

ABSTRACT
Impaired activities and abnormally enlarged structures of endolysosomes are frequently observed in Alzheimer disease (AD) brains. However, little is known about whether and how endolysosomal dysregulation is triggered and associated with AD. Here, we show that vacuolar ATPase (V-ATPase) is a hub that mediates proteopathy of oligomeric amyloid beta (Aβ) and hyperphosphorylated MAPT/Tau (p-MAPT/Tau). Endolysosomal integrity was largely destroyed in Aβ-overloaded or p-MAPT/Tau-positive neurons in culture and AD brains, which was a necessary step for triggering neurotoxicity, and treatments with acidic nanoparticles or endocytosis inhibitors rescued the endolysosomal impairment and neurotoxicity. Interestingly, we found that the lumenal ATP6V0C and cytosolic ATP6V1B2 subunits of the V-ATPase complex bound to the internalized Aβ and cytosolic PHF-1-reactive MAPT/Tau, respectively. Their interactions disrupted V-ATPase activity and accompanying endolysosomal activity in vitro and induced neurodegeneration. Using a genome-wide functional screen, we isolated a suppressor, HYAL (hyaluronidase), which reversed the endolysosomal dysfunction and proteopathy and alleviated the memory impairment in 3xTg-AD mice. Further, we found that its metabolite hyaluronic acid (HA) and HA receptor CD44 attenuated neurotoxicity in affected neurons via V-ATPase. We propose that endolysosomal V-ATPase is a bona fide proteotoxic receptor that binds to pathogenic proteins and deteriorates endolysosomal function in AD, leading to neurodegeneration in proteopathy.
Abbreviations: AAV, adeno-associated virus; Aβ, amyloid beta; AD, Alzheimer disease; APP, amyloid beta precursor protein; ATP6V0C, ATPase H+ transporting V0 subunit c; ATP6V1A, ATPase H+ transporting V1 subunit A; ATP6V1B2, ATPase H+ transporting V1 subunit B2; CD44.Fc, CD44-mouse immunoglobulin Fc fusion construct; Co-IP, co-immunoprecipitation; CTSD, cathepsin D; HA, hyaluronic acid; HMWHA, high-molecular-weight hyaluronic acid; HYAL, hyaluronidase; i.c.v, intracerebroventricular; LMWHA, low-molecular-weight hyaluronic acid; NPs, nanoparticles; p-MAPT/Tau, hyperphosphorylated microtubule associated protein tau; PI3K, phosphoinositide 3-kinase; V-ATPase, vacuolar-type H+-translocating ATPase; WT, wild-type
KEYWORDS: Alzheimer disease, amyloid beta, endolysosome, hyaluronidase-CD44, proteopathy, MAPT/Tau, V-ATPase
Introduction
Proteostasis is robustly maintained by eliminating defective proteins via two major routes; the ubiquitin-proteasome system (UPS) and the macroautophagy/autophagy-lysosomal system [1]. However, the proteostatic balance becomes impaired with aging and under various pathologic conditions [2]. In particular, sustaining neuronal proteostasis throughout the lifetime is a crucial aspect required for brain function, which is supported by the fact that the endolysosomal degradative pathway is highly correlated with AD pathology, generically known as proteopathy [3,4]. For example, the highest-ranking risk factors for late-onset AD, including APOE4, BIN1, CD2AP, PICALM, and SORL1, are all thought to influence the endolysosomal pathway [5]. Activating TFEB (transcription factor EB), a master regulator of endolysosomal biogenesis including V-ATPase subunits [6], promotes clearance of Aβ and p-MAPT and thereby alleviates pathology in various AD model mice [7]. Abnormally enlarged degradative vacuoles containing Aβ are observed in the brains of patients with early-stage AD [8]. Accumulating evidence showed that both pathogenic Aβ peptides and MAPT proteins, major misfolded proteins in AD, deteriorate endolysosomal homeostasis [5,9–13], including the failure of ESCRT machinery [14] and destruction of lysosomal integrity [15–17], which may finally lead to degeneration of affected neurons [18]. However, the mechanism by which Aβ and pathogenic MAPT proteins induce endolysosomal dysfunction remains unclear.
Maintaining the optimal lumenal pH within endolysosomes is crucial for many biological processes, including the activation of hydrolytic enzymes and vesicular transport or membrane translocation of small molecules [19]. The V-ATPase is a large multisubunit complex that plays a pivotal role in regulating the lumenal pH of intracellular compartments [20]. Many studies have highlighted the tight association between V-ATPase and neurodegeneration, including AD [19,21]. Reduced expression of V-ATPase components, altered modification, and single nucleotide polymorphism have been widely reported in the brains of AD patients, AD model mice (5xFAD, PSEN-APP, and 3xTg-AD), and in AD-like conditions [22–28]. Recently, ATP6V1A was identified as a top key regulator of a neuronal subnetwork most disrupted in late-onset AD through multi-omics profiling, RNAi-knockdown, or pharmacogenomic regulation [29]. The ATP6V1B2 subunit level decreases significantly during aging and particularly during the early development of AD pathology [30], while altered expression of the ATP6V0C subunit affects neurodegenerative features [31,32]. Moreover, proteomic analysis revealed that p-MAPT interacts with several V-ATPase subunits, including ATP6V0D1, ATP6V1B2, and ATP6V1H [33]. Despite these findings implying a close connection between AD and V-ATPase, little is known about the molecular link between endolysosomal V-ATPase and AD pathology.
Here, we report that V-ATPase activity in the endolysosomes is impaired in cultured neurons exposed to AD-pathogenic proteins and in AD model mice and that the V-ATPase ATP6V0C and ATP6V1B2 subunits bind to proteotoxic Aβ and MAPT species in vitro and in vivo. Furthermore, with a functional screen using cDNAs encoding endolysosomal proteins, we identified a HYAL-CD44 axis that regulates V-ATPase assembly, reverses the endolysosomal dysfunction, Aβ and MAPT proteotoxicity, and memory impairment in AD model mice.
Results
Lysosomal dysfunction triggered by endolysosomal Aβ mediates neurotoxicity
We evaluated whether the endolysosomal malfunction is crucial for Aβ toxicity by measuring cell viability after exposure to oligomeric Aβ1-42 (hereafter described as Aβ) together with acidic nanoparticles (NPs), which are trafficked to acidic vesicles, sustain acidic pH and improve degradative function [34]. To our surprise, Aβ neurotoxicity was largely reversed by acidic NPs treatment in HT22 mouse hippocampal cell line (Figure 1A), despite a low degree of toxicity per se in normal conditions [35]. To address whether toxic forms of Aβ exhibit an adverse impact on endolysosomal activity, we measured the fluorescence from DQ-Red-BSA, a BSA-derived protease substrate used as an indicator of endolysosomal activity [36]. We normalized it with the intensity of Cascade Blue-dextran, an endocytic uptake marker. The DQ-BSA:dextran ratio was significantly reduced by Aβ treatment and recovered by acidic NPs treatment (Figure 1B). The DQ-BSA:dextran ratio was time-dependently decreased by Aβ in primary cultured hippocampal neurons (Figure 1C), cortical neurons (Fig. S1A), and HT22 cells (Fig. S1B). Conditioned media of 7PA2 CHO cells stably expressing human APP751V717F is characterized to contain nanomolar concentrations of different soluble Aβ oligomers, including dimers, trimers, and zeta peptide [37,38], and increase neurotoxicity, especially in primary neurons (Fig. S1C and S1D) [38,39]. The endolysosomal degradative activity was also significantly reduced in mouse primary hippocampal neurons by treatment with naturally secreted Aβ in the conditioned media of 7PA2 CHO cells (Figure 1D). These impairments of endolysosomal activity were observed early (12 h and 24 h) under a toxic dose (5 μM) of Aβ treatment, prior to the onset of neurotoxicity seen later (36 h and 48 h) (Figure 1E).
Figure 1.

Aβ-induced endolysosomal dysfunction leads to neurotoxicity. (A, B) HT22 cells were treated with 5 μM Aβ in the absence or presence of 1 mg/ml acidic NPs for 48 h (A) or 24 h (B). Cytotoxicity was analyzed by PI and calcein-AM staining (n = 3, cells > 400 for each trial) (A). Proteolysis activity was analyzed by measuring DQ-red-BSA:Cascade Blue-dextran fluorescence ratio (n = 3, cells > 40 for each trial) (B), two-way ANOVA. (C) Primary hippocampal neurons (DIV 6–8) incubated with DQ-red-BSA and Cascade Blue-dextran for 30 min and treated with 5 μM Aβ Aβ for the indicated times (upper). DQ-red-BSA:Cascade Blue-dextran fluorescence ratio was measured (lower, n = 3, cells > 20 for each trial), two-way ANOVA. (D) Primary hippocampal neurons (DIV 6–8) were treated either with CHO-conditioned control media or 7PA2-conditioned media for 24 h. DQ-red-BSA:Cascade Blue-dextran fluorescence ratio was measured (n = 3, cells > 10 for each trial), two-tailed unpaired t-test. (E) HT22 cells were incubated with the indicated concentrations of Aβ for 12–48 h. Cytotoxicity was analyzed by PI and calcein-AM staining (n = 3, cells > 150 for each trial), two-way ANOVA. (F) Immunoblot (left) and the quantitative measurements of SQSTM1 (middle) and MAP1LC3B (right) in the primary hippocampal neurons (DIV 7) untreated (0 μM) or treated with the indicated concentrations of Aβ for 24 h (n = 3), one-way ANOVA. (G) Immunoblot (left) and the EGFP-CINCCKVL (GFP-8) levels (right) of GFP-8 in ectopically GFP-8-expressing HT22 cells untreated (0 μM) or treated with the indicated concentrations of Aβ for 24 h (n = 3), one-way ANOVA. (H) GFP puncta numbers per area were analyzed in HT22 cells expressing GFP-LGALS3 untreated (0 μM) or treated with the indicated concentrations of Aβ for 24 h (n = 3, cells >50 in total), one-way ANOVA. (I) Green fluorescence intensity of acridine Orange were measured in HT22 cells untreated (0 μM) or treated with the indicated concentrations of Aβ for 24 h (n = 3, cells >300 in total), one-way ANOVA. (J) HT22 cells was untreated (0 μM) or treated with the indicated concentrations of Aβ for 21 h, further incubated with 10 nM Baf.A1 for 3 h and stained with anti-LAMP1 antibody. The sizes of LAMP1-positive signals were analyzed (n = 3, cells > 30 for each trial), two-way ANOVA. (B, C, D, H, I, J) Scale bar: 10 μm.
We further characterized the endolysosomal impairments. Proteolytic maturation of endolysosomal CTSD/cathepsin D was dose-dependently impaired by Aβ (Fig. S1E). In addition, the endolysosomal substrates SQSTM1/p62, MAP1LC3B/LC3B, and GFP-8, a chimeric EGFP protein fused to the lysosomal targeting sequence CINCCKVL [40], were highly accumulated in Aβ-exposed neurons (Figure 1F and G). To test the impact of Aβ on endolysosomal membrane integrity, we used GFP-LGALS3 (galectin 3), a marker of damaged endolysosomes [41], and acridine orange, an acidotropic reagent that fluoresces green in nonacidic compartments [42]. The numbers of GFP-LGALS3 puncta in the endolysosomes (Figure 1H) and the green fluorescent signal from acridine orange in the cytosol (Figure 1I) all increased by a toxic dose of Aβ treatment. In particular, compared to control or low Aβ-treated cells, LAMP1-positive endolysosomes were abnormally enlarged in Aβ-overloaded cells (Figure 1J and Fig. S1F). These results suggest that endolysosomal integrity is impaired as an early event contributing to Aβ neurotoxicity.
Upon binding to Aβ, various receptors mediate its internalization and subsequent degradation along the endolysosomal trafficking route [18,43,44]. Accordingly, Fluorescein-labeled oligomeric Aβ (FITC-Aβ) was observed within RAB5, RAB7, and LAMP1-positive endolysosomal vesicles (Fig. S2A). Consistent with the previous findings [18], inhibiting Aβ uptake by the treatment with monodansylcadaverine (MDC), an inhibitor of clathrin-mediated endocytosis, reversed both cytotoxicity (Figure 2A) and endolysosomal damage (Figure 2B). Further, the single-cell level analysis confirmed that the intensity of the DQ-green-BSA signal remarkably declined in cells harboring high levels of DyLight 594-labeled Aβ within cellular vesicles (Figure 2C), which indicates an inverse relation between proteolytic activity and Aβ deposition within endolysosomal vesicles. Meanwhile, the Cascade Blue-dextran intensity was rarely changed in Aβ-containing vesicles. Together, these results indicate that retention of neurotoxic levels of Aβ along the endolysosomal pathway leads to proteolytic dysfunction and loss of endomembrane integrity.
Figure 2.

