0.7 | 0.22 | 0.76 | 0.64 | 0.1 | 0.97 | 1 | 0.33 | 0.19 | 0.51 | 0.4 | 0.86 | LWR | −0.93 | −1 | 0.85 | 0.94 | 0.84 | 0.8 | 0.98 | 1 | 0.87 | 0.55 | 0.91 | 0.86 | 0.91 | LWR | −1 | −1 | 0.64 | 0.93 | −0 | 0.94 | 0.97 | 1 | 0.97 | 0.94 | 0.98 | 0.99 | 0.88 | |||
| F-actin | −0.9 | −0.9 | −0.2 | 0.86 | −0.5 | 0.93 | 0.16 | 0.33 | 1 | 0.29 | 0.97 | 0.98 | 0.75 | F-actin | −0.98 | −0.9 | 0.9 | 0.85 | 0.79 | 0.95 | 0.82 | 0.87 | 1 | 0.7 | 0.97 | 0.98 | 0.98 | F-actin | −1 | −1 | 0.77 | 0.98 | −0.1 | 0.94 | 0.97 | 0.97 | 1 | 0.89 | 0.99 | 0.98 | 0.95 | |||
| BrdU | −0.4 | −0.4 | 0.83 | 0.29 | 0.07 | 0.45 | 0.22 | 0.19 | 0.29 | 1 | 0.31 | 0.32 | 0.4 | BrdU | −0.75 | −0.7 | 0.85 | 0.74 | 0.51 | 0.88 | 0.44 | 0.55 | 0.7 | 1 | 0.78 | 0.79 | 0.71 | BrdU | −0.9 | −0.9 | 0.4 | 0.87 | −0.1 | 0.96 | 0.9 | 0.94 | 0.89 | 1 | 0.92 | 0.96 | 0.78 | |||
| MBD1 | −1 | −0.9 | −0.1 | 0.93 | −0.3 | 0.86 | 0.34 | 0.51 | 0.97 | 0.31 | 1 | 0.99 | 0.85 | MBD1 | −0.99 | −0.9 | 0.97 | 0.91 | 0.85 | 0.97 | 0.85 | 0.91 | 0.97 | 0.78 | 1 | 0.99 | 0.97 | MBD1 | −1 | −1 | 0.71 | 0.97 | −0.1 | 0.95 | 0.96 | 0.98 | 0.99 | 0.92 | 1 | 0.99 | 0.92 | |||
| LC3B | −1 | −0.9 | −0.1 | 0.87 | −0.4 | 0.9 | 0.22 | 0.4 | 0.98 | 0.32 | 0.99 | 1 | 0.79 | LC3B | −0.98 | −0.9 | 0.96 | 0.85 | 0.83 | 0.98 | 0.79 | 0.86 | 0.98 | 0.79 | 0.99 | 1 | 0.96 | LC3B | −1 | −1 | 0.64 | 0.96 | −0.1 | 0.97 | 0.96 | 0.99 | 0.98 | 0.96 | 0.99 | 1 | 0.89 | |||
| H2AX | −0.9 | −1 | 0.18 | 0.96 | 0.21 | 0.6 | 0.76 | 0.86 | 0.75 | 0.4 | 0.85 | 0.79 | 1 | H2AX | −0.99 | −1 | 0.88 | 0.93 | 0.75 | 0.95 | 0.88 | 0.91 | 0.98 | 0.71 | 0.97 | 0.96 | 1 | H2AX | −0.9 | −0.9 | 0.77 | 0.94 | 0.07 | 0.87 | 0.95 | 0.88 | 0.95 | 0.78 | 0.92 | 0.89 | 1 | |||
| BPS | CellViab | G0/1 | S phase | G2/M | A | Area | P2A | LWR | F-actin | BrdU | MBD1 | LC3B | H2AX | CellViab | G0/1 | S phase | G2/M | A | Area | P2A | LWR | F-actin | BrdU | MBD1 | LC3B | H2AX | CellViab | G0/1 | S phase | G2/M | A | Area | P2A | LWR | F-actin | BrdU | MBD1 | LC3B | H2AX | |||||
| CellViab | 1 | −0.6 | 0.92 | −0.5 | −0.4 | −0.6 | 0.58 | 0.84 | −1 | 0.63 | −0.9 | −0.87 | −0.89 | CellViab | 1 | −0.9 | 0.96 | 0.83 | −0.3 | −0.6 | 0.44 | 0.37 | −1 | 0.51 | −0.97 | −0.96 | −0.84 | CellViab | 1 | 0.89 | 0.91 | −0.8 | 0.19 | −0.9 | 0.74 | 0.39 | −1 | 0.7 | −1 | −0.97 | −0.97 | |||
| G0/1 | −0.6 | 1 | −0.7 | −0.2 | 0.14 | 0.92 | −0.7 | −0.7 | 0.5 | −0.6 | 0.27 | 0.12 | 0.35 | G0/1 | −0.86 | 1 | −1 | −1 | 0.06 | 0.32 | −0.7 | −0.7 | 0.92 | −0.8 | 0.87 | 0.85 | 0.67 | G0/1 | 0.89 | 1 | 0.97 | −1 | 0.27 | −0.8 | 0.78 | 0.5 | −0.9 | 0.79 | −0.9 | −0.93 | −0.84 | |||
| S phase | 0.92 | −0.7 | 1 | −0.5 | −0.4 | −0.7 | 0.55 | 0.79 | −0.9 | 0.71 | −0.7 | −0.75 | −0.78 | S phase | 0.96 | −1 | 1 | 0.95 | −0.3 | −0.4 | 0.57 | 0.47 | −1 | 0.71 | −0.92 | −0.89 | −0.74 | S phase | 0.91 | 0.97 | 1 | −1 | 0.12 | −0.9 | 0.75 | 0.43 | −0.9 | 0.76 | −0.9 | −0.93 | −0.9 | |||
| G2/M | −0.5 | −0.2 | −0.5 | 1 | −0.1 | −0.2 | −0.1 | −0.4 | 0.46 | 0.02 | 0.48 | 0.74 | 0.56 | G2/M | 0.83 | −1 | 0.95 | 1 | −0.2 | −0.2 | 0.62 | 0.5 | −0.9 | 0.89 | −0.8 | −0.76 | −0.53 | G2/M | −0.8 | −1 | −1 | 1 | −0.3 | 0.76 | −0.75 | −0.5 | 0.85 | −0.8 | 0.89 | 0.89 | 0.79 | |||
| A | −0.4 | 0.14 | −0.4 | −0.1 | 1 | 0.24 | 0.17 | 0.08 | 0.55 | −0.3 | 0.59 | 0.44 | 0.58 | A | −0.28 | 0.06 | −0.3 | −0.2 | 1 | −0.1 | 0.34 | 0.43 | 0.15 | −0.3 | 0.07 | 0.05 | 0.03 | A | 0.19 | 0.27 | 0.12 | −0.3 | 1 | −0.2 | −0.19 | −0.3 | −0.2 | −0.2 | −0.2 | −0.16 | −0.15 | |||
| Area | −0.6 | 0.92 | −0.7 | −0.2 | 0.24 | 1 | −0.8 | −0.7 | 0.62 | −0.8 | 0.21 | 0.14 | 0.36 | Area | −0.64 | 0.32 | −0.4 | −0.2 | −0.1 | 1 | −0.3 | −0.3 | 0.63 | 0.31 | 0.7 | 0.75 | 0.9 | Area | −0.9 | −0.8 | −0.9 | 0.76 | −0.2 | 1 | −0.49 | −0.1 | 0.84 | −0.5 | 0.83 | 0.83 | 0.94 | |||
| P2A | 0.58 | −0.7 | 0.55 | −0.1 | 0.17 | −0.8 | 1 | 0.89 | −0.5 | 0.82 | −0.2 | −0.21 | −0.25 | P2A | 0.44 | −0.7 | 0.57 | 0.62 | 0.34 | −0.3 | 1 | 0.99 | −0.6 | 0.43 | −0.54 | −0.57 | −0.58 | P2A | 0.74 | 0.78 | 0.75 | −0.8 | −0.2 | −0.5 | 1 | 0.9 | −0.8 | 0.99 | −0.8 | −0.86 | −0.72 | |||
| LWR | 0.84 | −0.7 | 0.79 | −0.4 | 0.08 | −0.7 | 0.89 | 1 | −0.7 | 0.72 | −0.5 | −0.55 | −0.56 | LWR | 0.37 | −0.7 | 0.47 | 0.5 | 0.43 | −0.3 | 0.99 | 1 | −0.5 | 0.28 | −0.49 | −0.54 | −0.59 | LWR | 0.39 | 0.5 | 0.43 | −0.5 | −0.3 | −0.1 | 0.9 | 1 | −0.5 | 0.91 | −0.5 | −0.56 | −0.35 | |||
| F-actin | −1 | 0.5 | −0.9 | 0.46 | 0.55 | 0.62 | −0.5 | −0.7 | 1 | −0.7 | 0.79 | 0.84 | 0.88 | F-actin | −0.98 | 0.92 | −1 | −0.9 | 0.15 | 0.63 | −0.6 | −0.5 | 1 | −0.5 | 0.98 | 0.98 | 0.87 | F-actin | −1 | −0.9 | −0.9 | 0.85 | −0.2 | 0.84 | −0.82 | −0.5 | 1 | −0.8 | 0.98 | 0.98 | 0.95 | |||
| BrdU | 0.63 | −0.6 | 0.71 | 0.02 | −0.3 | −0.8 | 0.82 | 0.72 | −0.7 | 1 | −0.4 | −0.35 | −0.36 | BrdU | 0.51 | −0.8 | 0.71 | 0.89 | −0.3 | 0.31 | 0.43 | 0.28 | −0.5 | 1 | −0.45 | −0.39 | −0.08 | BrdU | 0.7 | 0.79 | 0.76 | −0.8 | −0.2 | −0.5 | 0.99 | 0.91 | −0.8 | 1 | −0.8 | −0.84 | −0.67 | |||
| MBD1 | −0.9 | 0.27 | −0.7 | 0.48 | 0.59 | 0.21 | −0.2 | −0.5 | 0.79 | −0.4 | 1 | 0.92 | 0.85 | MBD1 | −0.97 | 0.87 | −0.9 | −0.8 | 0.07 | 0.7 | −0.5 | −0.5 | 0.98 | −0.5 | 1 | 0.99 | 0.88 | MBD1 | −1 | −0.9 | −0.9 | 0.89 | −0.2 | 0.83 | −0.84 | −0.5 | 0.98 | −0.8 | 1 | 1 | 0.94 | |||
| LC3B | −0.9 | 0.12 | −0.8 | 0.74 | 0.44 | 0.14 | −0.2 | −0.6 | 0.84 | −0.4 | 0.92 | 1 | 0.88 | LC3B | −0.96 | 0.85 | −0.9 | −0.8 | 0.05 | 0.75 | −0.6 | −0.5 | 0.98 | −0.4 | 0.99 | 1 | 0.92 | LC3B | −1 | −0.9 | −0.9 | 0.89 | −0.2 | 0.83 | −0.86 | −0.6 | 0.98 | −0.8 | 1 | 1 | 0.95 | |||
| H2AX | −0.9 | 0.35 | −0.8 | 0.56 | 0.58 | 0.36 | −0.3 | −0.6 | 0.88 | −0.4 | 0.85 | 0.88 | 1 | H2AX | −0.84 | 0.67 | −0.7 | −0.5 | 0.03 | 0.9 | −0.6 | −0.6 | 0.87 | −0.1 | 0.88 | 0.92 | 1 | H2AX | −1 | −0.8 | −0.9 | 0.79 | −0.2 | 0.94 | −0.72 | −0.4 | 0.95 | −0.7 | 0.94 | 0.95 | 1 | |||
| TBBPA | CellViab | G0/1 | S phase | G2/M | A | Area | P2A | LWR | F-actin | BrdU | MBD1 | LC3B | H2AX | CellViab | G0/1 | S phase | G2/M | A | Area | P2A | LWR | F-actin | BrdU | MBD1 | LC3B | H2AX | CellViab | G0/1 | S phase | G2/M | A | Area | P2A | LWR | F-actin | BrdU | MBD1 | LC3B | H2AX | |||||
