

SUMMARY
The shedding of extracellular vesicles (EVs) represents an important but understudied means of cell-cell communication in cancer. Among the currently described classes of EVs, tumor-derived microvesicles (TMVs) comprise a class of vesicles released directly from the cell surface. TMVs contain abundant cargo, including functional proteins and miRNA, which can be transferred to and alter the behavior of recipient cells. Here, we document that a fraction of extracellular double-stranded DNA (dsDNA) is enclosed within TMVs and protected from nuclease degradation. dsDNA inclusion in TMVs is regulated by ARF6 cycling and occurs with the cytosolic DNA sensor, cGAS, but independent of amphisome or micronuclei components. Our studies suggest that dsDNA is trafficked to TMVs via a mechanism distinct from the multivesicular body-dependent secretion reported for the extracellular release of cytosolic DNA. Furthermore, TMV dsDNA can be transferred to recipient cells with consequences to recipient cell behavior, reinforcing its relevance in mediating cell-cell communication.
Graphical Abstract
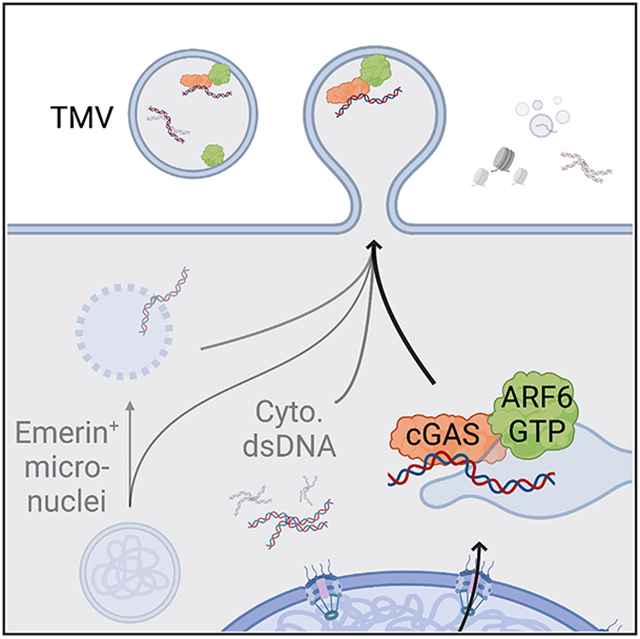
In brief
Clancy et al. show that a fraction of extracellular double-stranded DNA (dsDNA) is enclosed within tumor microvesicles (TMVs). dsDNA inclusion in TMVs is regulated by ARF6 GTP/GDP cycling and occurs with the cytosolic DNA sensor, cGAS. TMVs transfer dsDNA to recipient cells with consequences to recipient cell behavior. Created with BioRender.com
INTRODUCTION
It is now understood that cells of diverse origin release a range of nanometer- and micrometer-sized, membrane-bound sacs generically referred to as extracellular vesicles (EVs). The broad family of EVs shed from tumor cells, has expanded to include not only exosomes and microvesicles (MVs) but also large oncosomes, nanovesicles, and apoptotic bodies (Latifkar et al., 2019; Raposo and Stahl, 2019; Sheehan and D’Souza-Schorey, 2019; Becker et al., 2016). EVs are now understood to be crucial mediators of both cell-cell and cell-ECM interactions in both normal physiology and disease, in large part through the activity or transfer of their bioactive cargoes (Kalluri and LeBleu, 2020; D’Souza-Schorey and Schorey, 2018; Becker et al., 2016; Clancy et al., 2015b). EVs contain nucleic acids, functional proteases, oncoproteins, RNA binding proteins, guanosine triphosphatases (GTPases), integrins, cell surface receptors, lipids, and cytoskeletal components (Zhao et al., 2020; van Niel et al., 2018), although not all forms of EVs contain all types of cargo. Moreover, the type and quantity of cargo packaged within shed EVs is known to vary in accordance with the cell type from which the vesicles were derived (van Niel et al., 2018). Knowledge of the cargo content and mechanism of cargo delivery are important to understand the mode of biogenesis and function of the various EV subpopulations, yet they remain outstanding questions in the field.