Accumulation of Aβ leads to proteolytic dysfunction in endolysosomes. (A, B) HT22 cells were treated with 5 μM Aβ in the absence or presence of 100 μM MDC for 48 h (A) or 24 h (B). Cytotoxicity was analyzed by PI and calcein-AM staining (n = 3, cells > 400 for each trial) (A). Proteolysis activity was analyzed by measuring DQ-red-BSA:Cascade Blue-dextran fluorescence ratio (n = 3, cells > 50 for each trial) (B). Two-way ANOVA. (C) HT22 cells were co-treated with 5 μM DyLight 594-labeled Aβ, 10 μg/ml DQ-green-BSA and 10 μg/ml Cascade Blue-dextran for 24 h (upper). Pixel intensities in dual-channel images were analyzed and displayed as a scatter plot (lower). (D) Microsomes purified from mouse brains were incubated with 1 μM Baf.A1 or liposomes pre-loaded with 2.5 μM Aβ or 3 μM BSA (shown in Fig. S2C). The amount of phosphate released from the microsomes was determined by the malachite green method (n = 4), two-way ANOVA. (B, C) Scale bar: 10 μm.
V-ATPase activity is impaired by Aβ-binding to the ATP6V0C subunit
Using fluorescence protease protection assays, we confirmed that FITC-Aβ internalized into HT22 cells evades proteinase K digestion and resides within the intralumenal space of intracellular vesicles (Fig. S2B). Given that maintenance of the proton gradient in acidic vacuoles is one of the most critical factors for endolysosomal function and V-ATPase is a central player in lumenal acidification, we surmised that intralumenal Aβ might perturb V-ATPase activity. To test that idea, we performed in vitro V-ATPase activity assays following intralumenal insertion of Aβ, achieved through liposomal delivery (Fig. S2C). The free phosphate concentration indicates the proper ATP hydrolysis at catalytic sites of the V1, which is tightly coupled with the rotatory movement of the ring of proteolipid subunits (ATP6V0C and V0B) in the V0 complex. Microsomes, including endolysosomal vesicles, were prepared from mouse brains and fused with the liposomes preloaded with Aβ. Subsequent malachite green phosphate assays revealed that ATP hydrolysis was markedly lower in the microsomal fraction treated with Aβ-liposomes than in the fraction treated with BSA-liposomes (Figure 2D), suggesting that intralumenal Aβ disrupts ATP hydrolysis by the proton pump.
Through both a genome-wide protein chip analysis of 20,000 human recombinant proteins (Dataset S1) and an expressional screen of cDNA encoding endolysosomal proteins (Dataset S2), we found two integral subunits of V-ATPase, ATP6V0C and ATP6V0B, which mediate proton translocation, as potential targets of Aβ toxicity. In these screens, FITC-Aβ bound to V0 subunits, and ectopic expression of ATP6V0C or ATP6V0B suppressed Aβ neurotoxicity. While both ATP6V0C and ATP6V0B were effective, we hardly detected overexpressed HA- or GFP-tagged ATP6V0B in the transfected cells (Fig. S3A-C) and thus decided to characterize a role of ATP6V0C. To confirm the possible interaction between these subunits and Aβ, we first purified the V-ATPase complex using the high-affinity interaction between the V-ATPase ATP6V1A subunit and the Legionella pneumophila effector protein SidK [45]. With an affinity purification protocol employing SidK (residues 1 to 278) fused to a C-terminal 3× FLAG tag and M2-agarose, highly purified V-ATPases were prepared from mouse brains (Figure 3A). Co-IP assays using anti-Aβ antibodies (4G8 and 6E10) showed that Aβ binds to the ATP6V0C subunit but not with ATP6V1A or ATP6V1B2 despite their higher levels than ATP6V0C as seen in input in vitro (Figure 3B).
Figure 3.

V-ATPase ATP6V0C subunit is functionally associated with Aβ toxicity. (A) Silver staining of V-ATPase complexes purified from the brains of mice through a high-affinity purification strategy involving SidK. (B) Eight μg purified V-ATPases were incubated with 3 μg Aβ in PBS containing 1% Triton-X 100 for 3 h, and the affinity of V-ATPase subunits (ATP6V0C, ATP6V1A, and ATP6V1B2) for Aβ were analyzed by co-IP with anti-Aβ (4G8) antibody. (C) ATP6V0C-knockdown HT22 stable cells were established using a CRISPR/Cas9 system and the endogenous ATP6V0C level was analyzed by western blotting; HT22 control (left), sgRNA ATP6V0C #14 (middle), and #19 (right) stable cells. (D) HT22 control, sgRNA ATP6V0C #14, and #19 stable cells were transfected with EGFP or EGFP-ATP6V0C for 24 h and further treated with Aβ for 48 h. Cytotoxicity was analyzed by PI staining (n = 3, cells > 150 for each trial), two-way ANOVA. (E) HT22 control and sgRNA ATP6V0C #14 cells were treated with 5 μM Aβ for 24 h. Proteolysis activity was analyzed by measuring DQ-red-BSA:Cascade Blue-dextran fluorescence ratio (n = 3, cells > 35 for each trial), two-way ANOVA. Scale bar: 10 μm. (F) HT22 control and sgRNA ATP6V0C #14 cells stably expressing pHLARE were reconstituted with ATP6V0C gene and then treated with 5 μM Aβ for 24 h. The pH was measured by calibrating sfGFP:mCherry fluorescence ratio (n = 5, each dot presents the average pH of cells > 300 per trial), two-way ANOVA. (G) Schematic representation of the structure of the ATP6V0C subunit of V-ATPase. (H, I) HT22 cells were co-transfected with pEGFP, and pcDNA-HA (Control), indicated HA-ATP6V0C WT or mutants and further treated with 5 μM Aβ for 48 h. The apoptotic cell death was analyzed by PI staining. Two-way ANOVA (H, n = 3, cells > 100 for each trial; I, n = 4, cells > 100 for each trial) (upper). Expression of HA-ATP6V0C WT or mutants were analyzed by western blotting (lower).
We reasoned that if ATP6V0C binds to Aβ, overexpressed ATP6V0C might compete with endogenous ATP6V0C to bind to Aβ and thereby attenuate Aβ toxicity. Alternatively, overexpressed ATP6V0C might also increase the activity of the endolysosome to potentiate Aβ elimination. As expected, Aβ-induced neurotoxicity was significantly reduced by ectopic expression of ATP6V0C in HT22 cells (Figure 3D, 3H, 3I, and 4A). On the other hand, Aβ neurotoxicity or proteolytic defects was exacerbated in HT22 ATP6V0C knockdown cells that express endogenous ATP6V0C below the half level compared to the control (Figure 3C-E). Reconstitution of the protein in ATP6V0C knockdown cells reversed the Aβ damage (Figure 3D). Furthermore, pH impairment by Aβ was rescued by ectopic expression of ATP6V0C subunit (Figure 3F and Fig. S3D) in HT22 stable cells expressing pHLARE, a genetically encoded ratiometric pH sensor predominantly localized in endolysosomes [46]. It is interesting to note that ATP6V0C-depletion impairs endolysosomal acidification, but additional Aβ burden is required to cause cell death and proteolytic defects.
Figure 4.

Aβ interrupts endolysosomal function by binding to ATP6V0C. (A) HT22 cells were transfected with pEGFP (Control), EGFP-ATP6V0C WT, EGFP-ATP6V0C Δ82-85, or EGFP-ATP6V0CD82E,D83E,S85T for 12 h and further treated with 5 μM Aβ for 48 h. Cytotoxicity was analyzed by PI staining (n = 7, cells > 100 for each trial), two-way ANOVA. (B) HT22 cells were transfected with pcDNA-HA (Control), HA-ATP6V0C WT, HA-ATP6V0C Δ82-85, or HA-ATP6V0CD82E,D83E,S85T for 12 h and further treated with 5 μM Aβ for 24 h. Proteolysis activity was analyzed by measuring DQ-red-BSA:Cascade Blue-dextran fluorescence ratio (n = 3, cells >40 for each trial), two-way ANOVA. (C) HEK293T cells were transfected with EGFP (control), EGFP-ATP6V0C WT, Δ82-85, or EGFP-ATP6V0CD82E,D83E,S85T mutant for 24 h. Cell homogenates were subjected to in vitro binding assays after the addition of 5 μg Aβ and followed by co-IP using anti-Aβ (4G8) antibody. (D-H) (D) The hippocampal tissues from seven non-AD and five AD brains were immunostained for Aβ (6E10, red), ATP6V0C (green), and MAP2 (blue). (E-H) A dot presents the mean size of ATP6V0C-positive vesicles within neuron. Age, the numbers of cases and cells used in statistical analysis are represented below the graph. Two-tailed unpaired t-test (F, H) or two-way ANOVA (G, I). (B, D) Scale bar: 10 μm.
Considering that ATP6V0C contains three lumenal regions (Figure 3G), we speculated that ATP6V0C binds to lumenal Aβ at one of those intralumenal regions. To test that idea, we generated a series of ATP6V0C deletion mutants lacking one of the three lumenal regions, ∆1-10, ∆81-88, or ∆153-155 (Figure 3H and Fig. S3E), and found that expression of ATP6V0C ∆1-10 or ∆153-155 reversed Aβ-toxicity, while ATP6V0C ∆81-88 failed to do so. Among a collection of alanine-substitution mutants in the 81–88 residues, the ATP6V0C D82A, D83A, or S85A mutants partially lost their ability to reverse Aβ toxicity (Figure 3I and Fig. S3E). We further found that a deletion mutant lacking 82–85 residues (∆82-85) or a substitution mutant in which D82-D83-S85 were replaced with E82-E83-T85 (EGFP-ATP6V0CD82E,D83E,S85T) did not alleviate the Aβ-toxicity (Figure 4A). Unlike ATP6V0C WT, the ∆82-85 and ATP6V0CD82E,D83E,S85T mutants also failed to affect Aβ-induced impairment of endolysosomal activity (Figure 4B). Endogenous ATP6V0C is distributed along the endocytosis pathway and lysosomes (Fig. S1F and S4A). The distributions of WT and mutant proteins were similar, despite the significant signal due to overexpression. (Fig. S4B). Collectively, these results show that the ATP6V0C lumenal region (82–85) is important for Aβ pathologies, including neurotoxicity and endolysosomal impairment.
We then analyzed the ability of these mutants for binding to Aβ in cells. Co-IP assays following the incubation of post-mitochondrial supernatants with added Aβ revealed that Aβ bound to ATP6V0C with greater affinity than it bound to the ATP6V0C ∆82-85 or ATP6V0CD82E,D83E,S85T mutant (Figure 4C and Fig. S4C), which is consistent with their distinctive inhibitory effects on Aβ neurotoxicity (Figure 4A) and endolysosomal activity (Figure 4B). Moreover, ectopic expression of a chimeric protein containing the peptide spanning the ATP6V0C 81–88 region and the lysosomal targeting sequence (GFP-WT-8) reversed Aβ proteotoxicity, whereas a chimera containing the mutations (GFP-mt-8) was insufficient (Fig. S4D-F). Thus, the second lumenal region of ATP6V0C is critical for Aβ binding, which likely contributes to the failure of V-ATPase activity within endolysosomes. In addition, we found that Aβ was massively accumulated within the ATP6V0C-positive vesicles in the hippocampal neurons of human brains, and Aβ-containing ATP6V0C-positve vesicles were markedly enlarged in AD brains compared to non-AD (Figure 4D-H and Table S1), which suggests a possible interaction of Aβ and ATP6V0C in AD.
Phosphorylated MAPT interacts with ATP6V1B2 and disrupts V-ATPase function
Given that highly phosphorylated MAPT oligomers and aggregates cause neurodegeneration and often function as downstream mediators of Aβ signaling in AD [47,48], we assessed the role of MAPT in the endolysosomal axis. Interestingly, the Aβ-induced reduction of proteolytic activity was smaller in MAPT-depleted primary hippocampal neurons than in control neurons (Fig. S5A). To determine whether MAPT protein itself affects endolysosomal function, we transduced primary neurons with adeno-associated virus (AAV) expressing human MAPTP301L as a model system of MAPT pathogenesis. Consistent with the observation in the MAPT knockdown neurons, DQ-BSA:dextran ratio were decreased in neurons infected with AAV-MAPTP301L::GFP (Figure 5A). What is more, the signals in MAPT -expressing neurons were additively diminished by the Aβ treatment (Figure 5A). To visualize in vivo endolysosomal protease activity, we developed a new assay by co-injecting DQ-red-BSA and Cascade Blue-dextran, a control for cellular uptake, into the mouse brain tissues (Fig. S5B). Using this method, we found that endolysosomal activity was significantly impaired in rTg4510 Tauopathy model mice (Figure 5B), which express human MAPT under doxycycline control. This evidence enabled us to extend the V-ATPase hypothesis to MAPT.
