

ABSTRACT
Age-related human pathologies present with a multitude of molecular and metabolic phenotypes, which individually or synergistically contribute to tissue degeneration. However, current lack of understanding of the interdependence of these molecular pathologies limits the potential range of existing therapeutic intervention strategies. In our study, we set out to understand the chain of molecular events, which underlie the loss of cellular viability in macroautophagy/autophagy deficiency associated with aging and age-related disease. We discover a novel axis linking autophagy, a cellular waste disposal pathway, and a metabolite, nicotinamide adenine dinucleotide (NAD). The axis connects multiple organelles, molecules and stress response pathways mediating cellular demise when autophagy becomes dysfunctional. By elucidating the steps on the path from efficient mitochondrial recycling to NAD maintenance and ultimately cell viability, we highlight targets potentially receptive to therapeutic interventions in a range of genetic and age-related diseases associated with autophagy dysfunction.
Abbreviations: IMM: inner mitochondrial membrane; NAD: nicotinamide dinucleotide; OXPHOS: oxidative phosphorylation; PARP: poly(ADP-ribose) polymerase; ROS: reactive oxygen species.
KEYWORDS: Aging, autophagy, DNA damage, mitochondria, mitophagy, NAD, PARP, ROS, sirtuins
Aging is strongly associated with a decline in autophagic activity, impairment of mitochondrial function, and diminishing cellular NAD levels. It is well established that among its many cellular functions, autophagy maintains mitochondrial health by the selective recycling of dysfunctional organelles, via a process called mitophagy. Autophagy, and specifically mitophagy, are also stimulated in response to NAD supplementation due to increased activity of NAD-consuming SIRT (sirtuin) enzymes. In addition to its role as a co-factor for NADases, NAD supports cellular bioenergetics by shuttling electrons to the oxidative phosphorylation (OXPHOS) apparatus within the inner mitochondrial membrane (IMM). In turn, OXPHOS in mitochondria maintains a pro-survival H+ potential across the IMM which powers energy generation. Autophagy/mitophagy dysfunction has been implicated in the pathology of age-related neurodegenerative disorders such as Alzheimer and Parkinson diseases. Furthermore, inactivation of autophagy is sufficient to cause cell death, for example loss of neurons in the brain in vivo. However, the molecular events linking autophagy dysfunction and cell death remain incompletely understood. Therefore, in our study we sought to clarify exactly which of the many physiological functions of autophagy contribute to cell survival, and, remarkably, we identified a critical dependence on mitochondrial recycling and NAD levels [1].
To mimic in cell culture the metabolic conditions in vivo, we exposed mouse autophagy-deficient cells to galactose-based medium which enforces mitochondrial OXPHOS. Unlike in the normal culture media containing glucose and allowing cells to derive energy via glycolysis, we observed a rapid onset of apoptotic cell death in the populations of actively respiring cells lacking autophagy machinery. In this model, we explored the metabolic profile of cells prior to the activation of apoptosis and discovered that autophagy-deficient cells present with a depleted pool of nucleotides, of which the reduced form of NAD (NADH) was the most significantly affected. Importantly, boosting NAD levels by precursor supplementation rescues cell viability, demonstrating that NAD/NADH depletion is sufficient to trigger cell death downstream of autophagy impairment.
In an effort to understand the cause of NAD depletion, we focused our attention on two main classes of NAD-consuming enzymes. SIRTs and PARPs (poly(ADP-ribose) polymerases) are stress-response enzymes activated by elevated reactive oxygen species (ROS) and DNA damage, both of which we found to co-occur in respiring autophagy-deficient cells. Although SIRTs and PARPs are typically thought of as pro-survival enzymes, we found that both classes of enzymes are persistently activated in autophagy-deficient cells which contributes to the depletion of intracellular NAD pools and the loss of cell viability (Figure 1).
Figure 1.
