

Abstract
Objective
Brain organoids are miniaturized in vitro brain models generated from pluripotent stem cells, which resemble full‐sized brain more closely than conventional two‐dimensional cell cultures. Although brain organoids mimic the human brain's cell‐to‐cell network interactions, they generally fail to faithfully recapitulate cell‐to‐matrix interactions. Here, an engineered framework, called an engineered extracellular matrix (EECM), was developed to provide support and cell‐to‐matrix interactions to developing brain organoids.
Methods
We generated brain organoids using EECMs comprised of human fibrillar fibronectin supported by a highly porous polymer scaffold. The resultant brain organoids were characterized by immunofluorescence microscopy, transcriptomics, and proteomics of the cerebrospinal fluid (CSF) compartment.
Results
The interstitial matrix‐mimicking EECM enhanced neurogenesis, glial maturation, and neuronal diversity from human embryonic stem cells versus conventional protein matrix (Matrigel). Additionally, EECMs supported long‐term culture, which promoted large‐volume organoids containing over 250 μL of CSF. Proteomics analysis of the CSF found it superseded previous brain organoids in protein diversity, as indicated by 280 proteins spanning 500 gene ontology pathways shared with adult CSF.
Interpretation
Engineered EECM matrices represent a major advancement in neural engineering as they have the potential to significantly enhance the structural, cellular, and functional diversity that can be achieved in advanced brain models.
Introduction
Brain disorders are the leading cause of disability and the second leading cause of death globally. Stroke, dementias, including Alzheimer's disease and Parkinson's disease, multiple sclerosis, brain infections, and brain cancers underlie the high mortality rate, which contributes to substantial morbidity, along with headache, epilepsy, mental disability, and autism spectrum disorder. 1 Estimates of the global burden of brain diseases vary, but, on average, at least 20% of all living persons are afflicted with a brain condition. 1 The annual direct healthcare costs and indirect costs from loss of productivity enumerate well over 1 trillion dollars in both Europe and the United States. 2 , 3 , 4 Despite decades‐long investments in basic and clinical research, most brain diseases remain untreatable.
Mouse models are useful for research into some brain diseases, and many have been developed for the most common brain conditions, for example, Alzheimer's disease 5 or traumatic brain injury. 6 However, there are inherent differences between humans and mice, which limit their applicability for some areas of inquiry. 7 Human brain organoids offer an alternative research opportunity, 8 with applications ranging from studying normal brain development 9 , 10 , 11 to modeling neurological disorders 12 and testing therapeutics. 13 Brain organoids are typically derived from embryoid bodies of either embryonic stem cells (ESCs) or induced pluripotent stem cells (iPSCs), which are differentiated into the specific central nervous system (CNS) germ layers. The resultant brain organoids assume a multi‐layered, multi‐tissue, three‐dimensional (3D) structure with cell‐to‐cell and cell‐to‐matrix network interactions, 11 which more closely model the complex brain structure compared to conventional two‐dimensional cultures.
Traditionally, brain organoids are generated by the cell‐autonomous organization on xenogenic support matrices, such as Matrigel, which is a largely undefined protein cocktail derived from the basement membrane of Engelbreth–Holm–Swarm mouse sarcoma with significant batch‐to‐batch variability. 14 To control and improve brain organoid formation and functionality, the development of precisely engineered, xenofree microenvironments with controlled matrix composition is urgently needed. 15 Brain cells experience cell‐to‐extracellular matrix interactions in vivo during brain development; however, these interactions are not faithfully captured by brain organoid cultures. Our study addresses this challenge by developing engineered extracellular matrices (EECMs) comprised of fibrillar networks of human fibronectin supported by a highly porous polymer scaffold. Fibronectin is an extracellular matrix protein that, across tissues, serves as a bedrock macromolecule that supports the deposition of other extracellular matrix molecules, such as collagens I and III. 16 Fibronectin also serves as a reservoir and transport system for key growth factors and cytokine families implicated in several biological cascades. 17 These include protein families such as bone morphogenetic proteins (BMPs), and fibroblast growth factors (FGFs), which are implicated in neural development. 18 Moreover, adult brain neurons have shown increased neurite outgrowth on fibronectin relative to other substrates, including laminin. 19 Recent advances in controlling fibronectin assembly in situ enable the development of precisely bioengineered scaffolds composed of fibrillar fibronectin suitable for cell culture. 20 Compared to Matrigel cultures, EECMs enhanced neurogenesis and glial maturation over time, as determined by single‐cell RNA‐Seq and allowed for long‐term organoid growth of up to 7 months with cerebrospinal fluid (CSF) production. EECMs provide the controlled microenvironment required to improve brain organoid development and represent a major advancement with broad utility for brain disease research.
Materials and Methods
Fabrication of engineered extracellular matrices
EECMs are a composite material of synthetic polymer and human‐derived protein. EECMs were fabricated as previously described 20 , 21 and as fully outlined in the supplementary information. Briefly, a highly porous polymer structure was generated by 3D jet writing and then placed between two medical‐grade stainless steel frames with window cut‐outs and secured with 1 μL of acrylate adhesive (Loctite, Rocky Hill, CT). To create EECMs of fibrillar fibronectin, this scaffolding was placed in a plasma fibronectin solution (Corning, Glendale, AZ) diluted in DPBS with no magnesium or calcium (Gibco, Grand Island, NY) to a concentration of 0.111 mg/mL and then tumbled in a microcentrifuge tube at 8 RPM in a 30°C chamber for at least 2 h. The air‐polymer‐protein interface induces fibrillogenesis of the fibronectin to convert the soluble protein to insoluble protein fibrils. 20 , 22 The resulting structure is an ultraporous polymer scaffold laden with a fibrillar fibronectin protein matrix, referred to collectively as EECM.
Stem cell culture
Two human PSC (hPSC) sources were used in this study. H9 (WA09) embryonic stem cells were originally from WiCell (Madison, Wisconsin) and were a gift from the lab of Paul Krebsbach (The University of Michigan, Ann Arbor, MI). iPSC 19‐9‐11 cells were provided by the University of Michigan Cardiovascular and Regeneration Core (Ann Arbor, MI). Both cells were maintained and passaged on Matrigel‐coated plates and were below passage 40. H9 cells were cultured in StemFlex media (Thermo Fisher Scientific), and iPSC was maintained in StemMACS iPS‐Brew XF, human (Miltenyi Biotec). These cells were maintained at 37°C in a humidified incubator with 5% CO2. The cells were passaged weekly with picking to remove or keep unwanted cells to maintain highly pure populations of pluripotent stem cells. Desirable properties include cells that were polygonal in shape, homogenous with sharp borders, and a high nucleus‐to‐cytoplasm ratio. Differentiated cells were mechanically removed using a sterile pulled‐glass pipet under a stereomicroscope (LeicaMZ9.5, Leica Microsystems Inc., Buffalo Grove, IL).
Generation of Matrigel and EECM‐based neural cultures
Stem cells were seeded and expanded using two methods, either Matrigel or EECM expansion. hPSCs were dissociated from Matrigel to single cells using Accutase and cell‐scraping with 10 μM ROCK inhibitor Y27632 (Calbiochem). Cells were then seeded either back to Matrigel or on EECMs to carry out differentiation. To compare Matrigel and EECM protocols as consistently as possible, Matrigel neural progenitor cells were cultured in the absence of embryoid body formation as we did not generate embryoid bodies prior to EECM seeding. We followed the monolayer protocol provided by StemCell Technologies in products #05839 and #08522. The manufacturer media changing instructions were adhered to for both Matrigel and EECMs, with noted exceptions. For EECM‐supported organoids only, hPSC single cells were seeded onto EECMs that were placed in an ultra‐low attachment 24‐well plate (Corning product #CLS3473). The cells were allowed to attach overnight and then rinsed to remove unattached cells using the feeder‐free medium. The cells were allowed to expand until they reached confluency (about 5 days on average). Once a confluent monolayer of cells was achieved, the transition to differentiation media was performed. Cells were exposed to Neural Induction Medium for 7 days, Midbrain Neuron Differentiation media medium for 14 days (we note the lack of disassociating cells from their niches, per suggested in the manufacturer protocol), Midbrain Neuron Maturation Medium for 7 days, and Midbrain Neuron Maturation Medium until collection (minimum of 14 days, up to 154 days).
