

Abstract
Overactive inflammatory responses are central to the pathophysiology of many hemolytic conditions including sickle cell disease. Excessive hemolysis leads to elevated serum levels of heme due to saturation of heme scavenging mechanisms. Extracellular heme has been shown to activate the NLRP3 inflammasome leading to activation of caspase-1 and release of pro-inflammatory cytokines IL-1β and IL-18. Heme also activates the non-canonical inflammasome pathway which may contribute to NLRP3 inflammasome formation and leads to pyroptosis, a type of inflammatory cell death. Some clinical studies indicate there is a benefit to blocking the NLRP3 inflammasome pathway in patients with sickle cell disease and other hemolytic conditions. However, a thorough understanding of the mechanisms of heme-induced inflammasome activation is needed to fully leverage this pathway for clinical benefit. This review will explore the mechanisms of heme-induced NLRP3 inflammasome activation and the role of this pathway in hemolytic conditions including sickle cell disease.
Keywords: Heme, hemolysis, inflammatory caspase, inflammasome, sickle cell disease
Introduction
Excessive intravascular hemolysis is characteristic of many disease states including malaria and sickle cell disease. Hemolysis refers to the breakdown or rupture of red blood cells (erythrocytes) and, when it occurs in excess, this process can result in the activation of inflammatory responses within the vasculature and in tissues. Unchecked inflammation resulting from hemolysis is central to the pathophysiology of many hemolytic conditions. Inflammation can be induced by PAMPs (pathogen associated molecular patterns), which include bacterial, fungal, and viral components, or DAMPs (damage associated molecular patterns), which are generally endogenous signals released by dying cells [1]. Heme is a DAMP found in erythrocytes that is released following hemolysis [2]. Extracellular heme can induce sterile inflammation, or inflammation in the absence of infection, by activating a group of proteins known as the inflammatory caspases [3, 4]. The inflammatory caspases are a subset of the caspase (cysteinyl aspartate protease) family and induce cytokine release and inflammatory cell death following activation. The best characterized member of the inflammatory caspase subset is caspase-1 [5]. Caspase-1 is activated by inflammasomes, multi-protein complexes that assemble in response to PAMPs and DAMPs [6]. Patients with sickle cell disease (SCD) have elevated levels of inflammatory markers related to the inflammatory caspase pathway [7, 8]. Furthermore, these patients have high levels of extracellular heme [9, 10]. This suggests that heme-induced inflammatory caspase activation is a crucial mediator of inflammation in patients with SCD. This review will summarize the recent literature on how heme and other products of hemolysis promote inflammasome signaling in hemolytic conditions like SCD.
Heme: Definition, synthesis, and function
The term heme refers to a cyclic arrangement of four pyrrole rings (porphyrin) with an iron atom at its center (Figure 1). Heme is essential for a wide range of functions that include oxygen transport, oxygen storage, electron transport, and gene regulation [11, 12]. Based on the nature of heme’s substituents, heme can be classified into different variants with distinct biological functions. The most biologically relevant and abundant form of heme is heme B, which is the form found in hemoglobin (Hb) and myoglobin (Mb) proteins. The heme group in Hb contains an iron molecule in the Fe(II) ferrous state, which allows for reversible binding of iron with molecular oxygen [13]. The reactivity of this central iron ion is essential for many cellular processes; however, it also makes heme a potentially damaging molecule when released from hemoproteins, as it can catalyze the generation of reactive oxygen species [14]. One of the ways it does this is through the Fenton reaction which produces highly toxic free hydroxyl radicals from hydrogen peroxide and ferrous iron [15].
Figure 1. Heme production and catabolism.
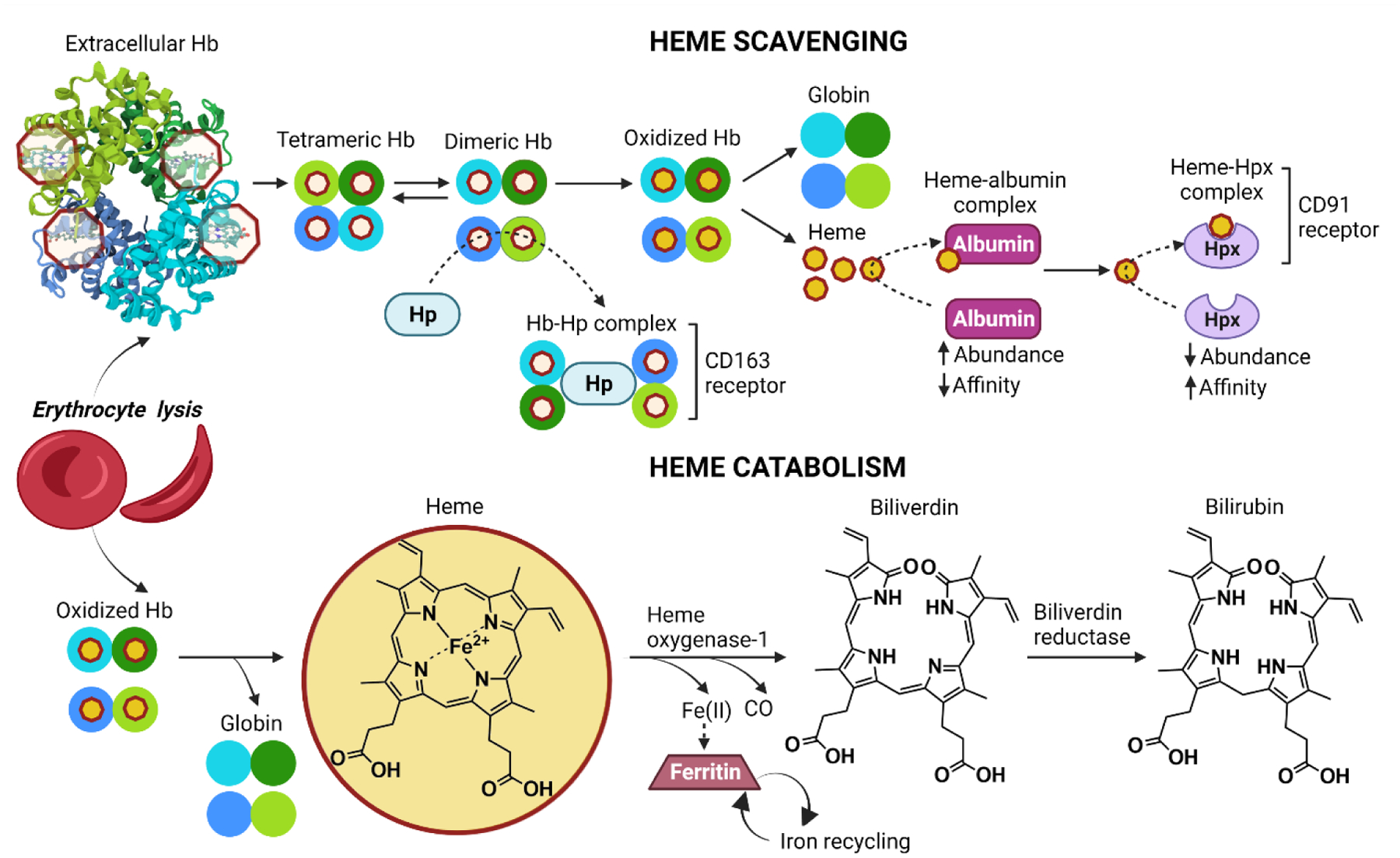
Upon erythrocyte lysis, extracellular tetrameric Hb dissociates into dimers that exist in a dynamic equilibrium with the tetramers. These dimers bind tightly to haptoglobin (Hp) leading to CD163-mediated clearance of the Hb-Hp complex from circulation by macrophages. Depletion of Hp leads to the accumulation and oxidation of extracellular Hb, which results in the release of heme. This heme binds to albumin and is subsquently sequestered by tight binding to hemopexin (Hpx). The heme-Hpx complex binds to the CD91 receptor on macrophages promoting internalization to clear heme from circulation. After uptake, the Hb-Hp and the heme-Hpx complexes are degraded releasing heme. Heme is then catabolized by heme oxygenase-1 (HO-1), producing carbon monoxide (CO), ferrous iron (Fe(II)), biliverdin, and its reduced form bilirubin. The resulting Fe(II) is sequestered by ferritin and recycled back into circulation.
About 80% of heme in the human body is produced and found in erythrocytes and there are approximately 2.5 × 1022 heme molecules in the human body [16–18]. Under physiological conditions, the majority of heme is bound to intracellular proteins. Therefore, the concentration of extracellular heme is low under normal circumstances [18]. However, given the high content of heme in erythrocytes, under conditions of excess hemolysis, the pool of extracellular heme increases significantly [19]. Hemolysis can occur extravascularly or intravascularly. Extravascular hemolysis occurs when damaged erythrocytes are cleared from the circulation by hepatic and splenic macrophages. In contrast, intravascular hemolysis occurs when erythrocytes lyse while in circulation and they release their contents into the vasculature [14, 20].
Upon intravascular hemolysis, tetrameric Hb dissociates into dimers and is quickly scavenged by the serum protein haptoglobin (Hp) in an irreversible complex to prevent extravascular translocation of the Hb dimers. The Hp-Hb complexes are then recognized and cleared from circulation by macrophages of the reticuloendothelial system via the CD163 receptor, followed by lysosomal degradation of the Hp-Hb complexes and heme catabolism (Figure 1) [21, 22]. Haptoglobin cannot be recycled so haptoglobin levels can be depleted following extensive hemolysis. This allows free Hb to accumulate. Hb in the plasma is readily oxidized to higher Hb iron oxidation states, such as metHb (FeIII) and ferrylHb (FeIV).These non-functional forms of Hb are unable to bind molecular oxygen and easily release associated heme groups [23]. Heme released from Hb is often referred to as free heme. However, due to its high hydrophobicity, extracellular heme is usually found loosely bound to plasma acceptor proteins and lipids, and not in its fully free form. This loosely bound form of heme is redox active, readily exchangeable with other proteins and is often called labile heme. Once released from Hb, heme loosely binds to albumin, the most abundant protein in plasma, to be subsequently transferred to hemopexin (Hpx), the most effective heme-binding protein, which is also present in plasma but at considerably lower concentrations [18, 24]. Hpx-heme complexes are recognized and cleared from circulation via binding to and internalization of the scavenger receptor CD91 on hepatocytes and macrophages. This is followed by heme catabolism, which results in heme breakdown by the heme oxygenase-1 (HO-1) system and ferrous iron recycling by ferritin [21, 22, 25, 26]. During excessive hemolysis, the heme scavenging proteins are saturated and depleted, allowing free heme to accumulate in the plasma and to form low affinity complexes with plasma proteins that fail to restrain its redox activity [17]. Free heme is toxic to cells due to its pro-oxidant and pro-inflammatory potential [27]. Heme’s cytotoxicity is aggravated by its extreme hydrophobicity, which allows it to easily enter the phospholipid bilayer and to intercalate into lipid membranes [19]. Heme toxicity contributes to the activation of the innate immune response, cytokine release, and tissue damage.