Figure 5.

Phosphorylated MAPT disrupts V-ATPase activity by interacting with ATP6V1B2. (A) Images of primary hippocampal neurons (DIV 7–9) that were transduced with AAV-GFP or AAV-MAPTP301L::GFP and further treated with 5 μM Aβ for 24 h (left). Cells were co-treated with DQ-red-BSA and Cascade Blue-dextran. Proteolysis activity was analyzed by measuring DQ-red-BSA:Cascade Blue-dextran fluorescence ratio (n = 3, cells = 22–27 in total), two-way ANOVA (right). (B) Representative images showing lysosomal activity in mouse hippocampal CA3 regions. Five-month-old tTA−/−MAPT± and tTA±MAPT± mice were i.c.v. injected with DQ-red-BSA and Cascade Blue-dextran (shown in Fig. S3B). Scale bar: 100 μm (left). DQ-BSA intensity was normalized to the Cascade Blue signal; tTA−/−MAPT± (n = 3); tTA±MAPT± (n = 3), two-tailed unpaired t-test (right). (C) Co-IP analysis of homogenates prepared from HEK293T cells co-expressing EGFP-ATP6V1B2 and either HA-MAPT WT or MAPTS396A,S404A mutant (shown in Fig. S5C) (upper). The relative levels of MAPT-bound to ATP6V1B2 were measured (n = 3), two-tailed unpaired t-test (lower). (D) Microsomes purified from mouse brains were incubated for 40 min with 1 μM Baf.A1, 3 μM BSA, 2 μM His-tagged MAPT WT, or MAPTS396A,S404A mutant protein purified from HEK293T cells, and the free phosphate levels were measured (n= 3), two-way ANOVA. (E) HT22 cells stably expressing pHLARE were transfected with either empty vector (-), HA-tagged MAPT WT, or HA-MAPTS396A,S404A mutant for 24 h. The pH was measured by calibrating sfGFP:mCherry fluorescence ratio (n = 3, each dot presents the average pH of cells > 300 for each trial), one-way ANOVA. (F) Domain structure of ATP6V1B2 WT and deletion (Δ) mutants; N, N-terminus; NBD, nucleotide-binding-domain; C, C-terminus and their binding ability to MAPT. (G) HEK293T cells were co-transfected with indicated EGFP-ATP6V1B2 constructs and HA-MAPT WT for 24 h. Cell homogenates were subjected to co-IP using anti-HA antibody. (H) Images of HT22 cells that were co-transfected with empty vector, or HA-MAPT and either with EGFP, EGFP-ATP6V1B2 WT or Δ120-172 constructs for 24 h. Cells were co-treated with DQ-red-BSA and Cascade Blue-dextran for 6 h. (A, H) Scale bar: 10 μm.
Two recent independent proteomic analyses of the human AD brain showed a high probability of p-MAPT interaction with ATP6V1B2 [33], the brain isoform of the V-ATPase V1B subunit. We performed co-IP assays and found that HA-MAPT bound to GFP-ATP6V1B2 when overexpressed in cells (Figure 5C). Notably, compared to MAPT WT, MAPTS396A,S404A mutant lacking the phosphorylated residues recognized by the PHF-1 antibody showed less affinity for ATP6V1B2 (Figure 5C and Fig. S5C), indicating that it is the phosphorylated forms of MAPT that could bind to ATP6V1B2 in cells. We also measured the effects of p-MAPT on V-ATPase activity in vitro. Overexpression of MAPT in human cells ensures its hyperphosphorylation at multiple epitopes, including PHF-1-detectable residues [49]. This enabled us to purify human p-MAPT WT and S396A, S404A double mutant (MAPTS396A,S404A) proteins from HEK293T cells after their overexpression. In vitro assays revealed that ATP hydrolysis was diminished in the microsomes incubated with p-MAPT compared to BSA control (Figure 5D). By contrast, V-ATPase activity was little affected by the MAPTS396A,S404A protein in the same assay. Similarly, pH impairment was shown only in MAPT WT-expressing HT22 cells (Figure 5E), suggesting p-MAPT exerts deleterious effects on V-ATPase activity. Then, in co-IP assays, we found that the deletion of the N-terminal (∆N), nucleotide-binding domain (∆NBD), or C-terminal (∆C) of ATP6V1B2 did not affect the MAPT interaction (Figure 5F and G). However, the deletion of residues 120–172 (∆120-172) abolished MAPT-binding, suggesting that the region adjacent to the ATP6V1B2 ATP-binding domain is crucial for MAPT -interaction. As seen in Aβ, MAPT expression also impaired endolysosomal proteolytic activity, which was restored by overexpression of ATP6V1B2 WT but not by ∆120-172 mutant (Figure 5H), indicating that the pattern of p-MAPT-ATP6V1B2 interaction coincides with the impaired endolysosomal activity.
Recovery of impaired V-ATPase activity: enhancing HA flux reverses Aβ neurotoxicity
The notion that endolysosomal and V-ATPase impairment is an early and critical event in Aβ pathology led us to perform a genome-wide functional screen to search for a novel regulator of the endolysosomal pathway that rescues Aβ neurotoxicity. Because we assumed that the regulator would be located primarily on endolysosomes, we collected about 300 cDNAs encoding endolysosomal proteins. We tested whether their ectopic expressions could rescue HT22 cells from Aβ toxicity (Figure 6A). Among the 300 cDNAs screened, a list of putative positive clones that lowered Aβ neurotoxicity to less than control was isolated from the transfected cells (Figure 6B). Notably, a group of genes encoding the HYAL (hyaluronidase) family (HYAL1, HYAL2 and HYAL3), which degrade hyaluronic acid [50], effectively suppressed Aβ toxicity to the control level (Figure 6C). Because HYAL1 is located within endolysosomes, we primarily characterized its function.
Figure 6.

Enhancing HA flux reverses Aβ neurotoxicity and V-ATPase malfunction. (A) Screen scheme to identify endolysosome-resident regulators of Aβ neurotoxicity with a cell-based assay using cDNAs expression. HT22 cells were transfected for 24 h with each cDNA, treated with 5 μM Aβ for 36 h, and apoptotic morphological changes were analyzed. (B) Effects of the overexpressed cDNAs on Aβ toxicity. The cDNAs showing resistance (green) or sensitization (red) to Aβ toxicity are shown. (C) HT22 cells were transfected with pEGFP (Control), EGFP-HYAL1, HYAL2, or HYAL3 for 12 h and further treated with 5 μM Aβ for 36 h. Cytotoxicity was analyzed by PI staining (n = 3, cells > 200 for each trial), two-way ANOVA. (D) HT22 sgRNA control (control), sgRNA Hyal1 #2 cells were co-transfected with EGFP and HYAL1 for 24 h and treated with 5 μM Aβ for 36 h. Cytotoxicity was analyzed by PI staining (n = 3, cells > 300 for each trial), two-way ANOVA. (E) Primary hippocampal neurons (DIV 6) were treated with 100 μg/ml LMWHA (8–12 kDa) or HMWHA (1.75–2 MDa) in the presence or absence of 5 μM Aβ for 36 h. Cytotoxicity was analyzed by PI and calcein-AM staining (n = 3, cells > 250 for each trial), two-way ANOVA. (F) Primary hippocampal neurons (DIV 5–7) were transfected with EGFP and treated with 100 μg/ml LMWHA in the presence or absence of 5 μM Aβ for 48 h. The quantitative analysis showing the numbers of dendritic spines per 10 μm in the Control (cells = 18 in total), Aβ (cells = 24 in total), and Aβ + 100 μg/ml LMWHA (cells = 32 in total) (n = 3), one-way ANOVA. (G) Immunoblot (left) and the quantitative measurements (right) of GFP-8 signal on the blots. HT22 cells were transfected with GFP-8 and treated with 5 μM Aβ in the presence or absence of 100 μg/ml LMWHA for 24 h (n = 3), two-way ANOVA. (H) HT22 cells were treated with 5 μM Aβ alone or with 100 μg/ml LMWHA for 24 h, and stained with anti-LAMP1 antibody. The sizes of LAMP1-positive signals were analyzed (n = 3, cells > 30 for each trial), two-way ANOVA. (I) HT22 cells stably expressing pHLARE were treated with 5 μM Aβ in the presence or absence of 100 μg/ml LMWHA for 24 h. The pH was measured by calibrating sfGFP:mCherry fluorescence ratio (n = 3, each dot presents the average pH of cells > 300 for each trial), two-way ANOVA. (J) HT22 cells stably expressing pHLARE were transfected with HA-MAPT and treated with 100 μg/ml LMWHA for 24 h. The pH was measured by calibrating sfGFP:mCherry fluorescence ratio (n = 3, each dot presents the average pH of cells > 300 for each trial), two-way ANOVA. (K) HT22 cells were transfected with HA-MAPT and treated with 100 μg/ml LMWHA for 24 h. Cell extracts were then fractionated into the membrane (memb.) and cytosolic (cyto.) fractions, and the fractions were subjected to western blotting (left). ATP6V1A (middle) and ATP6V1B2 (right) levels in the membrane fractions were quantified using LAMP1 loading controls (n = 3), two-way ANOVA. (F, H) Scale bar: 10 μm.
While knocking out HYAL1 expression itself did not affect HT22 cell viability, cell death among HYAL1-depleted cells exposed to Aβ was about 15% higher than that among control cells (Figure 6D and Fig. S6A), suggesting that HYAL1 exerts a protective effect against Aβ toxicity. An activity-dead mutant (HYAL1D129A,E131A) in which Asp129 and Glu131, critical residues for the enzyme’s activity [51,52], were replaced with Ala, however, exhibited considerably less ability to suppress Aβ toxicity (Fig. S6B and S6C), which prompted us to examine the effects of HYAL substrate, hyaluronic acid (HA), on Aβ pathology. Because different HYAL family enzymes preferentially degrade different sized HA isoforms [50], we tested low-molecular-weight HA (LMWHA, 0.8–1.2 kDa) and high-molecular-weight HA (HMWHA, 1.75–2 MDa). Aβ neurotoxicity in primary hippocampal neurons was inhibited largely by LMWHA treatment but was less affected by HMWHA (Figure 6E). Similarly, dendritic spine density decreased in Aβ-treated hippocampal neurons, which was blocked by LMWHA such that spine density remained at nearly the untreated control level (Figure 6F). We confirmed that HA treatment hardly affected the cellular uptake of Aβ, which usually occurred within 30 min (Fig. S6D). Interestingly, LMWHA effectively reversed all lysosomal degradation of GFP-8, decreased proteolytic activity, abnormally enlarged endolysosomes, and impaired pH in cells exposed to Aβ (Figure 6G-I and Fig. S6E). We also found that overexpressed HA synthase, HAS1 or HAS3, efficiently inhibited Aβ-toxicity as much as HYAL in HT22 cells (Fig. S6F). LMWHA also restored pH impairment in MAPT-overexpressing HT22 cells (Figure 6J). Moreover, we observed the increased recruitment of ATP6V1A and ATP6V1B2 to membrane-enriched fractions by LMWHA treatment in HT22 cells and MAPT-overexpressing cells (Figure 6K), suggesting a possibility for the promotion of V-ATPase assembly by LMWHA.
Since peripherally administrated HA is rapidly degraded by the liver [53], we tested the effect of LMWHA on Aβ pathology by directly injecting LMWHA and Aβ into the mouse brains. As reported [54], Aβ-injected mice showed profound recognition memory loss in novel object recognition (NOR) tests (Fig. S6G). By contrast, mice co-injected with Aβ plus LMWHA exhibited recognition performance comparable to that of control mice, and the improvement persisted into the following days. All these results indicate that facilitating HA flux in a direction that increases levels of LMWHA, either by supplying raw material for subsequent HA degradation or by cleaving the HMWHA to smaller fragments, contributes to overcoming Aβ and MAPT neurotoxicity, probably by facilitating V-ATPase assembly.
CD44 reverses endolysosomal dysfunction and neurotoxicity triggered by Aβ via PI3K
We reasoned that HA exerted the protective effect through CD44, a well-known HA receptor [55]. As expected, overexpression of CD44 itself overcame Aβ toxicity in HT22 cells (Figure 7B). Because HA is localized in the endolysosomes as well as at the extracellular matrix, we tried to confirm whether the HA-CD44 signaling, or the HA within the endolysosomes, contributes to the protective effects. To do so, we generated a CD44-IgG.Fc fusion construct (CD44.Fc) by replacing the HA-binding ectodomain of CD44 with the mouse immunoglobulin (IgG) Fc region, enabling it to form dimers in the absence of a ligand (Figure 7A). We found that the ectopic expression of CD44.Fc inhibited Aβ neurotoxicity as much as CD44 (Figure 7B, 7C, and 7E) and recovered the endolysosomal dysfunction triggered by Aβ as well (Figure 7D). CD44.Fc also reversed the impaired assembly of V-ATPase by MAPT (Fig. S7A). Thus, it is the HA-CD44 signaling that can attenuate Aβ pathology.