Immunofluorescence staining and microscopy
Immunofluorescence was performed as previously described 20 , 23 and as fully outlined in the supplementary information.
Western blot analysis
Western blot was performed as previously described 24 and as fully outlined in the supplementary information.
Single‐cell sequencing library generation
Drop‐Seq was performed as described in Macosko et al. 25 and as fully outlined in the supplementary information. Briefly, cells were resuspended in BSA at 100 cells/μL in 0.01% BSA in PBS. Oligo‐labeled beads from Chemgenes were resuspended in lysis buffer at 120 beads/μL. Labeled beads and cells were processed through the standard Drop‐Seq device (FlowJEM) for 10 min per sample, followed by reverse transcription, an Exonuclease I step, and then universal PCR.
Single‐cell sequencing data analysis
The computational processing pipeline from Macosko et al. 25 (version 1.2) was followed to map raw Illumina reads to the human genome (hg19) using STAR as the aligner to generate digital gene expression matrices. Details are fully outlined in the supplementary information.
Mass spectrometry
Mass spectrometry was performed as previously described 20 and as fully outlined in the supplementary information. Briefly, proteins were resolved by SDS‐PAGE. Gel bands were excised and processed by in‐gel digestion with trypsin. Digests were analyzed by nano LC–MS/MS with a Waters NanoAcquity HPLC system interfaced with a Thermo Fisher Fusion Lumos mass spectrometer.
Proteomics analysis
Signatures were compared to previously published cerebrospinal fluid data acquired provided in Pellegrini et al. 26 Details are fully outlined in the supplementary information.
Quantitative qPCR analysis
qPCR was performed as outlined in the supplementary information. Each qPCR analysis comprised at least three batches of brain organoids (n ≥ 3). A batch is defined as a biological replicate using the same differentiation protocol on different EECMs at different times.
Statistical analysis
Statistics were performed in Python (version 3.6.8) using the SciPy package. 27 Pairwise comparison tests were chosen based on tests for normality (Shapiro–Wilk's test) and variance (Levene's test). On this basis, unless otherwise specified, we selected the nonparametric Mann–Whitney U test (“Wilcoxon” in Scanpy) to evaluate the statistical significance between two groups. For pathways analysis, we used g:Profiler 28 to perform functional enrichment analysis on supplied gene lists which were selected on the basis of being upregulated relative to another group with an adjusted p‐value < 0.05 as assessed by the rank_genes_group function in Scanpy 29 with Benjamini–Hochberg correction unless noted otherwise. All statistics were run on data replicates derived from distinct samples.
Results
EECMs support human pluripotent stem cell culture and neural differentiation
We developed a novel microenvironment for hPSCs to expand and differentiate into neural lineages. This acellular microenvironment is composed of a tessellated poly(d,l‐lactic‐co‐glycolic acid) (PLGA) polymer framework, 21 which supports a suspended fibrillar network of 3D, full‐length, insoluble human fibronectin 20 (Fig. 1A, Methods). The major function of the PLGA scaffold is to mechanically support the fibronectin matrix and facilitate the handling of the structures. We generated large volume structures (7 mm × 7 mm × 100 μm) from stacked polymer fibers (10 μm diameter) separated by 500 μm open pores using solution‐based 3D jet writing 21 (Fig. 1B). The polymer PLGA material occupies 3.4% of the scaffold, leaving 96.4% of open volume. Hydrodynamically induced fibrillogenesis 20 generated an insoluble, fibrillar network of human‐derived fibronectin, which fills the entire scaffold‐free volume 20 (Fig. 1C). To evaluate appropriate seeding conditions, this EECM was then seeded with human ESCs and allowed to expand until the cells reached confluence (Fig. 1D, Methods). Immunofluorescence with confocal microscopy was used to visualize differentiation progression over time (Fig. 1E, Methods). ESCs express classical pluripotency markers, including nuclear SOX2, OCT4, and NANOG (Fig. 1F). We then differentiated ESCs using Neural Induction Medium for 7 days and stained for rosette formation indicative of early brain regionalization (ZO‐1, SOX1, beta‐catenin) after Neural Induction Medium (Fig. 1G). Maturation was achieved using the STEMdiff Midbrain Neuron differentiation kit (Fig. 1E, Methods). Following maturation, we stained for general neuronal process markers TUJ1 and synaptophysin and midbrain specification with OTX2 to reveal the successful emergence of neuronal populations at 47 days (Fig. 1H). In addition, qPCR analysis of organoids formed on EECM identified the presence of key markers of neuronal development, such as LMX1A, FOXA2, FGF8, TUJ1, OTX2, and SYP after 45 days of culture (Fig. 1I). Similarly, Western blot analysis of EECM organoids after 45 days confirmed the presence of hallmarks of neuronal development, such TUJ1, p‐YAP, Nestin, SOX1, Brn2, and N‐cad and the disappearance of embryonic stem cell markers, such as Nanog, OCT4, and E‐cad.
Figure 1.
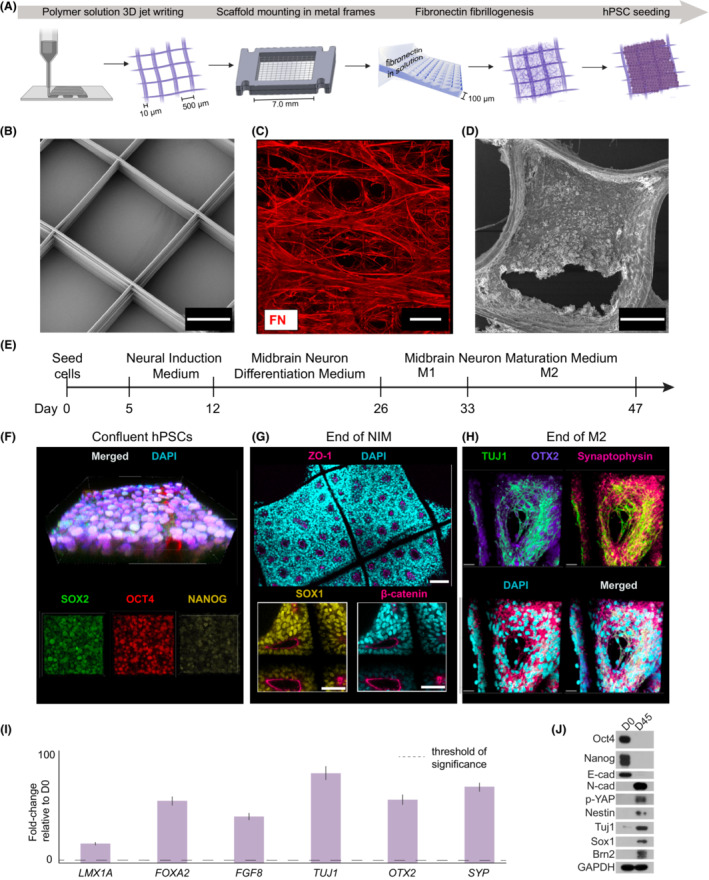
Engineered Extracellular Matrix fabrication and neural differentiation. (A) Fabrication of Engineered Extracellular Matrices (EECMs) used for seeding ESCs and downstream organoid generation. 3D jet writing is used to produce poly(d,l‐lactide‐co‐glycolide, PLGA) scaffolds, as previously described. 72 Polymer PLGA scaffolds are mounted onto medical‐grade stainless steel and then this structure is placed into a fibronectin solution that undergoes rotation to induce fibrillogenesis and deposit an insoluble fibronectin (FN) matrix. 20 , 22 Human ESCs are then seeded onto EECMs. (B) Scanning electron micrograph (SEM) of the PLGA scaffold structure (scale bar is 200 μm). (C) Deposited fibronectin matrix is stained using an amine‐reactive fluorophore (red channel; scale bar is 500 μm). (D) SEM of confluent embryonic stem cells (ESCs; H9) cultured on the EECM (scale bar is 100 μm). (E) Timeline for brain organoid production using a commercial media kit. (F) A confluent stem cell layer is generated prior to Neural Induction Medium (NIM), stained for pluripotency markers SOX2 (green channel), OCT4 (red channel), and NANOG (yellow channel) plus merged image (top; scale bar is 30 μm). (G) EECMs with differentiated H9 ESCs after NIM, stained for ZO‐1 (pink channel; scale bar is 200 μm), SOX1 (yellow channel), and beta‐catenin (red channel) markers (scale bar in insets is 100 μm), identifying neural progenitor cells and neural rosette formation. (H) Neuronal populations stained for neuronal markers TUJ1 (green channel), OTX2 (purple channel), and synaptophysin (red channel), plus merged image, at the end of maturation (scale bar is 20 μm). Panels F–H are all counterstained with DAPI (cyan channel). (I) qPCR results showing the fold‐change of expression for neuronal differentiation markers of EECM organoids at D45 of culture relative to day 0 (undifferentiated cells); n ≥ 3 biological replicates, that is, three distinct batches of brain organoids, for each gene of interest. All genes are normalized to GAPDH expression levels. (J) Western blot showing protein expression for hPSCs before differentiation (D0) and EECM organoids at D45 of culture.