Heme activates the NLRP3 inflammasome
Inflammasome formation is initiated by pattern recognition receptors (PRRs) that recognize fixed patterns in molecules that are associated with damage or infection [28]. Heme induces the assembly of the NLRP3 (Nod like receptor protein with a PYRIN domain 3) inflammasome, that is comprised of the PRR NLRP3, the adaptor protein ASC (Apoptosis-associated Speck-like protein containing a CARD [Caspase Activation and Recruitment Domain]) and a member of the inflammatory caspase subgroup, caspase-1 [3, 4, 29, 30].
In most contexts, NLRP3 activation occurs in two steps. First, a priming signal engages various PRRs, which are usually at the plasma membrane, including toll-like receptors (TLRs) and cytokine receptors. These receptors converge on the activation of the transcription factor NF-κB. NF-κB is responsible for the expression of a wide array of proteins involved in inflammatory responses, including proteins required for NLRP3 inflammasome signaling. After a priming signal, the NLRP3 inflammasome is activated by a secondary intracellular signal. The priming signal required for expression of inflammasome components is referred to as Signal 1 and the secondary signal that induces NLRP3 inflammasome formation is referred to as Signal 2. There is extensive structural diversity amongst the molecules that activate Signal 2 and the precise upstream pathways that lead to inflammasome activation remain unknown [31]. Potassium efflux and mitochondrial reactive oxygen species (mtROS) formation have both been proposed as universal activating signals for the NLRP3 inflammasome [31]. However, each has also been shown to be dispensable in certain contexts and no single signal has been conclusively shown to universally induce activation [31].
The protein NLRP3 is present in the cell in a closed conformation. NLRP3 activation signals induce a conformational change that exposes its oligomerization domain and a binding motif known as the PYRIN domain. This allows NLRP3 to oligomerize and bind to ASC via interactions between the PYRIN domains present in both molecules [32]. ASC then binds pro-caspase-1 via CARD-CARD interactions. Binding to this scaffold allows for proximity-induced pro-caspase-1 dimerization followed by auto-cleavage to form an active heterodimer enzyme [33].
Once activated, caspase-1 cleaves the inflammatory cytokines pro-interleukin(IL)-1β and pro-IL-18 into their active forms [34]. Through binding to its receptor IL-1R, IL-1β has a variety of functions including the recruitment of innate immune cells to sites of inflammation, the induction of pro-inflammatory enzymes, and the regulation of T cell responses [35]. IL-18 also contributes to diverse inflammatory processes including production of IFN-γ by various immune cells, mast cell activation, and protection against extracellular pathogens [36]. Active caspase-1 also cleaves and activates the pore-forming protein, gasdermin D (GSDMD) [37]. Cleaved GSDMD inserts into cell membranes to form pores, facilitating the release of processed cytokines [38]. Extensive pore formation also results in an influx of ions leading to cell swelling and culminating in a form of inflammatory cell death known as pyroptosis [38].
Heme has been shown to induce NLRP3 activation and IL-1β release in a variety of cell types including human and mouse macrophages as well as endothelial cells [3, 4, 29, 30]. Heme-induced IL-1β release is dependent on the NLRP3 inflammasome (Figure 2) [3, 29]. Deficiency in NLRP3, ASC, or caspase-1 prevents heme-induced IL-1β release in macrophages [3, 29]. While NLRP3-dependent IL-1β release has also been detected from endothelial cells, the levels of IL-1β released were significantly lower than levels released by macrophages after heme exposure [29]. Other components of the inflammasome pathway, like IL-18 release, may be more important in endothelial cells. Mouse models have also demonstrated a clear relationship between heme, hemolysis, and inflammasome activation [3]. In mice, heme injection into the peritoneal cavity resulted in extensive neutrophil recruitment to the peritoneal cavity, which was abrogated by ASC deficiency [3]. Additionally, Nlrp3−/−, Asc−/−, and Casp1−/− mice are all protected from lethal hemolysis induced by phenylhydrazine. IL-1R knockout also protects mice from hemolysis-induced lethality [3]. This evidence underscores how free heme and the NLRP3 inflammasome pathway are key players in adverse inflammatory responses following hemolysis.
Figure 2. Proposed mechanism of heme-induced NLRP3 activation.

Signal 1 is initiated by heme binding to RAGE or the MD-2/TLR4 complex to activate the NF-kB pathway. NF-κB induces expression of pro-IL-1β, pro-IL-18 and NLRP3. S100A8 amplifies heme-induced priming. This pathway constitutes Signal 1. In Signal 2, heme induces mitochondrial reactive oxygen species (mtROS) generation via spleen tyrosine kinase (Syk). The pathway most likely involves Syk activation of NADPH oxidase-2 (NOX2) via protein kinase C (PKC) or phosphoinositide 3 kinase (PI3K) signaling. Active NOX2 then triggers the production of mtROS. Mitochondrial ROS and potassium (K+) efflux induce the assembly of the NLRP3 inflammasome and recruitment of caspase-1. Caspase-1 then cleaves pro-IL-1β and pro-IL-18. In Signal 3, heme activates caspase-4 and caspase-5. Caspase-4 induces cleavage of GSDMD which forms the GSDMD pore and allows for release of active IL-1β and IL-18. Formation of GSDMD pores also facilitates the efflux of potassium ions (K+) that in turn can lead to the activation of the NLRP3 inflammasome and influx of ions inducing pyroptosis. Caspase-5 is required for IL-1β cleavage independent of GSDMD cleavage and may induce NLRP3 activation.
Heme provides a priming signal for inflammasome activation
In most contexts, NLRP3 activation requires priming to induce expression of NLRP3, pro-IL-1β, and other components of the inflammasome pathway [31]. Heme has been shown to initiate a priming signal by activating TLR4 signaling (Signal 1, Figure 2) [39]. TLR4 is a PRR best known for its role in inducing inflammation in response to LPS [40]. The first paper to demonstrate TLR4 activation by heme showed that in macrophages, heme-induced NF-κB activation and subsequent TNFα release was dependent on TLR4 [39]. TLR4 signaling induced by heme also requires the TLR4 co-receptor CD14, the TLR adaptor protein MyD88, and the TLR4 cofactor Myeloid Differentiation Factor-2 (MD-2) [39, 41, 42]. MD-2 is known to bind LPS and induce LPS-dependent TLR4 signaling [43]. Recent evidence suggests that heme induces TLR4 activation by binding to MD-2 [42]. It had been reported that an antibody targeting the TLR4/MD-2 complex blocked LPS-induced TLR4 signaling but did not block heme-induced TLR4 signaling [39]. This suggested that MD-2 is dispensable for heme-induced TLR4 activation in this context. This discrepancy is likely explained by the fact that the predicted heme binding site on MD-2 is distinct from the LPS binding site and may not have been blocked by the anti-TLR4/MD-2 antibody used in the study [39]. Using a luciferase-based reporter assay, a more recent study demonstrated that MD-2 was necessary for heme-induced NF-κB activity [42]. It remains unknown if heme binding to MD-2 alone is sufficient to induce TLR4 activation. The relevance of heme binding to MD-2 has been further demonstrated in samples from patients with SCD. These patients have significantly elevated levels of soluble MD-2 in their plasma, which was shown to help mediate TLR4-dependent cytokine release from endothelial cells [41]. Soluble MD-2 found in the plasma of these patients was also shown to bind exogenous heme [41]. Thus, heme binding to MD-2 is an important regulatory factor for heme-induced TLR4 activation and is likely essential for the downstream expression of inflammasome components. There appears to be exquisite specificity in heme-induced TLR4 signaling. TLR4 is not activated by any other porphyrins including protoporphyrin IX, which is structurally identical to heme except that it lacks iron in the central ring [39]. As expected based on heme’s predicted binding site on MD-2, heme-induced TLR4 activation is distinct from LPS-induced TLR4 activation. Protoporphyrin IX blocks TLR4 activation by heme but not TLR4 activation by LPS [39]. In addition, structural analogues of lipid A, the component of LPS that activates TLR4, prevent TNFα release in response to LPS but not in response to heme [39]. Thus, the activation of TLR4/MD-2 by heme is specific to heme’s molecular structure. These specific interactions between heme, MD-2, and TLR4 are likely important factors in the regulation of the downstream expression of inflammasome components.
Heme also activates the receptor for advanced glycation products (RAGE) [44]. Like TLR4, RAGE is a plasma membrane-associated receptor that binds several PAMPs and DAMPs. RAGE is expressed in most immune cells including neutrophils, macrophages, B cells, and T cells [45]. Additionally, RAGE expression is high in pulmonary tissue where it is expressed in alveolar macrophages, endothelial tissue, and type II pneumocytes [46]. Binding of RAGE to one of its ligands activates various inflammatory pathways including the NF-κB pathway. Heme binds directly to human RAGE in cell-free systems and RAGE-deficient mice release significantly less IL-1β in response to heme [44]. This suggests TLR4 is not the only receptor through which heme can induce a priming signal for inflammasome activation. Heme-induced RAGE signaling may be especially important in the lung where RAGE expression is high [44]. However, it has not been directly investigated if heme-induced activation of RAGE leads to expression of NLRP3. Therefore, it is unclear if RAGE activation is sufficient to prime for heme-induced inflammasome activation.
There is also evidence to suggest that the protein S100A8 is an important co-factor for heme-induced priming. S100A8 has a variety of roles in the immune system that include promoting leukocyte recruitment to sites of inflammation and mediating cytoskeletal rearrangements [47]. Interestingly, the induction of hemolysis in mice was shown to increase circulating S100A8 in a heme-dependent fashion [48]. Similarly, S100A8 levels are elevated in patients with SCD [48]. S100A8 has been shown to signal through both TLR4 and RAGE suggesting that it can function as a DAMP [48]. One study demonstrated that heme exposure increased S100A8 expression in human macrophages and that S100A8 alone significantly increased pro-IL-1β expression [48]. Heme-induced pro-IL-1β expression was also significantly reduced in the presence of an antibody against S100A8 [48]. This suggests that heme facilitates the production of S100A8, which in turn amplifies heme-induced priming. Furthermore, following hemolysis, elevated levels of S100A8 are detected even after free heme levels have normalized [48]. This suggests that S100A8 can continue to mediate priming signals secondary to heme exposure even after free heme has been scavenged.