Figure 7.
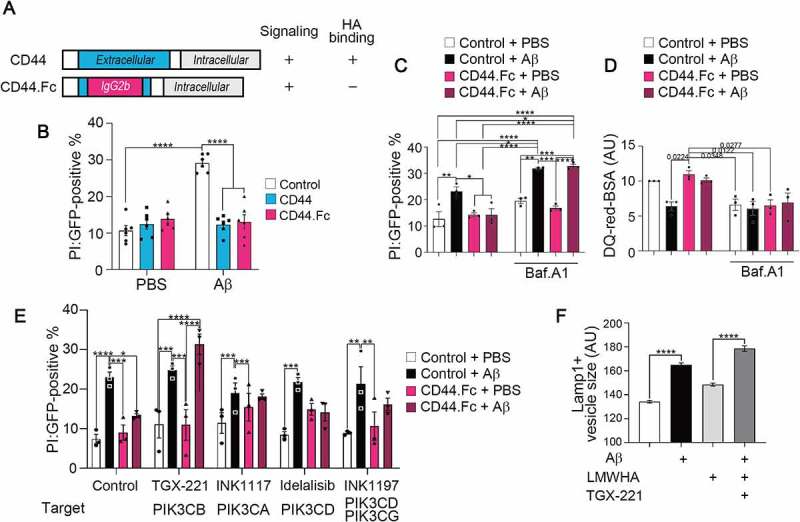
CD44-V-ATPase axis suppresses Aβ-induced lysosomal dysfunction and neurotoxicity via PI3Kβ. (A) Schematic representation of CD44.Fc fusion protein, a constitutively active form of CD44. (B) HT22 cells were co-transfected with pEGFP and pcDNA-HA (Control), CD44 (variant 2)-HA, or CD44.Fc-HA, and treated with 5 μM Aβ for 48 h. Cytotoxicity was analyzed by PI staining (n = 3, cells > 100 for each trial), two-way ANOVA. (C, D) HT22 cells were transfected with pEGFP (Control) or CD44.Fc-EGFP (CD44.Fc) and treated with 5 μM Aβ in the absence or presence of 10 μM Baf.A1 for 48 h (A) or 24 h (B). Cytotoxicity was analyzed by PI staining (n = 3, cells > 100 for each trial) (C). Proteolysis activity was analyzed by measuring DQ-red-BSA fluorescence intensity (n = 3, cells > 30 for each trial) (D), two-way ANOVA. (E) HT22 cells were transfected with pEGFP (Control) or CD44.Fc-EGFP (CD44.Fc) and treated with 5 μM Aβ in the presence or absence of 5 μM PI3K inhibitors for 48 h; TGX-221, PIK3CB/p110β; INK1117, PIK3CA/p110α; Idelalisib, PIK3CD/p110δ; INK1197, PIK3CD and PIK3CG/p110γ. Cytotoxicity was analyzed by PI staining (n = 3, cells > 100 for each trial), two-way ANOVA. (F) HT22 cells treated with 5 μM Aβ in the presence or absence of 100 μg/ml LMWHA and 5 μM TGX-221 for 24 h, and stained with anti-LAMP1 antibody. The sizes of LAMP1-positive signals were analyzed (n = 3, cells >30 for each trial), two-way ANOVA.
Further, the protective effect of CD44.Fc against cell death and endolysosomal dysfunction in Aβ-exposed cells was abolished by treating bafilomycin A1 (Baf.A1), an inhibitor of V-ATPase (Figure 7C and D), which suggests the HA-CD44 signaling protects against Aβ pathology via V-ATPase. To map the downstream signaling of CD44, we employed a pharmacogenomic approach utilizing bioactive compounds. Using 34 bioactive kinase inhibitors targeting 19 signaling pathways, we found that inhibitors of the PI3K-AKT and MAPK/p38 pathways most effectively abolished the protective activity of CD44.Fc against Aβ toxicity (Fig. S7B and Table S2). By then testing several inhibitors selective for PI3K subtypes, we found that only TGX-221, which targets PIK3CB/p110β, disrupted the protective effect of CD44.Fc against Aβ toxicity (Figure 7E). The protective effect of LMWHA against Aβ-induced enlargement of LAMP1-positive endolysosomes was also hindered by TGX-221 treatment (Figure 7F). CD44 thus mitigates Aβ pathology accompanied by endolysosomal dysfunction via PIK3CB.
Facilitating HA flux attenuates AD pathology in model mice
To address whether regulation of HA flux has pathological relevance, we employed 3xTg-AD mice, a rodent model of AD that carries three human genes associated with familial AD: APPKM670/671ML (Swedish), MAPTP301L, and PSEN1M146V [56]. The lentivirus harboring the HYAL1 gene was delivered into the dentate gyrus region of 6- to 7-month-old 3xTg-AD mice. After five weeks, the cognitive function of the mice was measured. In the Y-maze test, we found that the tendency to avoid entering previously visited arms was weaker in 3xTg-AD mice than age-matched WT mice. However, this memory deficit was reversed in the HYAL1-expressing 3xTg-AD mice (Figure 8A). Similar protective effects of HYAL1 on the behavioral deficits in 3xTg-AD mice were seen in the NOR test (Figure 8B) and the passive avoidance test (Figure 8C).
Figure 8.
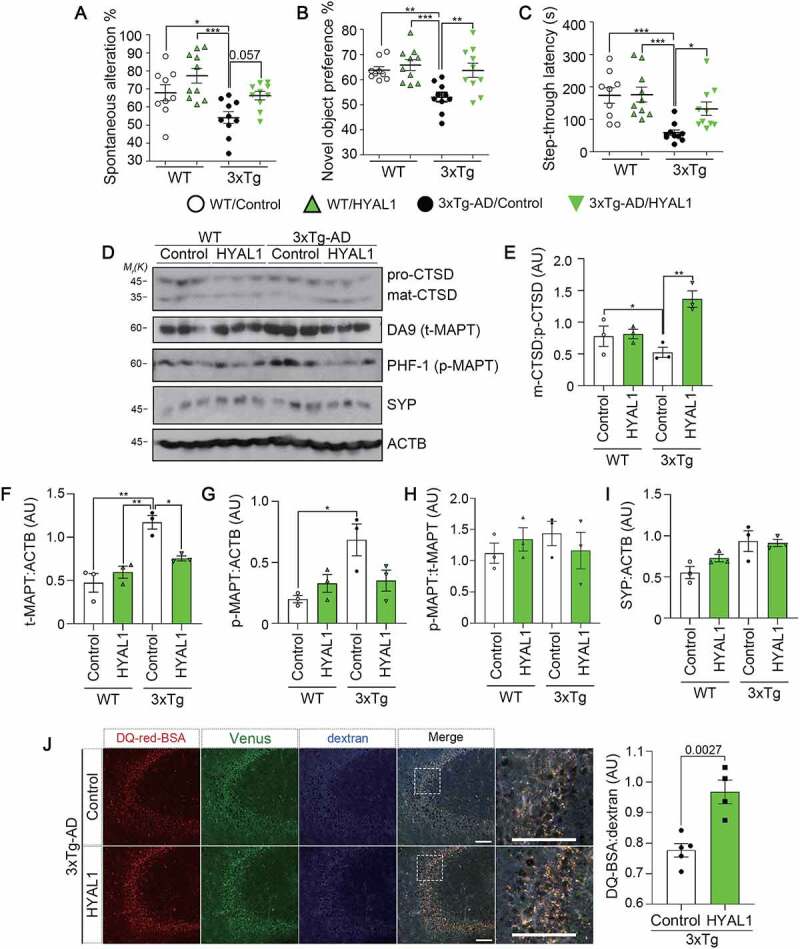
Facilitating HA flux attenuates behavioral deficits and lysosomal defects in AD model mice. (A-C) Alleviation of behavioral deficits by HYAL1 expression in 3xTg-AD mice. The 6- to 7-month-old WT and 3xTg-AD mice were stereotaxically injected with human HYAL1-carrying lentivirus (HYAL1) or empty control: WT/Control (n = 9); WT/HYAL1 (n = 10); 3xTg-AD/Control (n = 10); 3xTg-AD/HYAL1 (n = 10). Effects on spontaneous alteration in Y-maze task (A), exploration time in a novel object recognition task (B), and latency in a passive avoidance task (C) were analyzed. (D-I) Reversal of the impaired lysosomal activity by HYAL1 expression in 3xTg-AD mice. Immunoblots show mature-CTSD:pro-CTSD (D, E), total MAPT levels (D, F), phosphorylated (PHF1) MAPT levels (D, G), p-MAPT:t-MAPT (D, H), and synaptophysin (SYP) levels (D, I) in the mouse hippocampal tissues. (J) Representative images showing in vivo lysosomal activity in the mouse hippocampal tissues. The 3xTg-AD mice analyzed in (A) were i.c.v. injected with DQ-red-BSA and Cascade Blue-dextran. Scale bar: 100 μm (left). DQ-BSA signals were normalized to the Cascade Blue signal; 3xTg-AD/Control (n = 5); 3xTg-AD/HYAL1 (n = 4) (right).
Interestingly, we found that the proteolytic maturation of CTSD was impaired in the hippocampal tissues of 3xTg-AD mice compared to age-matched WT mice (Figure 8D and E). However, this impaired maturation of CTSD in 3xTg-AD mice was significantly restored by HYAL1-expression. Total levels of CTSD protein slightly decreased in the HYAL1-overexpressed mouse hippocampal tissues (Fig. S8A-C). On the other hand, levels of Ctsd mRNA were not affected by overexpressed HYAL1 in HT22 cells (Fig. S8D and Table S3). Accordingly, levels of total (DA9) and p-MAPT (PHF-1) proteins, but not SYP/synaptophysin in presynaptic neurons, were reduced in HYAL1-injected 3xTg-AD mouse brains compared to control 3xTg-AD mice (Figure 8D-I), suggesting the improved clearance of aggregate-prone MAPT, a well-known substrate of autophagy-lysosomal [57], by HYAL1 overexpression. Consistent with the CTSD processing, measuring in vivo endolysosomal protease activity revealed that the DQ-BSA:dextran ratio was remarkably enhanced by HYAL1-expression in 3xTg-AD mice (Figure 8J). These results support the idea that HYAL1 preserves endolysosome function against AD proteopathy. In conclusion, the HYAL-HA pathway was able to protect against AD pathologies in vivo.
Discussion
Lysosomal overload of misfolded or pathogenic proteins and abnormal endolysosomal structures and functions are often detected in degenerating neurons and neurodegenerative diseases [18,23]. In particular, evidence of a close link between V-ATPase deficiency and AD has accumulated over the past decade. For example, the misrouting of the V-ATPase ATP6V0A1 subunit received early attention as a core event in the disrupted lysosomal proteolysis seen under PSEN1-deficient conditions [22,23,58]. In the present study, the V-ATPase complex acts as a cellular receptor for pathogenic forms of Aβ and p-MAPT over threshold dose, leading to inhibition of V-ATPase activity and impairment of endolysosome-mediated proteostasis. While endolysosomal pH declines by ATP6V0C depletion in neuronal cells, cell viability in ATP6V0C KD cells is rarely affected but deteriorated when additionally loaded with Aβ. It is conceivable that endolysosomal weakening and the storage burden act together in Aβ pathology, leading to a significant change in cell function and viability.
Given that the ATP6V0C subunit forms a channel-like ring complex and its rotation is essential for the proton transport catalyzed by V-ATPase, it is conceivable that intralumenal Aβ may interfere with it the rotatory movement of the c-ring by interacting with ATP6V0C. How the ATP6V0C-Aβ or ATP6V1B2-MAPT interaction affects V-ATPase activity remains to be resolved. As delineated in this study, the ATP6V1B2 subunit on the cytosolic side of the V-ATPase complex binds to cytosolic p-MAPT, which may also inhibit V-ATPase assembly. Consequently, the presence of impaired V-ATPase in acidic vacuoles along the endocytosis pathway may cause defective vesicle trafficking in affected neurons. Thus, loss of proteostasis robustness in endolysosomes may reflect the increased vulnerability to stress in postmitotic neurons during aging and age-related diseases [2].
