

ABSTRACT
Mitophagy regulates cancer stem cell (CSC) populations affecting tumorigenicity and malignancy in various cancer types. Here, we report that cisplatin treatment led to the activation of higher mitophagy through regulating CLU (clusterin) levels in oral CSCs. Moreover, both the gain-of-function and loss-of-function of CLU indicated its mitophagy-specific role in clearing damaged mitochondria. CLU also regulates mitochondrial fission by activating the Ser/Thr kinase AKT, which triggered phosphorylation of DNM1L/Drp1 at the serine 616 residue initiating mitochondrial fission. More importantly, we also demonstrated that CLU-mediated mitophagy positively regulates oral CSCs through mitophagic degradation of MSX2 (msh homeobox 2), preventing its nuclear translocation from suppressing SOX2 activity and subsequent inhibition of cancer stemness and self-renewal ability. However, CLU knockdown disturbed mitochondrial metabolism generating excessive mitochondrial superoxide, which improves the sensitivity to cisplatin in oral CSCs. Notably, our results showed that CLU-mediated cytoprotection relies on SOX2 expression. SOX2 inhibition through genetic (shSOX2) and pharmacological (KRX-0401) strategies reverses CLU-mediated cytoprotection, sensitizing oral CSCs toward cisplatin-mediated cell death.
KEYWORDS: Cancer stem cells, clusterin, mitophagy, MSX2, SOX2
Introduction
Oral cancer is one of the 10 leading cancer types affecting global populations [1]. Oral squamous cell carcinoma (OSCC) is a crucial subtype covering approximately 90% of oral cancers [2]. At present, chemotherapy is the most commonly used treatment for OSCC. However, the primary concern with chemotherapy is the enrichment of cancer stem cells (CSCs) [3–5]. CSCs are one of the strong driving forces of tumorigenesis; these cells typically survive therapy and are enriched during post-therapeutic stress, leading to drug resistance and tumor relapse in OSCC [6,7]. The selective elimination of damaged mitochondria generated from successful mitochondrial fission is necessary for the survival of CSCs against chemotherapeutic stress [8]. Mitophagy is a selective form of autophagy through which cancer cells eliminate damaged mitochondria for a better adaptive response [9,10]. Similarly, mitophagy is also involved in maintaining stemness in various cancer types [11]. Recent reports suggest that mitophagy inhibition significantly improves chemosensitivity in cancer cells [12,13]. However, the extent to which mitophagy inhibition affects CSCs populations during chemotherapeutic stress remains an exciting area for targeted cancer therapy in oral cancer.
CLU (clusterin) is a secreted molecular chaperone that is activated in response to stress and transports to the cytosol, where it guides misfolded proteins to the proteasome and/or autophagy for degradation [14]. It has been found that CLU can function as either an oncoprotein or a tumor suppressor protein, depending on the cellular context, which has important implications from a cancer prevention standpoint [15]. A recent study showed that nucleolar localization of CLU is responsible for negatively regulating ribogenesis and maintaining nuclear morphology in human oral cancer cells [16]. CLU functions as an adaptor protein, enhancing cancer cell survival through autophagy activation [17]. In a recent study, we demonstrated that CLU regulates the AMPK-MTOR-ULK1 axis to trigger autophagy, providing apoptosis resistance in oral squamous cell carcinoma cells [18]. Even though several reports suggest the crucial role of CLU in regulating different cancer-associated signaling in oral cancer, very little is known about its function in regulating the stemness and CSC-associated chemoresistance in OSCC.
In the current study, we demonstrated that cisplatin-induced CLU regulates mitophagy status in cells that are CD44+ (a marker of cancer stem cells), which was further verified through both gain-of-function and loss-of-function studies. In addition, CLU regulates mitochondrial fission by activating AKT-mediated phosphorylation of DNM1L/DRP1 at serine 616. More importantly, CLU promotes mitophagic degradation of MSX2 after being translocated to mitochondria, enhancing SOX2-mediated stemness. SOX2 KD improves the therapeutic efficacy as indicated by the higher apoptosis rate in remarkably resistant CLU-overexpressing (OE) CD44+ cells. Moreover, CLU knock down (KD) impaired mitophagy, causing a higher accumulation of mitochondrial ROS, which sensitized CSCs to cisplatin treatment and could be crucial for better clinical outcomes in oral cancer.
Results
Oral CD44+ cells display enhanced mitophagic flux compared to CD44– cells
To investigate the autophagy status in oral CSC, we first monitored the expression of different autophagy-associated proteins in both developed orospheres (nonadherent colonies of cells sorted from primary cells) and MACS-sorted CD44+ cells (Figure S1A) derived from two oral cancer cell lines, FaDu and CAL-33. As shown in Figure 1A-B and S1Bi-Ciii, both orospheres and CD44+ cells showed enhanced expression of autophagy markers such as LC3-II (membrane-bound lipidated form) and the autophagy protein ATG5, along with a drastic reduction in the level of the autophagy receptor and substrate SQSTM1/p62 compared to adherent cancer cells and CD44− cells, respectively. In addition to the autophagy activation, both orospheres and CD44+ cells showed increased expression of LAMP1 (lysosomal associated membrane protein 1), indicating a higher number of lysosomes and enhanced degradation of mitochondrial proteins such as TOMM20 and COX4/COXIV than the comparison cells. However, there was no alteration in the levels of proteins marking other organelles, including the ER (HSP90B1/GRP94), nucleus (LMNB2), and endosome (RAB7) (Figure 1C-D, and S1Di-Eiii), suggesting the activation of selective mitochondrial autophagy (i.e., mitophagy) in oral CSCs.
Figure 1.

Cisplatin promotes mitophagy in oral CD44+ cells. (A-D) Oral cancer cells (FaDu, left panel; and CAL-33, right panel), the derived orospheres and MACS-sorted CD44− and CD44+ cells were analyzed for expression of (A, B) autophagic protein LC3-II, ATG5, and SQSTM1, (C, D) mitochondrial-, nuclear-, endoplasmic reticulum (ER)-, lysosome-, and endosome-related proteins through western blot. ACTB was used as a loading control to normalize band intensities. (E-F) Western blot analysis for mitochondrial proteins in CD44+ cells and CD44− cells of FaDu (left panel) and CAL-33 (right panel) in response to cisplatin treatment for dose-dependent (1, 5, and 10 µM) and time-course (6, 12 and 24 h) studies. ACTB was used as a loading control to normalize band intensities. (Gi-ii) Immunofluorescence-based quantification of the number of mitophagosomes in CSC compared to their non-stem counterparts exposed to 10 µM of cisplatin for 24 h using MitoTracker Green (MTG) and LysoTracker Red (LTR). The graph shows Pearson’s coefficient values ±S.D., calculated from images with multiple cells randomly taken for each sample of three different experiments. Scale bars: 10 μm. *p < 0.05, **p < 0.01.
Next, we examined the mitophagic status in MACS-sorted CD44+ and CD44− cells in response to cisplatin treatment. Interestingly, both dose-dependent (1, 5, and 10 µM) (Figure 1E and S2Ai-iv) and time-course analyses (6, 12 and 24 h) (Figure 1F and S2Bi-iv) of cisplatin treatment showed higher degradation of TOMM20, and COX4 in CD44+ cells relative to CD44− cells, suggesting that cisplatin triggered autophagy-dependent mitochondrial degradation in oral CD44+ cells. To confirm that lysosomes were involved in mitochondrial degradation, we performed colocalization analysis using LysoTracker Red (LTR; lysosome) and MitoTracker Green (MTG; mitochondria) dyes. Of note, a significant increase in colocalization of mitochondria with lysosomes was observed in CD44+ versus CD44− cells which further increased after cisplatin treatment (Figure 1Gi-Gii). These observations led us to conclude that cisplatin induces lysosome-mediated, autophagy-dependent degradation of mitochondria in oral CD44+ cells at a higher level than in oral CD44− cells.
CLU promotes mitophagy in oral CD44+ cells
Along with mitophagy activation, cisplatin treatment promoted CLU expression in CD44+ cells isolated from both FaDu and CAL-33 cell lines (Figure 2A and S2C). To establish the connection between cisplatin-induced CLU and mitophagy activation, we performed gain-of-function studies in both CD44− and CD44+ cells and found that ectopic expression of CLU in CD44+ cells led to a marked reduction in SQSTM1 and an enhanced expression of LC3-II levels compared to CD44− cells. Notably, CLU overexpression led to a higher degradation of mitochondrial proteins such as TOMM2O and COX4, suggesting enhanced mitophagy in CLU-OE CD44+ cells (Figure 2B and S2Di-Diii). To strengthen this point, we performed colocalization analysis in both CD44− and CD44+ cells under both gain and loss of function of CLU. As expected, CLU OE led to more LC3 puncta colocalized with mitochondria (TOMM20) (Figure 2Ci-ii), and higher colocalization of MTG with LTR (Figure S2Ei-ii) than MOCK transfection in CD44+ cells than CD44− cells. In contrast, loss of function of CLU using small hairpin RNAs (shRNA) targeting CLU reversed the effect showing reduced LC3-II expression with a more significant recovery of SQSTM1, TOMM20, and COX4, indicating mitophagy inhibition (Figure 2D and S3Ai-Aiii). Next, we performed the colocalization analysis in CLU KD CD44− and CD44+ cells to establish whether endogenous CLU could induce mitophagy. Notably, CLU KD significantly reduced LC3 puncta level and decreased the extent of overlapping (Figure 2Ei-ii) along with reduced colocalization of MTG with LTR (Figure S3Bi-ii) than seen with the control shNC (nontargeting shRNA) in both CD44− and CD44+ cells, suggesting that both CD44− and CD44+ cells have reduced mitophagy during CLU silencing. Moreover, CLU KD significantly rescued an autophagic substrate (SQSTM1), along with mitochondrial proteins (Figure 2F and S3Ci-ii) from degradation in response to cisplatin treatment. Similarly, treatment with Mdivi-1 (a DNM1L inhibitor that blocks mitochondrial division) also reversed CLU-mediated clearance of impaired mitochondria in CLU-OE CD44+ cells compared to the untreated cells (Figure 2G and S3D). Furthermore, we investigated whether PINK1 plays a role in regulating cisplatin-driven CLU-mediated mitophagy, and found that loss of function of CLU did not affect PINK1 expression (Figure S3Ei-ii), suggesting that PINK1 might not be involved in the current observations.
Figure 2.