EECMs enhance neurogenesis relative to Matrigel at the early stages of organoid differentiation
Because most protocols of brain organoid generation depend on Matrigel, 30 we compared EECM‐supported brain organoids to Matrigel cultures using otherwise identical differentiation protocols. Single‐cell RNA sequencing (scRNA‐seq) was used to characterize the samples after exposure to Neural Induction Medium (Fig. 2). Both groups were relatively homogenous in their cellular makeup in the early stages of development (Fig. S1A,B); however, EECM‐supported organoids exhibited stronger neurogenesis expression patterns, as determined by a composite score for select genes in the neurogenesis pathway (Fig. 2B). Analyzing the drivers of stronger neurogenesis revealed elevated expression for all genes in this pathway, including FGF8, PAX6, SOX2, OTX2, and CDH2 (Fig. 2C). Additional pathway analysis of the top 200 differentially expressed genes between both groups (p‐value < 0.05) showed that EECM‐supported organoids enriched pathways implicated in brain development and morphogenesis (Figs. 2D, S1C, Tables S1 and S2). These included “neurogenesis”, “neural precursor cell proliferation”, “neuron differentiation”, “forebrain development”, and “central nervous system development”. In contrast, Matrigel‐generated organoids showed significant upregulation of injury‐related pathways, including “Alzheimer's disease” and “Parkinson's disease” (Fig. 2E). Notably, pathway analysis also found that Matrigel increased the pathway “interspecies interaction between organisms”, gene ontology (GO) 0044419, which was driven by 53 of the 200 analyzed genes (Fig. S1D, Tables S1 and S3). These findings support the notion of interspecies interactions and suggest that the xenogenic Matrigel is detrimental for the development of human cells.
Figure 2.
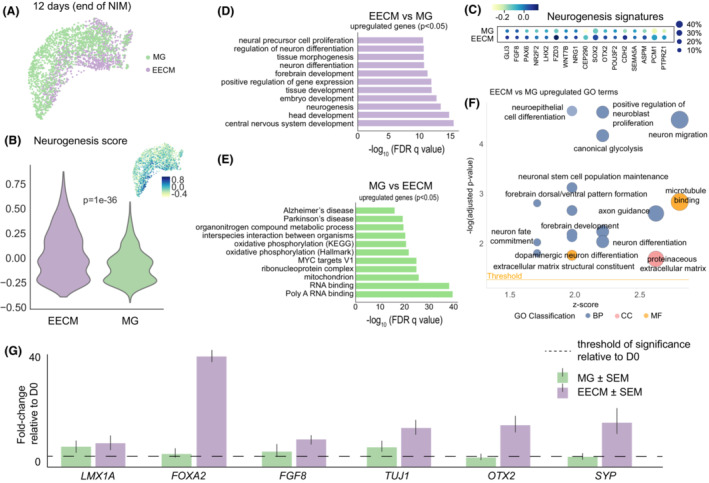
Comparison of neural lineages by scRNA‐seq in EECMs versus Matrigel after hiPSC‐induction by Neural Induction Medium. (A) Uniform Manifold Approximation and Projection (UMAP) plot visualizes scRNA‐seq of hiPSCs after culturing for up to 5 days in EECMs versus Matrigel (MG) followed by differentiation in NIM for 7 days. (B) A neurogenesis score was calculated for each cell by averaging the expression of a subset of neurogenesis pathway genes (shown in c) subtracted with the average expression of a randomly generated reference gene set. A higher score corresponds to a higher neurogenesis signature. (C) Dotplot of neurogenesis gene expression patterns by expression level (color intensity; blue, high; green, low) and percent of cells from the population expressing those signatures (dot size). (D) and (E) Pathways analysis was performed of genes with adjusted p‐values < 0.05 for the EECM versus Matrigel and Matrigel versus EECM comparisons. (F) Significantly upregulated GO terms for EECM versus Matrigel. Data represent 2843 high‐quality cells taken from pooled EECM or Matrigel sample replicates, n ≥ 3 samples each. (G) qPCR results showing the fold‐change of expression for neural differentiation markers of EECM organoids and Matrigel cultures after 7 days of NIM relative to day 0 (undifferentiated cells); n ≥ 3 biological replicates, that is, three distinct batches of brain organoids, for each gene of interest. All genes are normalized to GAPDH expression levels. BP, biological process; CC, cellular component; EECM, Engineered Extracellular Matrix; FDR, false discovery rate; GO, Gene Ontology; MF, molecular function; MG, Matrigel; NIM, Neural Induction Medium.
Additional GO pathway analysis found that biological processes associated with neural development constituted the largest class of pathways that were upregulated in EECM, followed by extracellular matrix constituent pathways (Fig. 2F). Overall, from the 200 top differentially expressed genes in EECM versus Matrigel, EECM‐generated brain organoid cells were elevated in 42 neurogenesis genes (subset shown in Fig. 2D, Table S2), 21 axonogenesis genes (Table S2), and 12 gliogenesis genes (Table S2). Further quantification of these expression profiles using an aggregated score across all GO subsets for neurogenesis (1867 genes), gliogenesis (342 genes), and interspecies interaction between organisms (1750 genes) corroborated our findings that EECM‐derived brain organoid cells were significantly upregulated in neurogenesis and gliogenesis versus cells cultured on Matrigel, which were associated with enhanced interspecies interactions due to its xenogenic nature (Fig. S1E, Table S4). We then compared cells expanded on EECM and Matrigel after culture in Neural Induction Medium with each other using qPCR analysis and found accelerated maturation in EECM organoids. Specifically, we found a significant increase in neuronal markers, such as LMX1A, FOXA2, FGF8, TUJ1, OTX2, and SYP (Fig. 2G). These data are consistent with the results of the pathway analysis of EECM organoids expanded in Neural Induction Media (Fig. 2D).
EECMs facilitate maturation of excitatory neurons and non‐neuronal organoid subpopulations relative to Matrigel
Next, we performed scRNA‐seq of EECM‐supported versus Matrigel‐generated brain organoids in later stages of differentiation (47 days total) (Fig. 3). In this analysis, we identified subpopulations based on standards in the field. 31 We used the Leiden clustering algorithm followed by a hierarchical marker‐based approach to classifying clusters. 32 Cells were first stratified into neuronal or non‐neuronal populations, and then further subclassified based on canonical markers (Tables [Link], [Link]). Cells were categorized from the EECM‐supported organoids into 11 populations (Fig. 3A), including a population of doublets, which was excluded from quantitative analyses. Doublets are an artifact of scRNA‐seq but can only be identified by clustering. Once identified, we excluded doublets from subsequent investigation to prevent them from contaminating quantitative analyses.
Figure 3.