Although heme can initiate TLR4 signaling, priming signals for inflammasome activation initiated by heme appear to be much weaker than priming signals initiated by other TLR4 ligands. Heme exposure of up to twenty hours did not result in significant IL-1β release without LPS priming in human macrophages and endothelial cells [4, 29, 48]. Although heme is able to induce some increases in IL-1β mRNA levels and pro-IL-1β expression in these cell types, these levels are much lower compared to cells primed with LPS or S100A8 [4, 29, 48]. However, there does seem to be some synergistic activity between heme and other priming agents. In endothelial cells, LPS and heme together induce increases in IL-1β mRNA levels that are much higher than levels induced by each signal alone [29]. Additionally, although the priming signal appears to be important for pro-IL-1β expression and IL-1β release, priming appears to be less important for other components of the inflammasome pathway. In human macrophages, heme exposure induced the same level caspase-1 activation with or without of LPS priming [4]. Thus, although heme is able to initiate TLR4 signaling, other DAMPs released from erythrocytes during hemolysis or from macrophages as a result of initial heme signaling may play larger roles in priming cells for subsequent inflammasome activation.
Mechanisms of heme-induced NLRP3 activation
Heme-induced inflammasome activation requires both potassium efflux and mtROS formation (Figure 2) [3]. Heme induces mtROS production and scavenging of mtROS prevents caspase-1 cleavage and subsequent IL-1β release [3, 29]. However, scavenging of mtROS does not interfere with levels of pro-caspase-1 and pro-IL-1β [3], suggesting that mtROS are necessary for Signal 2 but dispensable for Signal 1. Several proteins have been identified as necessary for heme-induced mtROS formation. One of these is spleen tyrosine kinase (Syk), a protein that initiates a variety of signal transduction pathways in cells of hematopoietic lineage. Syk is known to mediate NLRP3 activation following fungal infection [49]. Syk has been shown to be required for heme-induced mtROS formation and IL-1β release [3]. Syk likely mediates heme-induced mtROS formation through the downstream protein kinase C (PKC) and PI3K signaling pathways [49]. Both of these pathways result in the activation of NADPH oxidases, which are also required for heme-induced IL-1β release [50]. Specifically, activation of NADPH oxidase-2 (NOX2) produces ROS following heme exposure (Figure 2). Although there are many NOX2-independent ways heme can induce ROS formation [23], only NOX2-derived ROS seem to result in inflammasome activation as NOX2 deficiency completely abrogates cleavage of caspase-1 and IL-1β following heme exposure [3]. Other activators of the NLRP3 inflammasome, like ATP, also require mtROS formation in order to induce IL-1β cleavage. However, ATP-induced IL-1β cleavage is independent of Syk and NOX2 [3]. This suggests that these two factors are not conserved between heme and all other NLRP3 activators. It is not yet clear if heme-induced potassium efflux and mtROS formation are part of the same pathway or if heme is inducing two separate signals required for inflammasome formation. There is at least one study to suggest that cellular ROS exposure can lead to potassium efflux [51]. Conversely, potassium efflux may lead to activation of signaling pathways that cause ROS formation.
It remains to be shown if heme is mediating NLRP3 activation via binding to plasma membrane bound PRRs or if heme internalization is required to initiate signaling. Although heme can intercalate into plasma membranes, free heme is not able to efficiently cross membranes [52]. Other NLRP3 activators, including silica crystals, require internalization by endocytosis to initiate activation [3]. Treatment with an inhibitor of actin polymerization, which should block all forms of endocytosis including phagocytosis, did not prevent heme-induced IL-1β release in macrophages [3]. Furthermore, inhibition of phagosomal maturation, phagolysosomal fusion, and the lysosomal protease cathepsin all prevent silica-induced inflammasome activation but do not prevent heme-induced activation [3]. While endocytosis may be the primary way for foreign particulate matter to enter cells, several cells express membrane-bound proteins that may facilitate heme import into a cell [53]. Heme internalization could be mediated by the putative heme import proteins HCP1 (heme carrier protein 1) and FLVCR2 (Feline Leukemia Virus Subgroup C Cellular Receptor 2), both of which are expressed on the plasma membrane in macrophages [17]. HCP-1 is a low-affinity heme transporter known to play a role in intestinal cells [54] while the affinity of FLVCR2 for heme and the contexts in which it may be relevant remain unknown [17].
Heme activates the non-canonical inflammasome pathway
In addition to caspase-1, the inflammatory caspases include caspase-4 and caspase-5 in humans and caspase-11 in mice. Caspase-4, caspase-5, and caspase-11 are best known for inducing inflammatory responses after exposure to intracellular LPS [55–58]. Activation of these three proteins is termed non-canonical inflammasome activation because it does not require any known inflammasome components [56]. In fact, LPS binds directly to caspase-4, caspase-5, and caspase-11 to induce their oligomerization and activation in the absence of any adaptor or scaffold proteins [56]. Once activated, caspase-4, caspase-5, and caspase-11 induce GSDMD pore formation [56, 59]. Although none of these three caspases directly cleave IL-1β, they are required for IL-1β cleavage after LPS exposure [58, 60–62]. This is likely explained by potassium efflux through GSDMD pores, formed as a result of non-canonical pathway activation, promoting NLRP3 inflammasome formation and IL-1β cleavage [58, 61].
Heme is also able to activate the non-canonical inflammasome pathway, making it one of the only known DAMPs to stimulate this pathway (Figure 2) [4]. In response to heme, caspase-1, caspase-4, and caspase-5 all undergo activation in human macrophages [4]. Silencing of NLRP3 or ASC in macrophages prevents caspase-1 activation but does not disrupt heme-induced caspase-4 or −5 activation. Thus, similar to caspase-4 and −5 activation by LPS, activation of these proteins by heme is independent of canonical inflammasomes. However, it is unknown if heme can directly bind caspase-4 and caspase-5 or if activation of these proteins is due to engagement of unknown cytoplasmic factors.
Heme-induced IL-1β release in macrophages is dependent on caspase-4 and caspase-5 [4]. However, the role of each of these caspases in promoting IL-1β release appears to be different. Caspase-4 deficiency inhibits heme-induced GSDMD cleavage but does not prevent heme-induced IL-1β cleavage [4]. In contrast, caspase-5 deficiency does not significantly inhibit heme-induced GSDMD cleavage but inhibits heme-induced IL-1β cleavage. This suggests that caspase-4 and caspase-5 have different functions in response to heme: Heme activates caspase-4, resulting in GSDMD cleavage, pore formation, and subsequent IL-1β release, while caspase-5 facilitates caspase-1-mediated IL-1β cleavage and release independent of GSDMD cleavage. We propose that the activation of the non-canonical pathway represents a third signal (Signal 3) required for an intact inflammatory response to heme (Figure 2).
Heme-induced cell death and DAMP release
Out of the three inflammatory caspases, only caspase-4 deficiency significantly decreases the amount of heme-induced cell death observed in macrophages [4]. Coupled with the fact that only caspase-4 induces GSDMD cleavage in response to heme, caspase-4 appears to be the only inflammatory caspase responsible for inducing pyroptosis in response to heme.
There is evidence that heme can induce IL-1β release without causing cell death [29]. In human umbilical vein endothelial cells, concentrations of heme that do not cause changes in cell viability are still able to induce IL-1β release [29]. The release of IL-1β from living cells has been demonstrated in macrophages exposed to S. aureus mutants lacking the O-acetyltransferase gene. Cells exposed to these bacterial mutants undergo less extensive GSDMD pore formation compared to other NLRP3 activators, which may explain their ability to induce IL-1β release without causing pyroptosis [63]. Thus, it is possible that the ability of heme to induce pyroptosis and cytokine release can be similarly separated.
Importantly, pyroptosis is not the only form of cell death induced by heme. A pan-caspase inhibitor does not completely abrogate cell death after heme exposure demonstrating that heme is also able to induce caspase-independent cell death [4]. Heme has been shown to induce necroptosis, a distinct form of inflammatory cell death [64]. In this pathway, the kinases RIPK1 and RIPK3 form a complex that phosphorylates the effector protein MLKL, resulting in pore formation and cell death [65]. Heme is able to induce TNFα production and TNFR1 activation by TNFα can lead to either apoptosis or necroptosis. Heme-induced macrophage death was shown to be partially TNFR1-dependent [64]. Heme-induced macrophage death was also shown to be inhibited by the necroptosis inhibitor necrostatin-1 and by RIPK3 deficiency [64]. Normally, necroptosis following TNF signaling only occurs in the setting of caspase-8 inhibition since caspase-8 inactivates RIPK1 and RIPK3 and leads to apoptosis [66]. It remains unknown if or how heme exposure prevents or bypasses caspase-8-mediated inhibition of necroptosis. Given that the death was only halfway protected in the absence of TNFR1 [64], it is also possible that necroptosis can be directly activated by the inflammatory caspases but this has yet to be investigated.
Another form of cell death where heme may play a role is ferroptosis. Ferroptosis is distinct from other forms of cell death and is characterized by iron-induced formation of lipid ROS, likely via iron-dependent enzymes [67]. Because heme breakdown by the enzyme HO-1 results in the release of iron, it is likely that high levels of heme can induce ferroptosis. In platelets, neither inhibition of caspases nor inhibition of necroptosis prevents heme-induced cell death [68]. This suggests that heme does not induce apoptosis, pyroptosis, or necroptosis in platelets. Furthermore, inhibition of HO-1, iron chelation, and inhibition of ferroptosis with the inhibitor ferrostatin-1 all prevent heme-induced lipid peroxidation and heme-induced cell death in platelets [68]. This suggests that heme-induced cell death in platelets is occurring via ferroptosis. However, due to the ability of platelets to be activated by a range of stimuli in vitro, the specificity of heme in this context is unclear. In addition, it has not yet been investigated if heme-induced ferroptosis can occur in other cell types including macrophages and endothelial cells.