CLU is essential for mitophagy-mediated clearance of damaged mitochondria in oral CD44+ cells. (A) Western blot analysis for the expression of CLU in CD44− and CD44+ cells of FaDu (top panel) and CAL-33 (bottom panel). ACTB was used as a loading control to normalize band intensities. (B, D) Western blot analysis for the expression of autophagic markers and mitochondrial proteins such as TOMM20 and COX4 in both CD44+ and CD44− cells with gain and loss of function of CLU. ACTB was used as a loading control to normalize band intensities. (C, E) Representative confocal images of CLU (cyan), LC3 (green), TOMM20 (red) and DAPI (blue) in (Ci) MOCK or CLU-OE (Ei) shNC or shCLU CD44− and CD44+ cells. (Cii, Eii) Quantification for the number of LC3 puncta per cells ±S.D. calculated from images with multiple cells randomly taken for each sample of three different experiments and the colocalization analysis (Pearson’s coefficient) between LC3 and TOMM20 in the indicated conditions. Scale bars: 10 μm. Error bars: ±S.D. **p < 0.01, ***p < 0.001. (F) Western blot analysis for autophagy markers or mitochondrial protein turnover in shNC or shCLU CD44+ cells treated with cisplatin (10 µM, 24 h). ACTB was used as a loading control to normalize band intensities. (G) Western blot analysis for mitochondrial protein turnover in the presence of Mdivi-1 (2 µM, 3 h) in MOCK or CLU-OE CD44+ cells. ACTB was used as a loading control to normalize band intensities.
Next, we found that CLU overexpression led to a higher mitophagic flux as indicated by higher conversion of LC3-I to LC3-II and substantial reduction of autophagic substrate SQSTM1 along with mitochondrial proteins COX4 and TOMM20 when compared to MOCK-expressing CD44+ cells. Notably, simultaneous chloroquine (CQ) and bafilomycin A1 (BafA1) (a V-ATPase inhibitor that blocks general autophagy) treatment enhanced the levels of LC3-II as well as SQSTM1 along with mitochondrial protein COX4, indicating that the changes in LC3-II protein levels corresponded to a complete autophagic response rather than a block in autophagy (Figure 3A-B and S3F-G). To further confirm mitophagic flux, we performed confocal live-cell microscopy in MOCK, and CLU-OE CD44+ cells stably expressing mito-mKeima (mt-mKeima). Mt-mKeima is a pH-sensitive dual-excitation ratiometric fluorescent protein that exhibits shorter-wavelength excitation (488 nm) at the physiological pH of 8.0 in the mitochondria and longer-wavelength excitation (561 nm) within the acidic lysosome at pH 4.5 during mitophagy. The majority of MOCK CD44+ cells had green-fluorescent, extended networks of mitochondria (cytosolic mitochondria), whereas the majority of CLU-OE CD44+ cells had red-fluorescent, fragmented mitochondria (mitolysosomes), indicating basal mitophagy. Additionally, the mt-mKeima red:green ratio (mitolysosome:mitochondrion) increased in CLU-OE CD44+ cells after cisplatin treatment, indicating an increase in mitophagic flux. Further analysis of mitophagic flux in the presence of BafA1 confirmed the total blockage of mitophagic flux in MOCK and CLU-OE CD44+ cells, demonstrating a reversal of both basal and cisplatin-induced mitophagy as indicated by the predominance of green fluorescence (Figure 3Ci-ii). Likewise, genetic inhibition of CLU (shCLU) could reverse the mitophagy flux in both basal and cisplatin-treated CD44+ CSCs, as evidenced by a significant decrease in the mt-mKeima red:green ratio (Figure 3Di-ii). Collectively, these results showed the essential role of CLU in the selective elimination of impaired mitochondria.
Figure 3.

CLU promotes higher mitophagic flux in oral CD44+ cells. Western blot analysis for autophagic flux or mitochondrial protein turnover (A) in the presence of CQ (20 µM, 3 h) in MOCK or CLU-OE CD44+ cells and (B) in the presence of BafA1 (50 nM, 3 h) of MOCK or CLU-OE CD44+ cells. ACTB was used as a loading control to normalize band intensities. (Ci) Representative confocal images of mt-mKeima in MOCK or CLU-OE CD44+ cells treated with cisplatin (10 µM, 24 h) in combination with BafA1 (50 nM, 3 h). (Cii) The graph shows the quantification for the mitophagy index (red:green intensity), calculated from images with multiple cells randomly taken for each sample of three different experiments in the indicated treatment conditions. (Di) Representative confocal images of mt-mKeima in shNC or shCLU CD44+ cells treated with cisplatin (10 µM, 24 h). (Dii) The graph represents the quantification of the mitophagy index (red:green intensity), calculated from images with multiple cells randomly taken for each sample of three different experiments in the indicated treatment conditions. Scale bars: 10 μm. Error bars: ±S.D. *p < 0.05, **p < 0.01.
CLU promotes mitochondrial fission by regulating the AKT-DNM1L axis to activate mitophagy in oral CD44+ cells
Mitochondrial fission may play an important role in efficiently segregating damaged mitochondrial portions from the active mitochondrial network. Next, we examined the status of mitochondrial fission in oral CSCs, and, to test this, we performed immunofluorescence analysis, which showed that cisplatin treatment led to a higher number of fragmented mitochondria (TOMM2O-stained cells) in both CD44+ and CD44− cells, denoting activation of mitochondrial fission (Figure S4Ai-iii). Moreover, cisplatin treatment activated DNM1L as indicated by higher expression of p-DNM1L (Ser616) in CD44+ cells (Figure S4Bi-ii), suggesting the activation of DNM1L-mediated mitochondrial fission. Next, to establish the role of CLU in regulating DNM1L-dependent mitochondrial fission, we performed gain-of-function and loss-of-function studies in both CD44+ and CD44− cells. CLU overexpression resulted in enhanced expression of p-DNM1L (Ser616) in CD44+ cells compared to CD44− cells (Figure 4A and S4C). Moreover, immunofluorescence analysis of TOMM2O-stained cells showed a higher number of fragmented mitochondria (average branch length < 2.5 µm) in CLU-overexpressing CD44+ cells compared to those with the empty vector (Figure 4Bi, Bii). In contrast, CLU inhibition reduced the phosphorylation status of DNM1L (Figure 4A and S4D) and the number of fragmented mitochondria (Figure 4Bi, Biii) in both CD44+ and CD44− cells. We also examined the mitochondrial fission status of CLU-OE CD44+ cells after exposing them to Mdivi-1 (2 µM; 3 h) and found that Mdivi-1 treatment significantly increased the branch length compared to DMSO-treated CLU-OE CD44+ cells (Figure S4E). Mitochondrial ROS production was tested because Mdivi-1 is known to disrupt complex I of the electron transport chain, and we found that the concentrations used did not induce mtROS production (Figure S4F). Next, we evaluated the effect of cisplatin on mitochondrial fission in the absence of CLU. CLU KD not only reduced the phosphorylation status of DNM1L (Figure 4C and S4G) but also led to the accumulation of elongated mitochondria (average branch length >5 µm) (Figure S5Ai-ii), suggesting the role of CLU in regulating DNM1L-mediated mitochondrial fission in oral CD44+ cells.
Figure 4.

CLU promotes mitochondrial fission by regulating DNM1L activity in oral CD44+ cells. (A-E) Western blot, confocal microscopy and mitochondrial morphology analyses. Western blot analysis for DNM1L phosphorylation status in CD44+ and CD44− cells, with (A) MOCK or CLU expression (left panel), cells expressing shNC or shCLU (right panel), (C) cells expressing shNC or shCLU and treated with vehicle or cisplatin (10 µM, 24 h) and (D) DNM1L KD cells expressing MOCK or CLU transfected with wild-type DNM1L or DNM1LS616A mutant. ACTB was used as a loading control. (Bi) Representative confocal images of the corresponding cells stained with CLU (red), and TOMM20 (green; mitochondria) (Ei) with showing mitochondrial morphology. (Bii, Biii, Eii) ImageJ-based quantification of mitochondrial branch length, calculated from images with multiple cells randomly taken for each sample of three different experiments, which was further categorized into fragmented (<2.5 µm), intermediate (2.5–4 µm), and elongated (>4 µm) according to the indicated parameter. Scale bars: 10 μm. Error bars: ±S.D. *p < 0.05, **p < 0.01, ***p < 0.001.
Next, to define the mechanistic role of DNM1L in mitochondrial fragmentation in CLU-OE CD44+ cells, we overexpressed wild-type (WT) DNM1L and a dephosphorylation-mimic mutant DNM1LS616A in DNM1L KD CD44+ cells expressing MOCK and CLU. As a result of DNM1L KD, phosphorylation of DNM1L was completely suppressed in MOCK and CLU-OE CD44+ cells. Furthermore, we found that the phosphorylation of DNM1L was restored upon the reintroduction of WT DNM1L but not DNM1LS616A (Figure 4D and S5B). Again, DNM1L KD caused a greater abundance of mitochondria with an elongated shape in CLU-OE CD44+ CSCs, and the reintroduction of WT DNM1L, but not DNM1LS616A, resulted in a greater abundance of mitochondria with a fragmented shape (Figure 4Ei-ii).
In addition, cisplatin treatment increased the phosphorylation of Ser/Thr kinase AKT in CD44+ cells compared to CD44− cells (Figure 5A and S5C). Next, we used gain and loss of function of CLU in CD44− and CD44+ cells and found that CLU overexpression triggered phosphorylation of AKT, whereas CLU KD reversed the effect, establishing the role of CLU in activating the Ser/Thr kinase AKT (Figure 5B and S5D-E). Moreover, cisplatin treatment promoted the phosphorylation of AKT in CLU KD CD44+ cells (Figure 5C and S5F). We extended this analysis by performing rescue experiments in which we overexpressed wild-type AKT and the AKTS473A phosphorylation variant in AKT KD MOCK and CLU-OE CD44+ cells, demonstrating reduced phosphorylation was AKT dependent. As expected, DNM1L S616 phosphorylation was significantly downregulated in CLU-OE AKT KD CD44+ cells but was effectively recovered by expressing WT AKT, but not AKTS473A. Notably, the reintroduction of WT AKT induced DNM1L S616 phosphorylation in CLU-OE cells compared to MOCK-expressing CD44+ CSCs (Figure 5D and S5G-H). As a next step, we looked into how the phosphorylation of DNM1L at the S616 residue by AKT affects mitochondrial morphology. Intriguingly, higher levels of mitochondrial fragmentation were observed after the reintroduction of WT AKT but not AKTS473A in AKT KD MOCK and CLU-OE CD44+ CSCs (Figure 5Ei-ii). Moreover, we also treated CLU-OE CD44+ cells with KRX-0401 (a pharmacological inhibitor of AKT) and noticed that AKT inhibition reduced CLU-induced phosphorylation of DNM1L (Figure 5F and S6Ai-ii). Additionally, we performed subcellular fractionation analysis, which confirmed that KRX-0401 treatment reduced the expression of p-DNM1L (Ser616) in the mitochondrial fraction (Figure 5Gi-ii), indicating attenuation of mitochondrial translocation of p-DNM1L. Moreover, KRX-0401 treatment also altered CLU-induced shortening of mitochondrial branch length, leading to the accumulation of elongated mitochondria (Figure S6Bi-ii). Similarly, the colocalization analysis showed a remarkably strong association of p-DNM1L (Ser616) with mitochondria in CLU-OE CD44+ cells compared to empty vector-expressing CD44+ cells. However, KRX-0401 treatment decreased the colocalization of p-DNM1L (Ser616) with mitochondria (TOMM20) in CLU-OE CD44+ cells (Figure S6Ci-ii). Further, we used confocal-based colocalization analysis with DNM1L (red color) and TOMM20 (green color) in CLU-OE CD44+ cells and discovered that CLU OE triggered mitochondrial relocalization of DNM1L, which was significantly suppressed by KRX-0401 treatment (Figure S6Di-ii). As a result, the regulation of mitochondrial fission in CLU-OE CD44+ CSCs relies on CLU-mediated activation of the AKT-DNM1L axis.