| CellViab | 1 | 0.74 | 0.87 | −0.9 | −0.4 | −0.9 | −0.1 | −0.3 | −0.9 | 0.71 | −0.9 | −0.99 | −0.99 | CellViab | 1 | −0.5 | 0.39 | 0.47 | 0.76 | −0.5 | 0.47 | 0.53 | −0.5 | 0.09 | −0.02 | −0.04 | −0.33 | CellViab | 1 | 0.96 | 0.8 | −0.9 | −0.2 | −0.6 | 0.91 | 0.1 | −1 | −0.1 | −0.9 | −0.86 | −0.96 | |||
| G0/1 | 0.74 | 1 | 0.93 | −0.9 | −0.3 | −0.5 | −0.3 | −0.6 | −0.9 | 0.97 | −0.9 | −0.79 | −0.79 | G0/1 | −0.5 | 1 | −0.2 | −0.9 | −0.5 | 0.62 | −0.6 | −0.7 | 0.84 | −0.7 | −0.33 | −0.33 | 0.42 | G0/1 | 0.96 | 1 | 0.62 | −1 | −0 | −0.8 | 0.79 | 0.08 | −1 | −0.3 | −0.9 | −0.86 | −0.98 | |||
| S phase | 0.87 | 0.93 | 1 | −1 | −0.2 | −0.7 | −0.1 | −0.4 | −1 | 0.95 | −1 | −0.93 | −0.92 | S phase | 0.39 | −0.2 | 1 | 0.6 | 0.75 | 0.36 | −0.3 | −0.3 | 0.2 | −0.4 | −0.63 | −0.67 | 0.02 | S phase | 0.8 | 0.62 | 1 | −0.7 | −0.6 | −0.1 | 0.9 | 0.28 | −0.6 | 0.35 | −0.8 | −0.53 | −0.64 | |||
| G2/M | −0.9 | −0.9 | −1 | 1 | 0.32 | 0.75 | 0.18 | 0.46 | 0.97 | −0.9 | 0.95 | 0.93 | 0.93 | G2/M | 0.47 | −0.9 | 0.6 | 1 | 0.64 | −0.3 | 0.36 | 0.39 | −0.6 | 0.38 | 0.02 | −0.01 | −0.33 | G2/M | −0.9 | −1 | −0.7 | 1 | 0.07 | 0.61 | −0.85 | −0.2 | 0.89 | 0.06 | 0.8 | 0.75 | 0.92 | |||
| A | −0.4 | −0.3 | −0.2 | 0.32 | 1 | 0.13 | 0.77 | 0.76 | 0.1 | −0.1 | 0.17 | 0.28 | 0.3 | A | 0.76 | −0.5 | 0.75 | 0.64 | 1 | −0.1 | 0.16 | 0.18 | −0.2 | −0.2 | −0.44 | −0.43 | 0.03 | A | −0.2 | −0 | −0.6 | 0.07 | 1 | −0.2 | −0.37 | 0.22 | 0.09 | −0.4 | 0.32 | 0.12 | 0.1 | |||
| Area | −0.9 | −0.5 | −0.7 | 0.75 | 0.13 | 1 | −0.4 | −0.1 | 0.77 | −0.5 | 0.79 | 0.82 | 0.83 | Area | −0.53 | 0.62 | 0.36 | −0.3 | −0.1 | 1 | −1 | −1 | 0.93 | −0.9 | −0.8 | −0.8 | 0.68 | Area | −0.6 | −0.8 | −0.1 | 0.61 | −0.2 | 1 | −0.31 | 0.42 | 0.83 | 0.78 | 0.68 | 0.87 | 0.8 | |||
| P2A | −0.1 | −0.3 | −0.1 | 0.18 | 0.77 | −0.4 | 1 | 0.93 | 0 | −0.2 | 0.03 | 0.05 | 0.04 | P2A | 0.47 | −0.6 | −0.3 | 0.36 | 0.16 | −1 | 1 | 0.99 | −0.9 | 0.88 | 0.76 | 0.78 | −0.61 | P2A | 0.91 | 0.79 | 0.9 | −0.9 | −0.4 | −0.3 | 1 | 0.2 | −0.8 | 0.28 | −0.8 | −0.62 | −0.78 | |||
| LWR | −0.3 | −0.6 | −0.4 | 0.46 | 0.76 | −0.1 | 0.93 | 1 | 0.29 | −0.5 | 0.27 | 0.27 | 0.27 | LWR | 0.53 | −0.7 | −0.3 | 0.39 | 0.18 | −1 | 0.99 | 1 | −1 | 0.86 | 0.76 | 0.77 | −0.63 | LWR | 0.1 | 0.08 | 0.28 | −0.2 | 0.22 | 0.42 | 0.2 | 1 | 0.1 | 0.49 | 0.14 | 0.3 | 0.06 | |||
| F-actin | −0.9 | −0.9 | −1 | 0.97 | 0.1 | 0.77 | 0 | 0.29 | 1 | −0.9 | 0.98 | 0.93 | 0.93 | F-actin | −0.45 | 0.84 | 0.2 | −0.6 | −0.2 | 0.93 | −0.9 | −1 | 1 | −0.9 | −0.73 | −0.74 | 0.63 | F-actin | −1 | −1 | −0.6 | 0.89 | 0.09 | 0.83 | −0.76 | 0.1 | 1 | 0.39 | 0.93 | 0.95 | 0.99 | |||
| BrdU | 0.71 | 0.97 | 0.95 | −0.9 | −0.1 | −0.5 | −0.2 | −0.5 | −0.9 | 1 | −0.9 | −0.79 | −0.78 | BrdU | 0.09 | −0.7 | −0.4 | 0.38 | −0.2 | −0.9 | 0.88 | 0.86 | −0.9 | 1 | 0.9 | 0.91 | −0.67 | BrdU | −0.1 | −0.3 | 0.35 | 0.06 | −0.4 | 0.78 | 0.28 | 0.49 | 0.39 | 1 | 0.29 | 0.56 | 0.36 | |||
| MBD1 | −0.9 | −0.9 | −1 | 0.95 | 0.17 | 0.79 | 0.03 | 0.27 | 0.98 | −0.9 | 1 | 0.97 | 0.97 | MBD1 | −0.02 | −0.3 | −0.6 | 0.02 | −0.4 | −0.8 | 0.76 | 0.76 | −0.7 | 0.9 | 1 | 0.99 | −0.74 | MBD1 | −0.9 | −0.9 | −0.8 | 0.8 | 0.32 | 0.68 | −0.8 | 0.14 | 0.93 | 0.29 | 1 | 0.95 | 0.94 | |||
| LC3B | −1 | −0.8 | −0.9 | 0.93 | 0.28 | 0.82 | 0.05 | 0.27 | 0.93 | −0.8 | 0.97 | 1 | 1 | LC3B | −0.04 | −0.3 | −0.7 | −0 | −0.4 | −0.8 | 0.78 | 0.77 | −0.7 | 0.91 | 0.99 | 1 | −0.69 | LC3B | −0.9 | −0.9 | −0.5 | 0.75 | 0.12 | 0.87 | −0.62 | 0.3 | 0.95 | 0.56 | 0.95 | 1 | 0.95 | |||
| H2AX | −1 | −0.8 | −0.9 | 0.93 | 0.3 | 0.83 | 0.04 | 0.27 | 0.93 | −0.8 | 0.97 | 1 | 1 | H2AX | −0.33 | 0.42 | 0.02 | −0.3 | 0.03 | 0.68 | −0.6 | −0.6 | 0.63 | −0.7 | −0.74 | −0.69 | 1 | H2AX | −1 | −1 | −0.6 | 0.92 | 0.1 | 0.8 | −0.78 | 0.06 | 0.99 | 0.36 | 0.94 | 0.95 | 1 | |||
Discussion:
Spermatogenesis is regulated and supported by Sertoli cells, Leydig cells, and other cell types in the testis. Injury of these cell types will result in testicular damage and reproductive dysfunction. Intercellular communication among spermatogonial, Sertoli and Leydig cell in the various compartments of the testis are essential to sustain the normal spermatogenic process. The animal models showed that these cells could be selectively targeted by specific toxicants, resulting in germ-cell apoptosis and spermatogenesis dysfunction (Boekelheide 2005). For example, ubiquitous environmental toxicants, and phthalates can produce testicular atrophy by targeting Sertoli cells in laboratory animals (Lee, Richburg et al. 1999). Ethane-1,2-dimethanesulfonate is a cytotoxic alkylating agent that selectively impacts adult Leydig cells, while X-irradiation targets the germ cell resulting in alteration of cell cycle and apoptosis (Hasegawa, Wilson et al. 1997). Although these animal models have generated valuable evidence to support toxicology assessment, advanced in vitro models allow us to elucidate the toxic mechanisms, discern the cell-specific sensitivity in the testis at a low cost, and broaden biological pathways. Previous studies demonstrated that BPA and its analogs induced a broad spectrum of adverse effects on the reproductive system in animal models (Yamasaki, Noda et al. 2004, Feng, Yin et al. 2012, Peretz, Vrooman et al. 2014, Cope, Kacew et al. 2015, Chen, Shu et al. 2016). However, cell-specific sensitivity to a particular compound remains unclear. The co-culture system with germ cells, Sertoli cells, and Leydig cells allows for evaluating cellular interactions. However, it is technically challenging to assess the effect of BPAs on these individual types of cells. Thus, comparing chemical toxicity in different testicular cell types could help elucidate cell type-specific toxicity and the functional significance of each cell type although this approach loses the cell-cell interactions that were present in the co-culture system . We selected C18-4 spermatogonial cells, TM3 Leydig, and TM4 Sertoli cells since