Because heme is able to induce inflammatory cell death independently of inflammasome activation [64], it is possible that DAMPs other than heme are the primary mediators of NLRP3 inflammasome activation following heme exposure. ATP is one of many DAMPs released from cells during inflammatory cell death. The ATP receptor P2X7 is required for ATP-induced inflammasome activation [69]. In conflicting studies, knockout of P2X receptors including P2X7 have been shown to be dispensable and essential for heme-induced inflammasome activation and IL-1β release [3, 30]. The iron lacking heme analog protoporphyrin IX also induces cell death [70]. However, it is not able to induce inflammasome activation. It is unclear from these studies if heme-induced NLRP3 activation is to a certain extent a consequence of cell death and subsequent ATP release. Since extended exposure to heme can cause substantial cell death, the discrepancy between studies could be attributed to differences in the duration of heme exposure in each of these studies. In this scenario, a short duration of heme exposure would induce caspase-4-dependent death and caspase-1/−5-dependent IL-1β release independent of ATP [3] but prolonged exposure to heme would release ATP to activate P2X7 activity that would amplify NLRP3 activation and IL-1β release in response to heme [30]. In the body the time that heme is free is dependent on the efficiency of heme scavenging mechanisms. In patients with SCD, Hp and Hpx levels are often low [9, 25], which would increase the exposure time to free heme. Under these conditions, it is possible that release of ATP and other DAMPs by heme-induced cell death amplifies NLRP3 activation.
Regulation of inflammasomes by heme-related factors
Hemolysis results in the release of Hb and extracellular Hb is rapidly oxidized in the vasculature. Oxidized Hb can induce various types of inflammation including expression of adhesion factors on endothelial cells and production of IL-6 and IL-8 [71, 72]. One oxidized form of Hb, ferrylHb, is able to induce NLRP3-dependent IL-1β release [73]. This finding stemmed from the observation that following phenylhydrazine-induced hemolysis in mice, activated caspase-1 and cleaved IL-1β levels in liver tissue increased much earlier than free heme levels did. Levels of oxidized Hb increase earlier than levels of free heme in plasma implicating ferrylHb as the DAMP mediating early inflammasome activation. FerrylHb could equally be mediating NLRP3 inflammasome activation through the efficient release of heme molecules [73]. Interestingly, metHb, which also readily releases free heme, does not appear to induce NLRP3 inflammasome activation [73]. Thus ferrylHb-mediated NLRP3 activation does not appear to be related to release of free heme but rather the entire hemoglobin protein.
Other heme-related factors can contribute to inflammasome activation. During a malaria infection, malarial parasites degrade Hb to create space for parasite replication and to provide replicating parasites a source of amino acids [74]. Hb breakdown causes the release of heme, which is toxic to the parasite [74]. To avoid this toxicity, malarial parasites convert heme into a crystal structure known as hemozoin [74]. Hemozoin is internalized by phagocytes either following erythrocyte rupture during the life cycle of the parasite or via ingestion of whole infected erythrocytes [74]. Hemozoin has been shown to induce IL-1β release from macrophages through activation of the NLRP3 inflammasome [75–77]. Additionally, peritoneal injection of hemozoin in mice resulted in increased levels of IL-1β in peritoneal fluid [76]. Upstream pathways known to be necessary for inflammasome activation are shared by hemozoin and other NLRP3 activators. For example, inhibiting K+ efflux and ROS formation both result in reduced IL-1β release in response to hemozoin [75, 77].
Like heme, hemozoin appears to be able to induce priming but unlike heme, it triggers this signal through TLR9 activation [78]. TLR9 is an intracellular PRR that is best known for recognizing unmethylated CpG motifs in microbial DNA [79]. TLR9-deficient macrophages exposed to Plasmodium-infected erythrocytes release less IL-1β [80]. Furthermore, Tlr9−/− mice have improved survival and significantly lower rates of cerebral malaria following Plasmodium infection [81]. At least one group has proposed that hemozoin is not directly inducing TLR9 signaling but instead complexing with parasite DNA and assisting with trafficking parasite DNA to intracellular compartments with TLR9 for activation [82]. Regardless of the exact mechanism of activation, TLR9 is an essential mediator of hemozoin-induced inflammation.
Heme catabolism products are also potential regulators of inflammasome activation following heme exposure. Heme is catabolized by HO-1 to produce iron, carbon monoxide, and biliverdin (Figure 1) [83]. Iron alone is not sufficient to induce IL-1β release, but the heme ring must contain iron to activate IL-1β [3]. Similarly, carbon monoxide and bilirubin, the reduced form of biliverdin, do not induce inflammasome activation [84, 85]. In fact, there is some evidence to suggest they both inhibit inflammasome activity. In macrophages, CO exposure prevents IL-1β and IL-18 release in response to ATP [84]. This protective effect appears to be mediated by the prevention of mitochondrial dysfunction and mtROS formation [84]. Bilirubin can also inhibit inflammasome activity although via a different mechanism than CO-mediated inhibition. Bilirubin downregulates the NF-κB signaling pathway to prevent expression of inflammasome proteins [85]. Thus, the downstream products of heme catabolism appear to provide a negative feedback loop to limit inflammation induced by heme.
HO-1 also functions as a negative regulator of heme although it is not yet clear if this effect is solely occurring through increased production of carbon monoxide and bilirubin. Heme exposure induces HO-1 expression. Studies have shown that HO-1 induction downregulates inflammasome activity [86, 87]. However, HO-1 knockout in macrophages does not significantly increase heme-induced IL-1β release [3]. It is likely that inflammasome activation after heme exposure only occurs when the levels of free heme greatly exceed the capacity of HO-1 to break heme down. In this case, HO-1 knockout may have a relatively small effect on the total heme levels. Overall, the balance between heme levels, HO-1 activity, and the levels of heme catabolism products likely provides a rheostat to regulate inflammasome activation.
Heme-induced NLRP3 inflammasome activation in hemolytic disease
SCD is an inherited blood disorder that impacts over three million people worldwide and causes over 150,000 deaths each year [88]. It is characterized by a mutation in the β-globin gene resulting in hemoglobin that polymerizes after deoxygenation. As a result of hemoglobin polymerization, erythrocytes assume a sickled shape and become highly prone to hemolysis [89]. Some of the complications associated with SCD include recurrent pain crises caused by vaso-occlusion, acute chest syndrome, venous thromboembolism, and stroke [89, 90]. Many of these complications are the result of excess inflammation and levels of inflammation in patients with SCD are correlated with mortality [91]. Inflammatory processes in both endothelial cells and cells of hematopoietic origin have been shown to contribute to these complications [8, 92–96]. There is increasing evidence that heme is a major contributor to inflammation and organ damage in patients with SCD [95, 96]. Additionally, inflammasome signaling is upregulated in patients with SCD. Peripheral blood mononuclear cells from patients with SCD have elevated mRNA expression of NLRP3, caspase-1, IL-1β, and IL-18 [8]. Similarly, monocytes from patients with SCD express elevated levels of IL-1β and release more IL-1β after heme exposure [4, 93].
Chronic hemolysis is characteristic of SCD and plasma concentrations of heme are significantly elevated in patients with SCD [9]. Heme exposure in mouse models of SCD induces vaso-occlusion, lung injury, and mortality [94, 96, 97]. In patients, steady state levels of heme in protein-depleted plasma are associated with increased risk for vaso-occlusive crises and acute chest syndrome [98]. One of the main ways heme contributes to these complications is by inducing expression of pro-adhesion molecules --like P-selectin, VCAM-1, and von Willebrand factor – on endothelial cells [95, 99]. In patients with SCD, serum levels of non-Hpx bound heme are positively correlated with levels of soluble adhesion markers sVCAM-1 and sE-selectin [10]. These pro-adhesion molecules promote blockage of small blood vessels (vaso-occlusion) by binding neutrophils, platelets, and sickled erythrocytes to the activated endothelium (Figure 3). IL-1β signaling is known to cause adhesion molecule expression in endothelial cells [100, 101]. Thus, NLRP3 inflammasome activation may be one of the mechanisms by which heme induces expression of these molecules. IL-1β signaling also promotes neutrophil migration and survival [102, 103], another mechanism by which heme-induced NLRP3 activation may promote vaso-occlusion. Despite this, heme-induced TLR4 activation appears to be the main driver of vaso-occlusion in SCD. Inhibition of TLR4 significantly reduces the amount of vaso-occlusion observed in response to heme in a mouse model of SCD [95]. TLR4 inhibition appears to mediate this effect by reducing adhesion molecule expression on endothelial cells [95]. Blocking TLR4 by genetic knockout or pharmacological inhibition has also been shown to protect against heme-mediated acute lung injury in mouse models of SCD [104]. It is unclear from these studies if some of the beneficial effects of TLR4 inhibition are due to inhibited inflammasome priming.
Figure 3. Mechanisms of heme-induced vaso-occlusion.