Among the EVs derived from tumor cells, tumor MVs (TMVs) represent a distinct subtype characterized in part by their size (~≥200 nm), mode of biogenesis, and molecular cargo content (Clancy et al., 2021; Abels and Breakefield, 2016). TMV biogenesis begins with the direct outward budding and pinching of the plasma membrane, with tight control exerted over the utilization of intrinsic contractile machinery to facilitate pinching and release (Clancy et al., 2021; Sedgwick and D’Souza-Schorey, 2018). This actomyosin-based contraction is regulated by the activation of small GTPases, including ADP-ribosylation factor 6 (ARF6), RhoA, and Rab35 (Clancy et al., 2021). The activation of ARF6, for example, enables myosin light chain (MLC) phosphorylation via extracellular signal-regulated kinase (ERK) activation, to promote TMV release (Muralidharan-Chari et al., 2009). Similarly, Rab35 regulates the intracellular distribution of the actin bundling protein fascin to regulate TMV release (Clancy et al., 2019a).
Given the inclusion of proteins and nucleic acids in TMVs, it is likely that budding TMVs are a convergence point for multiple trafficking pathways (Clancy et al., 2021; Phinney et al., 2015). These pathways include the recently reported O-GlcNAcylation modification of the RNA binding protein hnRNPA2B1, resulting in incorporation into TMVs (Lee et al., 2019). Prior research had highlighted a ZIPcode-like sequence that targets specific miRNAs for delivery to MVs at the cell surface (Bolukbasi et al., 2012). In yet another study, the delivery of pre-microRNA (miRNA) to TMVs was shown to be facilitated via an endosome trafficking complex involving ARF6 and the nuclear export protein Exportin-5 (Clancy et al., 2019b). Together, these reports underscore the existence of pathways delivering both bulk miRNA, and targeted, sequence-specific trafficking and enrichment of certain miRNAs in MVs. ARF6, via its effect on endosomal membrane trafficking has also been found to facilitate the delivery of protein cargo to sites of TMV biogenesis. Membrane type 1-matrix metalloproteinase (MT1-MMP), for example, is directed to sites of nascent TMV formation through a specific association with the vSNARE VAMP-3 on ARF6-regulated endosomes (Clancy et al., 2015a). Thus, in addition to regulating actomyosin contraction and release of surface TMVs, endosomal ARF6 is able to direct multiple forms of cytoplasmic bioactive cargo to sites of TMV biogenesis by regulating chaperones, which themselves facilitate the movement and enrichment of specific cargo.
The degree to which DNA is included as EV cargo remains unsettled. Despite reports that EVs carry DNA cargo, including single-stranded and double-stranded molecules, retrotransposon elements, mitochondrial DNA, and genomic DNA (Vagner et al., 2018; Kalluri and LeBleu, 2016; Lee et al., 2014; Thakur et al., 2014; Balaj et al., 2011), DNA remains the least understood EV cargo. Several studies to date have provided evidence for DNA in exosomes (Diamond et al., 2018; Torralba et al., 2018; Takahashi et al., 2017; Kahlert et al., 2014; Lazaro-Ibanez et al., 2014; Thakur, et al., 2014), while additional studies demonstrated the internal location of DNA within EVs, by identifying a DNase-resistant pool of EV DNA that only becomes susceptible to degradation upon permeabilization of the lipid membrane (Torralba et al., 2018; Vagner et al., 2018). Moreover, Vagner et al. reported that the majority of cell-free DNA emitted by prostate cancer cells is carried by the larger EVs (Vagner et al., 2018). More recently, it was reported that small EVs do not contain DNA; rather, exosome-associated DNA is a consequence of active secretion of extracellular chromatin via multivesicular bodies (Jeppesen et al., 2019). Here, we show that following a high-resolution density-gradient centrifugation, a fraction of extracellular double-stranded genomic DNA is contained within TMVs, where it is protected from nuclease degradation and capable of being transferred to recipient cells and eliciting changes in gene expression. Furthermore, we demonstrate that the intracellular movement of DNA, together with the cytosolic DNA sensor cGAS, is dependent upon the cycling of the small GTPase ARF6. Dominant inhibition of ARF6, or depletion of cyclic guanosine monophosphate (GMP)-AMP synthase (cGAS) reduces the efflux of double-stranded DNA (dsDNA) from tumor cells. These results not only represent an important advance in the current understanding of DNA cargo recruitment mechanisms to TMVs but they are also significant in light of the uncertainty surrounding the inclusion of similar cargo in subpopulations of EVs.