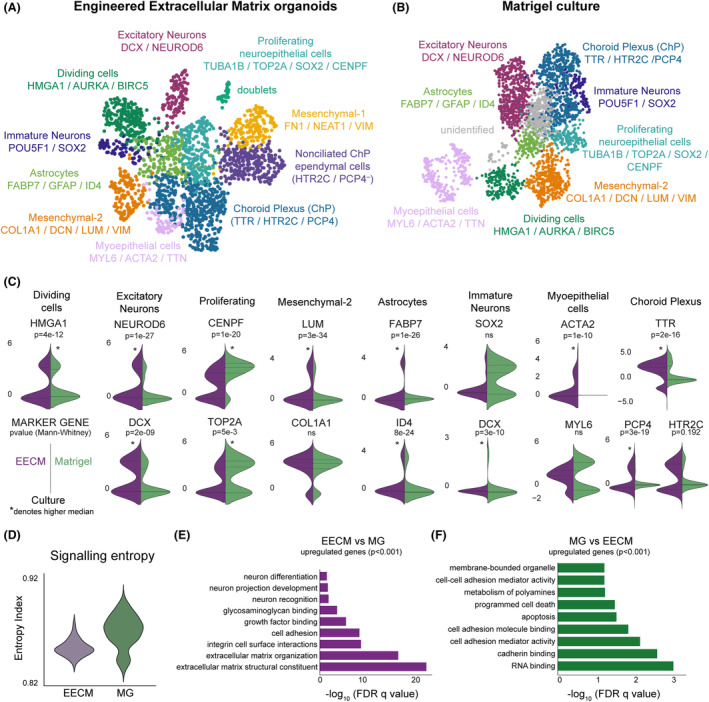
Comparison of organoid maturation and cellular diversity by scRNA‐seq in EECMs versus Matrigel at 47 days. UMAP plots show cellular diversity found in (A) EECM‐supported and (B) Matrigel‐generated brain organoids, using the same cell type annotations and gene markers (listed below cell type annotations). (C) For each cell type shared by EECM and Matrigel, the expression of one to three markers uniquely identifying that population was plotted using violin plots split by group. p‐Values are from a two‐tailed Mann–Whitney U test; the higher median sample values are denoted with an asterisk (*), as determined by alternative, one‐tailed hypothesis testing. (D) Signaling entropy was calculated as previously, using SCENT. 34 A higher entropy index corresponds to higher stemness, that is, lower differentiation. (E, F) Pathway analysis for overall genes upregulated in EECM compared to Matrigel (E) and Matrigel relative to EECM (F) at 47 days of culture. Differentially expressed genes were assessed by a t‐test and only upregulated genes with p < 0.001 were used in this analysis. n = 3 biological replicates per condition, with multiple organoids pooled per biological condition, representing 4393 high‐quality cells. EECM, Engineered Extracellular Matrix; MG, Matrigel; ns, not statistically significant.
There were nine clusters in the Matrigel sample, eight that were identified and one that was unidentifiable (Fig. 3B). EECM and Matrigel shared eight populations, including “Dividing cells”, “Excitatory Neurons”, “Immature Neurons”, “Proliferating neuroepithelial cells”, a mesenchymal population (“Mesenchymal‐2”), “Astrocytes”, “Myoepithelial cells”, and “Choroid Plexus (ChP)” (Fig. 3C). The Immature Neuron population was assigned based on the expression of stemness markers, POU5F1 and SOX2 (Table S5), as well as expression of DCX (Fig. 3C), a marker of migrating and differentiating neuronal precursor cells. Although the Immature Neuron population expressed DCX, it was not significantly upregulated in this cluster relative to all other populations combined and, thus, was not considered a defining marker (Table S5). The EECM samples had an additional ChP population, “Nonciliated ChP ependymal cells”, which expressed genes associated with the ciliation process but were not expressing PCP4 (PCP4 − negative; Fig 3A), as well as an additional mesenchymal population (“Mesenchymal‐1”), which was most prominently characterized by high fibronectin 1 (FN1) expression.
We quantified the expression of markers that uniquely identified populations in EECM versus Matrigel organoids in all shared populations (Fig. 3C). We found that Matrigel statistically enhanced gene expression defining the Proliferating (CENPF, TOP2A) and Dividing cells (HMGA1) populations. Expression patterns also indicated the strong distribution of SOX2 expression for the Immature Neuron population in Matrigel, though these differences were not statistically significant due to the slightly higher baseline expression in EECM organoids. In contrast, EECM‐supported organoids yielded significantly higher expression of marker genes for Excitatory Neurons (NEUROD6, DCX), Mesenchymal‐2 (LUM), Astrocytes (FABP7, ID4), Immature Neurons (DCX), and Myoepithelial cells (ACTA2) populations. Notably, EECMs also induced higher expression of markers for ciliated ependymal cells, as indicated by elevated coexpression of TTR and PCP4 expression (Fig. 3C). While the ChP population of EECMs did not have statistically higher HTR2C expression relative to Matrigel, a substantial population appeared to emerge with high HTR2C expression on EECMs.
Recent reports that brain organoids do not sufficiently capture late‐stage glial development relative to in vivo development identified lumican (LUM)‐expressing cells as potential mediators of these differences. 33 It is noteworthy, then, that LUM was highly expressed in the EECM‐supported brain organoid Mesenchymal‐2 population relative to Matrigel (Fig. 3C). Additionally, we used a non‐marker based approach to compare the extent of differentiation in EECM versus Matrigel groups in aggregate. We employed the SCENT algorithm, 34 which uses signaling entropy as a marker‐agnostic approach to estimate cellular potency. A higher entropy index corresponds to higher signaling promiscuity and thus higher pluripotency. 34 , 35 We found that EECMs enhanced differentiation relative to Matrigel, as indicated by a significantly lower entropy index (Fig. 3D). Finally, pathways analysis of the differentially expressed genes (p < 0.001) between the two conditions overall at this time point revealed that EECMs had elevated signatures associated with “neuron differentiation” and “neuron projection development”, as well as several extracellular matrix interaction pathways, including “glycosaminoglycan binding”, “growth factor binding”, “cell adhesion”, “integrin cell surface interactions”, “extracellular matrix organization”, and “extracellular matrix structural constituent” (Fig. 3E).
Meanwhile, the Matrigel samples exhibited upregulated pathways associated with “programmed cell death” and “apoptosis” (Figs. 3F and S2A). Together, these results suggest that the EECM environment facilitates stronger cell specification relative to Matrigel. Recent work by Bhaduri et al. 36 showed that cortical brain organoids ectopically induce cell stress responses, which negatively influence the ability of cells to differentiate. In this context, our findings are consistent with the notion that the more native‐like extracellular matrix that cells would encounter in an in vivo context enhances cell differentiation by ablating the cellular stress response to foreign matrix.
EECMs support long‐term brain organoid culture
Due to its fibrillar network structure, the EECM provides for a large surface area of fibronectin available for initial cell attachment (Fig. 1C). The adherent cells use this matrix as a template from which to expand, resulting in large‐volume brain organoids over time. However, the viability of these large organoids during long culturing remained unknown. To explore the long‐term survival of large‐scale organoids, we repeated the differentiation protocol on EECMs using human induced pluripotent stem cells (hiPSCs), allowing them to grow for 215 days (Fig. 4A). Remarkably, the vitality of each organoid generated was sustained throughout the timeline, as evidence by their continual size expansion (Fig. 4C) and analysis of apoptosis pathways (Fig. S2A). Organoids reached an average maximum continuous diameter of 11.88 mm (Fig. 4C). Fluid‐filled compartments emerged after extended culture of EECM‐supported brain organoids, similar to ChP organoids recently developed by Pellegrini et al., which used the small molecule GSK3 inhibitor CHIR99021 with bone morphogenetic protein 4 (BMP4) to pattern ChP production in Matrigel. 26 The cyst‐like structures observed in EECM‐templated brain organoids roughly match the global geometry of the overall EECM structure (Fig. 4B). Brain organoids generated using a bioreactor have previously been reported to survive up to a year, 37 however these organoids did not appear to continue expanding beyond 4 mm in diameter.
Figure 4.