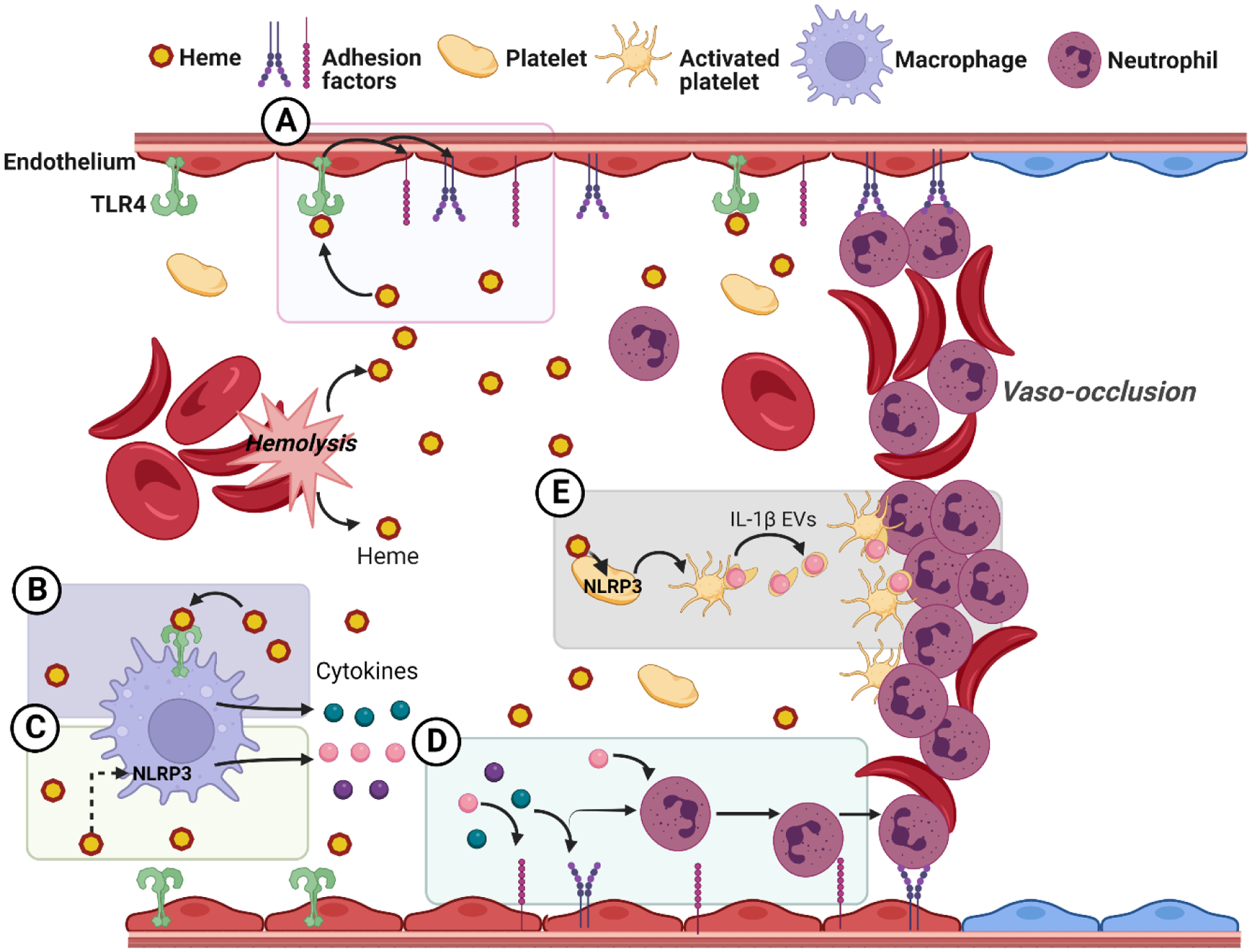
A) Activation of the endothelium. Heme triggers TLR4 signaling leading to endothelial cell activation and expression of endothelial adhesion molecules B) Activation of the TLR4 pathway in macrophages. Heme induces tumor necrosis factor-α (TNFα) (green circles) secretion in macrophages through the TLR4 signaling pathway. TNFα triggers endothelial cell activation and expression of adhesion factors. C) Activation of the NLRP3 pathway in macrophages. Heme acts both as a priming and as an intracellular signal that triggers NLRP3 inflammasome activation to produce the mature cytokines IL-1β and IL-18 (pink and purple circles respectively). These cytokines induce inflammatory activation, neutrophil infiltration, and adhesion factors expression. D) Cytokine-induced recruitment and binding of neutrophils. Neutrophil recruitment and activation leads to neutrophil adhesion to the endothelium and sickled erythrocytes E) Platelet activation and formation of platelet-neutrophil aggregates. NLRP3-inflammasome activation in platelets leads to the generation of extracellular vesicles (EVs) that carry IL-1β. These platelet extracellular vesicles promote the generation of platelet-neutrophil aggregates in a caspase-1 and IL-1β dependent manner
Inflammasome activation in platelets may be especially important in patients with SCD. Caspase-1 activity is elevated in platelets from patients with SCD and becomes further elevated in patients undergoing pain crises [105]. Furthermore, inhibition of NLRP3 reduces the amount of platelet aggregation observed in a mouse model of SCD [105]. These data suggest that inflammasome activation in platelets contributes to platelet aggregation and vaso-occlusion. It has been shown that inflammasome activation in platelets from patients with SCD is mediated by soluble factors in plasma. When healthy donor platelets were exposed to platelet-poor plasma from patients with SCD both at steady-state and during crisis, caspase-1 activity increased [105]. Heme is likely one of the soluble factors in plasma contributing to inflammasome signaling in platelets. Another soluble factor mediating this effect is the DAMP HMGB1, which is known to activate TLR4 [105]. Bruton’s Tyrosine Kinase (BTK) signaling also contributes to inflammasome activation in platelets after exposure to SCD plasma [105]. BTK has been suggested to promote inflammasome formation by interacting with NLRP3 and ASC and promoting ASC oligomerization [106, 107].
Platelet-neutrophil aggregates are also a common feature of occluded blood vessels in mouse models of SCD and in patients with SCD [108, 109]. Platelet extracellular vesicles are produced by platelets upon activation and can contribute to the formation of these aggregates. It has been shown that chemical inhibition of caspase-1 reduces the formation of platelet extracellular vesicles triggered by LPS exposure [109]. Furthermore, inhibition of caspase-1 or IL-1β prevents the formation of platelet-neutrophil aggregates and significantly reduces pulmonary vaso-occlusion induced by platelet extracellular vesicles in SCD mice [109]. Thus, inflammasome signaling appears to be contributing to two key steps that lead to pulmonary vaso-occlusion in this model: the formation of extracellular vesicles and the aggregation of platelets with neutrophils. Although this study did not investigate the specific DAMPs in SCD plasma contributing to inflammasome signaling, it is tempting to speculate that heme-induced NLRP3 activation directly contributes to platelet-neutrophil aggregate formation via inflammasome activation.
In addition to causing acute complications like vaso-occlusion, sustained heme exposure also contributes to the chronic manifestations of SCD. Chronically elevated heme levels may result in a prolonged pro-inflammatory state. Heme exposure induces polarization of macrophages into the M1 pro-inflammatory state [110]. This polarization appears to be dependent on TLR4 activation and ROS formation and can be reversed by hemopexin therapy in mice [110]. Heme exposure can also induce long-lasting epigenetic alterations in hematopoietic stem cells that lead to a more pro-inflammatory phenotype [111]. In mice exposed to heme, after differentiation of hematopoietic stem and progenitor cells (HSPCs) into macrophages ex vivo, macrophages displayed increased cytokine release and phagocytosis when stimulated with LPS [111]. These alterations were accompanied by changes in chromatin accessibility and binding patterns of various transcription factors. Together these data suggest heme can induce a state of persistently heightened inflammatory responses. Although it has not been directly studied, these data also suggest that prior heme exposure can induce increased inflammasome activation and IL-1β release by subsequent activating signals. This could be especially important for patients with inherited hemolytic anemias including SCD and thalassemias because high rates of hemolysis in these patients have exposed their cells to a lifetime of heme. Chronic heme-induced inflammation may also contribute to cardiac dysfunction and left ventricular hypertrophy (LVH) seen in patients with SCD [112]. One study found that exposure to heme increased cardiac expression of IL-6 as well as markers of cardiac hypertrophy in mouse models of SCD [113]. While it has not been directly investigated, it is likely that the inflammasome pathway is contributing to both of these findings. For example, binding of IL-1β to its receptor leads to activation of the PI3K/AKT signaling pathway and downstream transcription of IL-6 [114]. Furthermore, both IL-1β and IL-18 signaling can induce cardiac hypertrophy [115, 116]
Malaria is another disease state characterized by hemolysis in which the NLRP3 inflammasome plays a central role. Serum levels of IL-1β in patients with malaria are positively associated with disease severity [117]. Several groups have shown that Nlrp3−/− mice have increased survival and a lower risk of developing cerebral malaria [75, 77, 118]. Despite this finding, the levels of parasitemia in wild-type and Nlrp3−/− mice are not significantly different [77]. This suggests that while NLRP3 does not play a direct role in controlling the parasite burden during infection, hemozoin-induced NLRP3 activation and subsequent inflammation does play an important role in morbidity and mortality.
Recent evidence has suggested neutrophil extracellular trap (NET) formation is a key feature of both SCD and malaria. NETs are extracellular structures, composed of chromatin and proteins, that can trap microorganisms, induce expression of pro-adhesion markers on endothelial cells, and contribute to a variety of non-infectious conditions including atherosclerosis, tumor metastasis, and autoimmunity [119, 120]. Patients with SCD undergoing vaso-occlusive crisis and patients with malaria have high plasma levels of NET-associated markers [121–123]. Furthermore, inhibition of NET formation reduced lung injury and improved survival in mouse models of SCD and prevented organ damage in mouse models of malaria [123, 124]. Heme is implicated in the formation of NETs in both these conditions since heme can induce NET formation in vitro as well as in mice injected with heme [123–125]. Interestingly, it was recently suggested that both canonical NLRP3 inflammasome signaling and non-canonical inflammasome signaling contribute to NET formation [126, 127]. The engagement of the canonical pathway suggests that heme may be mediating NET formation in these conditions through GSDMD pore formation. Heme-induced NET formation is dependent on ROS production but independent of NOX2 [123, 124]. This further suggests that GSDMD cleavage, rather than NOX2-dependent cytokine release, is the main mediator of heme-induced NET formation. In fact, caspase-independent GSDMD cleavage also appears to play a role in NET formation [128]. Conversely, NET formation also contributes to inflammasome signaling since NET formation was shown to induce priming for inflammasome activation in macrophages [129].
Induction of inflammasome signaling by heme is one of many heme-induced inflammatory pathways relevant in hemolytic diseases. Heme has a diverse range of pro-inflammatory effects (reviewed in [23]) including activation of the complement cascade, enhancement of antibody activity, and induction of IL-8 release from neutrophils. Free heme is also involved in many deleterious processes in patients with SCD (reviewed in [130]). For example, heme induces expression of placenta growth factor, a protein associated with pulmonary hypertension and airway hyper-responsiveness in patients with SCD. Heme-induced activation of inflammasomes and other pro-inflammatory events likely occurs not only in SCD and malaria but in many conditions involving exposure of cells to free heme. Inherited red cell disorders, infections, liver cirrhosis, hemorrhage, autoimmune hemolytic conditions, and toxicity-induced hemolysis are among the many conditions that result in elevated extracellular heme and thus may involve heme-induced inflammasome activation [131]. For example, in a mouse model of intracerebral hemorrhage, TLR4 deficiency is associated with reduced serum levels of IL-1β and improved neurological outcomes [132].
Therapeutics targeting the inflammasome in hemolytic conditions
There is limited data on the efficacy of inhibiting inflammasome activity for the treatment of patients with SCD and other hemolytic conditions. Mouse models of SCD treated with the IL-1 receptor antagonist anakinra had reduced expression of the adhesion marker VCAM-1 on endothelial cells [133]. Following induction of ischemic stroke by middle cerebral artery occlusion in SCD mice, anakinra treatment reduced stroke volume and macrophage infiltration [134]. The heme scavenger Hpx significantly reduces the amount of vaso-occlusion observed in mouse models of SCD and an on-going clinical trial is investigating the efficacy of hemopexin in adults with SCD [135]. The protective effects of Hpx include inhibition of heme-induced TLR4 signaling, inhibition of adhesion factor expression on endothelial cells, and induction of heme oxygenase-1 and subsequent anti-inflammatory signaling [95, 135–138]. However, whether or not the protective effects of Hpx include inhibition of heme-mediated inflammasome activation has not been tested.