RESULTS
TMVs contain DNA that is enclosed within membranes and protected from nuclease degradation
As stated above, the inclusion of dsDNA as EV cargo remains unsettled (Lu et al., 2018; Dror et al., 2016; McKenzie et al., 2016; Sakha et al., 2016; Shurtleff et al., 2016; Melo et al., 2014; Bolukbasi et al., 2012). Since previously published research suggested that larger EVs contain the bulk of the DNA cargo, we probed TMVs for the presence DNA. TMVs can be distinguished from exosomes by their size (Figures 1A and 1B) and cargo content (Figure 1C). TMVs, like exosomes, are susceptible to detergent-mediated disruption (Figure 1D), highlighting their membrane-bound extracellular organelle nature. The ability to isolate TMVs is crucial for the subsequent identification and examination of their internal cargo components. To begin addressing the question of DNA as TMV cargo, we isolated total DNA from TMVs derived from LOX melanoma cells treated with the promiscuous endonuclease benzonase, chosen for its ability to completely digest all forms of DNA and RNA to 5′-monophosphate terminated oligonucleotides, 3–5 bases in length. Qubit fluorometer analysis of benzonase-treated TMVs reveals that the inclusion of dsDNA within TMVs as co-treatment with Triton X-100 reduces the quantity of dsDNA recovered from treated samples (Figure 1E). Using multiple quantification methods, including qRT-PCR-based absolute quantitation, the Quant-iT high-sensitivity dsDNA assay, and digital droplet PCR (ddPCR), we found an average dsDNA content obtained from benzonase-treated TMVs across analysis methods of 5.609 × 10−6 pg/TMV (Figure 1F). Taken together, these results confirm the presence of measurable quantities of dsDNA within isolated TMVs.
Figure 1. Tumor microvesicles (TMVs) are distinct from exosomes, and contain DNA.

(A) TMVs and exosomes were isolated from LOX melanoma cells and allowed to adhere to poly-l-lysine-coated coverslips. Following fixation and dehydration, the vesicles were sputter coated and subjected to scanning electron microscopy. Scale bars, 500 nm.
(B) TMVs and exosomes were isolated from conditioned media of LOX melanoma cells by serial centrifugation. The isolated EV pools were then resuspended in 0.1-μm filtered PBS, analyzed by flow cytometry, and the data displayed as histograms for the individual fractions.
(C) TMVs and exosomes were isolated from conditioned media as described in Method details. Equal amounts of protein lysates from whole cells, TMVs, and exosomes were then separated by SDS-PAGE and the cargo contents were examined by western blotting.
(D) Isolated TMVs were incubated at 37°C for 2 h with or without 0.1% Triton X-100. Vesicles were then isolated from the mixture as described in Method details, and particle concentrations measured by flow cytometry. The data are presented as means, with whiskers indicating maximum and minimum values. n = 18. The p value was obtained by an unpaired 2-tailed t test.
(E) dsDNA was isolated from TMVs treated with benzonase, or bBenzonase + 0.1% Triton X-100. DNA was then quantified by the Qubit fluorometer. Detergent disruption reduces the amount of recoverable DNA. The data are presented as means ± SDs. n = 6. The p value was obtained by an unpaired 2-tailed t test.
(F) Absolute quantification by qRT-PCR, Quant-IT high-sensitivity dsDNA assay, and digital PCR confirms the presence of dsDNA within benzonase-treated TMVs. The data are presented as means, with whiskers indicating maximum and minimum values. n = 6. The p value was obtained by 1-way ANOVA.
DNA is protected from nuclease degradation within membrane fractions associated with TMV markers
Given that dsDNA was identified as cargo within TMVs, we sought to extend this analysis, including the examination of dsDNA content in TMVs from multiple tumor cell types. We used flow cytometry of benzonase-treated TMVs first stained with the cell permeant, dsDNA intercalating dye DRAQ5, which showed a strong DNA+ signal above background, particularly within larger vesicles (Figure 2A). To rule out the possibility that residual external dsDNA not digested by the nuclease was in part responsible for the fluorescent signal, we conducted paired flow cytometric analysis wherein untreated or benzonase treated LOX-derived TMVs were stained with cell permeant (blue fluorescent) or impermeant (green fluorescent) dsDNA dyes. Treatment with benzonase had no effect on relative fluorescence from the cell permeant stain, but significantly reduced the impermeant, external signal (Figure 2B). Similar results were obtained with TMVs derived from a panel of tumor cell lines, including additional melanoma lines (A375P and A375-MA2), as well as breast (MDA-MB-468, MDA-MB-231, and T47D), prostate (PC3), colon (SW480), and lung (H460) cancer cell lines (Figure S1). Furthermore, we sought to examine whether progression toward a more invasive/metastatic status led to the release of increased numbers of TMVs with increased DNA content. To this end, examination of the paired primary and metastatic melanoma cell lines A375P and A375-MA2, respectively, revealed an increase in DNA per TMV in the metastatic cell line (Figure 2C).