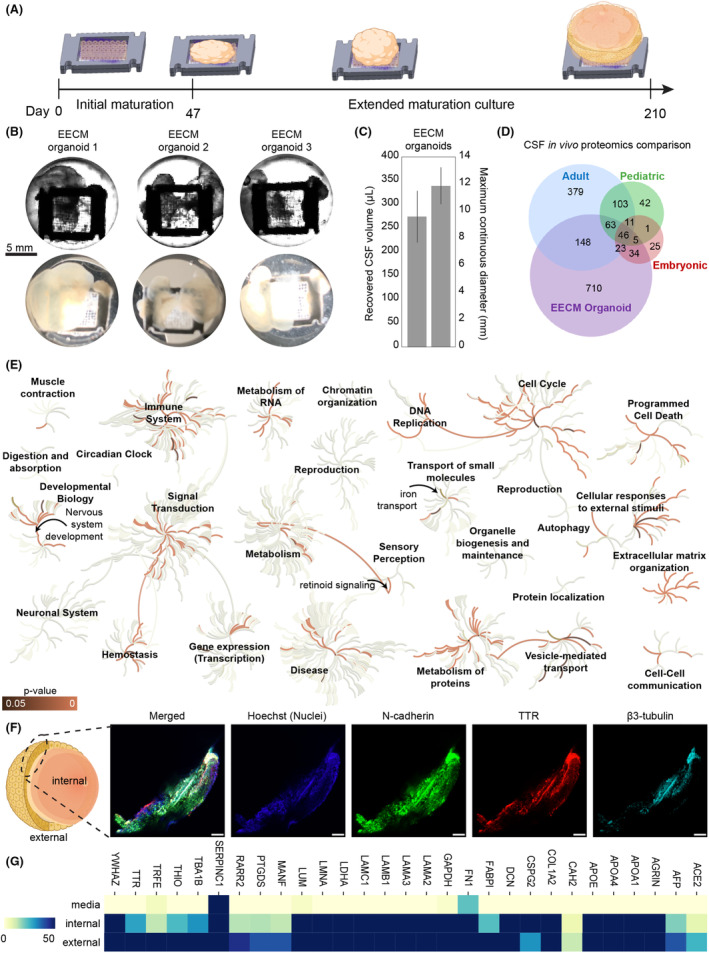
Extended organoid culture on EECMs for 7 months generated large CSF‐producing brain organoids. (A) Organoids were matured on EECMs in Midbrain Neuron Maturation Medium M2 for approximately 6 months and for a total duration of 215 days. Fluid‐filled compartments became increasingly larger over time. (B) Representative images were taken of three different EECM brain organoids using an IX83 Olympus microscope using phase contrast (top panel) and with a 12‐megapixel wide‐angle camera (bottom panel). Fluid‐filled compartments were punctured with a 22G needle and extracted for analysis. (C) Quantification of recovered cerebrospinal (CSF)‐mimicking fluid from the organoids and maximum continuous organoid diameters (n = 3 organoids). (D) Mass spectrometry was performed on the CSF fluid recovered from the organoids and compared to published proteomics signatures. 26 , 73 , 74 , 75 Values shown are for at least two organoids exceeding a relative abundance emPAI ≥ 1. (E) We used the Reactome database 38 to visualize the pathway and biological process coverage and significance from the recovered CSF‐like fluid from EECM‐supported organoids; the orange color indicates p‐value significance of protein coverage of pathway (dark = p ≤ 0.05, bright p ~ 0); gray color represents the binary status of coverage of pathway (lightest = not covered, darkest = covered). (F) Confocal images from EECM organoid section after 215 days of culture stained for N‐cadherin, TTR, and β3‐Tubulin. The scale is 200 μm. (G) Relative abundance (emPAI values) of selected proteins detected in media and internal and external organoid cultures represented as a color heatmap of lower (yellow, 0) to higher (blue, ≥ 50) relative abundance. Samples were extracted from three organoids per condition.
Extended organoid culture on EECMs reproducibly generates CSF‐filled compartments
Because we observed an elevated ChP signature in EECM‐supported organoids relative to Matrigel at the 47 day time point (Fig. 3C), we hypothesized this population could drive CSF‐like fluid production over the course of extended EECM brain organoid culture. To test this assumption, we recovered an average of 275 μL of CSF‐like fluid from the brain organoid internal sac structures. The fluid proteomics profile was subsequently characterized by mass spectrometry, and we compared the proteomics profile of the CSF from EECM‐based brain organoids to previously published datasets of human CSF (Fig. 4D). EECM‐supported brain organoids shared a total of 319 proteins with in vivo CSF signatures across embryonic, pediatric, and adult CSF. EECM organoids overlapped with 280 proteins in adult CSF (Table S8), particularly, spanning 500 GO pathways (Table S9). Characteristic proteins identified in the EECM group included neurological disease biomarkers, such as Alzheimer's (APP, MAPK3, and ADAM9), Parkinson's (YWHAE and SNCG), and Huntington's (CRYZ) diseases (Table S10, EECM column). Similarly, prominent endogenous metabolic pathways, such as opioid, oxytocin, cannabinoid, and serotonin signaling (GNB1, GNAI3, GNG12, ALDH1A3, ALDH9A1, and ALDH1A1), and insulin‐ and inflammation‐mediated cascades (IGF2, MAPK3, GRB2, IGF2R, and VWF) were observed. Not surprisingly, high levels of proteins implicated with cell‐extracellular matrix interaction pathways, including “cadherin binding”, “glycosaminoglycan binding”, and “integrin binding”, and “cell‐substrate junction”, “focal adhesion”, and “anchoring junction” were produced by cells cultured on EECM (Table S9).
Biological pathways and processes associated with the proteomics signatures present in EECM‐supported brain organoid CSF were visualized using the Reactome database 38 (Fig. 4E). Significant pathway expression occurred in “Extracellular matrix organization”, “Cellular responses to external stimuli”, “Metabolism of proteins”, “Signal transduction”, “Developmental biology” (particularly within “Nervous system development”), “Immune System”, and “Vesicle‐mediated transport”. Two sensory perception pathways were also upregulated, related to “Retinoid signaling” and “Sensory processing of sound by inner hair cells of the cochlea” (Fig. 4E). EECM brain organoids were abundant in expressing signatures associated with secreting extracellular vesicles, extracellular matrix organization, and cell‐extracellular matrix interactions (extended pathways analysis is shown in Fig. S3, Table S11).
Additionally, we placed our findings in the context of the current state of the field, by comparing the extracted EECM‐supported organoid CSF‐like fluid to the recently published CHIR99021/BMP4‐patterned ChP organoids (Fig. S4, Table S10). 26 Overall, our mass spectrometry identified 1036 protein signatures with an Exponentially Modified Protein Abundance Index (emPAI) value ≥1 in at least two samples, contributing to the 1.58‐fold higher overlap of EECM organoid CSF with adult in vivo CSF signatures relative to the ChP organoid, spanning 32 differentiated overlapped pathways identified by the Panther Pathway database (PantherDB 39 ) (Fig. S4A,B). Using a combination of PantherDB, 39 g:Profiler, 28 and Reactome, 38 we found these differences were largely reflected in pathways linked to signaling, such as integrin, TGFβ, and MAPK signaling, and the aforementioned cannabinoid, serotonin, and oxytocin signaling from GNB1, GNAI3, and GNG12 expression. Reactome database analysis showed the ChP organoids express pathways containing “Chromatin organization”, “Gene expression (Transcription)”, “Cell Cycle”, and “Metabolism of proteins” (Fig. S4C). Within the developmental biology pathways, fluid from both types of organoids highly expressed signatures of nervous system development.
To assess the reproducibility of our organoid generation approach, we tested three separate batches of organoids containing a total of nine replicates, which were segregated across multiple characterization workstreams. Our proteomics analysis showed high batch‐to‐batch consistency for the three organoids evaluated. Figure S5 shows the Spearman rank correlation calculation based on the quantitative mass spectrometry results. As a secondary analysis, we analyzed the secretome found both within the organoid compartment (“internal”) and outside of it (“external”), which we combined with hierarchical clustering. While there is overall a high similarity index between the two regions of the organoid, the internal and external regions are segregated into two distinct clusters, suggesting that the organoids are forming a tight barrier that preferentially filters some proteins.