these cells have been widely used to examine cell-specific toxicity in the testis (Mather 1980, Braydich-Stolle, Hussain et al. 2005, He, Jiang et al. 2008, Golestaneh, Beauchamp et al. 2009, Braydich-Stolle, Lucas et al. 2010, Zhang, Gong et al. 2013, Reis, Moreira et al. 2015, Liang, Yin et al. 2017). C18-4 cell line exhibited the morphological features of type A spermatogonia and expressed the germ cell-specific genetic markers, and has been applied in multiple studies to examine signaling pathways involved in germ cell development and characterize reproductive toxicity of nanoparticles (Braydich-Stolle, Hussain et al. 2005, Hofmann, Braydich-Stolle et al. 2005, He, Jiang et al. 2008, Golestaneh, Beauchamp et al. 2009, Braydich-Stolle, Lucas et al. 2010, Zhang, Gong et al. 2013). TM4 (Sertoli) and TM3 (Leydig) cells were isolated from pre-pubertal mouse gonads and expressed the various hormone receptors (Mather 1980). TM4 Sertoli cells express functional receptors for sollicle-stimulating hormone (FSH) in supporting spermatogenesis, whereas TM3 Leydig cells express luteinizing hormone (LH) receptors and stimulate androgen production. TM3 Leydig cells were used to assess chemical effects on steroidogenic activity (Komatsu, Tabata et al. 2008, Chen, Zhang et al. 2015). TM4 Sertoli cells have been used as a testicular toxicity model to investigate adverse effects of chemical exposure on blood-testis-barrier (BTB) stability (Reis, Moreira et al. 2015). Therefore, the cell-specific response within testis in response to specific compounds could reflect regulated differences in signaling molecules that underlie the discrete functional roles among these three cell types.
In this study, we selected different dose ranges for the tested compound, 0, 5, 50, and 100 μM for BPA and BPS, and 0, 1, 5, and 10 μM for BPAF and TBBPA since our previous study revealed about tenfold difference in LC50 between BPAF and BPA in the testicular co-culture model (Yin, Siracusa et al. 2020). Consistent with our previous study with the testicular cell co-culture model, we found BPAF induced cytotoxicity in all three types of testicular cells, showing significant dose-dependent decreases in cell number at both 24 and 48h. BPAF showed a significant reduction in cell number at a low dose of 5 μmM in spermatogonia cells compared to TM3 and TM4 cells, suggesting BPAF may target spermatogonial cells (C18-4). BPA mainly targeted at TM4, followed by TM3 cells, and less at spermatogonial cells. BPS was revealed to have similar cytotoxicity as BPA, more likely targeting Sertoli cell (TM4) and Leydig cell (TM3) and less at spermatogonial cells. TBBPA targeted spermatogonia, TM4 Sertoli cells, and less at TM3 Leydig cells. Toxicity and teratogenic effects of the bisphenols BPA, BPS, BPF, and BPAF were compared in zebrafish embryo-larvae on their estrogenic mechanisms in an estrogen-responsive transgenic fish (Moreman, Lee et al. 2017). Developmental deformities effects were assessed, including cardiac edema, spinal malformation, and craniofacial deformities, and the rank order for toxicity was BPAF > BPA > BPF > BPS (Moreman, Lee et al. 2017). The National Toxicology Program (Tox21) reported on the biological activity of BPA and several of its analogs, BPS, Bisphenol F (BPF), and BPAF, and suggested BPA analogs causing mammalian reproductive toxicity (Program 2021). Furthermore, BPAF was reported to have significantly more effects on the reproductive system than other BPA-related compounds (Program 2021). Like BPA, the BPA analogs were shown to have varying levels of estrogenic activity in the testis. Still, the majority had activity within the same order of magnitude as BPA and were less potent than positive control reference agonists such as -estradiol. However, BPAF was consistently one of the most potent BPA analogs in the estrogen agonist and antagonist assays. In Tox21, BPAF was the most potent (estrogen receptor) ER agonist of the BPA analogs and was more potent than BPA (Pelch, Wignall et al. 2017, Pelch, Wignall et al. 2019). It is interesting to explore further if observed BPAF toxicity is associated with the ER activity.
The underlying mechanism of the cell-specific cytotoxicity among BPA and its analogs are unclear. Therefore, we developed a multi-endpoint high-content analysis (HCA), including nuclear morphology, cell cycle, DNA synthesis, DNA damage, cytoskeletal structure and DNA methylation and phagocytosis based on our previous studies (Liang, Yin et al. 2017). These HCA-based endpoint assays are associated with adverse outcome pathways (AOPs), including 13 parameters from three types of testicular cells at two time periods. Multiplexed HCA parameters spectrum (Figure 7A) demonstrated differential profiles among BPA and its analogs, and revealed dynamic, dose-dependent, and cell-specific changes in these HCA markers. Each chemical displayed a unique dose-dependent profile within each type of cell. HCA index, the sum of the absolute fold changes of 13 endpoints, reflects dose and time dependent alterations. BPAF at 10 μM resulted in similar HCA in all three types of cells, but the spermatogonial cell has the highest HCA index value at 5 μM, indicating that C18-4 cells are more susceptible to BPAF than other cell types. types of cells. Our current observation was consistent with the previous study showed that BPAF was most cytotoxic compared to other analogs (Liang, Yin et al. 2017). We observed that BPAF was more toxic to C18-4 cells than Sertoli or Leydig cells, especially in nuclear structure (area, shapes) and cell cycle alteration (G2M phase). Most importantly, tthrough the comparison of these multi-endpoint parameters, we found a unique toxic signature for BPAF. We found cell viability was positively correlated with BrdU and negatively correlated with MBD1, LC3B, and H2AX in BPA-, BPS-, TBBPA-treated C18 cells. However, cell viability in BPAF treatment was negatively associated with BrdU in all testicular cells.