Figure 2. TMV genomic DNA is membrane protected and associated with TMV protein markers.

(A) TMVs were isolated from conditioned media and stained with the cell-permeant dsDNA dye DRAQ-5 or vehicle control. Fluorescent TMVs (containing dsDNA) were then detected by flow cytometry and plotted as size versus fluorescent intensity. Vehicle control used to determine baseline autofluorescence in DRAQ-5 channel.
(B) Control or benzonase-treated TMVs were stained with either the cell-permeable blue fluorescing dsDNA dye or the impermeable green fluorescing dye as described in Method details. Stained TMVs were then analyzed by flow cytometry, demonstrating the significant reduction in the number of vesicles carrying external DNA, stained with the impermeable dye, following treatment with benzonase. The data are presented as means ± SDs. n = 3. The p values were obtained by unpaired 2-tailed t tests for each independent condition.
(C) DNA isolated from an equivalent number of TMVs (measured by flow cytometry) shed by the paired primary and metastatic melanoma cell lines A375P and A375-MA2 was quantified by Quant-IT high-sensitivity dsDNA assay. The data are presented as means ± SDs. n = 3. The p values were obtained by unpaired 2-tailed t tests.
(D) LOX cells were fixed and stained as indicated. Confocal microscopy reveals the inclusion of dsDNA, stained with To-Pro-3 iodide, within budding TMVs.
(E and F) LOX cells were fixed and stained using a monoclonal antibody against dsDNA (E). Staining pattern reveals the inclusion of dsDNA as cargo within nascent TMVs, quantified in (F).
(G–I) TMVs isolated and treated with benzonase as described in Method details were overlaid onto poly-l-lysine-coated coverslips. These coverslips were then fixed, stained as indicated, and analyzed by confocal microscopy. The line plot in (G) shows the peak of To-Pro-3 iodide signal within the membrane.
(J) EVs isolated from iodixanol gradient fractions were analyzed by micro-flow cytometry. Ridge plot of histograms shown for each of the 11 fractions.
(K) EVs isolated from iodixanol gradient fractions were lysed, separated by SDS-PAGE, and EV content examined by western blotting.
(L) Genomic DNA was isolated from pooled EV fractions recovered following iodixanol gradient centrifugation and indicated nuclease treatments. DNA content was quantified by qRT-PCR as outlined in Method details. The data are presented as means ± SEMs. n = 5. The p value was obtained by ANOVA with Sidak’s correction for multiple comparisons.
Scale bars, 10 μm (D and E); 5 μm (F–I).
Immunofluorescent analysis of shedding cells revealed the inclusion of nucleic acids stained with the dsDNA dye To-Pro-3 iodide within budding TMVs (Figure 2D inset). Since To-Pro-3 iodide can exhibit low levels of binding to RNA, and to address the possibility of undetectable fluorescence when bound to short fragments of DNA, we confirmed the localization of dsDNA within budding TMVs using a dsDNA monoclonal antibody capable of binding fragments ≥16 bp in length. dsDNA can be found within nascent TMVs at the cell periphery, where it appears as putative cargo together with the known TMV cargo and regulator of TMV shedding, ARF6 (Muralidharan-Chari et al., 2009) (Figure 2E). Similar results were obtained when cells were counterstained with another TMV marker annexin-A1 (Jeppesen et al., 2019), both in LOX cells and in the aforementioned panel of melanoma, breast, prostate, and lung tumor cell lines (Figure S2). The quantification of dsDNA staining revealed detectable staining within 22.46% ± 2.54% (mean ± standard error) of budding TMVs (Figure 2F). DNA was also identified together with TMV markers by immunofluorescence within isolated and benzonase-treated TMVs using dsDNA dyes (Figures 2G and 2H) or dsDNA antibody (Figure 2I). The staining pattern, like the flow cytometry data, strongly suggests the presence of dsDNA within the membranes of shed TMVs (Figure 2G). We used a high-resolution iodixanol (Optiprep) gradient centrifugation procedure as previously described (Jeppesen et al., 2019) with an isolated TMV pellet as system input (Figure S3A). Flow cytometry of Optiprep gradient fractions revealed the appearance of a strong peak with a large-angle light scatter (LALS) profile similar to that of silica beads of 300–800 nm diameter. Scatter models have been shown to exhibit high correlation between the predicted scatter intensity of beads of known diameters and refractive index and the acquired scattered power when detected using a flow cytometer (Welsh et al., 2017). Flow cytometry detected a maximal concentration of TMV-sized particles in gradient fraction 5 (Figure 2J). Consistent with flow cytometry, western blot analysis of gradient fractions showed high concentrations of