Only one study to date has tested the efficacy of blocking the NLRP3 inflammasome pathway in patients with SCD. In this trial, patients with SCD were given placebo or canakinumab, a monoclonal antibody targeting IL-1β, every four weeks for a total of 24 weeks [139]. Patients receiving canakinumab experienced shorter hospital stays on average and significant reductions in fatigue and absences from school or work [139]. Only one patient in this study developed acute chest syndrome and this patient was in the placebo group [139]. There was also a downward trend in self-reported daily pain amongst patients receiving canakinumab but this endpoint did not meet the defined significance criterion. Given the multifactorial causes of chronic pain in patients with SCD, it is not unexpected that IL-1β blockade alone did not cause more substantial differences in self-reported pain [140]. While these clinical outcomes are promising, the molecular basis for these improvements is unclear. Patients treated with canakinumab had significantly lower levels of various immune cells, but there were no differences in the levels of pro-adhesion markers like soluble VCAM-1 or soluble P-selectin between the experimental groups [139]. This suggests that the main effects of preventing IL-1β release are blocking neutrophil infiltration and platelet aggregation at the endothelium rather than a direct impact on adhesion molecule expression from endothelial cells. Overall, the study showed there is a clinical benefit to inhibiting IL-1β signaling in patients with SCD. There is some evidence to suggest that combination therapy blocking both TNFα and IL-1β more effectively prevents the expression of adhesion molecules on endothelial cells [93]. It will be interesting to see if such a combinatorial approach further improves clinical symptoms. For example, in a mouse model of malaria, targeting IL-33, which downregulates components of the NLRP3 inflammasome, or chemically inhibiting NLRP3 in combination with anti-malarial drugs improved survival and lessened the severity of cerebral malaria [118]. It is thus possible that NLRP3 inhibition in combination with standard of care therapies may have beneficial effects in SCD and other hemolytic conditions.
Conclusion
Overall, there is an enormous need for preclinical studies investigating the role of inflammasome signaling in various complications of hemolytic conditions. Given the unique mechanisms of each SCD complication and the diverse triggers for acute complications, preclinical studies modeling each SCD manifestation in specific contexts will be most useful for informing viable therapeutic strategies. Similarly targeted approaches are likely required in other hemolytic conditions. It is our opinion that therapeutic approaches should not be restricted only to direct cytokine inhibition. The discovery that heme induces non-canonical inflammasome activation greatly expands the therapeutic options to combat hemolysis- and heme-induced inflammation. Caspase-1, caspase-4, and caspase-5 as well as any interacting proteins involved in the canonical and non-canonical inflammasome pathways are promising therapeutic targets. Uncovering how these inflammatory caspases interact and identifying other proteins involved in this pathway will be crucial to inform therapeutic strategies for patients with SCD and other hemolytic conditions.
Acknowledgements
Authors on this manuscript are funded by NIH/NIDDK F32DK121479 (BEB), NIH/NIGMS T32GM008231 (SS), NIH/NIGMS R01GM121389 (LBH), the Jeanne Marie Lusher Diversity Fellowship from the National Hemophilia Foundation (SJA), and NIH/NHLBI R01HL136415 (JMF). The authors declare no conflict of interest and all authors have read the journal’s conflict of interest disclosure policy. Figures were generated using Biorender. All authors have approved of this manuscript and have read the journal’s authorship statement.
References
- [1].Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. [DOI] [PubMed] [Google Scholar]
- [2].Dutra FF, Bozza MT. Heme on innate immunity and inflammation. Front Pharmacol. 2014;5:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dutra FF, Alves LS, Rodrigues D, Fernandez PL, de Oliveira RB, Golenbock DT, et al. Hemolysis-induced lethality involves inflammasome activation by heme. Proc Natl Acad Sci U S A. 2014;111:E4110–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bolivar BE, Brown-Suedel AN, Rohrman BA, Charendoff CI, Yazdani V, Belcher JD, et al. Noncanonical Roles of Caspase-4 and Caspase-5 in Heme-Driven IL-1beta Release and Cell Death. J Immunol. 2021;206:1878–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bolivar BE, Vogel TP, Bouchier-Hayes L. Inflammatory caspase regulation: maintaining balance between inflammation and cell death in health and disease. FEBS J. 2019;286:2628–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wun T, Cordoba M, Rangaswami A, Cheung AW, Paglieroni T. Activated monocytes and platelet-monocyte aggregates in patients with sickle cell disease. Clin Lab Haematol. 2002;24:81–8. [DOI] [PubMed] [Google Scholar]
- [8].Pitanga TN, Oliveira RR, Zanette DL, Guarda CC, Santiago RP, Santana SS, et al. Sickle red cells as danger signals on proinflammatory gene expression, leukotriene B4 and interleukin-1 beta production in peripheral blood mononuclear cell. Cytokine. 2016;83:75–84. [DOI] [PubMed] [Google Scholar]
- [9].Muller-Eberhard U, Javid J, Liem HH, Hanstein A, Hanna M. Brief Report: Plasma Concentrations of Hemopexin, Haptoglobin and Heme in Patients with Various Hemolytic Diseases. Blood. 1968;32:811–5. [PubMed] [Google Scholar]
- [10].Vinchi F, Sparla R, Passos ST, Sharma R, Vance SZ, Zreid HS, et al. Vasculo-toxic and pro-inflammatory action of unbound haemoglobin, haem and iron in transfusion-dependent patients with haemolytic anaemias. Br J Haematol. 2021;193:637–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gilles-Gonzalez MA, Gonzalez G. Heme-based sensors: defining characteristics, recent developments, and regulatory hypotheses. Journal of inorganic biochemistry. 2005;99:1–22. [DOI] [PubMed] [Google Scholar]
- [12].Dawson JH. Probing structure-function relations in heme-containing oxygenases and peroxidases. Science (New York, NY). 1988;240:433–9. [DOI] [PubMed] [Google Scholar]
- [13].Mansouri A, Lurie AA. Concise review: methemoglobinemia. American journal of hematology. 1993;42:7–12. [DOI] [PubMed] [Google Scholar]
- [14].Martins R, Knapp S. Heme and hemolysis in innate immunity: adding insult to injury. Current opinion in immunology. 2018;50:14–20. [DOI] [PubMed] [Google Scholar]
- [15].Sadrzadeh SM, Graf E, Panter SS, Hallaway PE, Eaton JW. Hemoglobin. A biologic fenton reagent. The Journal of biological chemistry. 1984;259:14354–6. [PubMed] [Google Scholar]
- [16].Aich A, Freundlich M, Vekilov PG. The free heme concentration in healthy human erythrocytes. Blood Cells, Molecules, and Diseases. 2015;55:402–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Donegan RK, Moore CM, Hanna DA, Reddi AR. Handling heme: The mechanisms underlying the movement of heme within and between cells. Free Radic Biol Med. 2019;133:88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hopp MT, Imhof D. Linking Labile Heme with Thrombosis. Journal of clinical medicine. 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sawicki KT, Chang HC, Ardehali H. Role of Heme in Cardiovascular Physiology and Disease. Journal of the American Heart Association.4:e001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rapido F The potential adverse effects of haemolysis. Blood Transfus. 2017;15:218–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Thomsen JH, Etzerodt A, Svendsen P, Moestrup SK. The haptoglobin-CD163-heme oxygenase-1 pathway for hemoglobin scavenging. Oxidative medicine and cellular longevity. 2013;2013:523652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Vallelian F, Buehler PW, Schaer DJ. Hemolysis, free hemoglobin toxicity and scavenger protein therapeutics. Blood. 2022. [DOI] [PubMed] [Google Scholar]
- [23].Bozza MT, Jeney V. Pro-inflammatory Actions of Heme and Other Hemoglobin-Derived DAMPs. Front Immunol. 2020;11:1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kumar S, Bandyopadhyay U. Free heme toxicity and its detoxification systems in human. Toxicology Letters. 2005;157:175–88. [DOI] [PubMed] [Google Scholar]
- [25].Schaer DJ, Buehler PW, Alayash AI, Belcher JD, Vercellotti GM. Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood. 2013;121:1276–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Soares MP, Bozza MT. Red alert: labile heme is an alarmin. Curr Opin Immunol. 2016;38:94–100. [DOI] [PubMed] [Google Scholar]
- [27].Immenschuh S, Vijayan V, Janciauskiene S, Gueler F. Heme as a Target for Therapeutic Interventions. Frontiers in pharmacology. 2017;8:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20. [DOI] [PubMed] [Google Scholar]
- [29].Erdei J, Toth A, Balogh E, Nyakundi BB, Banyai E, Ryffel B, et al. Induction of NLRP3 Inflammasome Activation by Heme in Human Endothelial Cells. Oxidative medicine and cellular longevity. 2018;2018:4310816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li Q, Fu W, Yao J, Ji Z, Wang Y, Zhou Z, et al. Heme induces IL-1beta secretion through activating NLRP3 in kidney inflammation. Cell Biochem Biophys. 2014;69:495–502. [DOI] [PubMed] [Google Scholar]
- [31].Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19:477–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lu A, Wu H. Structural mechanisms of inflammasome assembly. FEBS J. 2015;282:435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32. [DOI] [PubMed] [Google Scholar]
- [34].Malik A, Kanneganti TD. Inflammasome activation and assembly at a glance. J Cell Sci. 2017;130:3955–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gabay C, Lamacchia C, Palmer G. IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol. 2010;6:232–41. [DOI] [PubMed] [Google Scholar]
- [36].Yasuda K, Nakanishi K, Tsutsui H. Interleukin-18 in Health and Disease. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5. [DOI] [PubMed] [Google Scholar]
- [38].He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015;25:1285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, et al. Characterization of heme as activator of Toll-like receptor 4. J Biol Chem. 2007;282:20221–9. [DOI] [PubMed] [Google Scholar]