In our previous study, we developed an approach to constructing a cell cycle profile with discrete SubG1, G0/1, S, and G2/M phases in the spermatogonial cell line, which showed accurate segmentation of cell cycles with high-content image analysis (Liang, et al., 2017). In this study, we employed the same approach to examine the chemical effects on cell cycle progression in different cell lines. The data revealed that BPA and its analogs perturbed DNA synthesis and cell cycle progression in a cell- and chemical-specific manner. Our results demonstrated that BPA and its analogs induced G2/M phase arrest in both spermatogonial cells and TM4 Sertoli cells. However, in TM3 cells, BPA and BPS treatment induced G0/1 phase arrest. It has been reported that chemical modulation of cyclin-dependent kinase complexes was able to result in G2/M phase arrest. In contrast, for chemicals affecting the calmodulin-dependent protein kinase-II, the G1 phase arrest was observed in the cell population (Morris, DeLorenzo et al. 1998, Malumbres and Barbacid 2005). Thus, the differential alteration of cell cycle progression in testicular cells might suggest the distinct cellular mechanism underlying chemical toxicity.
Nuclear morphological features have been suggested as valuable biomarkers in various adverse cellular events (M, T et al. 1999, Eidet, Pasovic et al. 2014). Multinucleated germ cells formed from round spermatids, or occasionally spermatocytes were reported due to failed integrity of the intercellular bridges serving as partitions (Miething 1993, Miething 1995, Greenbaum, Iwamori et al. 2011, McClusky 2011).Our current study confirmed that BPAF induced unique multinucleated germ cells (MNGs) in the spermatogonia cell, not in the TM3 and TM4 cell. The above results are consistent with our previous studies in C18 spermatogonial cell culture (Liang, Yin et al. 2017) and the testicular cell co-culture model (Yin, Siracusa et al. 2020). It has been reported that MNGs occurred in cryptorchidism and may be linked to testicular cancers (Cho, Ahn et al. 1989, Nacov 1990, D, J et al. 2003, Nistal, Gonzalez-Peramato et al. 2006, Zhang, Lin et al. 2011) and decreased sperm count (D, J et al. 2003, DJ, SJ et al. 2014). The induction of multinucleated gonocyte was reported as a reproductive toxicity hallmark in animal models exposed to di-(n-butyl) phthalate (DBP) (T, W et al. 1999, Akbarsha and Murugaian 2000, Mylchreest, Sar et al. 2002, NJ, BS et al. 2004, Faridha, Faisal et al. 2007, G, J et al. 2010). Having anti-androgenic effects, DBP was found to induce high levels of MNG in correlation with the decrease of testosterone production in fetal rat testis and human fetal testis xenograft (Kleymenova, Swanson et al. 2005, Spade, Hall et al. 2015). DBP targets testicular cells by altering cytoskeletal features and creating aberrant mitosis at the basal lamina, which causes the disorganized localization of actin (Kleymenova, Swanson et al. 2005, Spade, Hall et al. 2015). Likewise, DBP created MNGs and disrupted testicular development in fetal rats (Gaido, Hensley et al. 2007). While Spade et al. discovered a nonproliferative mechanism of DBP in MNGs, our study using a single cell-based quantification of BrdU further demonstrated that multinucleated germ cells induced by BPAF were positively correlated with cell proliferation (BrdU positive staining The irregularity of multinucleation in the spermatogonial cells can result in genomic instability that affects various proteins, cell cycle checkpoints, and DNA responses that are necessary for proper cellular division (Hanahan and Weinberg 2000, Armit, O’Dea et al. 2002, Ariizumi, Ogose et al. 2009). Polyploidy and multinucleation have been identified as hallmarks of several malignancies, including testicular cancer (Makarovskiy, Siryaporn et al. 2002, D, J et al. 2003, Storchova and Pellman 2004, Nistal, Gonzalez-Peramato et al. 2006, Zhang, Lin et al. 2011). The molecular pathway of MNG formation is still poorly known (Spade, Hall et al. 2015), and the examination of the underlining mechanism of BPAF-induced multinucleation of germ cells will fill the knowledge gap and gain insight into the link between the formation of multinucleated spermatogonial cells and fertility and possibly testicular cancer cell growth.
Spermatogenesis encompasses multiple cellular events for generating mature sperm, including dramatic cell morphology change and cell movement within seminiferous tubules. As one of a major components of the cytoskeleton, F-actin has been shown to play an important role in cell movement, cell connection, cargo transportation, acrosome reaction, and nuclear modification during spermatogenesis (Vogl 1989, Sun, Kovacs et al. 2011). In Sertoli cells, F-actin forms parallel actin bundles in ectoplasmic specialization that are located between adjacent Sertoli cells and adhesion between Sertoli cells and germ cells, which regulate the movement and release of spermatids (Setchell 2008, Cheng and Mruk 2009, Sun, Kovacs et al. 2011). We observed the distinct pattern of F-actin cytoskeleton among three cell types and alteration of these structures in a dose-dependent manner. The significant induction of F-actin total intensity further suggested the aberrant F-actin accumulation within cells. In an in vivo study, BPA induced aberrant actin distribution, contributing to the cytokinesis failure in the porcine oocyte (Wang, Han et al. 2016). Exposure to BPA causes the truncation and depolymerization of actin via perturbing localization of actin regulatory proteins in Sertoli cells, which provide a cellular mechanism for BPA-induced BTB damage (Xiao, Mruk et al. 2014). DNA damage has been suggested to affect BPA reproductive toxicity (Meeker, Ehrlich et al. 2010, Ferris, Mahboubi et al. 2016) (Ganesan and Keating 2016). γ-H2AX, an early cellular DNA damage response marker, has recently been applied in HCA to screen genotoxicity of chemicals (Garcia-Canton, Anadon et al. 2013, Garcia-Canton, Anadon et al. 2013, Ando, Yoshikawa et al. 2014). In this study, we detected γ-H2AX expression level and found that BPA and its analogs differentially induced the DNA damage response in a cell type-specific manner. It could be explained by differential biotransformation of BPA and its analogs and DNA damage repair capacity among different cell models. In in vitro study BPA was able to induce γ-H2AX at a low dose of 10 nM in human breast cancer cells, while in HepG2 cell line, BPA did not induce γ-H2AX even at a dose of 100 μM, but bisphenol F did (Audebert, Dolo et al. 2011, Pfeifer, Chung et al. 2015). In addition, BPAF did not induce γ-H2AX in HepG2 and chicken DT40 cells, while it significantly increased γ-H2AX in spermatogonial cell lines and co-culture model (Fic, Zegura et al. 2013, Lee, Liu et al. 2013, Liang, Yin et al. 2017).
This study aimed to investigate how cell cycle and DNA damage are related to autophagy or epigenetic regulation in BPA and its analog exposure. DNA methylation, one of the significant processes of epigenetic alterations, has been reported to show a unique pattern in testis development (Oakes, La Salle et al. 2007). There is increasing evidence to indicate epigenetic alterations could be one of the possible mechanisms of BPA-induced reproductive dysfunction (Bromer, Zhou et al. 2010, Doshi, Mehta et al. 2011, Tang, Morey et al. 2012). It has been reported that neonatal exposure to BPA induced hypermethylation of the estrogen receptor promoter region in rat testis (Doshi, Mehta et al. 2011). In the current study, BPA and its analogs induced the expression level of MBD1, a transcriptional repressor that could bind to single methylated CpGs, in three testicular cell types, suggesting the aberrant methylation occurred. In an epidemiological study, DNA methylation on multiple loci has been associated with decreased sperm concentration and quality (Houshdaran, Cortessis et al. 2007). Autophagy and the DNA damage response are biological processes essential for cellular homeostasis. Notably, environmental chemical exposure could cause cellular damage, including DNA damage responses, resulting in numerous cellular processes, such as cell cycle checkpoint activation and DNA repair machinery. Along with the measurement of MBD1, we also examined the expression level of an autophagosome membrane protein, LC3B, to monitor autophagic activity. We found BPA and its analogs differentially activated autophagy in spermatogonial, Sertoli and Leydig cells. As a highly conserved process to degrade and recycle cytoplasmic components, autophagy is essential for cell proliferation and differentiation and serves as a cell survival mechanism in stressed cells (Baehrecke 2005, Mizushima and Levine 2010, Mizushima and Komatsu 2011). The epigenetic modulation regulates the autophagy process by altering the activity of transcriptional factors involved in signaling pathways (Yin, Ma et al. 2021). It has been reported that autophagy was activated by environmental chemical exposure during spermatogenesis (Liu, Zhang et al. 2014, Liu, Wang et al. 2015, Zhang, Ling et al. 2016). Activation of autophagy acted as a protective role against BPA-induced neurodegeneration via AMPK and mTOR pathways at doses of 40 and 400 μg (Agarwal, Tiwari et al. 2015). In contrast, BPA treatment induced autophagy and apoptosis concurrently in rat testis at a dose of 50 mg/kg (Quan, Wang et al. 2016). Uncovering the mechanisms by which autophagy, DNA damage response, and epigenetic regulation provide novel insight into the pathobiology of conditions associated with dynamic spermatogenesis, including cancer and aging, and novel concepts for the development of improved therapeutic strategies against these pathologies.