Figure 5.
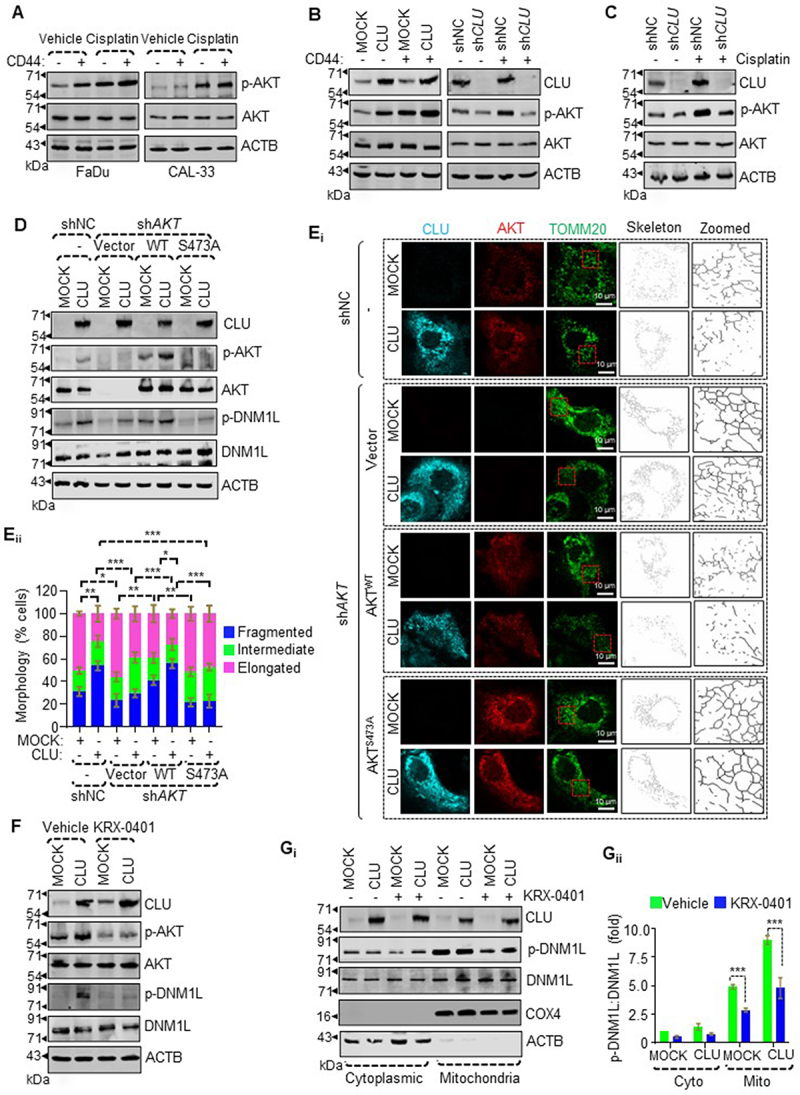
CLU-induced AKT is responsible for activating DNM1L-mediated mitochondrial fission in oral CD44+ cells. (A-D, F) Western blot analysis for the phosphorylation status of AKT in (A) CD44− or CD44+ cells of FaDu (left panel) and CAL-33 (right panel) treated with cisplatin (10 µM, 24 h), (B) CD44− or CD44+ cells with gain (left panel) and loss of function (right panel) of CLU, (C) shNC or shCLU CD44+ cells treated with cisplatin, (D) AKT KD cells expressing MOCK or CLU stably transfected with wild-type AKT or the AKTS473A mutant, and (F) MOCK or CLU-OE CD44+ cells treated with the AKT inhibitor (KRX-0401). ACTB was used as a loading control to normalize band intensities. (Ei) Representative confocal images of AKT KD cells expressing MOCK or CLU-OE transfected with wild-type AKT or AKTS473A mutant followed by staining with CLU (cyan), DNM1L (red), and TOMM20 (green) showing mitochondrial morphology. (Eii) Quantification of the categorized mitochondria according to their branch length, which was calculated from images with multiple cells randomly taken for each sample of three different experiments. Scale bars: 10 μm. Error bars: ±S.D. *p < 0.05, **p < 0.01, ***p < 0.001. (Gi-ii) Western blot analysis for the subcellular distribution of p-DNM1L in MOCK or CLU-OE CD44+ cells treated with AKT inhibitor (KRX-0401). The purity of the fraction was verified through monitoring ACTB (cytoplasm) and COX4 (mitochondria). The intensities of the proteins were adjusted with ACTB levels and normalized to the cytosolic fraction of untreated CD44+ cells expressing MOCK. Data were quantified from three independent experiments. Error bars: ±S.D. ***p < 0.001.
CLU promotes SOX2-mediated stemness through curtailing MSX2 protein half-life
Next, we performed an expression analysis of different stemness markers such as SOX2, POU5F1/OCT4, and NANOG in CLU-OE CD44+ cells. As shown in Figure 6A and S7A, SOX2 among other stemness-associated markers showed significantly higher expression in CD44+ cells compared to CD44− cells in CAL-33 and FaDu cell lines. Next, we established the role of CLU in regulating the stemness of oral CSCs and found that CLU overexpression promoted the enrichment of the CSC population relative to the empty vector. This effect was altered in the presence (increased) versus the absence (decreased) of mitophagy (Figure S7B). Next, to determine the cause of higher SOX2 expression, we checked the level of MSX2 (a SOX2 repressor protein) in CLU-OE CSCs. Interestingly, we noticed that CLU overexpression led to a decrease in MSX2 levels and a consequent increase in SOX2 levels (Figure 6Bi-ii). Likewise, CLU KD caused an accumulation of MSX2 levels and a resultant reduction of SOX2 levels (Figure 6Ci-ii) in CD44+ cells. Therefore, we assumed that CLU might regulate SOX2 activity in oral cancer by affecting the protein stability of MSX2. To test our hypothesis, both empty vector (MOCK) and CLU-expressing CD44+ cells were treated with cycloheximide (25 μM) for the designated times, and proteolytic turnover of MSX2 was measured through immunoblot analysis. Interestingly, CHX treatment markedly reduced the half-life of MSX2 protein in CLU-OE CD44+ cells compared to MOCK-expressing CD44+ cells (Figure 6Di-ii), suggesting that CLU accelerates MSX2 protein turnover. As the ubiquitin-proteasome system (UPS) and lysosome-dependent autophagy pathway are the two known degradation machinery responsible for regulating intracellular protein turnover [19], we next examined the pathways operating in CLU-mediated proteolysis of MSX2. In this setting, CLU-OE CD44+ cells were treated with MG132 (a proteasome inhibitor) for 8 h and CQ and Mdivi-1 (mitophagy inhibitors) for 3 h. Surprisingly, pre-treatment with proteasomal inhibitors did not affect CLU-driven dynamic alteration in the MSX2 level (Figure 6Ei-ii). Moreover, we also checked MCL1 expression, a known substrate of the proteasomal degradation pathway [20] in MG132-treated conditions and noticed a higher accumulation of MCL1 in MG132-treated groups (Figure 6Ei-ii). However, mitophagy inhibition (CQ, Mdivi-1, and siATG5) markedly rescued the MSX2 protein levels in CLU-expressing CD44+ cells (Figure 6F-H). Altogether, our results suggest that the CLU-mediated proteolysis of MSX2 requires mitophagy rather than proteasomal degradation pathways in oral CD44+ cells.
Figure 6.

Cisplatin promotes stemness by regulating the CLU-dependent degradation of MSX2. (A) Western blot analysis for stemness assessment in CD44+ and CD44− cells from FaDu (left panel) and CAL-33 (right panel) cell lines. ACTB was used as a loading control to normalize band intensities. (B-C) Western blot analysis of SOX2 and MSX2 in CLU-OE and CLU-KD CD44+ cells in response to cisplatin treatment. (Di) Western blot analysis showing the cycloheximide (CHX) chase analysis of MSX2 protein degradation at the indicated time points in MOCK or CLU-OE CD44+ cells. (Ei) Western blot analysis for degradation of MSX2 in MG132 treated MOCK or CLU-OE CD44+ cells. (F-H) Western blot analysis for stability and degradation of MSX2 in mitophagy compromised conditions (CQ [Fi], Mdivi-1 [Gi], and siATG5 [Hi]) of MOCK or CLU-OE CD44+ cells. (Bii, Cii, Dii, Eii, Fii, Gii, Hii) Quantification of the fold change through densitometric analysis followed by adjustment with ACTB levels and normalized to control untreated CD44+ cells expressing MOCK or shNC. Data were quantified from three independent experiments. Error bars: ±S.D. *p < 0.05, **p < 0.01, ***p < 0.001.
CLU promotes direct binding of MSX2 with LC3B, critical for its mitophagic degradation in oral CD44+ cells
As mitophagy-compromised conditions rescued the MSX2 protein level, we hypothesized that this effect resulted from impaired mitophagic degradation of MSX2. To test this hypothesis, we performed a colocalization analysis, which showed stronger colocalization of MSX2 with LC3, evidenced by a higher Pearson’s coefficient in CLU-OE CD44+ cells. Moreover, MSX2 was exclusively distributed in the cytoplasm with a significant level colocalized with mitochondria (MitoTrackerTM Deep Red [MTDR]) in CLU-OE CD44+ cells. Of note, cisplatin treatment further increased the colocalization as indicated by a higher Pearson’s coefficient in CLU-OE CD44+ cells (Figure 7Ai-Aii and S7C). In contrast, CLU KD abolished mitochondrial localization of MSX2 and its colocalization with LC3 as indicated by a lower Pearson’s coefficient, which increased after cisplatin treatment (Figure 7Bi-Bii and S7D), suggesting that CLU promotes mitochondrial localization of MSX2. To further confirm the CLU-dependent mitochondrial distribution of MSX2 in CD44+ cells, we performed subcellular fractionation and found that MSX2 was present in the nuclear, cytosolic, and mitochondrial fractions in CD44+ MOCK-expressing cells, whereas it was mainly associated with the mitochondrial fraction in CLU-OE cells (Figure 7Ci-iv). However, Mdivi-1 (mitophagy inhibitor) treatment abolished the mitochondrial translocation of MSX2, as indicated by higher accumulation in the nuclear fraction (Figure 7Ci-iv). Next, we performed in silico analysis of the MSX2 protein sequence through the iLIR database (https://ilir.warwick.ac.uk) [21] to identify any putative LC3-interacting region (LIR) motifs important for autophagic cargo. Notably, we identified one putative LIR motif in the MSX2 protein sequence (165 Y-x-x-l 169), which perfectly matched with the classical consensus sequence of the LIR motif (W/F/Y-X-X-V/L/I) [22] (Figure 7D), which might be essential to execute its binding to LC3. Next, we performed an immunoprecipitation assay to establish the potential binding ability of MSX2 with LC3B. Notably, a stronger association between endogenous MSX2 and LC3B was observed in CLU-OE cells compared to MOCK cells, which was further enhanced following treatment with the mitophagy flux inhibitor CQ (Figure 7E). The importance of the MSX2 LIR motif was further established by conducting the rescue experiment after overexpressing an empty vector, WT MSX2, and a LIR mutant of MSX2 (MSX2[mLIR]), in MSX2 KD CD44+ cells expressing MOCK or CLU. Intriguingly, MSX2 expression was significantly higher in CLU-OE cells with the MSX2[mLIR] variant than with WT MSX2, confirming that MSX2 degradation was mediated through CLU-dependent mitophagy (Figure 7F and S7E). Gain and loss of function of MSX2 in MOCK and CLU-OE cells allowed us to investigate MSX2ʹs direct effect on regulating the SOX2 level, and we found an inverse correlation between MSX2 overexpression and the reduced expression of SOX2, whereas MSX2 knockdown restored SOX2 levels to normal while preserving stemness (Figures 7G-H and S7F-G). Altogether, these data demonstrated that CLU overexpression recruits phagophores around MSX2 puncta for their selective degradation causing its downregulation in oral CSCs.