- [40].Vaure C, Liu Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front Immunol. 2014;5:316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhang P, Nguyen J, Abdulla F, Nelson AT, Beckman JD, Vercellotti GM, et al. Soluble MD-2 and Heme in Sickle Cell Disease Plasma Promote Pro-Inflammatory Signaling in Endothelial Cells. Front Immunol. 2021;12:632709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Belcher JD, Zhang P, Nguyen J, Kiser ZM, Nath KA, Hu J, et al. Identification of a Heme Activation Site on the MD-2/TLR4 Complex. Front Immunol. 2020;11:1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–5. [DOI] [PubMed] [Google Scholar]
- [44].May O, Yatime L, Merle NS, Delguste F, Howsam M, Daugan MV, et al. The receptor for advanced glycation end products is a sensor for cell-free heme. FEBS J. 2021;288:3448–64. [DOI] [PubMed] [Google Scholar]
- [45].Mulrennan S, Baltic S, Aggarwal S, Wood J, Miranda A, Frost F, et al. The role of receptor for advanced glycation end products in airway inflammation in CF and CF related diabetes. Sci Rep. 2015;5:8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Morbini P, Villa C, Campo I, Zorzetto M, Inghilleri S, Luisetti M. The receptor for advanced glycation end products and its ligands: a new inflammatory pathway in lung disease? Mod Pathol. 2006;19:1437–45. [DOI] [PubMed] [Google Scholar]
- [47].Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. S100A8/A9 in Inflammation. Front Immunol. 2018;9:1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Silveira AAA, Mahon OR, Cunningham CC, Corr EM, Mendonca R, Saad STO, et al. S100A8 acts as an autocrine priming signal for heme-induced human Mvarphi pro-inflammatory responses in hemolytic inflammation. J Leukoc Biol. 2019;106:35–43. [DOI] [PubMed] [Google Scholar]
- [49].Mocsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. 2010;10:387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Belambri SA, Rolas L, Raad H, Hurtado-Nedelec M, Dang PM, El-Benna J. NADPH oxidase activation in neutrophils: Role of the phosphorylation of its subunits. Eur J Clin Invest. 2018;48 Suppl 2:e12951. [DOI] [PubMed] [Google Scholar]
- [51].Hennige AM, Lembert N, Wahl MA, Ammon HP. Oxidative stress increases potassium efflux from pancreatic islets by depletion of intracellular calcium stores. Free Radic Res. 2000;33:507–16. [DOI] [PubMed] [Google Scholar]
- [52].Korolnek T, Hamza I. Like iron in the blood of the people: the requirement for heme trafficking in iron metabolism. Front Pharmacol. 2014;5:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chiabrando D, Vinchi F, Fiorito V, Mercurio S, Tolosano E. Heme in pathophysiology: a matter of scavenging, metabolism and trafficking across cell membranes. Front Pharmacol. 2014;5:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Le Blanc S, Garrick MD, Arredondo M. Heme carrier protein 1 transports heme and is involved in heme-Fe metabolism. Am J Physiol Cell Physiol. 2012;302:C1780–5. [DOI] [PubMed] [Google Scholar]
- [55].Kajiwara Y, Schiff T, Voloudakis G, Gama Sosa MA, Elder G, Bozdagi O, et al. A critical role for human caspase-4 in endotoxin sensitivity. J Immunol. 2014;193:335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–92. [DOI] [PubMed] [Google Scholar]
- [57].Casson CN, Yu J, Reyes VM, Taschuk FO, Yadav A, Copenhaver AM, et al. Human caspase-4 mediates noncanonical inflammasome activation against gram-negative bacterial pathogens. Proc Natl Acad Sci U S A. 2015;112:6688–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V. Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur J Immunol. 2015;45:2911–7. [DOI] [PubMed] [Google Scholar]
- [59].Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–71. [DOI] [PubMed] [Google Scholar]
- [60].Kamens J, Paskind M, Hugunin M, Talanian RV, Allen H, Banach D, et al. Identification and characterization of ICH-2, a novel member of the interleukin-1 beta-converting enzyme family of cysteine proteases. J Biol Chem. 1995;270:15250–6. [DOI] [PubMed] [Google Scholar]
- [61].Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–21. [DOI] [PubMed] [Google Scholar]
- [62].Baker PJ, Boucher D, Bierschenk D, Tebartz C, Whitney PG, D’Silva DB, et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur J Immunol. 2015;45:2918–26. [DOI] [PubMed] [Google Scholar]
- [63].Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity. 2018;48:35–44 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Fortes GB, Alves LS, de Oliveira R, Dutra FF, Rodrigues D, Fernandez PL, et al. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood. 2012;119:2368–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Newton K RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. 2015;25:347–53. [DOI] [PubMed] [Google Scholar]
- [66].Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–14. [DOI] [PubMed] [Google Scholar]
- [67].Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].NaveenKumar SK, SharathBabu BN, Hemshekhar M, Kemparaju K, Girish KS, Mugesh G. The Role of Reactive Oxygen Species and Ferroptosis in Heme-Mediated Activation of Human Platelets. ACS Chem Biol. 2018;13:1996–2002. [DOI] [PubMed] [Google Scholar]
- [69].Solle M, Labasi J, Perregaux DG, Stam E, Petrushova N, Koller BH, et al. Altered cytokine production in mice lacking P2X(7) receptors. J Biol Chem. 2001;276:125–32. [DOI] [PubMed] [Google Scholar]
- [70].Xu H, Sun Y, Zhang Y, Wang W, Dan J, Yao J, et al. Protoporphyrin IX induces a necrotic cell death in human THP-1 macrophages through activation of reactive oxygen species/c-Jun N-terminal protein kinase pathway and opening of mitochondrial permeability transition pore. Cell Physiol Biochem. 2014;34:1835–48. [DOI] [PubMed] [Google Scholar]
- [71].Liu X, Spolarics Z. Methemoglobin is a potent activator of endothelial cells by stimulating IL-6 and IL-8 production and E-selectin membrane expression. Am J Physiol Cell Physiol. 2003;285:C1036–46. [DOI] [PubMed] [Google Scholar]
- [72].Silva G, Jeney V, Chora A, Larsen R, Balla J, Soares MP. Oxidized hemoglobin is an endogenous proinflammatory agonist that targets vascular endothelial cells. J Biol Chem. 2009;284:29582–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Nyakundi BB, Toth A, Balogh E, Nagy B, Erdei J, Ryffel B, et al. Oxidized hemoglobin forms contribute to NLRP3 inflammasome-driven IL-1beta production upon intravascular hemolysis. Biochimica et biophysica acta Molecular basis of disease. 2019;1865:464–75. [DOI] [PubMed] [Google Scholar]
- [74].Coronado LM, Nadovich CT, Spadafora C. Malarial hemozoin: from target to tool. Biochim Biophys Acta. 2014;1840:2032–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Shio MT, Eisenbarth SC, Savaria M, Vinet AF, Bellemare MJ, Harder KW, et al. Malarial hemozoin activates the NLRP3 inflammasome through Lyn and Syk kinases. PLoS Pathog. 2009;5:e1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Griffith JW, Sun T, McIntosh MT, Bucala R. Pure Hemozoin is inflammatory in vivo and activates the NALP3 inflammasome via release of uric acid. J Immunol. 2009;183:5208–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Dostert C, Guarda G, Romero JF, Menu P, Gross O, Tardivel A, et al. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS One. 2009;4:e6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Coban C, Ishii KJ, Kawai T, Hemmi H, Sato S, Uematsu S, et al. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J Exp Med. 2005;201:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kalantari P, DeOliveira RB, Chan J, Corbett Y, Rathinam V, Stutz A, et al. Dual engagement of the NLRP3 and AIM2 inflammasomes by plasmodium-derived hemozoin and DNA during malaria. Cell Rep. 2014;6:196–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Coban C, Ishii KJ, Uematsu S, Arisue N, Sato S, Yamamoto M, et al. Pathological role of Toll-like receptor signaling in cerebral malaria. Int Immunol. 2007;19:67–79. [DOI] [PubMed] [Google Scholar]
- [82].Parroche P, Lauw FN, Goutagny N, Latz E, Monks BG, Visintin A, et al. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc Natl Acad Sci U S A. 2007;104:1919–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86:583–650. [DOI] [PubMed] [Google Scholar]
- [84].Jung SS, Moon JS, Xu JF, Ifedigbo E, Ryter SW, Choi AM, et al. Carbon monoxide negatively regulates NLRP3 inflammasome activation in macrophages. Am J Physiol Lung Cell Mol Physiol. 2015;308:L1058–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Lin Y, Wang S, Yang Z, Gao L, Zhou Z, Yu P, et al. Bilirubin alleviates alum-induced peritonitis through inactivation of NLRP3 inflammasome. Biomed Pharmacother. 2019;116:108973. [DOI] [PubMed] [Google Scholar]
- [86].Luo YP, Jiang L, Kang K, Fei DS, Meng XL, Nan CC, et al. Hemin inhibits NLRP3 inflammasome activation in sepsis-induced acute lung injury, involving heme oxygenase-1. Int Immunopharmacol. 2014;20:24–32. [DOI] [PubMed] [Google Scholar]
- [87].Li H, Zhou X, Zhang J. Induction of heme oxygenase-1 attenuates lipopolysaccharide-induced inflammasome activation in human gingival epithelial cells. Int J Mol Med. 2014;34:1039–44. [DOI] [PubMed] [Google Scholar]
- [88].Sundd P, Gladwin MT, Novelli EM. Pathophysiology of Sickle Cell Disease. Annu Rev Pathol. 2019;14:263–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, et al. Sickle cell disease. Nature reviews Disease primers. 2018;4:18010. [DOI] [PubMed] [Google Scholar]
- [90].Conran N, Belcher JD. Inflammation in sickle cell disease. Clin Hemorheol Microcirc. 2018;68:263–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].van Beers EJ, Yang Y, Raghavachari N, Tian X, Allen DT, Nichols JS, et al. Iron, inflammation, and early death in adults with sickle cell disease. Circ Res. 2015;116:298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J Clin Invest. 2000;106:411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Belcher JD, Marker PH, Weber JP, Hebbel RP, Vercellotti GM. Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vaso-occlusion. Blood. 2000;96:2451–9. [PubMed] [Google Scholar]