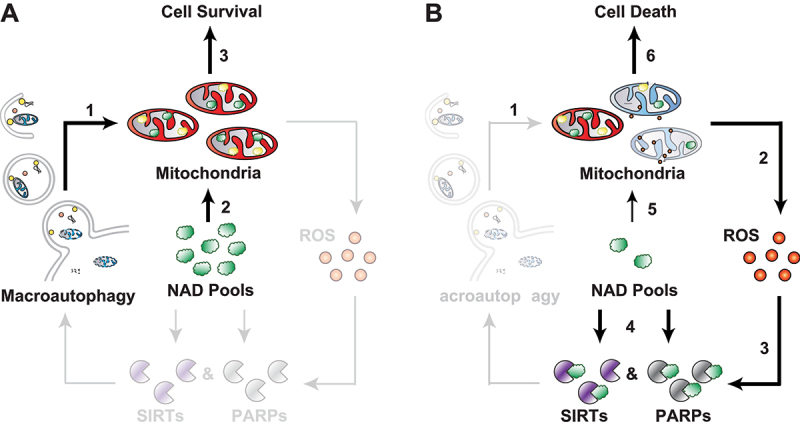
Autophagy promotes cell survival by maintaining intracellular NAD pools. Mitochondrial recycling by autophagy prevents a cascade of deleterious events downstream of mitochondrial dysfunction. (A) In autophagy-competent cells, recycling of mitochondria maintains a healthy population of organelles (1), which can draw on plentiful intracellular NAD pools (2) to support their metabolism and maintain cell survival (3). (B) In autophagy deficiency (1), accumulation of dysfunctional mitochondria results in elevated levels of ROS (2), which persistently activate stress-response enzymes of the SIRT and PARP families (3). Uncontrolled cleavage of NAD by these enzymes drives the exhaustion of intracellular NAD pools (4). Loss of available NAD negatively affects mitochondrial metabolism (5), which triggers IMM depolarization and apoptosis (6).
A large proportion of the NADH metabolite is found in mitochondria, the cellular hubs of metabolic and apoptotic signaling, and contributors to intracellular ROS levels. Mitochondrial structure, function and ROS release are all altered in respiring autophagy-deficient cells. Directing our focus toward the mitochondrial OXPHOS, we discovered that these cells suffer from a complex I dysfunction and membrane depolarization, which we found to be the trigger of apoptosis. Autophagy regulates mitochondrial quality control by removing damaged mitochondria via several known mitophagy pathways. By using mitophagy-defective pink1 knockout mouse embryonic fibroblasts and the PentaKO HeLa cells lacking five autophagy receptor proteins, we showed that impairment of mitophagy underlies NAD depletion and subsequent cell death. These studies allowed us to link the central role of autophagy in mitochondrial recycling to its pro-survival activity via protection of the intracellular NAD pools from exhaustion (Figure 1).
Throughout our study, we demonstrate that interventions at any step downstream of autophagy dysfunction can be sufficient to improve cell viability outcomes. Lowering ROS levels produced by dysfunctional complex I, or deploying ROS scavengers, prevents DNA damage and the subsequent hyperactivation of SIRTs and PARPs. Direct inhibition of these enzymes blocks the excessive cleavage of NAD. Finally, boosting intracellular NAD levels with precursors is sufficient to maintain mitochondrial membrane potential which is necessary for cell survival.
Autophagy is important for organismal survival, particularly in nutrient deprivation conditions, from yeast to humans. To test the evolutionary conservation of the autophagy-NAD axis, we investigated two models. In one we exposed autophagy-deficient (atg1Δ and atg5Δ) Saccharomyces cerevisiae to nitrogen starvation known to increase dependence of yeast on both autophagy and mitochondrial respiration. The second model represented human neurons affected by the neurodegenerative lysosomal storage Niemann-Pick type C1 disease and associated with an autophagy dysfunction. Both models present with a depletion of NAD levels and decreased survival, which are efficiently ameliorated by supplementation with nicotinamide, a precursor of NAD.
In summary our study highlights the interconnectedness of organellar and metabolic phenotypes in cells. We show that loss of autophagic recycling of mitochondria triggers a cascade of deleterious events that spans subcellular compartments, involves at least two stress-response pathways and the loss of the NAD metabolite, which negatively affects mitochondrial metabolism to the point of no return. As a subject for further study, we point to several therapeutic or lifestyle interventions that could potentially help to prevent cellular pathology in diseases associated with mitochondrial, autophagic and lysosome dysfunction. Overall, our hope is that further understanding the cross-talk between different, and often seemingly unrelated, molecular and cellular age-related phenotypes will lead to the development of innovative therapeutic strategies applicable to a range of diseases.
Acknowledgments
We appreciate the encouragement and helpful comments from the members of the Korolchuk and Sarkar laboratories.
Funding Statement
This work was supported by MRC studentship (BH174490) to L.S and V.I.K.; Fellowships from Uehara Memorial Foundation, the International Medical Research Foundation and Japan Society for the Promotion of Science (19J12969) to T.K.; Wellcome Trust. (109626/Z/15/Z, 1516ISSFFEL10), LifeArc (P2019-0004), UKIERI (UK–India Education and Research Initiative 2016-17-0087) and Birmingham Fellowship to S.S. Both V.I.K. and S.S. are Former Fellows for life at Hughes Hall, University of Cambridge.
Disclosure statement
V.I.K is a Scientific Advisor for Longaevus Technologies.
Reference
- [1].Kataura T, Sedlackova L, EG O, et al. Autophagy promotes cell survival by maintaining NAD levels. Dev Cell. 2022. Nov 21;57(22):2584–2598.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]