Figure 7.

MSX2 interacts with LC3 for mitophagic clearance in CLU-OE CD44+ cells. (Ai-ii, Bi-ii) Quantification of Pearson’s coefficient for the colocalization analysis between MSX2-LC3 and MSX2-mitochondria (MTDR) calculated from images with multiple cells randomly taken for each sample of three different experiments. Scale bars: 25 μm. Error bars: ±S.D. *p < 0.05, **p < 0.01, ***p < 0.001. (Ci-iv) Western blot analysis for subcellular fractionation in MOCK or CLU-OE CD44+ cells treated with Mdivi-1 (2 µM, 3 h) followed by expression analysis of MSX2 in the total cell lysate or different cellular fractions. The purity of respective fractions was determined using LMNB2 (nucleus), GAPDH (cytoplasm), and COX4 (mitochondria). (D) In-silico sequence analysis for identification of putative LIR motifs in the MSX2 protein conserved in previously reported mitophagy receptors. (E) Immunoprecipitation (IP) with anti-LC3 beads in the presence of CQ (20 µM, 3 h), followed by detection of MSX2 and LC3 by western blot in MOCK and CLU-OE CD44+ cells. Immunoglobulin G (IgG) was used as a negative control for IPs. (F) Western blot analysis for MSX2 expression in MSX2 KD cells expressing MOCK or CLU stably expressing wild-type MSX2 or MSX2[mLIR]. (G-H) Western blot analysis shows the expression of SOX2 in cells expressing MOCK or CLU with gain (G) and loss of function (H) of MSX2.
CLU-dependent mitophagy maintains stemness through SOX2 to inhibit chemosensitivity in oral CD44+ cells
CLU maintains SOX2 through mitophagic degradation of MSX2, and inhibition of mitophagy suppresses the expression of SOX2 in oral CD44+ cells. Here, we have examined the role of CLU and CLU-mediated enhanced SOX2 in stemness to display chemoresistance in oral CD44+ cells. Initially, we noticed that CLU inhibition drastically reduced the cell viability (Figure 8A) and the percentage of CD44+ cells following treatment with cisplatin compared to shNC (Figure 8B). Furthermore, the number of orospheres in oral CD44+ cells also followed the same trends (Figure 8Ci-ii). CLU KD also reduced the clonogenicity (Figure 8Di-ii) of CD44+ cells in response to cisplatin treatment. To delineate the possible mechanism for the growth inhibitory effects in CLU-KD CD44+ cells following cisplatin treatment, we monitored the apoptosis rate through ANXA5-PI staining. Interestingly, CLU KD significantly enhanced the apoptosis rate in CD44+ cells compared with shNC in response to cisplatin treatment (Figure 8Ei-ii). We also noticed significant changes in the expression of cleaved CASP3, as indicated by the Caspase 3/7 Glo assay (Figure S8A). Next, we sought to determine whether CLU-mediated cytoprotection is due to mitophagy activation. To accomplish this, we treated CLU-OE CD44+ cells with mitophagy inhibitors and monitored the stemness and cell death-inducing effect. As expected, cotreatment with BafA1+ cisplatin drastically reduced the number of orospheres (Figure S8Bi-ii). Similarly, cotreatment with mitophagy inhibitors (CQ, BafA1, and Mdivi-1)+cisplatin substantially decreased the cell viability (Figure S8C-E). Furthermore, ANXA5-PI staining confirmed that cotreatment groups (BafA1+ cisplatin) showed maximum response against cisplatin treatment with more apoptotic cells in CLU-OE CD44+ cells (Figure S8Fi-ii). Collectively, our results suggested that inhibition of CLU and CLU-dependent mitophagy triggers cell death in the presence of cisplatin in oral CD44+ cells.
Figure 8.

SOX2 is responsible for CLU-mediated cytoprotection in oral CD44+ cells during chemotherapeutic stress. (A) Cell viability assay to determine the estimated growth rate in shNC or shCLU CD44+ cells treated with cisplatin. Data were normalized to the untreated shNC CD44+ cells (mean ± SD, n = 3). Error bars: ±S.D. **p < 0.01, and ***p < 0.001. (B) Flow cytometry analysis for CD44 staining to estimate alteration in CD44+ cells after transfection with shNC (scrambled) and shCLU and treatment with cisplatin (10 µM, 24 h). (Ci) Representative images of orospheres formed after transfection with shNC (scrambled) and shCLU and treated with cisplatin (10 µM, 24 h conditions were used for all cisplatin treatments), then cultured for 7 days. Scale bar: 200 μm. (Cii) Quantifications of orosphere numbers (1000 cells/well) formed by oral CD44+ cells after treatment (mean ± SD, n = 3). Error bars: ±S.D. **p < 0.01, and ***p < 0.001. (Di) A colony formation assay was performed to monitor the colony-forming ability of CLU-KD CD44+ cells in response to cisplatin treatment. n = 3. Scale bars: 200 μm. (Dii) Quantifications of colony numbers (1000 cells/well) formed by oral CD44+ cells after treatment (mean ± SD, n = 3). Error bars: ±S.D. **p < 0.01, and ***p < 0.001. (Ei) Apoptosis was assessed by ANXA5-PI staining using flow cytometry in shNC or shCLU CD44+ cells in response to cisplatin treatment for 24 h. (Eii) Quantifications of apoptotic populations (ANXA5+ PI− and ANXA5+ PI+) after cisplatin treatment in shNC or shCLU CD44+ cells (mean ± SD, n = 3). Error bars: ±S.D. **p < 0.01, and ***p < 0.001. (F) Western blot analysis to confirm the knockdown efficiency of SOX2 in MOCK or CLU-OE CD44+ cells. ACTB was used as a loading control to normalize band intensities. (G) Flow cytometry analysis for CD44 staining to estimate alteration in CD44+ cells after transfection with shNC (scrambled) and shSOX2 in MOCK or CLU-OE CD44+ cells. (Hi) Representative images of orospheres formed after transfection with shNC (scrambled) and shSOX2 in MOCK or CLU-OE CD44+ cells, which were then cultured for 7 days. Scale bar: 200 μm. (Hii) Quantification of orosphere numbers (1000 cells/well) formed after transfection and cultured for the next 7 days (mean ± SD, n = 3). (I) Cell viability assay to determine the estimated growth rate in shNC- or shSOX2-transfected MOCK or CLU-OE CD44+ cells treated with cisplatin (10 µM, 24 h). Data were normalized to the untreated shNC CD44+ cells (mean ± SD, n = 3). (Ji) Cell apoptosis was assessed by ANXA5-PI staining using flow cytometry in shNC- or shSOX2-transfected MOCK or CLU-OE CD44+ cells in response to cisplatin treatment for 24 h. (Jii) Quantification of apoptotic populations (ANXA5+ PI− and ANXA5+ PI+) in shNC- or shSOX2-transfected MOCK or CLU-OE CD44+ cells in response to cisplatin treatment for 24 h (mean ± SD, n = 3).
Next, we evaluated the contribution of SOX2 in CLU-mediated cytoprotection against chemotherapy. To achieve this, we first knocked down SOX2 using shRNA in CLU-OE CD44+ cells and tested its effect on cell proliferation and orosphere-forming ability. The knockdown efficiency was verified through western blotting analysis, as indicated by marked downregulation in the expression of endogenous SOX2 (Figure 8F and S9A). SOX2 KD significantly reduced the enrichment of CD44+ cells (Figure 8G) and orospheres number (Figure 8Hi-ii) compared to shNC in MOCK or CLU-OE CD44+ cells after cisplatin treatment. To further establish the role of CLU-induced AKT in upregulating SOX2 protein levels, we treated cells with KRX-0401, which resulted in a substantial reduction of endogenous SOX2 protein levels in CLU-OE CD44+ cells (Figure 8I and S9Bi-ii). Similarly, the KRX-0401 treatment showed effective inhibition in orosphere formation (Figure S9Ci-ii), indicating that CLU primarily promotes orosphere formation through AKT-dependent pathways in CD44+ cells. More importantly, SOX2 KD showed effective inhibition of cell viability (Figure 8I). Furthermore, we assessed the effect of SOX2 inhibition on apoptosis rate and found that SOX2 inhibition significantly increased apoptotic populations (ANXA5+ PI− and ANXA5+ PI+) in cisplatin-treated and untreated CLU-OE CD44+ cells (Figure 8Ji-ii). SOX2 KD also exaggerated apoptosis upon cisplatin exposure as characterized by the higher expression of apoptosis markers such as cleaved PARP, and CYCS (pro-apoptotic) and a significant reduction in BIRC/cIAP (anti-apoptotic) proteins (Figure S9Di-v). To further confirm the apoptosis induction we performed a CASP3-CASP7 assay and detected enhanced caspase activity, suggesting the involvement of a caspase-dependent pathway (Figure S9E). Altogether, these results showed that SOX2 plays a crucial role in CLU-mediated chemoresistance.
Inhibition of CLU triggers mitochondrial superoxide to activate cell death in oral CD44+ cells
Reactive oxygen species (ROS) are essential by-products of cellular metabolism, and a low level of ROS is crucial for the maintenance of CSCs and their ability to resist therapeutic stress [23]. As cisplatin induces mitochondrial ROS in bulk tumor cells [24], we wanted to explore the possible role of CLU in managing ROS levels during cisplatin treatment in CD44+ CSCs. In this regard, we measured mitochondrial superoxide levels in both gain- and loss of function conditions of CLU. Interestingly, CLU overexpression markedly scavenged mtROS while CLU KD significantly increased mitochondrial superoxide (Figure 9A). Next, we evaluated the effect of mitophagy inhibition on mitochondrial superoxide production and found that mitophagy-compromised conditions (BafA1 treatment) markedly increased mitochondrial superoxide levels in CLU-OE CD44+ cells in response to cisplatin treatment (Figure 9B), suggesting that CLU-mediated clearance of mtROS is mitophagy dependent. Moreover, consistent with these findings, live-cell imaging analyses of the mitochondrial network with cell-permeant MitoTracker and TMRM showed a significant reduction in mitochondrial membrane potential in the presence of cisplatin in CLU-KD CD44+ cells (Figure S10Ai-ii). Further, we treated CLU-KD CD44+ cells with mitoTEMPO, a mitochondria-targeted SOD (superoxide dismutase) mimetic and found that mitoTEMPO abolished cisplatin-induced mtROS production in CLU-KD CD44+ cells (Figure 9C). Confocal-based live-cell imaging analysis using MitoTracker and mitochondrial-derived ROS (MitoSOX) showed a high production of mitochondrial superoxide in response to cisplatin treatment, which was reduced upon mitoTEMPO treatment (Figure S10Bi-ii), suggesting that mtROS acts as the precursor for inducing cell death in CLU-KD CD44+ cells.