We isolated an HYAL family member as a rescuer of endolysosomal activity from Aβ pathology in a functional screen and showed that the HYAL-HA pathway reverses the Aβ and MAPT-induced endolysosomal defects. Interestingly, the HA content in the central nervous system changes with aging [59–61], and the LMWHA-CD44 signaling was recently shown to be crucial for the anti-aging process [62]. It was previously reported that CD44 is critical for regulating the functional and structural plasticity of dendritic spines at mature synapses in the adult brain despite the low expression in neurons [63]. It is, therefore, encouraging that stimulating HA metabolism may rescue endolysosomal function through V-ATPase in these age-related conditions. In this study, we found the probability that the HA-CD44 signaling might regulate V-ATPase assembly along the endolysosomal trafficking route. An inhibitor of PI3K, a plausible downstream mediator of CD44 in the HA-CD44 signaling, was previously shown to block V-ATPase assembly [64]. That finding increases the likelihood that the HA-CD44 signaling is involved in V-ATPase regulation in neurons through PI3K, while CD44 often induces pro- or anti-inflammatory pathways, depending on the cell type [65,66]. In addition, downregulation of total CTSD protein in the HYAL1-expressing mouse hippocampal tissues without significant change in their mRNA suggest a possibility that HYAL1 overexpression and subsequent modulation of V-ATPase assembly may alter the exocytotic release of m-CTSD [67–69]. Recently, we identified the phosphorylation sites of the ATP6V1A subunit by LMWHA treatment (data not shown), which may contribute to regulating V-ATPase functions. We are now implementing proteomic analysis to delineate further detailed mechanisms on the beneficial effect of HA-CD44 signaling on V-ATPase regulation by analyzing affinity-purified V-ATPase complexes purified from the brains of AD model mice and AD patients.
The noteworthy feature of V-ATPase subunits, their binding to Aβ and p-MAPT, enables us to envision a potential mechanism underlying the proteopathy contributing to the pathogenesis of AD. Many top-ranked AD risk factors, including PICALM, BIN1, SORLA, and CD2AP, are strongly associated with regulating intracellular Aβ or MAPT levels and endolysosomal dysregulations [5,70–75]. Our understanding of their molecular mechanisms can increase by employing our “intracellular Aβ, p-MAPT and V-ATPase” hypothesis, which enlarges the intracellular pool of pathologic factors that adversely affect V-ATPase and, in turn, facilitate the pathogenesis of AD. Our findings also provide a novel therapeutic strategy for AD pathology. Genetic and pharmacological trials so far employing TFEB [76], MTOR inhibition [77,78], or lysosomal enzyme glucocerebrosidase or CTSB(cathepsin B) [79,80] are insufficient to maintain lysosomal functions without altering other biological processes, such as lipid catabolism or exocytosis [81–83]. The present study is the first to propose the beneficial effects of enhancing HA flux and activating CD44 signaling on endolysosomal activity in Aβ-MAPT pathology. Blocking the interaction between V-ATPase and Aβ, or MAPT could serve as specific means of V-ATPase regulation in mitigating AD pathology.
In conclusion, we envision that the proteotoxic activities of Aβ and p-MAPT proteins, which bind to V-ATPase and interfere with its activity, lead to endolysosomal dysfunction and neurodegeneration in vitro and proteopathy in vivo. Our findings provide insight into the potential therapeutic functionality of the HA-CD44-V-ATPase axis in AD pathology.
Materials and methods
Chemicals and reagents
Recombinant human Aβ1-42 peptides (A-1163), and synthetic fluorescein-labeled Aβ1-42 (A-1119) were from rPeptide; DQ-red-BSA (D12051), and DQ-green-BSA (D12050) were from Invitrogen; Dextran, Cascade Blue, 10,000 MW (D1976), and DyLight 594 NHS Ester (46,413) were from Thermo Scientific; acridine orange (A6014), Resomer® RG 503 H, poly(D,L-lactide-co-glycolide) (acidic nanoparticle, 719870), hyaluronic acid sodium salt from Streptococcus equi MW 8,000–15,000 (LMWHA, 40583), hyaluronic acid sodium salt from Streptococcus equi MW 1,750,000–2,000,000 (HMWHA, 02737), and Malachite Green Phosphate Assay Kit (MAK307) were from Sigma-Aldrich; Bioactive compound library (L4000) was from TargetMol; Lipofector-pMAX (AB-LF-M100) was from AptaBio; siMapt (17762–1 and 17762–2)was from Bioneers; HYAL1 lentiviral particle was from VectorBuilder.
Preparation of synthetic Aβ oligomers
Oligomeric Aβ1-42 (oAβ1-42) were generated as described [18,39]. Briefly, recombinant Aβ1-42 or synthetic Fluorescein-Aβ1-42 peptide lyophilized with HFIP (rPeptide) were solubilized in DMSO at 2 mM and diluted in PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.76 mM KH2PO4, pH 7.4) to final 125 μM. Aβ1-42 incubated at 4°C for 48 h was centrifuged at 12,000 g for 10 min, and the resulting supernatant was stored at −80°C until used as Aβ oligomers (Aβ in the text and Figure). For generating DyLight 594-Aβ1-42, Aβ oligomers were labeled by DyLight 594 NHS Ester according to the supplier’s instructions. Briefly, 125 μM oAβ1-42 in 100 μl were incubated with 50 μg DyLight 594 at room temperature for 1 h. The samples were dialyzed using a dialysis membrane with a molecular–weight cutoff ≥ 10 K for 2 h and stored at −80°C. Naturally secreted oAβ1-42 was acquired as previously described [54]. In brief, control Chinese hamster ovary (CHO) cells and CHO cells expressing human APP (amyloid beta precursor protein) 751 with the Val717Phe mutation (7PA2) were grown to 90% confluence and were washed with PBS and further cultured for 16 h in serum-free Dulbecco’s modified Eagle medium (DMEM; Welgene Inc., LM 001–05). The conditioned medium was collected and centrifugated at 1,000 g to remove cell debris. Supernatants were concentrated ten-fold using Vivaspin® Turbo 4, 3,000 MWCO (Sartorius, VS04T91). The concentration of Aβ1-42 in the culture medium of 7PA2 CHO cells was 5.14 ng/ml as previously described [54].
Antibodies
The following antibodies were used for western blotting, immunoprecipitation and immunocytochemistry: Aβ (4G8 [800703], 6E10 [803003]) and MAP2 (822501) from BioLegend; GFP (sc-9996), HA (sc-7392), CTSD (sc-6486), SYP (sc-9116), His (sc-803), HYAL1 (sc-101340), LAMP1 (sc-19992), and ACTB/β-actin (sc-47778) from Santa Cruz Biotechnology; SQSTM1/p62 (NBP1-48320), and MAP1LC3B/LC3B (NB100-2220) from Novus Biologicals; ANTI-FLAG M2 Affinity Gel (Sigma-Aldrich, A2220); ATP6V0C (ABclonal, A16350). TG5 (total MAPT/Tau), DA9 (total MAPT/Tau), and PHF-1 (p-MAPT Ser396, Ser404) were generously gifted by Dr. P. Davies, Albert Einstein College of Medicine, NY, USA.
Human brain samples and ethical statement
Hippocampal tissues from non-AD or AD patients were provided by the Brain Bank of Seoul National University Hospital Biomedical Research Institute (Table S1). This study was approved by the Institutional Review Board of Seoul National University.
Mice
The 3xTg-AD mice were previously described [84]. Age-matched C57BL/6 WT mice were used as controls for 3xTg-AD. The rTg4510 mice were obtained by crossing the human MAPT/TauP301L responder line (The Jackson Laboratory 015815) to a tetracycline-controlled transactivator (tTA) line (The Jackson Laboratory, 016198). Mice not carrying the Camk2/CaMKII-tTA transgene crossed to MAPT/TauP301L heterozygote were used as controls for rTg4510. All animal experiments were approved and performed under the guidelines of the Seoul National University Institutional Animal Care and Use Committee (SNU-170504-3-2; SNU-201013-6-1; SNU-201230-2-1; SNUIBC-200827-1).
Cell culture, DNA transfection, and Aβ treatment
Mouse primary hippocampal and cortical neurons were isolated from embryonic days 16–17 (E16-E17) of C57BL/6J mice as described [18,39]. Cells were gently triturated by trypsin-EDTA and were grown in Neurobasal Medium (Gibco, 21103049) supplemented with 2% (v:v) B-27 supplement (Gibco, 17504044), 10 μg/ml gentamycin (Gibco, 15710060), 100 units/ml penicillin-streptomycin (Gibco, 15140122), and 1% (v:v) GlutaMAX (Gibco, 35050061). HT22 and HEK293T cells (ATCC, CTL-3216) were cultured in DMEM containing 10% (v:v) fetal bovine serum (Gibco, 12483020), 100 units/ml Penicillin-Streptomycin. Primary neurons were transfected using Lipofectamine 2000 Transfection Reagent (Invitrogen, 52887), and HT22 and HEK293T cells were transfected using Lipofector-pMax according to the manufacturer’s instructions. Cells were treated with 5 μM oligomeric Aβ1-42 for cytotoxicity, DQ-red-BSA, or endolysosomal enlargement measurement assays.
DNA construction
Deletion or substitution mutants were produced by the site-directed mutagenesis. EGFP-tagged human LGALS3 was kindly gifted by Dr. D.H. Cho (Kyungpook National University, Korea). CTSB-GFP vector was generously provided by Dr. H.G. Lee (Korea Research Institute of Bioscience and Biotechnology, Korea) [85]. For the generation of GFP-8, EGFP was amplified by PCR using primers (5′- GAG TAG AAG CTT ATG GTG AGC AAG GGC GAG −3′ and 5′- CCG AAT TCT AGA GGA CCT TGC AAC AGT TGA TAC ACT TGT ACA GCT CGT CCA TGC −3′) and inserted to the HindIII/EcoRI site in pcDNA3.0-HA vector. GFP-WT-8 and GFP-mt-8 were produced by inserting given sequence into GFP-8 through the site-directed mutagenesis using primers (WT, 5′-ACG ACA TCA GCC TCT ACA AGT GTA TCA ACT GTT GCA AGG TCC-3′ and 5′-AGA GGC TGA TGT CGT CAT TCT TGT ACA GCT CGT CCA TGC C-3′; mt, 5′-CAG CAA TCG CAC TCT ACA AGT GTA TCA ACT GTT GCA AGG TCC-3′ and 5′-AGA GTG CGA TTG CTG CAT TCT TGT ACA GCT CGT CCA TGC C-3′). EGFP-ATP6V0C was generated by amplifying human ATP6V0C cDNA by PCR using primers (5′-CTC AAG CTT CGA ATT CAA TGT CCG AGT CCA AGA GCG G-3′ and 5′-TAG ATC CGG TGG ATC CCT ACT TTG TGG AGA GGA TGA GGG C-3′) and inserted into HindIII/BamHI site of pEGFP-C1 (Clontech, 6084–1). EGFP-ATP6V1B2 was generated by amplifying human ATP6V1B2 cDNA by PCR using primers (5′-CTC AAG CTT CGA ATT CAA TGG CGC TGC GGG CGA TGC-3′ and 5′-TAG ATC CGG TGG ATC CCT AAT GCT TTG CAG AGT CTC GAG GG-3′) and inserted into EcoRI/BamHI site of pEGFP-C1. EGFP-HYAL1 generated by amplifying human HYAL1 cDNA by PCR using primers (5′-CGG AAT TCC ATG GCA GCC CAC CTG CTT CC-3′ and 5′-CGC GGA TCC TCA CCA CAT GCT CTT CCG C-3′) and inserting into EcoRI/BamHI site of pEGFP-C1. CD44-HA was generated by amplifying human CD44 (variant 2) cDNA by PCR using (5′-CGT CAG AAT TAA GCT TAT GGA CAA GTT TTG GTG GCA CG-3′ and 5′-TCG CGG CCG CAA GCT TCA CCC CAA TCT TCA TGT CCA C-3′) and inserted into HindIII site of pcDNA-HA. CD44.Fc-HA, mouse IgG2b was amplified from ALK.Fc [86] with primers (5′-GGC CGC TGA CCT CTG CAA GGC TTG CAG CGT TGC TCA CCC-3′ and 5′-ATT CAG ATC CAT GAG TGG TAT GGG ATT TAC CCG GAG ACC GGG-3′) and inserted into CD44-HA, which was removed of extracellular HA binding domain by EcoNI/XcmI. CD44.Fc-EGFP was generated by amplifying CD44.Fc (CD44-IgG) by PCR using primers (5′-ATT CTG CAG TCG ACG GTA CCA TGG ACA AGT TTT GGT GGC ACG-3′ and 5′-GGC GAC CGG TGG ATC CGC CAC CCC AAT CTT CAT GTC C − 3 GC CAC CCC AAT CTT CAT-3′ and inserted into KpnI/BamHI site of pEGFP-N1 (Clontech, 6085–1).
Generation of KO cell line
The lentiCRISPR v2 vector was used to construct hyal1 KO (target sequence: TTG TCG TGT CAT AGA TC) or atp6v0c KO (target sequence: GCC ACT CTT GGC TGT GCC AT) constructs. After transfection for 24 h, HT22 cells were selected in DMEM with 2 μg/ml puromycin for 72 h. Monoclonal cells were isolated by limiting dilution, and the level of HYAL1 or ATP6V0C expressions was analyzed by western blotting. Validated cell lines were used for further experiments.