As compared to the previous study, a similar toxicity ranking of BPA and its analogs was observed in spermatogonial cells, Sertoli cells, and Leydig cells, respectively. However, BPAF and TBBPA exhibited toxicity at lower doses (Liang, Yin et al. 2017). Fetal bovine serum (FBS) contains multiple hormones, including the FSH, testosterone, progesterone, and LH, to support cell growth, which could potentially exert agonist or antagonist effects on BPA or its analogs toxicity in testicular cells. Reduced serum or serum-free cell culture conditions are increasingly required in current studies to remove the effect of the hormone. In this study, we employed Nu-serum, a cost-effective FBS alternative, in the cell culture system. As compared to FBS serum, Nu-serum reduced the various hormone levels and protein content and has successfully supported a large variety of cell types (Culty, Nguyen et al. 1992, Medh, Santell et al. 1992). Interestingly, among all three cell types, spermatogonial cells appear to be the target for BPAF, and Sertoli cell is highly resistant to TBBPA toxicity. Although we previously observed high testicular toxicity of TBBPA in in vitro spermatogonial cells and testicualr co-culture model (Liang, Yin et al. 2017), its in vivo toxicity still remains controversial (Cope, Kacew et al. 2015). Cope, et al. only observed the decrease in thyroid hormone T4 levels in SD rats exposed to TBBPA at a dose of 1000 mg/kg. In contrast, no changes in sperm motility, concentration, or abnormal sperm percentage were observed in multiple generations (Cope, Kacew et al. 2015). Furtehr study is needed to undersatnd the mechisim of TBBPA induced toxicity.
In summary, by utilizing HCA assays in spermatogonial cells, Sertoli cells, and Leydig cells, we characterized multiple adverse effects of BPA and its analog on different testicular cells . The HCA toxicity specttrum and HCA index generated from multiparametric HCA data were able to evaluate cell-type specific toxicity profiling and identify targeted cell types, which provide substantial evidence for chemical testicular toxicity and multidimensional toxicity profiling for toxicity classification. In the future, we will include more endpoints that are closely related to in vivo reproductive function, such as steroidogenesis and hormone receptor expression, and more compounds to construct reproductive toxicological profiles and provide mechanistic information for the prediction of biological targets of reproductive toxins.
Acknowledgments
We acknowledge the NIH funding supports (NIEHS R44 ES027374 and R43 ES031890) for Dr. Yin and NIEHS P30ES032755 for Dr. Yu
Declaration of interests
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
Lei Yin reports financial support was provided by National Institute of Environmental Health.
References
- Agarwal S, Tiwari SK, Seth B, Yadav A, Singh A, Mudawal A, Chauhan LKS, Gupta SK, Choubey V, Tripathi A, Kumar A, Ray RS, Shukla S, Parmar D and Chaturvedi RK (2015). “Activation of Autophagic Flux against Xenoestrogen Bisphenol-A-induced Hippocampal Neurodegeneration via AMP kinase (AMPK)/Mammalian Target of Rapamycin (mTOR) Pathways.” Journal of Biological Chemistry 290(34): 21163–21184. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Akbarsha MA and Murugaian P (2000). “Aspects of the male reproductive toxicity/male antifertility property of andrographolide in albino rats: effect on the testis and the cauda epididymidal spermatozoa.” Phytother Res 14(6): 432–435. [DOI] [PubMed] [Google Scholar]
- Ando M, Yoshikawa K, Iwase Y and Ishiura S (2014). “Usefulness of monitoring gamma-H2AX and cell cycle arrest in HepG2 cells for estimating genotoxicity using a high-content analysis system.” J Biomol Screen 19(9): 1246–1254. [DOI] [PubMed] [Google Scholar]
- Ariizumi T, Ogose A, Kawashima H, Hotta T, Umezu H and Endo N (2009). “Multinucleation followed by an acytokinetic cell division in myxofibrosarcoma with giant cell proliferation.” J Exp Clin Cancer Res 28: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armit CJ, O’Dea S, Clarke AR and Harrison DJ (2002). “Absence of p53 in Clara cells favours multinucleation and loss of cell cycle arrest.” BMC Cell Biol 3: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audebert M, Dolo L, Perdu E, Cravedi JP and Zalko D (2011). “Use of the gamma H2AX assay for assessing the genotoxicity of bisphenol A and bisphenol F in human cell lines.” Archives of Toxicology 85(11): 1463–1473. [DOI] [PubMed] [Google Scholar]
- Baehrecke EH (2005). “Autophagy: dual roles in life and death?” Nat Rev Mol Cell Biol 6(6): 505–510. [DOI] [PubMed] [Google Scholar]
- Barbonetti A, Castellini C, Di Giammarco N, Santilli G, Francavilla S and Francavilla F (2016). “In vitro exposure of human spermatozoa to bisphenol A induces pro-oxidative/apoptotic mitochondrial dysfunction.” Reproductive Toxicology 66: 61–67. [DOI] [PubMed] [Google Scholar]
- Berger A, Ziv-Gal A, Cudiamat J, Wang W, Zhou CQ and Flaws JA (2016). “The effects of in utero bisphenol A exposure on the ovaries in multiple generations of mice.” Reproductive Toxicology 60: 39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boekelheide K (2005). “Mechanisms of toxic damage to spermatogenesis.” J Natl Cancer Inst Monogr(34): 6–8. [DOI] [PubMed] [Google Scholar]
- Braydich-Stolle L, Hussain S, Schlager JJ and Hofmann MC (2005). “In vitro cytotoxicity of nanoparticles in mammalian germline stem cells.” Toxicological Sciences 88(2): 412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braydich-Stolle LK, Lucas B, Schrand A, Murdock RC, Lee T, Schlager JJ, Hussain SM and Hofmann MC (2010). “Silver Nanoparticles Disrupt GDNF/Fyn kinase Signaling in Spermatogonial Stem Cells.” Toxicological Sciences 116(2): 577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromer JG, Zhou Y, Taylor MB, Doherty L and Taylor HS (2010). “Bisphenol-A exposure in utero leads to epigenetic alterations in the developmental programming of uterine estrogen response.” FASEB J 24(7): 2273–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calafat AM, Ye X, Wong LY, Reidy JA and Needham LL (2008). “Exposure of the U.S. population to bisphenol A and 4-tertiary-octylphenol: 2003–2004.” Environ Health Perspect 116(1): 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho L, Lewis SM, Vanlandingham MM, Olson GR, Davis KJ, Patton RE, Twaddle NC, Doerge DR, Churchwell MI, Bryant MS, McLellen FM, Woodling KA, Felton RP, Maisha MP, Juliar BE, Gamboa da Costa G and Delclos KB (2019). “A two-year toxicology study of bisphenol A (BPA) in Sprague-Dawley rats: CLARITY-BPA core study results.” Food Chem Toxicol 132: 110728. [DOI] [PubMed] [Google Scholar]
- CDC (2022). https://www.cdc.gov/exposurereport/.
- Chen GL, Zhang SB, Jin YX, Wu Y, Liu L, Qian HF and Fu ZW (2015). “TPP and TCEP induce oxidative stress and alter steroidogenesis in TM3 Leydig cells.” Reproductive Toxicology 57: 100–110. [DOI] [PubMed] [Google Scholar]
- Chen YC, Shu L, Qiu ZQ, Lee DY, Settle SJ, Hee SQ, Telesca D, Yang X and Allard P (2016). “Exposure to the BPA-Substitute Bisphenol S Causes Unique Alterations of Germline Function.” Plos Genetics 12(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY and Mruk DD (2009). “An intracellular trafficking pathway in the seminiferous epithelium regulating spermatogenesis: a biochemical and molecular perspective.” Critical Reviews in Biochemistry and Molecular Biology 44(5): 245–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho NH, Ahn HJ and Park C (1989). “Large mononuclear cells in seminoma--an immunohistochemical analysis of 21 cases.” Yonsei Med J 30(2): 125–132. [DOI] [PubMed] [Google Scholar]
- Cope RB, Kacew S and Dourson M (2015). “A reproductive, developmental and neurobehavioral study following oral exposure of tetrabromobisphenol A on Sprague-Dawley rats.” Toxicology 329: 49–59. [DOI] [PubMed] [Google Scholar]
- Culty M, Nguyen HA and Underhill CB (1992). “The hyaluronan receptor (CD44) participates in the uptake and degradation of hyaluronan.” J Cell Biol 116(4): 1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D C, J T and J V (2003). “Multinucleated spermatogonia in cryptorchid boys: a possible association with an increased risk of testicular malignancy later in life?” APMIS : acta pathologica, microbiologica, et immunologica Scandinavica 111(1). [DOI] [PubMed] [Google Scholar]
- Deceuninck Y, Bichon E, Marchand P, Boquien CY, Legrand A, Boscher C, Antignac JP and Le Bizec B (2015). “Determination of bisphenol A and related substitutes/analogues in human breast milk using gas chromatography-tandem mass spectrometry.” Anal Bioanal Chem 407(9): 2485–2497. [DOI] [PubMed] [Google Scholar]
- Delclos KB, Camacho L, Lewis SM, Vanlandingham MM, Latendresse JR, Olson GR, Davis KJ, Patton RE, da Costa GG, Woodling KA, Bryant MS, Chidambaram M, Trbojevich R, Juliar BE, Felton RP and Thorn BT (2014). “Toxicity Evaluation of Bisphenol A Administered by Gavage to Sprague Dawley Rats From Gestation Day 6 Through Postnatal Day 90.” Toxicological Sciences 139(1): 174–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DJ S, SJ H, CM S, SM H, EV M and K B (2014). “Differential response to abiraterone acetate and di-n-butyl phthalate in an androgen-sensitive human fetal testis xenograft bioassay.” Toxicological sciences : an official journal of the Society of Toxicology 138(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi T, Mehta SS, Dighe V, Balasinor N and Vanage G (2011). “Hypermethylation of estrogen receptor promoter region in adult testis of rats exposed neonatally to bisphenol A.” Toxicology 289(2-3): 74–82. [DOI] [PubMed] [Google Scholar]
- Eidet JR, Pasovic L, Maria R, Jackson CJ and Utheim TP (2014). “Objective assessment of changes in nuclear morphology and cell distribution following induction of apoptosis.” Diagnostic Pathology 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EU. (2016). amending Annex XVII to Regulation (EC) No 1907/2006 of the European Parliament and of the Council concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) as regards bisphenol A. 2016/2235.