Figure 9.
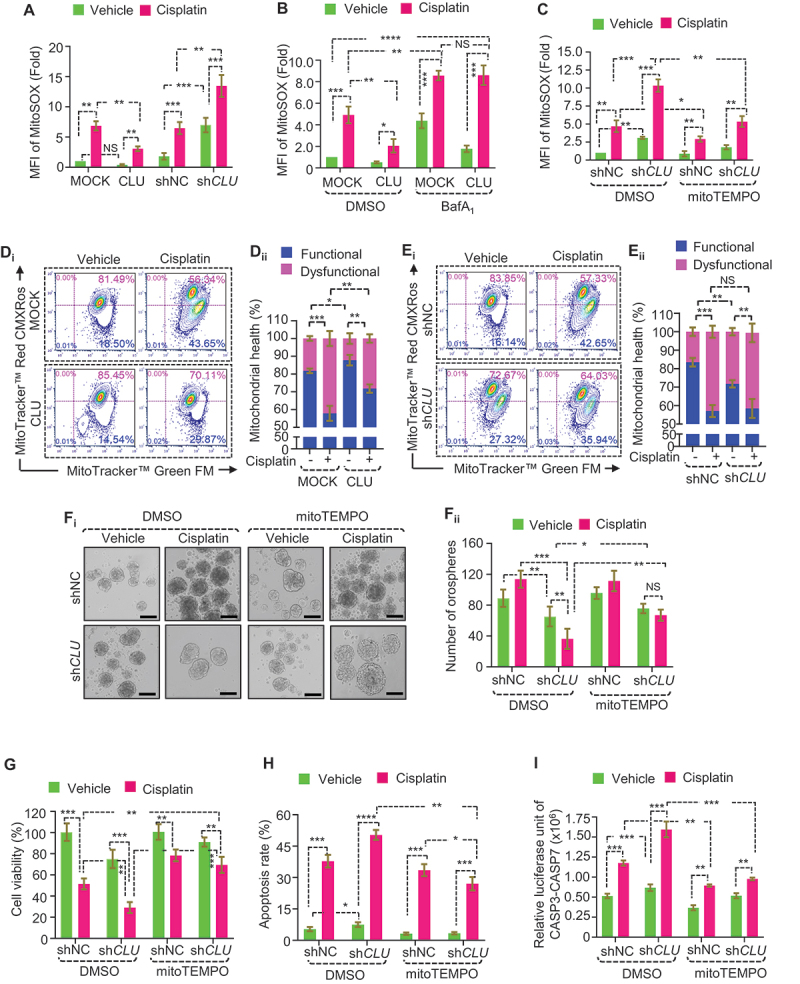
Mitochondrial ROS production through inhibition of CLU suppresses stemness and triggers cisplatin sensitivity in oral CD44+ cells. (A-C) Flow cytometry analysis for quantifying the mitochondrial superoxide levels, as indicated by MitoSOX fluorescence in (A) CD44+ cells with gain and loss of function of CLU followed by cisplatin treatment (mean ± SD, n = 3). (B) MOCK or CLU overexpressed CD44+ cells treated with cisplatin in combination with BafA1 (mitophagy inhibitor) (mean ± SD, n = 3). (C) shNC or shCLU CD44+ cells treated with cisplatin in combination with mitoTEMPO (mtROS mimetic) (mean ± SD, n = 3). (D, E) Flow cytometry analysis for estimating mitochondrial health status using MitoTracker Green (FL1-A) and MitoTracker Red (FL2-A) in (Di) MOCK or CLU-OE CD44+ cells or (Ei) shNC or shCLU CD44+ cells after cisplatin treatment (10 µM, 24 h conditions were used for all cisplatin treatments). Quantifications of functional and dysfunctional mitochondria in (Dii) MOCK or CLU-OE and (Eii) shNC or shCLU CD44+ cells after cisplatin treatment (mean ± SD, n = 3). (Fi) Representative images of orospheres formed after co-treatment with cisplatin and mitoTEMPO (20 nM, 3 h conditions were used for all mitoTEMPO treatments) in shNC or shCLU CD44+ cells, followed by culturing for 7 days. Scale bar: 200 μm. (Fii) Quantifications of orosphere numbers (1000 cells/well) formed after transfection and treatment and cultured for the next 7 days (mean ± SD, n = 3). (G) Cell viability assay to the estimated growth rate in shNC or shCLU CD44+ cells treated with cisplatin in combination with mitoTEMPO. Data were normalized to the untreated shNC CD44+ cells (mean ± SD, n = 3). (H) Quantifications of apoptotic populations (ANXA5+ PI− and ANXA5+ PI+) in shNC or shCLU CD44+ cells in response to treatments (mean ± SD, n = 3). (I) CASP3-CASP7 dual luciferase assay estimates the relative expression of caspases in shNC or shCLU CD44+ cells in response to cisplatin treatment combined with mitoTEMPO. Error bars: (mean ± SD, n = 3) **p < 0.05, **p < 0.01, and ***p < 0.001.
As clearance of damaged mitochondria is crucial for ROS management, we next assessed how CLU affects mitochondrial health in CD44+ cells in response to cisplatin treatment. In this setting, two mitochondria-specific dyes, MitoTracker Red CMXRos (MTR) and MTG were used to monitor functional and total mitochondria levels, respectively [25]. Interestingly, CLU expression led to the clearance of dysfunctional mitochondria as indicated by the decrease in the functional (MTG-positive, MTR-positive) and increased dysfunctional (MTG-positive, MTR-negative) mitochondria (Figure 9Di-ii) in response to cisplatin treatment. However, CLU KD led to an increase in the pool of cells with dysfunctional mitochondria relative to shNC CD44+ cells as indicated by the decrease in the functional (MTG-positive, MTR-positive) and increased dysfunctional (MTG-positive, MTR-negative) mitochondria (Figure 9Ei-ii), suggesting that accumulation of damaged mitochondria is responsible for mtROS production. Next, to understand how mtROS induced by CLU KD sensitizes oral CD44+ cells to cisplatin treatment, we treated CLU-KD CD44+ cells with mitoTEMPO (50 nM, 3 h) followed by cisplatin treatment (10 µM, 24 h) and found that the growth inhibitory effect of loss of function of CLU in response to cisplatin treatment was significantly rescued after mitoTEMPO treatment as indicated by orosphere number (Figure 9Fi-ii), and cell viability (Figure 9G). Moreover, mitoTEMPO treatment significantly reduced apoptotic populations (ANXA5+ PI− and ANXA5+ PI+) in response to cisplatin treatment (Figure 9H and S10C). Similarly, cleaved CASP3-CASP7 expression was also markedly reduced in cotreatment groups (Figure 9I). Collectively, our data suggest CLU is critical for maintaining the low mitochondrial ROS status in oral CD44+ cells, which promotes stemness in oral cancer.
Discussion
Mitophagy is essential for the clearance of damaged mitochondria that would otherwise injure the cell, promoting CSC survival in response to chemotherapeutic stress [26,27]. Dysregulation in mitochondrial dynamics and mitophagy can severely affect cancer stemness and therapy resistance [28]. Extensive studies on mitophagy in the context of tumorigenesis highlights a list of mitophagy receptors involved in autophagy regulation (such as BNIP3L/NIX [27], FUNDC1, HIF1A/HIF1α) exclusively required for successful mitophagy [29]. More importantly, many oncogenic signaling pathways activate mitophagy through these regulator proteins promoting cancer cell survival [30]. The ER and Golgi are the primary sites of CLU synthesis outside of the secretory pathway [14]. CLU can increase stress tolerance to anticancer treatment by encouraging autophagosome formation, where it acts as a scaffold for LC3-I and ATG3, priming PE conjugation to occur, and the formation of LC3-II and autophagosomes. In addition, the α-chain of CLU contains five LIR-like sequences (at positions 235, 341, 350, 366, and 383 aa), with the 341YNEL region serving as the most efficient site for the interaction with LC3 to promote autophagy [17]. In this connection, a previous study showed that CLU regulates cancer cell survival through activating AKT-GSK3B-CTNNB1/β-catenin signaling to promote the growth of liver CSCs [31]. Our results suggest that CLU promotes pro-survival mitophagy, and inhibition of CLU and CLU-mediated mitophagy reduces self-renewal, potently slows cell proliferation and clonogenicity and improves sensitization toward cisplatin treatment in oral CSCs (Figure 10).
Figure 10.

Proposed mechanism by which CLU promotes mitophagic degradation of MSX2 in oral CD44+ cells. CLU facilitates AKT-dependent phosphorylation of DNM1L at S616 triggering mitochondrial fission. Inhibition of CLU-mediated mitophagy suppresses MSX2 turnover, leading to suppression of SOX2-mediated stemness and generating excessive mitochondrial superoxide, sensitizing oral CSCs toward cisplatin-mediated cell death.
As noted above, mitochondrial fission may be important in efficiently segregating damaged parts of the mitochondria from the active mitochondrial network [32,33]. Our data suggest that cisplatin-induced CLU triggers mitochondrial fission by activating DNM1L, a well-established regulator of mitochondrial fission [34]. In support of our findings, a previous report indicated that CLU inhibition also alters mitochondrial dynamics in breast CSCs, increasing camptothecin sensitivity through necrosis activation [35]. DNM1L primarily resides in the cytoplasm and is translocated to the mitochondrial outer membrane once activated (by phosphorylation at serine 616) to trigger mitochondrial fission by constricting the mitochondrial membrane using its GTPase activity [25]. Our results showed that CLU promotes phosphorylation of AKT, a serine/threonine-protein kinase, which phosphorylates different downstream substrates critically crucial for cell survival and proliferation [36]. Moreover, AKT inhibition reduces the phosphorylation status of DNM1L and inhibits the shuttling of p-DNM1L (Ser616) from the cytoplasm to mitochondria in CLU-OE CD44+ cells. These results agree with the previous findings, where AKT-mediated phosphorylation of DNM1L promotes neuronal apoptosis through excessive mitochondrial fission [37].