Discussion
Brain organoids have emerged as a promising in vitro tool to study neurological phenomena related to development and disease. 40 In particular, brain organoids can be generated from human iPSCs and ESCs; thus, they can be useful in research areas that have inherent differences between humans and mice, such as brain development, brain disorders, or infection of human‐specific pathogens. 7 , 41 Compared to conventional two‐dimensional neuronal or glial cultures, the 3D architecture and cellular heterogeneity of these miniaturized brain organoid structures better typifies in vivo brain composition. Concentrated research efforts have generated protocols that differentiate stem cells into forebrain, midbrain, and hindbrain with remarkable biological complexity and reproducibility. 42 However, in the decade since the inception of modern cerebral organoids, 10 few advancements have been made with respect to bioengineering matrices to support organoid growth. With few exceptions, 43 conventional brain organoid protocols use murine‐derived Matrigel as an extracellular matrix, 44 which suffers significant disadvantages. Thus, overreliance on Matrigel is often viewed as holding the organoid field back, exemplified by recent calls to action to reframe organoid generation through materials engineering. 15 , 45
Here, we heeded these calls by implementing novel EECMs comprised of both synthetic PLGA and defined human‐derived fibronectin materials. Our EECMs enhanced neurogenesis and glial maturation versus Matrigel, maintaining neuronal diversity commensurate with the cerebral organoid literature. 32 Additionally, our EECMs facilitated long‐term culture up to 7 months, which promoted the emergence of large‐volume CSF‐filled organoid structures. The EECM‐derived CSF proteomics signature shared a significant number of matches with human adult CSF and spanned 75 biological pathways of relevance to development. We designed EECMs to overcome several Matrigel weaknesses. 46 Matrigel, a basement membrane hydrogel derived from mouse tumors, is rich in certain proteins and polysaccharides relevant to tissue support and is a serviceable material; however, it is of relatively undefined composition 47 and contains xenogenic material. This is evidenced by the enrichment of “Interspecies interaction between organisms” in Matrigel‐ versus EECM‐derived brain organoid cells in our analysis. To counter this, our EECMs are precisely engineered from well‐defined PLGA and full‐length fibronectin materials. PLGA is a commercially available biocompatible material, which has been approved for use in biomedical devices by the FDA. 48 The fibronectin is of human origin and thus xeno‐free, rendering our EECMs superior for human‐derived organoid growth.
Additionally, Matrigel possesses biological activity 49 because it contains cytokines as well as growth and transcription factors. 47 , 50 Indeed, we found a significantly larger proliferating population in the Matrigel‐ versus EECM‐grown brain organoids, even at 47 days once they had matured, which may arise from the presence of exogenous growth factors. By contrast, our EECMs, being derived from chemically defined materials, are free of biologically active exogenous molecules, which could influence organoid development outside of the neural differentiation program. Another important factor for reproducible organoid development is matching the matrix stiffness to the native dynamics of the target tissue for lineage‐specific differentiation of stem cells, 46 , 51 , 52 including for neurogenesis. 53 , 54 Although soft matrices, such as Matrigel, which has an elastic modulus of ~400 Pa, have been found to be neurogenic, 53 the intrinsic heterogeneity of xenogeneic matrices can cause locoregional differences in stiffness, which can vary up to ~3 kPa. 55 Since the biophysical properties of the extracellular matrix influences organoid development, 56 this variability in Matrigel stiffness could impair development leading to immature brain organoids. By contrast, the engineered nature of our EECM platform provides opportunities for higher batch‐to‐batch consistency and facilitates the potential for large‐scale brain organoid development. Finally, our fibronectin‐based EECMs pose several practical advantages, including implantable capability, manufacturing scalability, and tunable design parameters (e.g., scaffold geometry and additional matrix composition changes).
Our findings join a growing body of evidence, which advocates considering synthetic, precisely‐designed extracellular matrices as Matrigel alternatives. Sorrentino et al. generated liver organoids using enzymatically crosslinked poly(ethylene glycol) (PEG) hydrogels infused with laminin‐111, collagen IV, and fibronectin. 57 They found that PEG hydrogel stiffness controlled liver organoid growth, which could even be tuned to model fibrotic liver by increasing matrix stiffness. Funfak et al. compared bile duct epithelial morphogenesis in PEG hydrogels versus Matrigel 58 ; although the efficiency of cyst formation was similar in both scaffolds, larger cysts formed in Matrigel. Nevertheless, varying PEG stiffness and integrin ligand density could support the development of functional cholangiocyte organoids with good multidrug resistance protein‐1 activity. Ranga et al., also utilizing PEG constructs with modular additives influencing degradability, extracellular components, and growth factors, could precisely control neural tube morphogenesis by tuning scaffold properties. 54 Similar control or amelioration of organoid development using PEG has been achieved for intestinal organoids, 59 , 60 lung organoids, 61 mammary epithelial cell acini, 62 and renal tubules, 63 albeit producing smaller‐sized organoids. 58 , 60 Polycaprolactone, alone 64 or blended with chitosan, 65 can also support organoid growth, as can PLGA for brain 66 and islet β‐like cell 67 organoids. Lancaster et al. used floating PLGA fiber microfilaments seeded with hiPSCs to generate neuroectoderm, which was then embedded in Matrigel. 66 The two‐step process incorporating PLGA microfilaments improved neuroectoderm formation and cortical development versus Matrigel. Here, we demonstrate that fibronectin‐coated PLGA scaffolds, devoid of any Matrigel, can support the development of highly differentiated, CSF‐containing brain organoids.
Beyond the technical advancements realized by our EECMs, the resultant brain organoids, as closer brain mimics, could improve upon several current applications. By stimulating greater differentiation of hiPSCs into diverse neuronal and non‐neuronal cell types, our brain organoids could provide deeper insight into neurodevelopmental biology, 9 including human‐specific characteristics. 7 Furthermore, given the ability for long‐term culturing, 8 our brain organoids could also constitute a viable model for neurodegenerative diseases, such as Alzheimer's disease, 68 and serve as a test‐bed for screening potential therapeutics. The presence of rich CSF compartments similarly advocates our EECM‐supported brain organoids for studies of neurological disease biomarkers, such as amyloid‐β, tau, 69 and neurofilaments. 70 , 71 Additionally, neurological illnesses dictated by impaired inhibitory or excitatory neuron function, such as Down syndrome, could similarly be modeled well by our EECM‐supported brain organoids, 8 which contain a larger population of excitatory neurons than Matrigel‐derived organoids.
In conclusion, we present a path forward for advancing brain and other organoid development through EECMs, which advocates exploring novel approaches. Enhanced brain organoid development could also improve their applications, more faithfully mirroring neurological diseases.
Author Contributions
A.J.M. conceived of the study design conducted the experiments, analyzed the data, produced the figures, and prepared the manuscript. T.T. conducted the culture and differentiation experiments related to H9 embryonic stem cells and immunofluorescence microscopy for Fig. 1. M.D.B. generated the sequencing libraries. A.S. contributed to scaffold manufacturing. D.H.K. assisted with cell culture. J.J. provided scaffolds for the cell culture. J.N. contributed to microscopy and cell biology. M.G.S. and E.L.F. contributed to data interpretation and manuscript preparation. P.H.K. and J.L. contributed to the study design, data interpretation, and manuscript preparation.
Funding Information
A.J.M. is funded by the National Science Foundation with grant no. DGE 1256260. E.L.F. thanks the Robert and Katherine Jacobs Health Environmental Initiative Fund, the Andrea and Lawrence A. Wolfe Brain Health Initiative Fund, Robert E. Nederlander Sr. Program for Alzheimer's Research, and NeuroNetwork for Emerging Therapies. We acknowledge funding from the University of Michigan Biointerfaces Institute (E.L.F. and J.L.).
Conflicts of Interest
The authors have nothing to disclose.