TMV markers centered around fraction 5 (Figure 2K). dsDNA was then isolated from pooled fractions 4–7 and 8–11 and analyzed by qRT-PCR. In both pools, treatment with benzonase reduced the detectable level of dsDNA. However, only in the fractions with detectable TMV markers is a portion of the DNA membrane protected and subsequently degraded upon detergent disruption of the encapsulating TMV (Figure 2L). Analysis of TMVs isolated from our panel of tumor cell lines revealed similar results when DNA was quantified by Quant-iT high-sensitivity dsDNA assay (Figure S3B). Furthermore, qRT-PCR-based analysis of more than 180 genes in 3 biologically independent samples of LOX TMV dsDNA revealed a heterogeneous mixture of genes, including some that were commonly detected (Figures S3C; Table S1). Taken together, these results indicate that dsDNA is bona fide cargo within TMVs and that tumor cells incorporate dsDNA as TMV cargo.
DNA does not traffic to nascent TMVs with mitochondria or amphisome components
The strong, punctate, cytoplasmic immunostaining for dsDNA, coupled with prior reports of mitochondrial fragments being shed in EVs (Zhao et al., 2020), suggested that mitochondria were a possible source of DNA within shed TMVs. Immunofluorescent analysis, however, revealed only a partial overlap between dsDNA and MitoTracker signals, with no signal contained within budding TMVs (Figure S3D). In addition, the identification of dsDNA within CD63-containing hybrid organelles, called amphisomes (Jeppesen et al., 2019), raised the possibility that this may represent a novel means through which dsDNA is trafficked to the cell periphery, and subsequently included within TMVs. Immunofluorescent localization of ARF6 and CD63, however, suggests that amphisomes are not the source of TMV dsDNA. ARF6 is readily detected within TMV lysates and can be identified within nascent TMVs by immunofluorescence. In contrast to ARF6, and in line with previous reports, CD63 is below detectable levels in TMVs (Clancy et al., 2019b; Sedgwick et al., 2015) (Figures 1C and 3A). Furthermore, analysis of the intracellular pools of ARF6 and CD63 reveals no robust colocalization, but rather residence on distinct pools within the endolysosomal system (Figure 3B). ARF6 within budding TMVs, however, can be localized together with dsDNA by immunofluorescence (Figure 3C). To further examine the possible contribution of amphisome components to TMV dsDNA content, we examined the TMV levels of histone H3. Only small amounts of histone H3 were detected in TMV or exosome lysates relative to lysates generated from the shedding cells (Figure 3D). Analysis of lysates generated by iodixanol fractionation showed that, in line with known TMV markers, histone H3 was predominantly found in fraction 5, with smaller amounts detected in the heavier fraction 6 (Figure 3E). Immunofluorescent localization of histone H3 shows significant co-staining for dsDNA, as one would expect within the nucleus. However, only a portion of the extrachromosomal, cytoplasmic dsDNA co-labels for histone H3 (Figure 3F, arrowheads), while other cytoplasmic dsDNA is not associated with histone H3 signal (Figure 3F, arrows) and only low levels of TMV histone H3 remained associated with dsDNA within budding TMVs (Figure 3G, arrowhead). Furthermore, we were unable to detect H3K9 trimethylated histone H3 in TMVs isolated by serial centrifugation alone, or following iodixanol gradient centrifugation (Figures 3D and 3E). Tri-methylation at H3K9 is a modification associated with heterochromatin structure and gene silencing (Hyun et al., 2017; Barski et al., 2007). Immunofluorescent analysis of melanoma cells revealed strong H3K9-trimethyl histone H3 staining in the nucleus but no detectable signal at the cell periphery (Figure S3E), suggesting that the DNA contained within TMVs is likely available for subsequent transcription and gene expression, with minimal binding to histones. Furthermore, the introduction of exogenous double stranded, PCR amplified cDNA encoding EGFP, resulted in the robust trafficking of this DNA for inclusion in shed TMVs, where it is protected from nuclease degradation and readily detected by qRT-PCR (Figure S3F). Similar to dsDNA, CD63 was found tightly apposed with cytoplasmic histone H3, in line with previous reports (Jeppesen et al., 2019) that describe chromatin fragments residing within similar multivesicular endosome-like (MVE) compartments, but not as a part of the same intraluminal vesicle (Figure 3H). These closely juxtaposed histone H3- and CD63-containing structures can be seen being released from the cell surface in areas absent of budding TMVs (Figure 3I, arrowheads). Taken together, these results indicate that amphisomes are not the source of TMV dsDNA.