Western blot
Western blot analysis was performed as described previously [54]. Cell extracts or mouse hippocampal brain samples were dissolved with ice-cold RIPA buffer (50 mM Tris-Cl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1.1% [v:v] Nonidet P40 substitute [USB Corporation, 19628], 0.25% [w:v] sodium deoxycholate [Sigma-Aldrich, D6750], and 1 mM phenylmethylsulfonyl fluoride [PMSF; Amresco, M145-25 G]) and briefly sonicated. After centrifugation at 12,000 g for 10 min, the supernatant was subjected to SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to polyvinylidene fluoride (PVDF; Atto, AE-6667-P) membrane using Semi-Dry transfer system WSE-4025 HorizeBLOT 2 M-R (ATTO, Tokyo, Japan). Blots were blocked by 3% (w:v) BSA (GenDEPOT, A0100-010) solution in TBS-T (10 mM Tris-Cl, pH 8.0, 150 mM NaCl, and 0.5% [v:v] Tween-20 [Anatrace, T1003]) and subjected to western blot analysis. Immunoreactive blots were detected by using the chemiluminescent HRP method.
Cell fractionation
Cell fractionation was performed according to a modified version of the established protocol [64]. Briefly, 3 × 106 cells were washed and harvested with ice-cold PBS. Cells were resuspended into 300 μl homogenization buffer (250 mM sucrose [USB Corporation, 21938], 1 mM EDTA, 10 mM HEPES, 1 mM PMSF, and proteinase inhibitor cocktail [Quartett, PPI 1015]) and lysed by passing through a 26-gauge needle. Nuclei and unbroken cells were cleared by centrifuging the samples at 500 g for 10 min. The supernatant was ultracentrifuged at 100,000 g for 60 min to pellet the membrane fraction. The membrane pellet was rinsed three times with homogenization buffer and resuspended in homogenization buffer supplemented with 1% SDS. The supernatant was collected to give the cytosolic fraction.
Co-immunoprecipitation (co-IP)
Co-IP was performed as described previously [54]. Generally, cells were lysed with 0.5X RIPA buffer (25 mM Tris-Cl, pH 7.5, 15 mM NaCl, 0.5% [v:v] Triton® X-100 [USB Corporation, 22686], 0.25% [w:v] sodium deoxycholate, 0.05% [w:v] SDS, and 1 mM PMSF), followed by centrifugation at 10,000 g for 10 min. For Aβ in vitro binding assay, Aβ peptides were incubated with supernatant from cell extracts, lysed in homogenizing buffer (50 mM Tris-Cl, pH 7.5, 15 mM NaCl, 1 mM EDTA, 0.05% [w:v] SDS, and 1 mM PMSF) by passing the lysate 50x through a 26-gauge syringe. Mixtures were pre-cleared by Protein G Sepharose (GE Healthcare, 17–0618-05), and the resulting supernatant was incubated with 2 μg proper antibodies overnight at 4°C. Samples were incubated with 30 μl Protein G bead for an additional 3 h at 4°C. Proteins bound to Protein G bead were pulled down by centrifugation after five to ten times washes and analyzed by western blotting.
Cell live:dead assessment
Cell Live:Dead ratios were determined as described previously [18]. Viable cells were stained with calcein-AM (Invitrogen, C1430), originally non-fluorescent but converted to fluorescent form under hydrolysis by intracellular esterases in live cells. Dead cells were stained with propidium iodide (PI; Sigma-Aldrich, P4170), observed under a fluorescence microscope (Olympus, Tokyo, Japan), and the Live:Dead ratios were calculated by dividing the number of dead cells by the number of total cells (calcein-AM-positive cells plus PI-positive cells). Cell death in HT22 cells transfected with GFP-tagged effector genes was assessed by counting GFP-positive cells showing apoptotic fractured nuclei characterized by PI.
Dendritic spine density analysis
Dendritic spine density was determined as described previously [39]. Briefly, hippocampal neurons were transfected at 5–7 days in vitro (DIV) with pEGFP-N1. Two days after transfection, neurons were incubated with oAβ1-42, and cultures were fixed in 4% (w:v) paraformaldehyde (PFA) for 10 min. Images of more than 10 randomly chosen GFP-expressing neurons were acquired by Zeiss LSM700 laser scanning confocal microscope. The number of spines within 10 μm in length was manually counted based on morphology.
Immunocytochemistry and fluorescence microscopy
For immunostaining in vitro, cells were fixed in 4% (w:v) PFA. After washing twice with PBS, cells were incubated in PBS with 5% (v:v) fetal bovine serum to block nonspecific antibody adsorption at room temperature for 1 h. Then, the cells were incubated with appropriate primary (LAMP1 1:200, TG5 1:2000) and secondary antibodies in a blocking buffer containing 0.003% (w:v) CHAPS (GenDEPOT, C0660-005). Images were captured on a Zeiss LSM700 laser scanning confocal microscope. For the DQ-BSA assay, cells were co-incubated with 10 μg/ml DQ-red-BSA and 10 μg/ml Cascade Blue-dextran at 37°C 30 min prior to drug treatment. Next, the cells were washed with PBS to remove the excess probe, fixed, and imaged by confocal microscopy. For the lysosomal membrane permeabilization (LMP) assay, cells were preloaded with 1 μg/ml acridine orange for 5 min in complete culture medium, rinsed. Cells were then further treated with other compounds. The green fluorescence was monitored under fluorescence microscopy and analyzed by photoshop or ImageJ software [87]. The size of LAMP1-positive vacuoles was analyzed using StarDist plugin [88].
pH measurement
Endolysosomal pH was measured according to the previous report [46]. Briefly, HT22 control cells or sgRNA ATP6V0C #14 cells, stably expressing pHlys biosensor, were generated by the lentiviral infection of pHLARE with Eef1a/Ef1α promoter. Fluorescence ratios of pHLARE in cells were acquired under fluorescence microscopy after cDNA transfection or LMWHA treatment. Calibration curves were acquired by incubating cells for 5 min before imaging in a potassium-phosphate buffer (50 mM potassium phosphate, 80 mM potassium chloride, 1 mM magnesium chloride) containing 20 μM nigericin sodium salt (BioVision, 2096–5) at pH 4.0 to 7.0. The sfGFP and mCherry fluorescence were determined at 488 and 561 nm excitation and calculated by subtracting the mean background intensity from the endolysosomal fluorescence intensity.
Immunohistochemistry of AD patient brain
Immunohistochemistry (IHC) was performed as described [49]. Briefly, hippocampal sections from AD patients were retrieved with 10% (v:v) formic acid for 15 min at 37°C and then blocked with 5% (w:v) BSA. Blocked sections were stained with antibodies against Aβ and ATP6V0C overnight. Antibodies 6E10 (1:1000), ATP6V0C (1:500), MAP2 (1:3000) were used to detect human Aβ, ATP6V0C, and MAP2, respectively. Secondary antibodies used in IHC assays are as follows: Alexa Fluor 488 goat anti-rabbit IgG (H + L) (Invitrogen, A11008), Alexa Fluor 594 goat anti-mouse IgG (H + L) (Invitrogen, A11005), DyLight405-AffiniPure Goat Anti-Chicken IgY (IgG) (H + L) (Jackson ImmunoResearch, 103-475-155).
In vivo endolysosomal activity assay
Mice were intracerebroventricularly (i.c.v.) injected with 50 μg DQ-red-BSA (2 mg/kg) and 50 μg Cascade Blue-dextran 10,000 MW dye (2 mg/kg). After 6–8 h, mice brains were fixed with 4% (w:v) PFA overnight and further dehydrated with 30% (w:v) sucrose. Forty-μm-thick slices from frozen sections were observed under a confocal microscope LSM700. The intensity ratio of DQ-red-BSA:Cascade Blue-dextran in the CA3 region was analyzed by ImageJ.
Purification of His-MAPT protein from mammalian cells
HEK293T cells transformed with His-tagged MAPT plasmid were resuspended in His-binding buffer (20 mM Tris-HCl, pH 7.5, 500 mM NaCl, 20 mM imidazole, 1 mM sodium orthovanadate, 10 mM sodium fluoride, 1 mM PMSF, and proteinase inhibitor cocktail), disrupted by sonication, and centrifuged at 10,000 g for 10 min. The supernatant was filtered and loaded onto a Ni-NTA Agarose (QIAGEN, 30210) pre-equilibrated with binding buffer. The column was washed, and the bound protein was eluted using a linear 300–1,000 μM imidazole gradient. The purified protein was stored at −20°C.
Isolation of microsomes
Microsomes were isolated from the mouse brain according to a modified version of the established protocol [89]. A brain was washed twice in 10 ml of homogenization buffer (20 mM HEPES-KOH, 1 mM EDTA, 2 mM dithiothreitol, 250 mM sucrose, pH 7.4 at 4°C), and the collected were passed 20 times through a 26-gauge needle. The homogenate was centrifuged for 10 min at 5,000 g at 4°C. The unbroken cells and mitochondria pellet was discarded, and the supernatant was centrifuged at 100,000 g for 60 min in a Beckman TLA100.2 rotor. The pellet was suspended in buffer A (l50 mM KCl, 20 mM HEPES-KOH, 2 mM dithiothreitol, pH 7.4), and glycerol was added to the suspension to 25% (v:v) of the final volume.
V-ATPase activity assay
The microsomes were used for the malachite green phosphate assay according to the manufacturer’s instructions. The protocol was modified from the established protocol [90]. Briefly, microsomes were preincubated for 40 min at room temperature with or without the testing chemicals or proteins. To test the effect of intralumenal Aβ, 2.5 μM Aβ was preloaded to pMax-based liposomes for 30 min. BSA preloaded to liposomes as that of Aβ was used as a negative control. To test the effect of MAPT, His-MAPT WT or MAPTS396A,S404A mutant purified from HEK293T cells was added to the reactions for 40 min before the addition of ATP. The reaction was initiated by adding 1 mM Mg-ATP, and samples were withdrawn at indicated time points to measure the production of inorganic. Briefly, 20 µl malachite green reagent was added to 5 µl samples (1:20 volume) withdrawn, and absorbance at OD 620 nm was measured. A standard curve simultaneously acquired with a series of phosphate solutions of known concentrations was used to determine the amount of phosphate released.
AAV-MAPT viral production
A recombinant AAV vector was generously gifted from Dr. H. Lee, University of Ulsan College of Medicine. GFP-MAPT/TauP301L was subcloned into the pJDK viral vector [91]. Viral production was performed using HEK293T cells and monitored under fluorescence microscopy. Cells were harvested, lysed by freeze-thawing, and viral particles were purified using AAVpro® Purification Kit Maxi (Takara Bio, 6666) according to the manufacturer’s instruction.
Acidic nanoparticle treatment
Acidic NPs were freshly prepared at 1 mg/ml in culture medium. Solutions were uniformly dispersed via sonication, vortex, passing through a 0.45-µm filter, and treated directly to cells [34,92].
V-ATPase complex purification
The V-ATPase complex was purified according to the modified protocol described previously [45].
SidK peptide purification
SidK1-278-3× FLAG peptides were prepared in advance. Briefly, BL21 Codon+ cells were transformed with plasmid pSAB35 bearing SidK1-278 with an N-terminal 6× histidine (generously gifted by Dr. John L. Rubinstein, The Hospital for Sick Children Research Institute, Toronto, Canada) and grown at 37°C with shaking in 2 L LB media supplemented with 50 mg/ml kanamycin. At an OD600 of 0.65, protein expression was induced with 1 mM isopropyl-beta-D-thiogalactopyranoside (IPTG; Gold Biotechnology, 12481C25) at 16°C. Cells were harvested and lysed by sonication in HisTrap Buffer (50 mM Tris-HCl, pH 7.4, 25 mM imidazole, and 300 mM NaCl). The cell lysate was centrifuged at 38,000 g, and the supernatant was loaded onto a 5 ml Ni-NTA Agarose (QIAGEN, 30210). The column was washed with HisTrap Buffer, and protein eluted with a linear imidazole gradient from 25 to 300 mM in HisTrap Buffer over 10 column volumes (CV). Fractions containing protein were pooled and stored at −80°C. Shortly before the flow-through of membrane solution, thawed SidK1-278-3× FLAG (1 mg) was diluted in 3 ml TBSG (50 mM Tris-HCl, pH 7, 300 mM NaCl, and 10% [v:v] glycerol), and 1 ml of this solution was applied to each of three columns (Bio-Rad, 7311550) containing 800 µL ANTI-FLAG M2 Affinity Gel (Sigma-Aldrich, A2220), previously equilibrated with TBSG. The columns were washed with 5 ml DTBSG (TBSG supplemented with 0.03% (w:v) dodecylmaltoside [DDM; Thermo Scientific, 89902]).