Mitophagic degradation of tumor suppressor proteins is associated with cancer growth and progression. For example, mitophagy controls the proteolytic turnover of TP53/p53 through PINK1-mediated phosphorylation promoting NANOG-mediated stemness in hepatic CSCs, which is abolished during mitophagy inhibition [38]. Our data showed that CLU promotes proteolytic turnover of MSX2 to maintain SOX2 activity in oral CD44+ cells. MSX2 is one of the homeobox transcription factors of the msh family, which represses SOX2 transcription through direct association with its promoter [39,40]. More importantly, mitophagy-compromised conditions cause MSX2 accumulation in the nuclear fractions to repress SOX2 expression, severely affecting stemness. SOX2 regulates stemness and therapeutic resistance in different cancer types, where downregulation of SOX2 diminishes CSC properties and radio-chemoresistance [41,42]. Interestingly, we also noticed that CLU-induced AKT enhances SOX2 levels, and SOX2 KD through shSOX2 and KRX-0401 treatment significantly reduces the CSC populations and increases apoptosis in CLU-OE CD44+ cells. In this connection, a recent study showed that autophagic degradation of FOXO3 promotes self-renewal potential in CSCs through transcriptional activation of SOX2 in head and neck cancer [43].
Previous studies reported that mitophagy reduces mtROS levels, essential for stemness maintenance and their ability to resist therapy. For example, PRDX2 (peroxiredoxin 2) is a redox regulatory protein that promotes stemness by inhibiting ROS levels in hepatocellular carcinoma cells [44]. In another study, PHB (prohibitin) maintains a low level of mtROS by stabilizing PRDX3 to maintain stemness in glioblastoma stem cells, determining therapeutic outcomes [23]. In this setting, CLU maintains a low level of mtROS through mitophagy. CLU KD leads to the accumulation of damaged mitochondria, which significantly increases mtROS, accelerating cell death in response to cisplatin treatment. Similarly, a previous report also suggested that successful inhibition of SOD2 and CAT (catalase) ROS scavenging proteins increases the ROS level leading to higher chemosensitivity against cisplatin treatment [45]. These results conclusively summarize the importance of CLU in the maintenance of SOX2-mediated stemness and growth of oral CSCs through controlling the activities of MSX2 (Figure 10) and explain why targeting CLU is more effective and promising than conventional chemotherapy.
Conclusion
In summary, findings from this investigation provide evidence that CLU acts as a positive regulator of oral CSCs by activating mitophagy in response to cisplatin treatment. More importantly, CLU-mediated mitophagy degrades the MSX2 protein, enhancing SOX2-dependent chemoresistance. However, CLU KD reverses the mitophagic cytoprotection causing mtROS-induced apoptosis in oral CSCs. While our current findings suggest that activation of CLU-mediated mitophagy directly regulates MSX2 degradation, further investigation is required to identify the undisclosed target(s) involved in the selective elimination of MSX2 after mitophagy activation. Moreover, the precise mechanism behind the mitophagy-dependent regulation of CSCs in vivo could provide better treatment outcomes in oral cancers.
Materials and methods
Cell culture
FaDu was obtained from American Type Culture Collection (ATCC; HTB-43, RRID: CVCL_1218). CAL-33 (DSMZ, ACC-447, RRID: CVCL_1108) was generously provided by Dr Goutam Sethi (National University of Singapore). These cell lines were cultured in their respective media (FaDu: Minimum Essential Medium [MEM; Himedia, AL047]; CAL-33: Dulbecco’s Modified Eagle’s Medium [DMEM; Himedia, AL151A]) using standard cell culture techniques. FBS (10%; Gibco, 10,270,106) and antibiotic-antimycotic solution (Himedia, A002A) were added to each media before culturing the cells.
Reagents and antibodies
The bafilomycin A1 (BafA1; Calbiochem, 196,000), chloroquine (CQ; Sigma-Aldrich, C6628), cisplatin (cis-diammineplatinum [II] dichloride; Sigma-Aldrich, P4394), cycloheximide (Calbiochem®, 239,765), Mdivi-1 (Sigma-Aldrich, M0199), mitoTEMPO (Sigma-Aldrich, SML0737), NAC (N-acetyl-L-cysteine; Sigma-Aldrich, 1,009,005), and KRX-0401/Perifosine (Sigma-Aldrich, SML0612) are commercially available.
The ACTB/β-Actin (BD Biosciences, 612656; 1:10,000), ATG5 (Cell Signaling Technology, 2630S;1:1,000), BCL2 (Santa Cruz Biotechnology, sc-7382; 1:1,000), BIRC/cIAP1 (Cell Signaling Technology, 7065S; 1:1,000), LC3 (Cell Signaling Technology, 83506S; 1:1000 for WB and 1:500 for IF), LC3 (Sigma-Aldrich, L7543; 1:500 for IF), SQSTM1/p62 (Cell Signaling Technology, 88588S; 1:1,000), AKT (pan; Cell Signaling Technology, 4691S; 1:2,000), BAX (Santa Cruz Biotechnology, sc-20,067; 1:500), CD44 (ABclonal, A19020; 1:2,000), Clusterin-α (Santa Cruz Biotechnology, sc-6419; 1:1,000), CLU/clusterin (Santa Cruz Biotechnology, sc-166907; 1:1,000), COX4/COXIV (Cell Signaling Technology, 4844S; 1:6,000), CYCS/cytochrome C (BD Biosciences, 556433; 1:1,000), DNM1L/DRP1 (Santa Cruz Biotechnology, sc-271583; 1:1000 for WB and 1:250 for IF), GAPDH (Abgenex India Pvt. Ltd., 10–10011; 1:10,000), LMNB2 (Invitrogen, 33–2100; 1:2000), MSX2 (Santa Cruz Biotechnology, sc-393986; 1:1000 for WB and 1:250 for IF), MCL1 (Santa Cruz Biotechnology, sc-12756;1:1000), NANOG (BD Biosciences, 560109; 1:1000), PARP (Cell Signaling Technology, 9542S; 1:1000), p-AKT (Ser473; Cell Signaling Technology, 4060S; 1:1,000), p-DNM1L/DRP1 (Ser616; Cell Signaling Technology, 3455S; 1:1000 for WB and 1:500 for IF), PU05F1/Oct3/4A (BD Biosciences, 561555; 1:1000), SOX2 (BD Biosciences, 561469; 1:1000 for WB and 1:500 for IF), TOMM20 (BD Biosciences, 612278; 1:6000 for WB and 1:3000 for IF), anti-mouse IgG Alexa Fluor 568 (Thermo Fisher Scientific, A-11004; 1:500), anti-rabbit IgG Alexa Fluor 568 (Thermo Fisher Scientific, A-11011; 1:500), anti-rabbit IgG Alexa Fluor 488 (Thermo Fisher Scientific, A-11008; 1:500), anti-mouse IgG Alexa Fluor 488 (Thermo Fisher Scientific, A-11001; 1:500), anti-mouse IgG Alexa Fluor 647 (Abcam, ab150115; 1:500), and anti-rabbit IgG Alexa Fluor 700 (Thermo Fisher Scientific, A-21038; 1:500) antibodies are commercially available.
MACS-based separation of CD44+ cells
CD44, the most useful marker for identifying oral CSCs [46], was used to isolate the CSCs from both FaDu, and CAL-33 cells using the CD44 MicroBeads (Miltenyi Biotec, 130–095-194), LS column (Miltenyi Biotec, 130–042-401), and a MidiMACS™ Separator (Miltenyi Biotec, 130–042-302). Briefly, FaDu and CAL-33 cells (1x107 cells/mL) were incubated with 20 µl of the anti-human CD44 monoclonal antibody-conjugated magnetic microbeads for 30 min in the dark at 4°C. Cells were then resuspended in 1 ml of buffer (PBS [Himedia, TL1101] supplemented with 0.5% FBS) and centrifuged for 5 min at 300xg, followed by resuspension in 500 µl of the buffer. For separation, after placing the LS column in the MidiMACS™ Separator, 3 ml of buffer followed by cell suspension was passed through it. The unlabeled cells were collected and combined with the cell suspension to pass through the column. After the column reservoir was empty, the column was washed with buffer (3x3 mL). The unlabeled washout cells were collected and considered CD44− cells. Next, the column was removed from the separator and placed on a 15-ml collection tube, followed by placing 5 mL of resuspension buffer into the column reservoir. The magnetically labeled CD44+ cells were immediately flushed out and collected. The purified CD44+ and CD44− cells were expanded for the next 45 days, similar to previously described culture methods [47], and the purity was checked at regular intervals through flow cytometric analysis using PE Mouse Anti-Human CD44 (BD Bioscience, 550989) (Figure S1A). This enrichment method allowed us to obtain a sufficient number of CD44+ CSCs for the experiment.
Overexpression and knockdown experiments
For overexpression studies, CD44− and CD44+ cells (70% confluency) were transfected with the indicated constructs corresponding to the empty vector (MOCK) or encoding CLU, (pBABE-DNM1LWT, and pBABE-DNM1LS616A were kindly provided by Dr Jerry Edward Chipuk [Icahn School of Medicine at Mount Sinai]). Similarly, for knockdown studies, cells (70% confluency) were transfected with siRNA specifically targeting human ATG5 (Santa Cruz Biotechnology, sc-41445), or siRNA controls (Santa Cruz Biotechnology, sc-37007), using Lipofectamine® 3000 reagent (ThermoFisher Scientific, L3000015) following the manufacturer’s instruction.
Lentivirus-mediated knockdown and overexpression studies for stable expression
The lentiviral construct carrying pLenti6.3/V5-DEST-GFP (empty vector; Addgene, 40125; deposited by Lynda Chin), wild-type MSX2 (DNASU, HsCD00852264), MSX2-mLIR (generated using QuikChange II XL Site-Directed Mutagenesis Kit [Agilent, 200521]), and lentiviral shRNAs such as shCLU (Sigma-Aldrich, SHCLND; targeting CLU), shAKT1 (Sigma-Aldrich, TRCN0000039797), shDNM1L (Sigma-Aldrich, TRCN0000318424), shMSX2 (Sigma-Aldrich, TRCN0000234848), shSOX2 (Addgene, 26353; deposited by Matthew Meyerson, targeting SOX2) and pLKO-control shRNA (Addgene, 1864; deposited by Dr David M. Sabatini) were used along with the lentiviral packaging psPAX2 (Addgene, 12260; deposited by Dr Didier Trono) and envelop vectors pMD2G (Addgene, 12259; deposited by Dr Didier Trono). In brief, lentiviruses were packaged in HEK293 FT cells by transfecting constructs with psPAX2 and pMD2G plasmid using Lipofectamine® 3000 reagent (ThermoFisher Scientific, L3000015) following the manufacturer’s instruction. Next, lentiviral supernatants were harvested after 48- and 72-h transfection using a 0.45-µm syringe filter and mixed with fresh media (1:1) and polybrene (8 µg/ml; Sigma Aldrich, 107689) and subsequently used for transduction of CD44+ cells. Finally, the transduced cells were grown in the presence of puromycin (500 ng/ml; Sigma Aldrich, P8833) and blasticidin (5 µg/ml; Sisco Research Laboratories Pvt. Ltd., 92370) and screened for further studies.