- Faridha A, Faisal K and Akbarsha MA (2007). “Aflatoxin treatment brings about generation of multinucleate giant spermatids (symplasts) through opening of cytoplasmic bridges: Light and transmission electron microscopic study in Swiss mouse.” Reproductive Toxicology 24(3-4): 403–408. [DOI] [PubMed] [Google Scholar]
- FDA. (2012). Indirect Food Additives: Polymers. 21 CFR 177: 41899−41902. [Google Scholar]
- FDA. (2013). Indirect Food Additives: Adhesives and Components of Coatings. 21 CFR 175: 41840−41843. [Google Scholar]
- Feng YX, Yin J, Jiao ZH, Shi JC, Li M and Shao B (2012). “Bisphenol AF may cause testosterone reduction by directly affecting testis function in adult male rats.” Toxicology Letters 211(2): 201–209. [DOI] [PubMed] [Google Scholar]
- Ferris J, Mahboubi K, MacLusky N, King WA and Favetta LA (2016). “BPA exposure during in vitro oocyte maturation results in dose-dependent alterations to embryo development rates, apoptosis rate, sex ratio and gene expression.” Reprod Toxicol 59: 128–138. [DOI] [PubMed] [Google Scholar]
- Fic A, Zegura B, Sollner Dolenc M, Filipic M and Peterlin Masic L (2013). “Mutagenicity and DNA damage of bisphenol A and its structural analogues in HepG2 cells.” Arh Hig Rada Toksikol 64(2): 189–200. [DOI] [PubMed] [Google Scholar]
- G G-A, J A-R, A N-G, M O-M and G J-R (2010). “Multinucleation of spermatozoa and spermatids in infertile men chronically exposed to carbofuran.” Reproductive toxicology (Elmsford, N.Y.) 29(4). [DOI] [PubMed] [Google Scholar]
- Gaido KW, Hensley JB, Liu D, Wallace DG, Borghoff S, Johnson KJ, Hall SJ and Boekelheide K (2007). “Fetal mouse phthalate exposure shows that Gonocyte multinucleation is not associated with decreased testicular testosterone.” Toxicol Sci 97(2): 491–503. [DOI] [PubMed] [Google Scholar]
- Gallagher LE, Williamson LE and Chan EY (2016). “Advances in Autophagy Regulatory Mechanisms.” Cells 5(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan S and Keating AF (2016). “Bisphenol A-Induced Ovotoxicity Involves DNA Damage Induction to Which the Ovary Mounts a Protective Response Indicated by Increased Expression of Proteins Involved in DNA Repair and Xenobiotic Biotransformation.” Toxicological Sciences 152(1): 169–180. [DOI] [PubMed] [Google Scholar]
- Garcia-Canton C, Anadon A and Meredith C (2013). “Assessment of the in vitro γH2AX assay by High Content Screening as a novel genotoxicity test.” Mutation Research/Genetic Toxicology and Environmental Mutagenesis 757(2): 158–166. [DOI] [PubMed] [Google Scholar]
- Garcia-Canton C, Anadon A and Meredith C (2013). “Genotoxicity evaluation of individual cigarette smoke toxicants using the in vitro gammaH2AX assay by high content screening.” Toxicol Lett 223(1): 81–87. [DOI] [PubMed] [Google Scholar]
- Geens T, Roosens L, Neels H and Covaci A (2009). “Assessment of human exposure to Bisphenol-A, Triclosan and Tetrabromobisphenol-A through indoor dust intake in Belgium.” Chemosphere 76(6): 755–760. [DOI] [PubMed] [Google Scholar]
- Gladfelter AS, Hungerbuehler AK and Philippsen P (2006). “Asynchronous nuclear division cycles in multinucleated cells.” J Cell Biol 172(3): 347–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golestaneh N, Beauchamp E, Fallen S, Kokkinaki M, Uren A and Dym M (2009). “Wnt signaling promotes proliferation and stemness regulation of spermatogonial stem/progenitor cells.” Reproduction 138(1): 151–162. [DOI] [PubMed] [Google Scholar]
- Greenbaum MP, Iwamori T, Buchold GM and Matzuk MM (2011). “Germ cell intercellular bridges.” Cold Spring Harb Perspect Biol 3(8): a005850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D and Weinberg RA (2000). “The hallmarks of cancer.” Cell 100(1): 57–70. [DOI] [PubMed] [Google Scholar]
- Hargarten JC and Williamson PR (2018). “Epigenetic Regulation of Autophagy: A Path to the Control of Autoimmunity.” Front Immunol 9: 1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Wilson G, Russell LD and Meistrich ML (1997). “Radiation-induced cell death in the mouse testis: relationship to apoptosis.” Radiat Res 147(4): 457–467. [PubMed] [Google Scholar]
- He ZP, Jiang JJ, Kokkinaki M, Golestaneh N, Hofmann MC and Dym M (2008). “GDNF upregulates c-fos transcription via the Ras/ERK1/2 pathway to promote mouse spermatogonial stem cell proliferation.” Stem Cells 26(1): 266–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindel JJ, Newbold RR, Bucher JR, Camacho L, Delclos KB, Lewis SM, Vanlandingham M, Churchwell MI, Twaddle NC, McLellen M, Chidambaram M, Bryant M, Woodling K, Gamboa da Costa G, Ferguson SA, Flaws J, Howard PC, Walker NJ, Zoeller RT, Fostel J, Favaro C and Schug TT (2015). “NIEHS/FDA CLARITY-BPA research program update.” Reprod Toxicol 58: 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann MC, Braydich-Stolle L, Dettin L, Johnson E and Dym M (2005). “Immortalization of mouse germ line stem cells.” Stem Cells 23(2): 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houshdaran S, Cortessis VK, Siegmund K, Yang A, Laird PW and Sokol RZ (2007). “Widespread Epigenetic Abnormalities Suggest a Broad DNA Methylation Erasure Defect in Abnormal Human Sperm.” Plos One 2(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenardhanana P, Panneerselvama M and Mathur PP (2016). “Effect of environmental contaminants on spermatogenesis.” Seminars in Cell & Developmental Biology 59: 126–140. [DOI] [PubMed] [Google Scholar]
- Jeng HA (2014). “Exposure to endocrine disrupting chemicals and male reproductive health.” Front Public Health 2: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JH, Kondo F and Katayama Y (2006). “Human exposure to bisphenol A.” Toxicology 226(2-3): 79–89. [DOI] [PubMed] [Google Scholar]
- Kitamura S, Suzuki T, Sanoh S, Kohta R, Jinno N, Sugihara K, Yoshihara S, Fujimoto N, Watanabe H and Ohta S (2005). “Comparative study of the endocrine-disrupting activity of bisphenol A and 19 related compounds.” Toxicological Sciences 84(2): 249–259. [DOI] [PubMed] [Google Scholar]
- Kleymenova E, Swanson C, Boekelheide K and Gaido KW (2005). “Exposure in utero to di(n-butyl) phthalate alters the vimentin cytoskeleton of fetal rat Sertoli cells and disrupts Sertoli cell-gonocyte contact.” Biol Reprod 73(3): 482–490. [DOI] [PubMed] [Google Scholar]
- Knez J (2013). “Endocrine-disrupting chemicals and male reproductive health.” Reprod Biomed Online 26(5): 440–448. [DOI] [PubMed] [Google Scholar]
- Komatsu T, Tabata M, Kubo-Irie M, Shimizu T, Suzuki K, Nihei Y and Takeda K (2008). “The effects of nanoparticles on mouse testis Leydig cells in vitro.” Toxicology in Vitro 22(8): 1825–1831. [DOI] [PubMed] [Google Scholar]
- Lakind JS and Naiman DQ (2011). “Daily intake of bisphenol A and potential sources of exposure: 2005–2006 National Health and Nutrition Examination Survey.” J Expo Sci Environ Epidemiol 21(3): 272–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Richburg JH, Shipp EB, Meistrich ML and Boekelheide K (1999). “The Fas system, a regulator of testicular germ cell apoptosis, is differentially up-regulated in Sertoli cell versus germ cell injury of the testis.” Endocrinology 140(2): 852–858. [DOI] [PubMed] [Google Scholar]
- Lee S, Liu X, Takeda S and Choi K (2013). “Genotoxic potentials and related mechanisms of bisphenol A and other bisphenol compounds: a comparison study employing chicken DT40 cells.” Chemosphere 93(2): 434–440. [DOI] [PubMed] [Google Scholar]
- Leung MCK, Phuong J, Baker NC, Sipes NS, Klinefelter GR, Martin MT, McLaurin KW, Setzer RW, Darney SP, Judson RS and Knudsen TB (2016). “Systems Toxicology of Male Reproductive Development: Profiling 774 Chemicals for Molecular Targets and Adverse Outcomes.” Environmental Health Perspectives 124(7): 1050–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S, Yin L, Shengyang Yu K, Hofmann M-C and Yu X (2017). “High-Content Analysis Provides Mechanistic Insights into the Testicular Toxicity of Bisphenol A and Selected Analogues in Mouse Spermatogonial Cells.” Toxicological Sciences 155(1): 43–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S, Yin L, Shengyang Yu K, Hofmann MC and Yu X (2016). “High-Content Analysis Provides Mechanistic Insights into the Testicular Toxicity of Bisphenol A and Selected Analogues in Mouse Spermatogonial Cells.” Toxicol Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S, Yin L, Shengyang Yu K, Hofmann MC and Yu X (2017). “High-Content Analysis Provides Mechanistic Insights into the Testicular Toxicity of Bisphenol A and Selected Analogues in Mouse Spermatogonial Cells.” Toxicol Sci 155(1): 43–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao CY, Liu F, Alomirah H, Loi VD, Mohd MA, Moon HB, Nakata H and Kannan K (2012). “Bisphenol S in Urine from the United States and Seven Asian Countries: Occurrence and Human Exposures.” Environmental Science & Technology 46(12): 6860–6866. [DOI] [PubMed] [Google Scholar]