- [94].Beckman JD, Abdullah F, Chen C, Kirchner R, Rivera-Rodriguez D, Kiser ZM, et al. Endothelial TLR4 Expression Mediates Vaso-Occlusive Crisis in Sickle Cell Disease. Front Immunol. 2020;11:613278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood. 2014;123:377–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Ghosh S, Adisa OA, Chappa P, Tan F, Jackson KA, Archer DR, et al. Extracellular hemin crisis triggers acute chest syndrome in sickle mice. The Journal of clinical investigation. 2013;123:4809–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Vercellotti GM, Zhang P, Nguyen J, Abdulla F, Chen C, Nguyen P, et al. Hepatic Overexpression of Hemopexin Inhibits Inflammation and Vascular Stasis in Murine Models of Sickle Cell Disease. Molecular medicine. 2016;22:437–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Adisa OA, Hu Y, Ghosh S, Aryee D, Osunkwo I, Ofori-Acquah SF. Association between plasma free haem and incidence of vaso-occlusive episodes and acute chest syndrome in children with sickle cell disease. British journal of haematology. 2013;162:702–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Wagener FA, Feldman E, de Witte T, Abraham NG. Heme induces the expression of adhesion molecules ICAM-1, VCAM-1, and E selectin in vascular endothelial cells. Proc Soc Exp Biol Med. 1997;216:456–63. [DOI] [PubMed] [Google Scholar]
- [100].Mako V, Czucz J, Weiszhar Z, Herczenik E, Matko J, Prohaszka Z, et al. Proinflammatory activation pattern of human umbilical vein endothelial cells induced by IL-1beta, TNF-alpha, and LPS. Cytometry A. 2010;77:962–70. [DOI] [PubMed] [Google Scholar]
- [101].Hawrylowicz CM, Howells GL, Feldmann M. Platelet-derived interleukin 1 induces human endothelial adhesion molecule expression and cytokine production. J Exp Med. 1991;174:785–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Miller LS, Pietras EM, Uricchio LH, Hirano K, Rao S, Lin H, et al. Inflammasome-mediated production of IL-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. J Immunol. 2007;179:6933–42. [DOI] [PubMed] [Google Scholar]
- [103].Prince LR, Allen L, Jones EC, Hellewell PG, Dower SK, Whyte MK, et al. The role of interleukin-1beta in direct and toll-like receptor 4-mediated neutrophil activation and survival. Am J Pathol. 2004;165:1819–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Ghosh S, Adisa O, Yang Y, Tan F, Ofori-Acquah SF. Toll-Like Receptor 4 Mediates Heme Induced Acute Lung Injury: Preclinical Study of Resatorvid in Sickle Cell Disease. Blood. 2011;118:2113-. [Google Scholar]
- [105].Vogel S, Arora T, Wang X, Mendelsohn L, Nichols J, Allen D, et al. The platelet NLRP3 inflammasome is upregulated in sickle cell disease via HMGB1/TLR4 and Bruton tyrosine kinase. Blood Adv. 2018;2:2672–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, et al. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun. 2015;6:7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Liu X, Pichulik T, Wolz OO, Dang TM, Stutz A, Dillen C, et al. Human NACHT, LRR, and PYD domain-containing protein 3 (NLRP3) inflammasome activity is regulated by and potentially targetable through Bruton tyrosine kinase. J Allergy Clin Immunol. 2017;140:1054–67 e10. [DOI] [PubMed] [Google Scholar]
- [108].Bennewitz MF, Jimenez MA, Vats R, Tutuncuoglu E, Jonassaint J, Kato GJ, et al. Lung vaso-occlusion in sickle cell disease mediated by arteriolar neutrophil-platelet microemboli. JCI Insight. 2017;2:e89761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Vats R, Brzoska T, Bennewitz MF, Jimenez MA, Pradhan-Sundd T, Tutuncuoglu E, et al. Platelet Extracellular Vesicles Drive Inflammasome-IL-1beta-Dependent Lung Injury in Sickle Cell Disease. Am J Respir Crit Care Med. 2020;201:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Vinchi F, Costa da Silva M, Ingoglia G, Petrillo S, Brinkman N, Zuercher A, et al. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood. 2016;127:473–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Jentho E, Ruiz-Moreno C, Novakovic B, Kourtzelis I, Megchelenbrink WL, Martins R, et al. Trained innate immunity, long-lasting epigenetic modulation, and skewed myelopoiesis by heme. Proc Natl Acad Sci U S A. 2021;118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Faro GB, Menezes-Neto OA, Batista GS, Silva-Neto AP, Cipolotti R. Left ventricular hypertrophy in children, adolescents and young adults with sickle cell anemia. Rev Bras Hematol Hemoter. 2015;37:324–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Gbotosho OT, Kapetanaki MG, Ghosh S, Villanueva FS, Ofori-Acquah SF, Kato GJ. Heme Induces IL-6 and Cardiac Hypertrophy Genes Transcripts in Sickle Cell Mice. Front Immunol. 2020;11:1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Cahill CM, Rogers JT. Interleukin (IL) 1beta induction of IL-6 is mediated by a novel phosphatidylinositol 3-kinase-dependent AKT/IkappaB kinase alpha pathway targeting activator protein-1. J Biol Chem. 2008;283:25900–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].O’Brien LC, Mezzaroma E, Van Tassell BW, Marchetti C, Carbone S, Abbate A, et al. Interleukin-18 as a therapeutic target in acute myocardial infarction and heart failure. Mol Med. 2014;20:221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Bujak M, Frangogiannis NG. The role of IL-1 in the pathogenesis of heart disease. Arch Immunol Ther Exp (Warsz). 2009;57:165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Prakash D, Fesel C, Jain R, Cazenave PA, Mishra GC, Pied S. Clusters of cytokines determine malaria severity in Plasmodium falciparum-infected patients from endemic areas of Central India. J Infect Dis. 2006;194:198–207. [DOI] [PubMed] [Google Scholar]
- [118].Strangward P, Haley MJ, Albornoz MG, Barrington J, Shaw T, Dookie R, et al. Targeting the IL33-NLRP3 axis improves therapy for experimental cerebral malaria. Proc Natl Acad Sci U S A. 2018;115:7404–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Papayannopoulos V Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18:134–47. [DOI] [PubMed] [Google Scholar]
- [120].Folco EJ, Mawson TL, Vromman A, Bernardes-Souza B, Franck G, Persson O, et al. Neutrophil Extracellular Traps Induce Endothelial Cell Activation and Tissue Factor Production Through Interleukin-1alpha and Cathepsin G. Arterioscler Thromb Vasc Biol. 2018;38:1901–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Schimmel M, Nur E, Biemond BJ, van Mierlo GJ, Solati S, Brandjes DP, et al. Nucleosomes and neutrophil activation in sickle cell disease painful crisis. Haematologica. 2013;98:1797–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Hounkpe BW, Chenou F, Domingos IF, Cardoso EC, Costa Sobreira MJV, Araujo AS, et al. Neutrophil extracellular trap regulators in sickle cell disease: Modulation of gene expression of PADI4, neutrophil elastase, and myeloperoxidase during vaso-occlusive crisis. Res Pract Thromb Haemost. 2021;5:204–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Knackstedt SL, Georgiadou A, Apel F, Abu-Abed U, Moxon CA, Cunnington AJ, et al. Neutrophil extracellular traps drive inflammatory pathogenesis in malaria. Sci Immunol. 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Chen G, Zhang D, Fuchs TA, Manwani D, Wagner DD, Frenette PS. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood. 2014;123:3818–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Kono M, Saigo K, Takagi Y, Takahashi T, Kawauchi S, Wada A, et al. Heme-related molecules induce rapid production of neutrophil extracellular traps. Transfusion. 2014;54:2811–9. [DOI] [PubMed] [Google Scholar]
- [126].Munzer P, Negro R, Fukui S, di Meglio L, Aymonnier K, Chu L, et al. NLRP3 Inflammasome Assembly in Neutrophils Is Supported by PAD4 and Promotes NETosis Under Sterile Conditions. Front Immunol. 2021;12:683803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol. 2018;3. [DOI] [PubMed] [Google Scholar]
- [128].Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. 2018;3. [DOI] [PubMed] [Google Scholar]
- [129].Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015;349:316–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Gbotosho OT, Kapetanaki MG, Kato GJ. The Worst Things in Life are Free: The Role of Free Heme in Sickle Cell Disease. Front Immunol. 2020;11:561917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Mendonca R, Silveira AA, Conran N. Red cell DAMPs and inflammation. Inflamm Res. 2016;65:665–78. [DOI] [PubMed] [Google Scholar]
- [132].Lin S, Yin Q, Zhong Q, Lv FL, Zhou Y, Li JQ, et al. Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J Neuroinflammation. 2012;9:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Solovey A, Somani A, Belcher JD, Milbauer L, Vincent L, Pawlinski R, et al. A monocyte-TNF-endothelial activation axis in sickle transgenic mice: Therapeutic benefit from TNF blockade. Am J Hematol. 2017;92:1119–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Venugopal J, Wang J, Mawri J, Guo C, Eitzman D. Interleukin-1 receptor inhibition reduces stroke size in a murine model of sickle cell disease. Haematologica. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Gentinetta T, Belcher JD, Brugger-Verdon V, Adam J, Ruthsatz T, Bain J, et al. Plasma-Derived Hemopexin as a Candidate Therapeutic Agent for Acute Vaso-Occlusion in Sickle Cell Disease: Preclinical Evidence. J Clin Med. 2022;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Belcher JD, Chen C, Nguyen J, Abdulla F, Zhang P, Nguyen H, et al. Haptoglobin and hemopexin inhibit vaso-occlusion and inflammation in murine sickle cell disease: Role of heme oxygenase-1 induction. PLoS One. 2018;13:e0196455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Belcher JD, Mahaseth H, Welch TE, Otterbein LE, Hebbel RP, Vercellotti GM. Heme oxygenase-1 is a modulator of inflammation and vaso-occlusion in transgenic sickle mice. J Clin Invest. 2006;116:808–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Vinchi F, De Franceschi L, Ghigo A, Townes T, Cimino J, Silengo L, et al. Hemopexin therapy improves cardiovascular function by preventing heme-induced endothelial toxicity in mouse models of hemolytic diseases. Circulation. 2013;127:1317–29. [DOI] [PubMed] [Google Scholar]
- [139].Rees DC, Kilinc Y, Unal S, Dampier C, Pace BS, Kaya B, et al. A randomized, placebo-controlled, double-blind trial of canakinumab in children and young adults with sickle cell anemia. Blood. 2022;139:2642–52. [DOI] [PubMed] [Google Scholar]
- [140].Osunkwo I, O’Connor HF, Saah E. Optimizing the management of chronic pain in sickle cell disease. Hematology Am Soc Hematol Educ Program. 2020;2020:562–9. [DOI] [PMC free article] [PubMed] [Google Scholar]