Orsophere-formation assay
For orospheres formation, two oral cancer cell lines, FaDu and CAL-33 (2000 cells/well) were transfected with the gene of interest and cultured in ultra-low attachment 6-well plates (Corning®, CLS3473). The cells were then grown in stem-cell media (serum-free media + B27 [1:50; Gibco,17504044] + N2 supplement [1%; Gibco, 17502048] + human FGF2 [10 ng/ml; Gibco, PHG0024] + human EGF [20 ng/ml; Gibco, PHG0311]) formulated as previously described [48] for 7 days. The number of orospheres in each well was counted by microscopy. The orosphere images were captured, followed by their size measurement through the use of cell Sens imaging software of an Olympus IX71 fluorescence inverted microscope with a 20X objective (bright-field mode).
Western blot analysis
After treatment, cells were harvested and mixed with cell lysis buffer (Cell Signaling Technology, 9803), supplemented with phosphatase (Sigma-Aldrich, P0044) and protease (Sigma-Aldrich, P8340) inhibitors and ruptured through vigorous mixing for 2 h over ice. Then the whole content was subjected to centrifugation (12,000 x g) for 10 min and the supernatant was collected. The Bio-Rad Protein Assay kit (Bio-Rad, 5000006) was used to quantify the protein concentration in the collected supernatant. The whole-cell lysates (35 μg of protein) were mixed with 4x Laemmli buffer (Bio-Rad, 1610747) supplemented with β-mercaptoethanol [0.1%; HIMEDIA, RM2895] (v:v) and boiled for 5 min. SDS-PAGE was used to separate the proteins, then transferred onto nitrocellulose membranes (PALL Life Sciences, 66485) at 100 V for 2 h. Then nitrocellulose membrane was incubated with nonfat milk (5%; HIMEDIA, M530; w:v) for membrane blocking (1 h; RT) with continuous shaking with subsequent probing at 4°C with the respective primary antibodies. Following overnight incubation, the membranes were washed with PBST (PBS supplemented with TWEEN 20 [0.001%; Sigma-Aldrich, P9416; v:v]; 3 × 10 min) followed by probing with HRP-conjugated secondary antibodies (2 h in the dark; RT). Finally, the protein bands were detected using an ECL detection reagent (Bio-Rad, 1705062).
Subcellular fractionation
Cells were lysed using a Cell Fractionation Kit (Abcam, ab109719), following all the instructions given in the kit. In brief, cells were first trypsinized and pelleted down by centrifugation at 300 x g for 5 min. Then cells were resuspended in an equal volume of Buffer A and Buffer B and incubated for 7 min at RT with constant mixing, followed by sequential centrifugation at 5000 x g and 10,000 x g for 1 min each at 4°C to obtain the cytosolic fraction. For the mitochondrial fraction, the cytosolic pellet was again resuspended in Buffer C to the equal volume as Buffer A, followed by constant mixing for 10 min at RT. The pellets were then sequentially centrifuged at 5000 x g and 10,000 x g for 1 min each at 4°C to obtain the mitochondrial fraction. The respective fractions were then used for western blot analysis with GAPDH as a cytosolic and COX4 as a mitochondrial purity marker.
Confocal microscopy-based colocalization analysis
For colocalization studies, cells were first fixed with formaldehyde (Sigma-Aldrich, 252,549; 4% v:v; 15 min at room temperature [RT]), and then blocked and permeabilized using BSA (5% w:v; HIMEDIA, MB083) + Triton X-100 (0.03% v:v; Sigma-Aldrich, X100) for 1 h. Then cells were probed with respective primary antibodies prepared in BSA (1%, w:v) + Triton X-100 (0.03%, v:v). Following overnight incubation at 4°C, cells were rinsed with 1X PBS (5 x 5 min) and reprobed with specific secondary antibodies for 6 h with constant shaking in the dark. Last, cells were counterstained with DAPI for 5 min followed by PBS rinsing (5 x 5 min) and imaged immediately. For live-cell colocalization studies, cells were marked with MitoTracker Green (50 nM; Invitrogen, M7514) and LysoTracker Red (50 nM; Invitrogen, L7528), followed by 30 min incubation (37°C; 5% CO2) and counterstained with Hoechst 33342 solution (1 µg/ml; Sigma-Aldrich, B2261) for 5 min. Then cells were rinsed with PBS (5 x 5 min) and immediately imaged. For mitochondrial fission assessment, cells were tagged with MitoTracker Red (50 nM; Invitrogen, M7512) for 30 min (37°C; 5% CO2) and counterstained with Hoechst 33342 solution for 5 min to delineate the nuclei. All images were analyzed through ImageJ software using a recommended plugin tool. The Mitochondrial Network Analysis/MiNA toolset (additional plugin of ImageJ) was used to analyze average mitochondrial fragment length per cell and categorized into three subgroups (fragmented [<2.5 µm], intermediate [2.5–4.5 µm], and elongated [>4.5 µm]). All the images were randomly taken for each sample using a 63X objective lens of a Leica confocal microscope (Leica Microsystems; Wetzlar, Germany).
Immunoprecipitation assay
For the immunoprecipitation assay, FaDu-MOCK and FaDu-CLU CD44+ cells were treated with CQ (Sigma-Aldrich, C6628), harvested and treated per the instructions given in the Dynabeads™ Protein A Immunoprecipitation Kit (Invitrogen™, 10006D). In brief, 25 µg/ml of LC3B antibody in PBS were incubated for 30 min with Dynabeads™ Protein A with constant shaking. Then, protein A-antibody complexes were incubated with whole cell lysate (350 µg of protein) overnight at 4°C. Samples were then washed three times using washing buffer followed by sample elution using elution buffer (20 µl) supplied with the kit. Next, the eluted samples were mixed with 4x Laemmli sample buffer, boiled for 10 min at 70°C for denaturation, and subjected to western blot analysis. For negative controls, 350 µg of protein was mixed with normal IgG.
Estimation of mitochondrial health
For estimating mitochondrial health, cells were labeled with two mitochondrial-specific dyes, MitoTracker Green (100 nM) and MitoTracker Red CMXRos (100 nM) and incubated for 30 min at 37°C in the dark. Similarly, after treatment, cells were stained with tetramethylrhodamine methyl ester perchlorate (TMRM, 50 nm; Tokyo Chemical Industry Co. Ltd., T3608) for 25 min at 37°C to measure mitochondrial membrane potential. After incubation, cells were harvested and resuspended in PBS solution (1% FBS) for flow cytometry analysis. The mean fluorescence intensity of the respective dye was measured using the flow cytometer (BD Accurri C6 Software (RRID: SCR_001456).
Estimation of intracellular and mitochondrial ROS generation
Cells were harvested after drug treatments and resuspended with 100 μl of PBS containing 2′,7′-dichlorofluorescin diacetate (10 µM; Sigma-Aldrich, D6283), followed by incubation (37°C, 30 min) in the dark for monitoring intracellular ROS generation. Similarly, DHE (1 μM; Sigma-Aldrich, 37291) and MitoSOX Red (5 μM; Invitrogen, M36008) were used for monitoring extra-mitochondrial and intra-mitochondrial superoxide production, respectively. After adding the respective fluorescent dye, cells were incubated for 30 min at 37°C in the dark, followed by washing (PBS) and data acquisition. The amount of both intracellular and mitochondrial ROS generation was determined using a flow cytometer and FCS Express™ software 7 (De Novo Software, Glendale, CA, USA).
Apoptosis estimation using ANXA5/annexin V-propidium iodide (PI) staining
For apoptosis analysis, cells were harvested after treatments and resuspended in 1 ml of pre-cooled PBS followed by centrifugation at (300 x g) for 5 min. The pellet was then resuspended in 100 µl of 1X binding buffer and incubated for 10 min at RT. Next, the pellet was washed twice with 1 ml of pre-cooled PBS (1X) and centrifuged at 300 x g for 5 min. Then cells were again resuspended with 100 µl of ice-cold 1X binding buffer, followed by the addition of ANXA5 (ImmunoTools, 31490013X2) and PI (Sigma-Aldrich, P4170) (4 µl each) and incubated for 20 min in the dark. Finally, 100 µl of ice-cold PBS was added to the mixture and data were acquired through a flow cytometer using the FL2 channel (PI) and FL1 channel (ANXA5-FITC).
Measurement of caspase activity
For measuring caspase activity, the Caspase-Glo® 3/7 Assay kit (Promega Corp, G8090) was used according to the manufacturer’s protocol. In brief, after treatment, cells were harvested and lysed similar to the western blot protocol, and 25 µg of whole cell lysate was mixed with Caspase-Glo 3/7 reagent (1:1, v:v). The mixture was then gently mixed and incubated for 2 h at RT while being mixed in the dark and then subjected to measurement of the luminescence using a GloMax® 20/20 Luminometer (Promega Corp, Fitchburg, WI, USA). The estimated values for CASP3-CASP7 activity were represented as a mean of relative light units.
Monitoring cell viability using the MTT assay
Approximately 3000 cells/well were cultured in 96-well plates and placed in a humidified CO2 incubator (37°C; 5% CO2) overnight, followed by respective treatments for the desired time points. At specific time points, MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide; 5 mg/ml, 20 µl; Sigma-Aldrich, M2128) was added to each well and further incubated in a CO2 incubator for an additional 2 h. After incubation, the media was cautiously decanted from each well, followed by the addition of DMSO (200 µl; Sigma-Aldrich, D8418) to solubilize formazan crystals and further incubated for an additional 20 min in the dark at RT followed by an absorbance reading at 595 nm.
Clonogenicity assay
Approximately 25,000 cells/well were cultured in 12-well plates and placed in a humidified CO2 incubator (37°C; 5% CO2) overnight, followed by respective treatments for the required time points. Then, cells were harvested, and approximately 1000 cells/well were again seeded in another 6-well plate for the next 10 days. The cells were then exposed to paraformaldehyde (4%) for fixation (30 min, at RT), followed by crystal violet (HIMEDIA, GRM961; 0.1%) staining (30 min, at RT) and rinsing with tap water. The stained colonies were imaged through an IX71 inverted microscope (Olympus, Japan) and counted manually.
Statistical analysis
GraphPad Prism 9 software analyzed all the data generated from experiments performed in triplicate (independently). All confocal microscopy experiments were performed in triplicate, and, for each experiment, ten images with multiple cells were randomly selected for analysis. A two-tailed paired t-test was performed to compare two groups, and for more than two groups, one-way ANOVA (followed by Bonferroni posttest analysis) was performed. All the data are represented as mean ± S.D. (standard deviation) with significance levels denoted as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Supplementary Material
Acknowledgments
The authors are thankful to members of the CCD lab for the critical reading of the manuscript.
Funding Statement
This work was supported by the Department of Biotechnology, Government of India [BT/PR23304/MED/30/1823/2017]; and the NIH [GM131919].
Abbreviations
ACTB: actin beta; BafA1: bafilomycin A1; BIRC: baculoviral IAP repeat-containing protein; CSC: cancer stem cell; CLU: clusterin; CQ: chloroquine; COX4: cytochrome c oxidase subunit 4; CHX: cycloheximide; CYCS: cytochrome c, somatic; DMSO: dimethyl sulfoxide; DNM1L: dynamin 1 like; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; KD: knockdown; LMNB2: lamin B2; LIR: LC3-interacting region; LTR: LysoTracker Red; MTG: MitoTracker green; MTDR: MitoTrackerTM Deep Red; MSX2: msh homeobox 2; NAC: N-acetyl-L-cysteine; OE: overexpression; SOX2: SRY-box transcription factor 2; TMRM: tetramethylrhodamine methyl ester perchlorate; TOMM20: translocase of outer mitochondrial membrane 20.