- Liao CY, Liu F, Guo Y, Moon HB, Nakata H, Wu Q and Kannan K (2012). “Occurrence of Eight Bisphenol Analogues in Indoor Dust from the United States and Several Asian Countries: Implications for Human Exposure.” Environmental Science & Technology 46(16): 9138–9145. [DOI] [PubMed] [Google Scholar]
- Liu K, Zhang G, Wang Z, Liu Y, Dong J, Dong X, Liu J, Cao J, Ao L and Zhang S (2014). “The protective effect of autophagy on mouse spermatocyte derived cells exposure to 1800MHz radiofrequency electromagnetic radiation.” Toxicol Lett 228(3): 216–224. [DOI] [PubMed] [Google Scholar]
- Liu ML, Wang JL, Wei J, Xu LL, Yu M, Liu XM, Ruan WL and Chen JX (2015). “Tri-ortho-cresyl phosphate induces autophagy of rat spermatogonial stem cells.” Reproduction 149(2): 163–170. [DOI] [PubMed] [Google Scholar]
- M I, T S, K E, M M and N K (1999). “Computerized nuclear morphometry is a useful technique for evaluating the high metastatic potential of colorectal adenocarcinoma.” Cancer 86(10). [DOI] [PubMed] [Google Scholar]
- Makarovskiy AN, Siryaporn E, Hixson DC and Akerley W (2002). “Survival of docetaxel-resistant prostate cancer cells in vitro depends on phenotype alterations and continuity of drug exposure.” Cell Mol Life Sci 59(7): 1198–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M and Barbacid M (2005). “Mammalian cyclin-dependent kinases.” Trends Biochem Sci 30(11): 630–641. [DOI] [PubMed] [Google Scholar]
- Mather JP (1980). “Establishment and characterization of two distinct mouse testicular epithelial cell lines.” Biol Reprod 23(1): 243–252. [DOI] [PubMed] [Google Scholar]
- McClusky LM (2011). “Testicular degeneration during spermatogenesis in the blue shark, Prionace glauca: nonconformity with expression as seen in the diametric testes of other carcharhinids.” J Morphol 272(8): 938–948. [DOI] [PubMed] [Google Scholar]
- Medh RD, Santell L and Levin EG (1992). “Stimulation of Tissue Plasminogen-Activator Production by Retinoic Acid - Synergistic Effect on Protein-Kinase-C Mediated Activation.” Blood 80(4): 981–987. [PMC free article] [PubMed] [Google Scholar]
- Meeker JD, Ehrlich S, Toth TL, Wright DL, Calafat AM, Trisini AT, Ye XY and Hauser R (2010). “Semen quality and sperm DNA damage in relation to urinary bisphenol A among men from an infertility clinic.” Reproductive Toxicology 30(4): 532–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miething A (1993). “Multinucleated spermatocytes in the aging human testis: formation, morphology, and degenerative fate.” Andrologia 25(6): 317–323. [DOI] [PubMed] [Google Scholar]
- Miething A (1995). “Multinuclearity of germ cells in the senescent human testis originates from a process of cell-cell fusion.” J Submicrosc Cytol Pathol 27(1): 105–113. [PubMed] [Google Scholar]
- Mizushima N and Komatsu M (2011). “Autophagy: Renovation of Cells and Tissues.” Cell 147(4): 728–741. [DOI] [PubMed] [Google Scholar]
- Mizushima N and Levine B (2010). “Autophagy in mammalian development and differentiation.” Nature Cell Biology 12(9): 823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore-Ambriz TR, Acuna-Hernandez DG, Ramos-Robles B, Sanchez-Gutierrez M, Santacruz-Marquez R, Sierra-Santoyo A, Pina-Guzman B, Shibayama M and Hernandez-Ochoa I (2015). “Exposure to bisphenol A in young adult mice does not alter ovulation but does alter the fertilization ability of oocytes.” Toxicology and Applied Pharmacology 289(3): 507–514. [DOI] [PubMed] [Google Scholar]
- Moreman J, Lee O, Trznadel M, David A, Kudoh T and Tyler CR (2017). “Acute Toxicity, Teratogenic, and Estrogenic Effects of Bisphenol A and Its Alternative Replacements Bisphenol S, Bisphenol F, and Bisphenol AF in Zebrafish Embryo-Larvae.” Environ Sci Technol 51(21): 12796–12805. [DOI] [PubMed] [Google Scholar]
- Morris TA, DeLorenzo RJ and Tombes RM (1998). “CaMK-II inhibition reduces cyclin D1 levels and enhances the association of p27kip1 with Cdk2 to cause G1 arrest in NIH 3T3 cells.” Exp Cell Res 240(2): 218–227. [DOI] [PubMed] [Google Scholar]
- Mylchreest E, Sar M, Wallace DG and Foster PMD (2002). “Fetal testosterone insufficiency and abnormal proliferation of Leydig cells and gonocytes in rats exposed to di(n-butyl) phthalate.” Reproductive Toxicology 16(1): 19–28. [DOI] [PubMed] [Google Scholar]
- Nacov E (1990). “Tumor angiogenesis formation of vessels de novo at germ cell tumors.” Cancer 66(5): 916–922. [DOI] [PubMed] [Google Scholar]
- Nistal M, Gonzalez-Peramato P, Regadera J, Serrano A, Tarin V and De Miguel MP (2006). “Primary testicular lesions are associated with testicular germ cell tumors of adult men.” Am J Surg Pathol 30(10): 1260–1268. [DOI] [PubMed] [Google Scholar]
- NJ B, BS M and PM F (2004). “Male reproductive tract lesions at 6, 12, and 18 months of age following in utero exposure to di(n-butyl) phthalate.” Toxicologic pathology 32(1). [DOI] [PubMed] [Google Scholar]
- Nordkap L, Joensen UN, Jensen MB and Jorgensen N (2012). “Regional differences and temporal trends in male reproductive health disorders: Semen quality may be a sensitive marker of environmental exposures.” Molecular and Cellular Endocrinology 355(2): 221–230. [DOI] [PubMed] [Google Scholar]
- O’Brien PJ, Irwin W, Diaz D, Howard-Cofield E, Krejsa CM, Slaughter MR, Gao B, Kaludercic N, Angeline A, Bernardi P, Brain P and Hougham C (2006). “High concordance of drug-induced human hepatotoxicity with in vitro cytotoxicity measured in a novel cell-based model using high content screening.” Archives of Toxicology 80(9): 580–604. [DOI] [PubMed] [Google Scholar]
- Oakes CC, La Salle S, Smiraglia DJ, Robaire B and Trasler JM (2007). “A unique configuration of genome-wide DNA methylation patterns in the testis.” Proceedings of the National Academy of Sciences of the United States of America 104(1): 228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owczarek K, Kubica P, Kudlak B, Rutkowska A, Konieczna A, Rachon D, Namiesnik J and Wasik A (2018). “Determination of trace levels of eleven bisphenol A analogues in human blood serum by high performance liquid chromatography-tandem mass spectrometry.” Sci Total Environ 628-629: 1362–1368. [DOI] [PubMed] [Google Scholar]
- Pelch K, Wignall JA, Goldstone AE, Ross PK, Blain RB, Shapiro AJ, Holmgren SD, Hsieh JH, Svoboda D, Auerbach SS, Parham FM, Masten SA, Walker V, Rooney A and Thayer KA (2019). “A scoping review of the health and toxicological activity of bisphenol A (BPA) structural analogues and functional alternatives.” Toxicology 424: 152235. [DOI] [PubMed] [Google Scholar]
- Pelch KE, Wignall JA, Goldstone AE, Ross PK, Blain RB, Shapiro AJ, Holmgren SD, Hsieh JH, Svoboda D, Auerbach SS, Parham FM, Masten SA and Thayer KA (2017). NTP Research Report on Biological Activity of Bisphenol A (BPA) Structural Analogues and Functional Alternatives: Research Report 4. Research Triangle Park (NC). [PubMed] [Google Scholar]
- Peretz J, Vrooman L, Ricke WA, Hunt PA, Ehrlich S, Hauser R, Padmanabhan V, Taylor HS, Swan SH, VandeVoort CA and Flaws JA (2014). “Bisphenol A and Reproductive Health: Update of Experimental and Human Evidence, 2007-2013.” Environmental Health Perspectives 122(8): 775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer D, Chung YM and Hu MCT (2015). “Effects of Low-Dose Bisphenol A on DNA Damage and Proliferation of Breast Cells: The Role of c-Myc.” Environmental Health Perspectives 123(12): 1271–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Program NT (2021). DART-08: Developmental and Reproductive Toxicity Growth and Clinical Finding Tables (I), Pathology Tables (PA), Developmental and Reproductive Tables (R) from NTP Modified One Generation Dose Range Finding Study and Modified One Generation Main Study Studies.