Isolation of V-ATPase from mouse brains
Membrane enriched for synaptic vesicles was collected from old nine 3xTg-AD mice brains (~4.5 g). Brains were washed 3 times with ice-cold Homogenization Buffer (320 mM sucrose, 4 mM HEPES, pH 7.4), diced into cubes, and transferred into Homogenization Buffer supplemented with 0.2 mM PMSF. Brains were 50x passed a 26-gauge syringe and centrifuged twice at 1,000 g for 10 min. The supernatant was centrifuged at 110,000 g for 40 min to collect membranes. The membrane pellet was resuspended in ~14 ml Solubilization Buffer (50 mM Tris-HCl, pH 7, 320 mM sucrose, 300 mM NaCl, 10% [v:v] glycerol, 5 mM e-amino-n-caproic acid [Tokyo Chemical Industry Co., Ltd., D2254], 5 mM p-aminobenzamidine [Tokyo Chemical Industry Co., Ltd., A2115], 5 mM EDTA, and 0.2 mM PMSF), flash-frozen in liquid N2, and stored at −80°C. Frozen membranes were thawed and diluted with 40 ml of Solubilization Buffer supplemented with 20% (w:v) DDM, and the mixture was gently shaken for 30 min. Insoluble material was removed by centrifugation at 130,000 g for 70 min. The solubilized membranes were filtered with a 0.45 µm syringe filter, divided into three pools, and applied to the previously prepared three SidK 1–278-3× FLAG-bound M2 Agarose columns. The flow-through of each column was collected, pooled, and passed over the columns repeatedly (a total of five column applications). Each column was washed with 10 ml DTBSG and V-ATPase with SidK1-278-3× FLAG was eluted with 3 ml DTBSG with 150 µg/ml 3× FLAG peptide (Apexbio, A6001), followed by 1 ml DTBSG.
Cell-based functional screen for endolysosomal regulators
The cell-based functional screen was performed based on a previous study [18]. HT22 cells were co-transfected with pEGFP-N1 and each of 300 cDNAs encoding endolysosomal proteins for 12 h and then treated with 5 μM Aβ for 48 h. Cells with apoptotic morphological features such as shrinkage or membrane rupture were considered as cell death. Two independent experiments were performed, and the mean values were plotted in Figure 6B.
Human protein chip assay for Aβ interactors
A protein chip assay was performed by Gene On Biotech (Daejeon, Korea). Briefly, a human protein microarray (CDI Labs, HuProt v3.1) which contains over 20,000 full-length GST-tagged recombinant human proteins was used. The protein microarray was incubated with blocking buffer (2% [w:v] BSA in PBS with 0.1% [v:v] Tween 20) for 2 h, and 3 µg of fluorescently labeled Aβ was treated onto the array for 8 h at 4°C. Subsequently, the array was washed with washing buffer (PBS with 0.1% [v:v] Tween 20). Finally, the microarray result was detected by scanning with a GenePix 4100A microarray laser scanner (Molecular Devices, USA).
Stereotaxic injection
The stereotaxic injection was performed as described previously [18]. Human HYAL1 ORF was amplified by PCR using synthetic primers: HYAL1 (5′-AAT TCT GCA GCG GCC GCA TGG CAG CCC ACC TGC TT-3′ and 5′-GGA GAG GGG CGG ATC CTC ACC ACA TGC TCT TCC GCT-3′). The PCR products were inserted into NotI/BamHI sites of CSII-EF-MCS-IRES-Venus, and HYAL1 lentivirus (Vectorbuilder, CA, USA) were produced. The stereotaxic injection of lentivirus (0.39 × 107 TU/ml, TU; transduction unit) into the dentate gyrus (10 μl per hemisphere) was performed with the coordinates: anteroposterior (AP) = 2.1 mm, mediolateral (ML) = ± 1.8 mm, and dorsoventral (DV) = 2.0 mm from bregma. Behavior tests were performed after 4 weeks of injection, and the brain extracts were analyzed by western blotting. All experiments involving animals were performed according to the protocols approved by the SNU IACUC guidelines.
Intracerebroventricular injection of Aβ
Intracerebroventricular (i.c.v.) injection of Aβ was performed as described previously [39,54]. Aβ (2.5 μg) was administrated into i.c.v. region of 5-week-old C57BL/6J (male) mice using a Hamilton microliter 702 syringe (Hamilton company, 80400) equipped with a 26-gauge needle.
Behavior tests
Behavior tests were performed as described previously [18,49].
Y maze
Mice were laid to the end of one arm and then explored freely in the apparatus (32.5-cm long x 15-cm high) for 7 min. The arm entry was valid when the whole body was entered entirely into each arm. The spontaneous alteration was assessed as the ratio of the number of effective alterations, an event that mouse enters three different arms consecutively, to the number of total arm entries.
Novel object recognition
Novel object recognition was composed of two days of habituation, one-day training, and two days of the test session. Mice were allowed to roam in the chamber freely (22-cm wide x 27-cm long x 30-cm high) for 7 min. At training, two identical objects were placed on the diagonal of the chamber. One object was replaced with a novel object once a day during test phases. The discrimination ratio was calculated by dividing the time spent on a novel object by the sum spent on a novel object and familiar object.
Passive avoidance
Passive avoidance was composed of one-day habituation, one-day training, and one-day test session. The apparatus (40-cm wide x 20-cm long x 20-cm high) is divided into a bright compartment with an overhead 8 W lamp and a dark compartment of which the floor is made of electrical grids. Mice were allowed to explore freely in a bright compartment for 2 min with the door closed for conditioning. At the training session, mice were initially placed into the bright compartment and shocked by the foot grid (0.25 mA, 2 s) when both hind limbs entered the dark compartment. The latency to enter the dark compartment at the test session was observed with a 7-min cutoff point.
Quantification and statistical analysis
Statistical analysis was performed using GraphPad Prism software (Version 8, GraphPad, La Jolla, CA). All values are presented as the mean ± standard error (SEM). Statistical significance was determined by unpaired two-tailed t-test (for groups of 2), by one-way (for groups of ≥ 3, one independent variable), or by two-way ANOVA (for two independent variable) followed by Bonferroni post hoc analysis. Statistical significance levels are indicated as follows: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Supplementary Material
Acknowledgments
We thank Dr. D. Selkoe (Harvard Medical School, Boston, MA, USA) for CHO and 7PA2 cells, Dr. P. Davies (Albert Einstein College of Medicine, NY) for TG5, DA9, and PHF-1 antibodies, Dr. H. Lee (University of Ulsan College of Medicine, Korea) for AAV vector, Dr. D.H. Cho (Kyungpook National University, Korea) for EGFP-LGALS3, Dr. H.G. Lee (Korea Research Institute of Bioscience and Biotechnology, Korea) for CTSB-GFP, Dr. J. Rubinstein and Dr. Y. Abbas (The Hospital for Sick Children, Canada) for technical assistance on V-ATPase purification and SidK plasmid, Dr. Bradley Webb (University of California San Francisco, CA) for pHLARE lentiviral plasmid, and Dr. S.H. Park (Seoul National University Hospital Biomedical Research Institute) for providing AD tissues. S.H. Kim was supported by a Global Ph.D. Fellowship Program, and Y.S. Cho and J. Gwak were in part supported by the BK21 program. This work was supported by a CRI grant (2022R1A2B5B03001249) from the National Research Foundation of Korea (NRF), and by a grant HU20C0334 from the Korea Health Industry Development Institute (KHIDI) and Korea Dementia research center (KDRC) from the Ministry of Health & Welfare & Ministry of Science and ICT, the Korea government.
Funding Statement
This work was supported by the National Research Foundation of Korea [2014H1A2A1021369]; National Research Foundation of Korea [2022R1A2B5B03001249]; Korea Health Industry Development Institute (KHIDI) and Korea Dementia research center (KDRC) [HU20C0334].
Disclosure statement
The authors declare that they have no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2181614
References
- [1].Hipp MS, Kasturi P, Hartl FU.. The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol. 2019. Jul;20(7):421–435. [DOI] [PubMed] [Google Scholar]
- [2].Kaushik S, Cuervo AM. Proteostasis and aging. Nat Med. 2015. Dec;21(12):1406–1415. [DOI] [PubMed] [Google Scholar]
- [3].Hetz C. Adapting the proteostasis capacity to sustain brain healthspan. Cell. 2021. Mar 18;184(6):1545–1560. [DOI] [PubMed] [Google Scholar]
- [4].Boland B, Yu WH, Corti O, et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat Rev Drug Discov. 2018;17(9):660–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Van Acker ZP, Bretou M, Annaert W. Endo-lysosomal dysregulations and late-onset Alzheimer’s disease: impact of genetic risk factors. Mol Neurodegener. 2019. Jun 3;14(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sardiello M, Palmieri M, Di Ronza A, et al. A gene network regulating lysosomal biogenesis and function. Science 2009. Jul 24;325(5939):473–477. [DOI] [PubMed] [Google Scholar]
- [7].Raben N, Puertollano R. TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu Rev Cell Dev Biol. 2016;32:255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cataldo AM, Peterhoff CM, Troncoso JC, et al. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000. Jul;157(1):277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Orr ME, Oddo S. Autophagic/lysosomal dysfunction in Alzheimer’s disease. Alzheimers Res Ther. 2013;5(5):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Almeida CG, Takahashi RH, Gouras GK. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci. 2006. Apr 19;26(16):4277–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tammineni P, Jeong YY, Feng T, et al. Impaired axonal retrograde trafficking of the retromer complex augments lysosomal deficits in Alzheimer’s disease neurons. Hum Mol Genet. 2017. Nov 15;26(22):4352–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Schützmann MP, Hasecke F, Bachmann S, et al. Endo-lysosomal Aβ concentration and pH trigger formation of Aβ oligomers that potently induce Tau missorting. Nat Commun. 2021. Jul 30;12(1):4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lee JH, Yang DS, Goulbourne CN, et al. Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nat Neurosci. 2022. Jun;25(6):688–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vaz-Silva J, Gomes P, Jin Q, et al. Endolysosomal degradation of Tau and its role in glucocorticoid-driven hippocampal malfunction. Embo J. 2018. Oct 15;37(20): e99084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yang AJ, Chandswangbhuvana D, Margol L, et al. Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Abeta1-42 pathogenesis. J Neurosci Res. 1998. Jun 15;52(6):691–698. [DOI] [PubMed] [Google Scholar]
- [16].Ji ZS, Miranda RD, Newhouse YM, et al. Apolipoprotein E4 potentiates amyloid beta peptide-induced lysosomal leakage and apoptosis in neuronal cells. J Biol Chem. 2002. Jun 14;277(24):21821–21828. [DOI] [PubMed] [Google Scholar]
- [17].Johansson AC, Appelqvist H, Nilsson C, et al. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis. 2010. May;15(5):527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gwon Y, Kam TI, Kim SH, et al. TOM1 regulates neuronal accumulation of amyloid-β oligomers by FcγRIIb2 variant in Alzheimer’s disease. J Neurosci. 2018. Oct 17;38(42):9001–9018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Song Q, Meng B, Xu H, et al. The emerging roles of vacuolar-type ATPase-dependent lysosomal acidification in neurodegenerative diseases. Transl Neurodegener. 2020. May 11;9(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Vacuolar FM. ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol. 2007. Nov;8(11):917–929. [DOI] [PubMed] [Google Scholar]
- [21].Colacurcio DJ, Nixon RA. Disorders of lysosomal acidification-The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res Rev. 2016. Dec;32:75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lee JH, Yu WH, Kumar A, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010. Jun 25;141(7):1146–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Avrahami L, Farfara D, Shaham-Kol M, et al. Inhibition of glycogen synthase kinase-3 ameliorates β-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. J Biol Chem. 2013. Jan 11;288(2):1295–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wolfe DM, Lee JH, Kumar A, et al. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur J Neurosci. 2013. Jun;37(12):1949–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Butterfield DA, Poon HF, St Clair D, et al. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer’s disease. Neurobiol Dis. 2006. May;22(2):223–232. [DOI] [PubMed] [Google Scholar]