Code Availability
Custom Python scripts were used to process these data and will be provided upon request.
Supporting information
Data S1.
Supplementary Table S1.
S.upplementary Table S2.
Supplementary Table S3.
Supplementary Table S4.
Supplementary Table S5.
Supplementary Table S6.
Supplementary Table S7.
Supplementary Table S8.
Supplementary Table S9.
Supplementary Table S10.
Supplementary Table S11.
Acknowledgments
We thank the Cardiovascular and Regeneration Core for providing access to 19‐9‐11 iPSC cells. We thank Dr. Lynn Beene for her expert assistance with manuscript editing.
Funding Statement
This work was funded by Robert and Katherine Jacobs Health Environmental Initiative Fund; Andrea and Lawrence A. Wolfe Brain Health Initiative Fund; National Science Foundation grant DGE 1256260; NeuroNetwork for Emerging Therapies; Robert E. Nederlander Sr. Program for Alzheimer's Research; University of Michigan Biointerfaces Institute.
Contributor Information
Eva L. Feldman, Email: efeldman@umich.edu.
Joerg Lahann, Email: lahann@umich.edu.
Data Availability Statement
These data will be made available through GEO accession upon publication.
References
- 1. Global, regional, and national burden of neurological disorders, 1990‐2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol. 2019;18(5):459‐480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. DiLuca M, Olesen J. The cost of brain diseases: a burden or a challenge? Neuron. 2014;82(6):1205‐1208. [DOI] [PubMed] [Google Scholar]
- 3. Feigin VL, Vos T, Alahdab F, et al. Burden of neurological disorders across the US from 1990‐2017: a global burden of disease study. JAMA Neurol. 2021;78(2):165‐176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Trautmann S, Rehm J, Wittchen HU. The economic costs of mental disorders: do our societies react appropriately to the burden of mental disorders? EMBO Rep. 2016;17(9):1245‐1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Puzzo D, Gulisano W, Palmeri A, Arancio O. Rodent models for Alzheimer's disease drug discovery. Expert Opin Drug Discov. 2015;10(7):703‐711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang YP, Cai J, Shields LB, Liu N, Xu XM, Shields CB. Traumatic brain injury using mouse models. Transl Stroke Res. 2014;5(4):454‐471. [DOI] [PubMed] [Google Scholar]
- 7. Marshall JJ, Mason JO. Mouse vs man: organoid models of brain development & disease. Brain Res. 2019;1724:146427. [DOI] [PubMed] [Google Scholar]
- 8. Shou Y, Liang F, Xu S, Li X. The application of brain organoids: from neuronal development to neurological diseases. Front Cell Dev Biol. 2020;8:579659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chan WK, Fetit R, Griffiths R, Marshall H, Mason JO, Price DJ. Using organoids to study human brain development and evolution. Dev Neurobiol. 2021;81(5):608‐622. [DOI] [PubMed] [Google Scholar]
- 10. Lancaster MA, Renner M, Martin CA, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501(7467):373‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lancaster MA, Knoblich JA. Organogenesis in a dish: modeling development and disease using organoid technologies. Science. 2014;345(6194):1247125. [DOI] [PubMed] [Google Scholar]
- 12. Wang H. Modeling neurological diseases with human brain organoids. Front Synaptic Neurosci. 2018;10:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Struzyna LA, Watt ML. The emerging role of neuronal organoid models in drug discovery: potential applications and hurdles to implementation. Mol Pharmacol. 2021;99(4):256‐265. [DOI] [PubMed] [Google Scholar]
- 14. Paşca AM, Sloan SA, Clarke LE, et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods. 2015;12(7):671‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garreta E, Kamm RD, Chuva de Sousa Lopes SM, et al. Rethinking organoid technology through bioengineering. Nat Mater. 2021;20(2):145‐155. [DOI] [PubMed] [Google Scholar]
- 16. Sottile J, Shi F, Rublyevska I, Chiang HY, Lust J, Chandler J. Fibronectin‐dependent collagen I deposition modulates the cell response to fibronectin. Am J Physiol Cell Physiol. 2007;293(6):C1934‐C1946. [DOI] [PubMed] [Google Scholar]
- 17. Zollinger AJ, Smith ML. Fibronectin, the extracellular glue. Matrix Biol. 2017;60‐61:27‐37. [DOI] [PubMed] [Google Scholar]
- 18. Martino MM, Hubbell JA. The 12th‐14th type III repeats of fibronectin function as a highly promiscuous growth factor‐binding domain. FASEB J. 2010;24(12):4711‐4721. [DOI] [PubMed] [Google Scholar]
- 19. Tonge DA, de Burgh HT, Docherty R, Humphries MJ, Craig SE, Pizzey J. Fibronectin supports neurite outgrowth and axonal regeneration of adult brain neurons in vitro. Brain Res. 2012;1453:8‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jordahl S, Solorio L, Neale DB, et al. Engineered fibrillar fibronectin networks as three‐dimensional tissue scaffolds. Adv Mater. 2019;31(46):e1904580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jordahl JH, Solorio L, Sun H, et al. 3D jet writing: functional microtissues based on tessellated scaffold architectures. Adv Mater. 2018;30(14):e1707196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Neale DB, Muñiz AJ, Jones MS, et al. Aligned networks of engineered fibrillar fibronectin guide cellular orientation and motility. Small Structures. 2021;2(6):2000137. [Google Scholar]
- 23. Humphries BA, Cutter AC, Buschhaus JM, et al. Enhanced mitochondrial fission suppresses signaling and metastasis in triple‐negative breast cancer. Breast Cancer Res. 2020;22(1):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gregory JV, Kadiyala P, Doherty R, et al. Systemic brain tumor delivery of synthetic protein nanoparticles for glioblastoma therapy. Nat Commun. 2020;11(1):5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Macosko EZ, Basu A, Satija R, et al. Highly parallel genome‐wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161(5):1202‐1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pellegrini L, Bonfio C, Chadwick J, Begum F, Skehel M, Lancaster MA. Human CNS barrier‐forming organoids with cerebrospinal fluid production. Science. 2020;369(6500), eaaz5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Virtanen P, Gommers R, Oliphant TE, et al. SciPy 1.0: fundamental algorithms for scientific computing in python. Nat Methods. 2020;17(3):261‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Raudvere U, Kolberg L, Kuzmin I, et al. G:profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019;47(W1):W191‐w8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wolf FA, Angerer P, Theis FJ. SCANPY: large‐scale single‐cell gene expression data analysis. Genome Biol. 2018;19(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. de Souza N. Organoid culture. Nat Methods. 2017;14(1):35. [Google Scholar]
- 31. Luecken MD, Theis FJ. Current best practices in single‐cell RNA‐seq analysis: a tutorial. Mol Syst Biol. 2019;15(6):e8746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tanaka Y, Cakir B, Xiang Y, Sullivan GJ, Park IH. Synthetic analyses of single‐cell transcriptomes from multiple brain organoids and fetal brain. Cell Rep. 2020;30(6):1682‐9.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eze UC, Bhaduri A, Haeussler M, Nowakowski TJ, Kriegstein AR. Single‐cell atlas of early human brain development highlights heterogeneity of human neuroepithelial cells and early radial glia. Nat Neurosci. 2021;24(4):584‐594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Teschendorff AE, Enver T. Single‐cell entropy for accurate estimation of differentiation potency from a cell's transcriptome. Nat Commun. 2017;8:15599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen W, Teschendorff AE. Estimating differentiation potency of single cells using single‐cell entropy (SCENT). Methods Mol Biol. 2019;1935:125‐139. [DOI] [PubMed] [Google Scholar]