- Qi SQ, Fu WJ, Wang CM, Liu CJ, Quan C, Kourouma A, Yan MS, Yu TT, Duan P and Yang KD (2014). “BPA-induced apoptosis of rat Sertoli cells through Fas/FasL and JNKs/p38 MAPK pathways.” Reproductive Toxicology 50: 108–116. [DOI] [PubMed] [Google Scholar]
- Quan C, Wang C, Duan P, Huang W, Chen W, Tang S and Yang K (2016). “Bisphenol a induces autophagy and apoptosis concurrently involving the Akt/mTOR pathway in testes of pubertal SD rats.” Environ Toxicol. [DOI] [PubMed] [Google Scholar]
- Reis MMS, Moreira AC, Sousa M, Mathur PP, Oliveira PF and Alves MG (2015). “Sertoli cell as a model in male reproductive toxicology: Advantages and disadvantages.” Journal of Applied Toxicology 35(8): 870–883. [DOI] [PubMed] [Google Scholar]
- Rochester JR (2013). “Bisphenol A and human health: A review of the literature.” Reproductive Toxicology 42: 132–155. [DOI] [PubMed] [Google Scholar]
- Rubin BS (2011). “Bisphenol A: an endocrine disruptor with widespread exposure and multiple effects.” J Steroid Biochem Mol Biol 127(1-2): 27–34. [DOI] [PubMed] [Google Scholar]
- Santoro A, Chianese R, Troisi J, Richards S, Nori SL, Fasano S, Guida M, Plunk E, Viggiano A, Pierantoni R and Meccariello R (2019). “Neuro-toxic and Reproductive Effects of BPA.” Curr Neuropharmacol 17(12): 1109–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setchell BP (2008). “Blood-Testis Barrier, Junctional and Transport Proteins and Spermatogenesis.” Molecular Mechanisms in Spermatogenesis 636: 212–233. [DOI] [PubMed] [Google Scholar]
- Shi ZX, Jiao Y, Hu Y, Sun ZW, Zhou XQ, Feng JF, Li JG and Wu YN (2013). “Levels of tetrabromobisphenol A, hexabromocyclododecanes and polybrominated diphenyl ethers in human milk from the general population in Beijing, China.” Science of the Total Environment 452: 10–18. [DOI] [PubMed] [Google Scholar]
- Siracusa JS, Yin L, Measel E, Liang S and Yu X (2018). “Effects of bisphenol A and its analogs on reproductive health: A mini review.” Reprod Toxicol 79: 96–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skakkebaek NE, Rajpert-De Meyts E, Louis GMB, Toppari J, Andersson AM, Eisenberg ML, Jensen TK, Jorgensen N, Swan SH, Sapra KJ, Ziebe S, Priskorn L and Juul A (2016). “Male Reproductive Disorders and Fertility Trends: Influences of Environment and Genetic Susceptibility.” Physiological Reviews 96(1): 55–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spade DJ, Hall SJ, Wilson S and Boekelheide K (2015). “Di-n-Butyl Phthalate Induces Multinucleated Germ Cells in the Rat Fetal Testis Through a Nonproliferative Mechanism.” Biol Reprod 93(5): 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storchova Z and Pellman D (2004). “From polyploidy to aneuploidy, genome instability and cancer.” Nat Rev Mol Cell Biol 5(1): 45–54. [DOI] [PubMed] [Google Scholar]
- Stossi F, Bolt MJ, Ashcroft FJ, Lamerdin JE, Melnick JS, Powell RT, Dandekar RD, Mancini MG, Walker CL, Westwick JK and Mancini MA (2014). “Defining Estrogenic Mechanisms of Bisphenol A Analogs through High Throughput Microscopy-Based Contextual Assays.” Chemistry & Biology 21(6): 743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Kovacs T, Hu YJ and Yang WX (2011). “The role of actin and myosin during spermatogenesis.” Molecular Biology Reports 38(6): 3993–4001. [DOI] [PubMed] [Google Scholar]
- T T, W N, I N, K A, K K and K H (1999). “Exposure with the environmental estrogen bisphenol A disrupts the male reproductive tract in young mice.” Life sciences 65(22). [DOI] [PubMed] [Google Scholar]
- Tang WY, Morey LM, Cheung YY, Birch L, Prins GS and Ho SM (2012). “Neonatal Exposure to Estradiol/Bisphenol A Alters Promoter Methylation and Expression of Nsbp1 and Hpcal1 Genes and Transcriptional Programs of Dnmt3a/b and Mbd2/4 in the RatProstate Gland Throughout Life.” Endocrinology 153(1): 42–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigezzi L, Bosquiazzo VL, Kass L, Ramos JG, Munoz-De-Toro M and Luque EH (2015). “Developmental exposure to bisphenol A alters the differentiation and functional response of the adult rat uterus to estrogen treatment.” Reproductive Toxicology 52: 83–92. [DOI] [PubMed] [Google Scholar]
- Vogl AW (1989). “Distribution and Function of Organized Concentrations of Actin-Filaments in Mammalian Spermatogenic Cells and Sertoli Cells.” International Review of Cytology-a Survey of Cell Biology 119: 1–56. [DOI] [PubMed] [Google Scholar]
- Wang T, Han J, Duan X, Xiong B, Cui XS, Kim NH, Liu HL and Sun SC (2016). “The toxic effects and possible mechanisms of Bisphenol A on oocyte maturation of porcine in vitro.” Oncotarget 7(22): 32554–32565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Mruk DD, Tang EI, Wong CKC, Lee WM, John CM, Turek PJ, Silvestrini B and Cheng CY (2014). “Environmental toxicants perturb human Sertoli cell adhesive function via changes in F-actin organization mediated by actin regulatory proteins.” Human Reproduction 29(6): 1279–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki K, Noda S, Imatanaka N and Yakabe Y (2004). “Comparative study of the uterotrophic potency of 14 chemicals in a uterotrophic assay and their receptor-binding affinity.” Toxicology Letters 146(2): 111–120. [DOI] [PubMed] [Google Scholar]
- Yang Y, Shi Y, Chen D, Chen H and Liu X (2022). “Bisphenol A and its analogues in paired urine and house dust from South China and implications for children’s exposure.” Chemosphere 294: 133701. [DOI] [PubMed] [Google Scholar]
- Ye X, Wong LY, Kramer J, Zhou X, Jia T and Calafat AM (2015). “Urinary Concentrations of Bisphenol A and Three Other Bisphenols in Convenience Samples of U.S. Adults during 2000-2014.” Environmental Science & Technology 49(19): 11834–11839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin B, Ma Q, Zhao L, Song C, Wang C, Yu F, Shi Y and Ye L (2021). “Epigenetic Control of Autophagy Related Genes Transcription in Pulpitis via JMJD3.” Front Cell Dev Biol 9: 654958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L, Siracusa JS, Measel E, Guan X, Edenfield C, Liang S and Yu X (2020). “High-Content Image-Based Single-Cell Phenotypic Analysis for the Testicular Toxicity Prediction Induced by Bisphenol A and Its Analogs Bisphenol S, Bisphenol AF, and Tetrabromobisphenol A in a Three-Dimensional Testicular Cell Co-culture Model.” Toxicol Sci 173(2): 313–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Ling X, Liu K, Wang Z, Zou P, Gao J, Cao J and Ao L (2016). “The p-eIF2alpha/ATF4 pathway links endoplasmic reticulum stress to autophagy following the production of reactive oxygen species in mouse spermatocyte-derived cells exposed to dibutyl phthalate.” Free Radic Res 50(7): 698–707. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhang Y, Li J and Yang M (2019). “Occurrence and exposure assessment of bisphenol analogues in source water and drinking water in China.” Sci Total Environ 655: 607–613. [DOI] [PubMed] [Google Scholar]
- Zhang W, Lin Q, Ramoth AJ, Fan D and Fidler IJ (2011). “Formation of solid tumors by a single multinucleated cancer cell.” Cancer 117(17): 4092–4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZZ, Gong YH, Guo Y, Hai YN, Yang H, Yang S, Liu Y, Ma M, Liu LH, Li Z, Gao WQ and He ZP (2013). “Direct transdifferentiation of spermatogonial stem cells to morphological, phenotypic and functional hepatocyte-like cells via the ERK1/2 and Smad2/3 signaling pathways and the inactivation of cyclin A, cyclin B and cyclin E.” Cell Communication and Signaling 11. [DOI] [PMC free article] [PubMed] [Google Scholar]