Disclosure statement
The authors disclose that the research was conducted without any commercial or financial conflict of interest.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2178876
References
- [1].Siegel RL, Miller KD, Fuchs HE, et al. Cancer Statistics, 2021. CA Cancer J Clin. 2021. Jan;71(1):7–33. [DOI] [PubMed] [Google Scholar]
- [2].Coletta RD, Yeudall WA, Salo T.. Grand challenges in oral cancers [Specialty Grand Challenge]. Frontiers in Oral Health. 2020. June 9;1(3). DOI: 10.3389/froh.2020.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wang L, Liu X, Ren Y, et al. Cisplatin-enriching cancer stem cells confer multidrug resistance in non-small cell lung cancer via enhancing TRIB1/HDAC activity. Cell Death Dis. 2017. Apr 13;8(4):e2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nör C, Zhang Z, Warner KA, et al. Cisplatin induces Bmi-1 and enhances the stem cell fraction in head and neck cancer. Neoplasia (New York, NY). 2014. Feb;16(2):137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jiang P, Xu C, Zhou M, et al. RXRα-enriched cancer stem cell-like properties triggered by CDDP in head and neck squamous cell carcinoma (HNSCC). Carcinogenesis. 2018. Feb 9;39(2):252–262. [DOI] [PubMed] [Google Scholar]
- [6].Naik PP, Das DN, Panda PK, et al. Implications of cancer stem cells in developing therapeutic resistance in oral cancer. Oral Oncol. 2016. Nov;62:122–135. [DOI] [PubMed] [Google Scholar]
- [7].MacDonagh L, Gray SG, Breen E, et al. BBI608 inhibits cancer stemness and reverses cisplatin resistance in NSCLC. Cancer Lett. 2018. Aug 1;428:117–126. [DOI] [PubMed] [Google Scholar]
- [8].Praharaj PP, Panigrahi DP, Bhol CS, et al. Mitochondrial rewiring through mitophagy and mitochondrial biogenesis in cancer stem cells: a potential target for anti-CSC cancer therapy. Cancer Lett. 2021. Feb;1(498):217–228. [DOI] [PubMed] [Google Scholar]
- [9].Praharaj PP, Naik PP, Panigrahi DP, et al. Intricate role of mitochondrial lipid in mitophagy and mitochondrial apoptosis: its implication in cancer therapeutics. Cell Mol Life Sci. 2019. May;76(9):1641–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Panigrahi DP, Praharaj PP, Bhol CS, et al. The emerging, multifaceted role of mitophagy in cancer and cancer therapeutics. Semin Cancer Biol. 2020. Nov;66:45–58. [DOI] [PubMed] [Google Scholar]
- [11].Pei S, Minhajuddin M, Adane B, et al. AMPK/FIS1-mediated mitophagy is required for self-renewal of human AML stem cells. Cell Stem Cell. 2018. Jul 5;23(1):86–100.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Villa E, Proïcs E, Rubio-Patiño C, et al. Parkin-independent mitophagy controls chemotherapeutic response in cancer cells. Cell Rep. 2017. Sep 19;20(12):2846–2859. [DOI] [PubMed] [Google Scholar]
- [13].Yao N, Wang C, Hu N, et al. Inhibition of PINK1/Parkin-dependent mitophagy sensitizes multidrug-resistant cancer cells to B5G1, a new betulinic acid analog. Cell Death Dis. 2019. March 8;10(3):232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Satapathy S, Wilson MR. The dual roles of clusterin in extracellular and intracellular proteostasis. Trends Biochem Sci. 2021. Aug;46(8):652–660. [DOI] [PubMed] [Google Scholar]
- [15].Praharaj PP, Patra S, Panigrahi DP, et al. Clusterin as modulator of carcinogenesis: a potential avenue for targeted cancer therapy. Biochim Biophys Acta. 2021. Apr;1875(2):188500. [DOI] [PubMed] [Google Scholar]
- [16].Kadam R, Harish M, Dalvi K, et al. Novel nucleolar localization of clusterin and its associated functions in human oral cancers: an in vitro and in silico analysis. Cell Biochem Funct. 2021. Apr;39(3):380–391. [DOI] [PubMed] [Google Scholar]
- [17].Zhang F, Kumano M, Beraldi E, et al. Clusterin facilitates stress-induced lipidation of LC3 and autophagosome biogenesis to enhance cancer cell survival. Nat Commun. 2014. Dec 12;5:5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Naik PP, Mukhopadhyay S, Praharaj PP, et al. Secretory clusterin promotes oral cancer cell survival via inhibiting apoptosis by activation of autophagy in AMPK/mTOR/ULK1 dependent pathway. Life Sci. 2020;5:118722. [DOI] [PubMed] [Google Scholar]
- [19].Dikic I. Proteasomal and Autophagic Degradation Systems. Annu Rev Biochem. 2017. Jun;20(86):193–224. [DOI] [PubMed] [Google Scholar]
- [20].Wu X, Luo Q, Liu Z. Ubiquitination and deubiquitination of MCL1 in cancer: deciphering chemoresistance mechanisms and providing potential therapeutic options. Cell Death Dis. 2020. Jul 22;11(7):556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jacomin AC, Samavedam S, Promponas V, et al. iLIR database: a web resource for LIR motif-containing proteins in eukaryotes. Autophagy. 2016. Oct 2;12(10):1945–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Johansen T, Lamark T. Selective autophagy: ATG8 family proteins, lir motifs and cargo receptors. J Mol Biol. 2020. Jan 3;432(1):80–103. [DOI] [PubMed] [Google Scholar]
- [23].Huang H, Zhang S, Li Y, et al. Suppression of mitochondrial ROS by prohibitin drives glioblastoma progression and therapeutic resistance. Nat Commun. 2021. Jun 17;12(1):3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kleih M, Böpple K, Dong M, et al. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis. 2019. Nov 7;10(11):851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chen H, Chan DC. Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab. 2017. Jul 5;26(1):39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Li Y, Li Y, Yin J, et al. A mitophagy inhibitor targeting p62 attenuates the leukemia-initiation potential of acute myeloid leukemia cells. Cancer Lett. 2021. Jul 10;510:24–36. [DOI] [PubMed] [Google Scholar]
- [27].Jung J, Zhang Y, Celiku O, et al. Mitochondrial NIX promotes tumor survival in the hypoxic niche of glioblastoma. Cancer Res. 2019. Oct 15;79(20):5218–5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Praharaj PP, Patro BS, Bhutia SK. Dysregulation of mitophagy and mitochondrial homeostasis in cancer stem cells: novel mechanism for anti-cancer stem cell-targeted cancer therapy. Br J Pharmacol. 2021 Feb 1;179(22):5015–5035. [DOI] [PubMed] [Google Scholar]
- [29].Poole LP, Macleod KF. Mitophagy in tumorigenesis and metastasis. Cell Mol Life Sci. 2021. Apr;78(8):3817–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Humpton TJ, Alagesan B, DeNicola GM, et al. Oncogenic KRAS induces NIX-mediated mitophagy to promote pancreatic cancer. Cancer Discov. 2019. 9;Sep(9):1268–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zheng W, Yao M, Wu M, et al. Secretory clusterin promotes hepatocellular carcinoma progression by facilitating cancer stem cell properties via AKT/GSK-3β/β-catenin axis. Journal of Translational Medicine. 2020. Feb 14;18(1):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Prasad P, Ghosh S, Roy SS. Glutamine deficiency promotes stemness and chemoresistance in tumor cells through DRP1-induced mitochondrial fragmentation. Cell Mol Life Sci. 2021. May;78(10):4821–4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sessions DT, Kashatus DF. Mitochondrial dynamics in cancer stem cells. Cell Mol Life Sci. 2021. Apr;78(8):3803–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Xie Q, Wu Q, Horbinski CM, et al. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci. 2015. Apr;18(4):501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Arumugam P, Samson A, Ki J, et al. Knockdown of clusterin alters mitochondrial dynamics, facilitates necrosis in camptothecin-induced cancer stem cells. Cell Biol Toxicol. 2017. Jun;33(3):307–321. [DOI] [PubMed] [Google Scholar]
- [36].Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017. Apr 20;169(3):381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kim DI, Lee KH, Gabr AA, et al. Aβ-Induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochim Biophys Acta. 2016. Nov;1863(11):2820–2834. [DOI] [PubMed] [Google Scholar]
- [38].Liu K, Lee J, Kim JY, et al. Mitophagy controls the activities of tumor suppressor p53 to regulate hepatic cancer stem cells. Mol Cell. 2017. Oct 19;68(2):281–292.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wu Q, Zhang L, Su P, et al. MSX2 mediates entry of human pluripotent stem cells into mesendoderm by simultaneously suppressing SOX2 and activating NODAL signaling. Cell Res. 2015. Dec;25(12):1314–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yin Y, Xie CM, Li H, et al. The FBXW2-MSX2-SOX2 axis regulates stem cell property and drug resistance of cancer cells. Proc Natl Acad Sci U S A. 2019. Oct 8;116(41):20528–20538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chou MY, Hu FW, Yu CH, et al. Sox2 expression involvement in the oncogenicity and radiochemoresistance of oral cancer stem cells. Oral Oncol. 2015. Jan;51(1):31–39. [DOI] [PubMed] [Google Scholar]
- [42].Liang X, Deng M, Zhang C, et al. Combined class I histone deacetylase and mTORC1/C2 inhibition suppresses the initiation and recurrence of oral squamous cell carcinomas by repressing SOX2. Cancer Lett. 2019. Jul 10;454:108–119. [DOI] [PubMed] [Google Scholar]
- [43].Chen Y, Zhao H, Liang W, et al. Autophagy regulates the cancer stem cell phenotype of head and neck squamous cell carcinoma through the noncanonical FOXO3/SOX2 axis. Oncogene. 2022. Jan;41(5):634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Son YW, Cheon MG, Kim Y, et al. Prx2 links ROS homeostasis to stemness of cancer stem cells. Free Radic Biol Med. 2019. Apr;134:260–267. [DOI] [PubMed] [Google Scholar]
- [45].Chang CW, Chen YS, Chou SH, et al. Distinct subpopulations of head and neck cancer cells with different levels of intracellular reactive oxygen species exhibit diverse stemness, proliferation, and chemosensitivity. Cancer Res. 2014. Nov 1;74(21):6291–6305. [DOI] [PubMed] [Google Scholar]
- [46].Prince ME, Sivanandan R, Kaczorowski A, et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A. 2007. Jan 16;104(3):973–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yan C, Luo L, Guo CY, et al. Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett. 2017. Mar 1;388:34–42. [DOI] [PubMed] [Google Scholar]
- [48].Naik PP, Mukhopadhyay S, Panda PK, et al. Autophagy regulates cisplatin-induced stemness and chemoresistance via the upregulation of CD44, ABCB1 and ADAM17 in oral squamous cell carcinoma. Cell Prolif. 2018. Feb;51:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.