- [26].Johnson ECB, Dammer EB, Duong DM, et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med. 2020. May;26(5):769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Coffey EE, Beckel JM, Laties AM, et al. Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience. 2014. Mar 28;263:111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sherva R, Baldwin CT, Inzelberg R, et al. Identification of novel candidate genes for Alzheimer’s disease by autozygosity mapping using genome wide SNP data. J Alzheimers Dis. 2011;23(2):349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang M, Li A, Sekiya M, et al. Transformative network modeling of multi-omics data reveals detailed circuits, key regulators, and potential therapeutics for alzheimer’s disease. Neuron. 2021;109(2):257–272.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Woody SK, Zhou H, Ibrahimi S, et al. Human ApoE ɛ2 promotes regulatory mechanisms of bioenergetic and synaptic function in female brain: a focus on V-type H+-ATPase. J Alzheimers Dis. 2016. Jun 18;53(3):1015–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mangieri LR, Mader BJ, Thomas CE, et al. ATP6V0C knockdown in neuroblastoma cells alters autophagy-lysosome pathway function and metabolism of proteins that accumulate in neurodegenerative disease. PLoS One. 2014;9(4):e93257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Liu QY, Lei JX, Sikorska M, et al. A novel brain-enriched E3 ubiquitin ligase RNF182 is up regulated in the brains of Alzheimer’s patients and targets ATP6V0C for degradation. Mol Neurodegener. 2008. Feb 25;3:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Drummond E, Pires G, MacMurray C, et al. Phosphorylated tau interactome in the human Alzheimer’s disease brain. Brain. 2020. Sep 1;143(9):2803–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Baltazar GC, Guha S, Lu W, et al. Acidic nanoparticles are trafficked to lysosomes and restore an acidic lysosomal pH and degradative function to compromised ARPE-19 cells. PloS one. 2012;7(12):e49635–e49635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zeng J, Shirihai OS, Grinstaff MW. Degradable nanoparticles restore lysosomal pH and autophagic flux in lipotoxic pancreatic beta cells. Adv Healthc Mater. 2019. Jun;8(12):e1801511. [DOI] [PubMed] [Google Scholar]
- [36].Marwaha R, Sharma M. DQ-red BSA trafficking assay in cultured cells to assess cargo delivery to lysosomes. Biol Protoc. 2017. Oct 5;7(19): e2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cleary JP, Walsh DM, Hofmeister JJ, et al. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005. Jan;8(1):79–84. [DOI] [PubMed] [Google Scholar]
- [38].Kam TI, Park H, Gwon Y, et al. FcγRIIb-SHIP2 axis links Aβ to tau pathology by disrupting phosphoinositide metabolism in Alzheimer’s disease model. Elife. 2016. Nov 11;5: e18691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kam TI, Song S, Gwon Y, et al. FcγRIIb mediates amyloid-β neurotoxicity and memory impairment in Alzheimer’s disease. J Clin Invest. 2013. Jul;123(7):2791–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Oeste CL, Pinar M, Schink KO, et al. An isoprenylation and palmitoylation motif promotes intraluminal vesicle delivery of proteins in cells from distant species. PLoS One. 2014;9(9):e107190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Maejima I, Takahashi A, Omori H, et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. Embo J. 2013. Aug 28;32(17):2336–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Thomé MP, Filippi-Chiela EC, Villodre ES, et al. Ratiometric analysis of acridine orange staining in the study of acidic organelles and autophagy. J Cell Sci. 2016. Dec 15;129(24):4622–4632. [DOI] [PubMed] [Google Scholar]
- [43].Ovsepian SV, Herms J. Drain of the brain: low-affinity p75 neurotrophin receptor affords a molecular sink for clearance of cortical amyloid β by the cholinergic modulator system. Neurobiol Aging. 2013. Nov;34(11):2517–2524. [DOI] [PubMed] [Google Scholar]
- [44].Kanekiyo T, Bu G. The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer’s disease. Front Aging Neurosci. 2014;6:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Abbas YM, Wu D, Bueler SA, et al. Structure of V-ATPase from the mammalian brain. Science. 2020. Mar 13;367(6483):1240–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Webb BA, Aloisio FM, Charafeddine RA, et al. pHLARE: a new biosensor reveals decreased lysosome pH in cancer cells. Mol Biol Cell. 2021. Jan 15;32(2):131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Nisbet RM, Polanco JC, Ittner LM, et al. Tau aggregation and its interplay with amyloid-β. Acta Neuropathol. 2015. Feb;129(2):207–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Vossel KA, Zhang K, Brodbeck J, et al. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010. Oct 8;330(6001):198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Park J, Choi H, Kim YD, et al. Aberrant role of ALK in tau proteinopathy through autophagosomal dysregulation. Mol Psychiatry. 2021. Jan 15;26(10):5542–5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Stern R, Kogan G, Jedrzejas MJ, et al. The many ways to cleave hyaluronan. Biotechnol Adv. 2007. Nov-Dec;25(6):537–557. [DOI] [PubMed] [Google Scholar]
- [51].Orimoto AM, Dumaresq-Doiron K, Jiang JY, et al. Mammalian hyaluronidase induces ovarian granulosa cell apoptosis and is involved in follicular atresia. Endocrinology. 2008. Nov;149(11):5835–5847. [DOI] [PubMed] [Google Scholar]
- [52].Zhang L, Bharadwaj AG, Casper A, et al. Hyaluronidase activity of human Hyal1 requires active site acidic and tyrosine residues. J Biol Chem. 2009. Apr 3;284(14):9433–9442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gupta RC, Lall R, Srivastava A, et al. Hyaluronic acid: molecular mechanisms and Therapeutic trajectory. Front Vet Sci. 2019;6:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gwon Y, Kim SH, Kim HT, et al. Amelioration of amyloid β-FcγRIIb neurotoxicity and tau pathologies by targeting LYN. Faseb J. 2019. Mar;33(3):4300–4313. [DOI] [PubMed] [Google Scholar]
- [55].Misra S, Hascall VC, Markwald RR, et al. Interactions between hyaluronan and its receptors (CD44, RHAMM) regulate the activities of inflammation and cancer. Front Immunol. 2015;6:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sterniczuk R, Antle MC, Laferla FM, et al. Characterization of the 3xTg-AD mouse model of Alzheimer’s disease: part 2. Behavioral and cognitive changes. Brain Res. 2010. Aug;12(1348):149–155. [DOI] [PubMed] [Google Scholar]
- [57].Jiang S, Bhaskar K. Degradation and transmission of tau by autophagic-endolysosomal networks and potential therapeutic targets for tauopathy. Front Mol Neurosci. 2020;13:586731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lee JH, McBrayer MK, Wolfe DM, et al. Presenilin 1 maintains lysosomal Ca(2+) homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep. 2015. Sep 1;12(9):1430–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Morita T, Kamada A. Age-related changes in central nervous system hyaluronic acid and chondroitin sulfate in senescence-accelerated mice. J Osaka Dent Univ. 1993. Apr;27(1):37–49 [PubMed] [Google Scholar]
- [60].Jenkins HG, Bachelard HS. Developmental and age-related changes in rat brain glycosaminoglycans. J Neurochem. 1988. Nov;51(5):1634–1640. [DOI] [PubMed] [Google Scholar]
- [61].Reed MJ, Damodarasamy M, Pathan JL, et al. The effects of normal aging on regional accumulation of hyaluronan and chondroitin sulfate proteoglycans in the mouse brain. J Histochem Cytochem. 2018. Oct;66(10):697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Schinzel RT, Higuchi-Sanabria R, Shalem O, et al. The hyaluronidase, TMEM2, promotes ER homeostasis and longevity independent of the UPR(ER). Cell. 2019. Nov 27;179(6):1306–1318.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Roszkowska M, Skupien A, Wójtowicz T, et al. CD44: a novel synaptic cell adhesion molecule regulating structural and functional plasticity of dendritic spines. Mol Biol Cell. 2016. Dec 15;27(25):4055–4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].McGuire CM, Forgac M. Glucose starvation increases V-ATPase assembly and activity in mammalian cells through AMP kinase and phosphatidylinositide 3-kinase/Akt signaling. J Biol Chem. 2018. Jun 8;293(23):9113–9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Petrey AC, de la Motte CA. Hyaluronan, a crucial regulator of inflammation. Front Immunol. 2014;5:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Schultz K, Grieger Lindner C, Li Y, et al. Gamma secretase dependent release of the CD44 cytoplasmic tail upregulates IFI16 in cd44-/- tumor cells, MEFs and macrophages. PLoS One. 2018;13(12):e0207358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Vidoni C, Follo C, Savino M, et al. The role of cathepsin D in the pathogenesis of human neurodegenerative disorders. Med Res Rev. 2016. Sep;36(5):845–870. [DOI] [PubMed] [Google Scholar]
- [68].Yadati T, Houben T, Bitorina A, et al. The ins and outs of cathepsins: physiological function and role in disease management. Cells. 2020. Jul 13;9(7): 1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bodzęta A, Kahms M, Klingauf J. The presynaptic v-ATPase reversibly disassembles and thereby modulates exocytosis but is not part of the fusion machinery. Cell Rep. 2017. Aug 8;20(6):1348–1359. [DOI] [PubMed] [Google Scholar]
- [70].van der Kant R, Goldstein LSB, Ossenkoppele R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat Rev Neurosci. 2020. Jan;21(1):21–35. [DOI] [PubMed] [Google Scholar]
- [71].Zhao N, Liu CC, Van Ingelgom AJ, et al. Apolipoprotein E4 impairs neuronal insulin signaling by trapping insulin receptor in the endosomes. Neuron. 2017. Sep 27;96(1):115–129.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Miyagawa T, Ebinuma I, Morohashi Y, et al. BIN1 regulates BACE1 intracellular trafficking and amyloid-β production. Hum Mol Genet. 2016. Jul 15;25(14):2948–2958. [DOI] [PubMed] [Google Scholar]
- [73].Ubelmann F, Burrinha T, Salavessa L, et al. Bin1 and CD2AP polarise the endocytic generation of beta-amyloid. EMBO Rep. 2017. Jan;18(1):102–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Caglayan S, Takagi-Niidome S, Liao F, et al. Lysosomal sorting of amyloid-β by the SORLA receptor is impaired by a familial Alzheimer’s disease mutation. Sci Transl Med. 2014. Feb 12;6(223):223ra20. [DOI] [PubMed] [Google Scholar]
- [75].Furusawa K, Takasugi T, Chiu YW, et al. CD2-associated protein (CD2AP) overexpression accelerates amyloid precursor protein (APP) transfer from early endosomes to the lysosomal degradation pathway. J Biol Chem. 2019. Jul 12;294(28):10886–10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci. 2016. Jul 1;129(13):2475–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Spilman P, Podlutskaya N, Hart MJ, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One. 2010. Apr 1;5(4):e9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Caccamo A, De Pinto V, Messina A, et al. Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer’s disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature. J Neurosci. 2014. Jun 4;34(23):7988–7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Wang C, Sun B, Zhou Y, et al. Cathepsin B degrades amyloid-β in mice expressing wild-type human amyloid precursor protein. J Biol Chem. 2012. Nov 16;287(47):39834–39841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Choi S, Kim D, Kam TI, et al. Lysosomal enzyme glucocerebrosidase protects against Aβ1-42 oligomer-induced neurotoxicity. PLoS One. 2015;10(12):e0143854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Settembre C, De Cegli R, Mansueto G, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013. Jun;15(6):647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Medina DL, Fraldi A, Bouche V, et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell. 2011. Sep 13;21(3):421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Koh JY, Kim HN, Hwang JJ, et al. Lysosomal dysfunction in proteinopathic neurodegenerative disorders: possible therapeutic roles of cAMP and zinc. Mol Brain. 2019. Mar 12;12(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003. Jul 31;39(3):409–421. [DOI] [PubMed] [Google Scholar]
- [85].Kim JT, Lee SJ, Kang MA, et al. Cystatin SN neutralizes the inhibitory effect of cystatin C on cathepsin B activity. Cell Death Dis. 2013. Dec 19;4(12):e974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Souttou B, Carvalho NB, Raulais D, et al. Activation of anaplastic lymphoma kinase receptor tyrosine kinase induces neuronal differentiation through the mitogen-activated protein kinase pathway. J Biol Chem. 2001. Mar 23;276(12):9526–9531. [DOI] [PubMed] [Google Scholar]
- [87].Schneider CA, Rasband WS, Eliceiri KWNIH. Image to ImageJ: 25 years of image analysis. Nat Methods. 2012. Jul;9(7):671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Caicedo JC, Goodman A, Karhohs KW, et al. Nucleus segmentation across imaging experiments: the 2018 data science bowl. Nat Methods. 2019. Dec;16(12):1247–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Ying H, Qin A, Cheng TS, et al. Disulfiram attenuates osteoclast differentiation in vitro: a potential antiresorptive agent. PLoS One. 2015;10(4):e0125696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Xu L, Shen X, Bryan A, et al. Inhibition of host vacuolar H+-ATPase activity by a Legionella pneumophila effector. PLoS Pathog. 2010. Mar 19;6(3):e1000822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Lee B, Lee H, Nam YR, et al. Enhanced expression of glutamate decarboxylase 65 improves symptoms of rat parkinsonian models. Gene Ther. 2005. Aug;12(15):1215–1222. [DOI] [PubMed] [Google Scholar]
- [92].Jiang Y, Sato Y, Im E, et al. Lysosomal dysfunction in down syndrome is APP-dependent and mediated by APP-βCTF (C99). J Neurosci. 2019;39(27):5255–5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.