- 36. Bhaduri A, Andrews MG, Mancia Leon W, et al. Cell stress in cortical organoids impairs molecular subtype specification. Nature. 2020;578(7793):142‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Quadrato G, Nguyen T, Macosko EZ, et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature. 2017;545(7652):48‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jassal B, Matthews L, Viteri G, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020;48(D1):D498‐d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mi H, Ebert D, Muruganujan A, et al. PANTHER version 16: a revised family classification, tree‐based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2021;49(D1):D394‐d403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Clevers H. Modeling development and disease with organoids. Cell. 2016;165(7):1586‐1597. [DOI] [PubMed] [Google Scholar]
- 41. Bartfeld S. Modeling infectious diseases and host‐microbe interactions in gastrointestinal organoids. Dev Biol. 2016;420(2):262‐270. [DOI] [PubMed] [Google Scholar]
- 42. Kanton S, Boyle MJ, He Z, et al. Organoid single‐cell genomic atlas uncovers human‐specific features of brain development. Nature. 2019;574(7778):418‐422. [DOI] [PubMed] [Google Scholar]
- 43. Chen C, Rengarajan V, Kjar A, Huang Y. A matrigel‐free method to generate matured human cerebral organoids using 3D‐printed microwell arrays. Bioact Mater. 2021;6(4):1130‐1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang L, Ng HH. Lab‐grown mini‐brains upgraded. Nat Cell Biol. 2017;19(9):1010‐1012. [DOI] [PubMed] [Google Scholar]
- 45. Takebe T, Wells JM. Organoids by design. Science. 2019;364(6444):956‐959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aisenbrey EA, Murphy WL. Synthetic alternatives to Matrigel. Nat Rev Mater. 2020;5(7):539‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hughes CS, Postovit LM, Lajoie GA. Matrigel: a complex protein mixture required for optimal growth of cell culture. Proteomics. 2010;10(9):1886‐1890. [DOI] [PubMed] [Google Scholar]
- 48. Manavitehrani I, Fathi A, Badr H, Daly S, Negahi Shirazi A, Dehghani F. Biomedical applications of biodegradable polyesters. Polymers (Basel). 2016;8(1):1‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kleinman HK, Martin GR. Matrigel: basement membrane matrix with biological activity. Semin Cancer Biol. 2005;15(5):378‐386. [DOI] [PubMed] [Google Scholar]
- 50. Talbot NC, Caperna TJ. Proteome array identification of bioactive soluble proteins/peptides in Matrigel: relevance to stem cell responses. Cytotechnology. 2015;67(5):873‐883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vining KH, Mooney DJ. Mechanical forces direct stem cell behaviour in development and regeneration. Nat Rev Mol Cell Biol. 2017;18(12):728‐742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sun Y, Yong KM, Villa‐Diaz LG, et al. Hippo/YAP‐mediated rigidity‐dependent motor neuron differentiation of human pluripotent stem cells. Nat Mater. 2014;13(6):599‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126(4):677‐689. [DOI] [PubMed] [Google Scholar]
- 54. Ranga A, Girgin M, Meinhardt A, et al. Neural tube morphogenesis in synthetic 3D microenvironments. Proc Natl Acad Sci U S A. 2016;113(44):E6831‐e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Soofi SS, Last JA, Liliensiek SJ, Nealey PF, Murphy CJ. The elastic modulus of Matrigel as determined by atomic force microscopy. J Struct Biol. 2009;167(3):216‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang W, Chu G, Wang H, Chen S, Li B, Han F. Effects of matrix stiffness on the differentiation of multipotent stem cells. Curr Stem Cell Res Ther. 2020;15(5):449‐461. [DOI] [PubMed] [Google Scholar]
- 57. Sorrentino G, Rezakhani S, Yildiz E, et al. Mechano‐modulatory synthetic niches for liver organoid derivation. Nat Commun. 2020;11(1):3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Funfak A, Bouzhir L, Gontran E, Minier N, Dupuis‐Williams P, Gobaa S. Biophysical control of bile duct epithelial morphogenesis in natural and synthetic scaffolds. Front Bioeng Biotechnol. 2019;7:417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gjorevski N, Lutolf MP. Synthesis and characterization of well‐defined hydrogel matrices and their application to intestinal stem cell and organoid culture. Nat Protoc. 2017;12(11):2263‐2274. [DOI] [PubMed] [Google Scholar]
- 60. Hernandez‐Gordillo V, Kassis T, Lampejo A, et al. Fully synthetic matrices for in vitro culture of primary human intestinal enteroids and endometrial organoids. Biomaterials. 2020;254:120125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cruz‐Acuña R, Quirós M, Farkas AE, et al. Synthetic hydrogels for human intestinal organoid generation and colonic wound repair. Nat Cell Biol. 2017;19(11):1326‐1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nowak M, Freudenberg U, Tsurkan MV, Werner C, Levental KR. Modular GAG‐matrices to promote mammary epithelial morphogenesis in vitro. Biomaterials. 2017;112:20‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Weber HM, Tsurkan MV, Magno V, Freudenberg U, Werner C. Heparin‐based hydrogels induce human renal tubulogenesis in vitro. Acta Biomater. 2017;57:59‐69. [DOI] [PubMed] [Google Scholar]
- 64. Nayak B, Balachander GM, Manjunath S, Rangarajan A, Chatterjee K. Tissue mimetic 3D scaffold for breast tumor‐derived organoid culture toward personalized chemotherapy. Colloids Surf B Biointerfaces. 2019;180:334‐343. [DOI] [PubMed] [Google Scholar]
- 65. Li YE, Wang JH, Wang YH, Shao HJ, Young LC, Young TH. PCL‐blended chitosan substrates for patterning the heterotypic cell distribution in an epithelial and mesenchymal coculture system. ACS Biomater Sci Eng. 2020;6(7):4225‐4235. [DOI] [PubMed] [Google Scholar]
- 66. Lancaster MA, Corsini NS, Wolfinger S, et al. Guided self‐organization and cortical plate formation in human brain organoids. Nat Biotechnol. 2017;35(7):659‐666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sun S, Cui W, Dong Y, Wang Q. Liraglutide immobilized on poly(lactic‐co‐glycolic acid) polymer films induced the differentiation of islet β‐like cells from bone marrow mesenchymal stem cells. Macromol Res. 2019;27(5):454‐459. [Google Scholar]
- 68. Venkataraman L, Fair SR, McElroy CA, Hester ME, Fu H. Modeling neurodegenerative diseases with cerebral organoids and other three‐dimensional culture systems: focus on Alzheimer's disease. Stem Cell Rev Rep. 2022;18(2):696‐717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Leuzy A, Ashton NJ, Mattsson‐Carlgren N, et al. 2020 update on the clinical validity of cerebrospinal fluid amyloid, tau, and phospho‐tau as biomarkers for Alzheimer's disease in the context of a structured 5‐phase development framework. Eur J Nucl Med Mol Imaging. 2021;48(7):2121‐2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ferreira‐Atuesta C, Reyes S, Giovanonni G, Gnanapavan S. The evolution of neurofilament light chain in multiple sclerosis. Front Neurosci. 2021;15:642384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Abu‐Rumeileh S, Parchi P. Cerebrospinal fluid and blood neurofilament light chain protein in prion disease and other rapidly progressive dementias: current state of the art. Front Neurosci. 2021;15:648743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Moon S, Jones MS, Seo E, et al. 3D jet writing of mechanically actuated tandem scaffolds. Sci Adv. 2021;7(16):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dayon L, Núñez Galindo A, Wojcik J, et al. Alzheimer disease pathology and the cerebrospinal fluid proteome. Alzheimers Res Ther. 2018;10(1):66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Guo L, Ren H, Zeng H, Gong Y, Ma X. Proteomic analysis of cerebrospinal fluid in pediatric acute lymphoblastic leukemia patients: a pilot study. Onco Targets Ther. 2019;12:3859‐3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zappaterra MD, Lisgo SN, Lindsay S, Gygi SP, Walsh CA, Ballif BA. A comparative proteomic analysis of human and rat embryonic cerebrospinal fluid. J Proteome Res. 2007;6(9):3537‐3548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Supplementary Table S1.
S.upplementary Table S2.
Supplementary Table S3.
Supplementary Table S4.
Supplementary Table S5.
Supplementary Table S6.
Supplementary Table S7.
Supplementary Table S8.
Supplementary Table S9.
Supplementary Table S10.
Supplementary Table S11.
Data Availability Statement
These data will be made available through GEO accession upon publication.