Figure 3. TMV DNA is not associated with amphisome markers.

(A) LOX melanoma cells were fixed and stained to examine the intracellular localization of ARF6 (green) and CD63 (blue). CD63 is not detected at the cell periphery in nascent TMVs (inset).
(B) Intracellular pools of ARF6 (red) and CD63 (green) were imaged by confocal microscopy and colocalization measured using Pearson’s R (n = 20 cells from n = 3 biological replicates).
(C) LOX cells were fixed and stained as indicated. Confocal microscopy reveals the inclusion of dsDNA, together with ARF6, within budding TMVs.
(D) TMVs and exosomes were isolated from conditioned media as described previously. Equal amounts of protein lysates from whole cells, TMVs, and exosomes were then separated by SDS-PAGE and histone H3 content was examined by western blotting.
(E) EVs isolated from iodixanol gradient fractions were lysed, separated by SDS-PAGE, and histone H3 and trimethyl H3K9 content examined by western blotting.
(F) Histone H3 and dsDNA contents were examined by confocal microscopy in shedding melanoma cells. Histone near the nucleus co-labels with dsDNA (arrowheads), while punctate histone H3 nearer the cell periphery does not (arrows).
(G) Histone H3 and dsDNA contents were examined in nascent TMVs by immunofluorescence. Only a small pool of TMV-associated histone co-labels for dsDNA (arrowhead inset).
(H) Amphisome markers histone H3 and CD63 content were examined by immunofluorescence. Similar to previously published reports, histone H3 and CD63 can be identified juxtaposed where they are likely to reside within the same late endosomal structure (arrowheads).
(I) Orthogonal view of histone H3 and CD63 immunofluorescent image reveals co-trafficking of histone H3 and CD63 to the cell periphery where they are released without containment within TMVs.
Scale bars, 10 μm.
ARF6 activation regulates DNA efflux via TMVs
One possible mechanism for the generation of cytosolic dsDNA that has also been proposed as a possible source of EV dsDNA is the formation of rupture-prone micronuclei (Reis-Sobreiro et al., 2018; Hatch et al., 2013). In line with previous reports, the constitutive activation of the ARF6 small GTP-binding protein led to an increase in TMV shedding when quantified by flow cytometry (Figure S4). However, immunofluorescence analysis did not reveal an increase in the formation of micronuclei in response to ARF6 activation, and furthermore, the nuclear envelope appears largely intact as defined by the inner nuclear membrane protein emerin (Figures 4A and 4B). Western blot analysis showed no significant changes to the very low levels of emerin as TMV cargo in response to ARF6 activation, suggesting that intact micronuclei are unlikely to traffic into budding TMVs (Figure 4C). In line with these results, the activation of ARF6 did not increase the quantity of cytosolic DNA (Figure 4D). Interestingly, however, the comparison of TMVs released by LOXARF6–GTP cells with those from the parental cell line revealed a consistent decrease in dsDNA per TMV upon ARF6 activation (Figure 4E). These results suggest that ARF6 activation regulates the efflux of DNA through TMVs, and the lower dsDNA per TMV in LOXARF6–GTP cells may be a consequence of increased TMV shedding observed in these cells. Next, we conducted a time course experiment in which we measured the rate of exogenous dsDNA efflux following transfection with EGFP cDNA. Examination of DNA isolated from paired whole-cell (WCL) and TMV fractions over time revealed that ARF6 activation results in a more rapid decrease in double-stranded EGFP cDNA in cells and a corresponding more rapid increase in EGFP cDNA detected within shed TMVs (Figure 4F).
Figure 4. TMVs represent a mechanism of DNA efflux that is independent of micronuclei formation.

(A) LOX cells were fixed and stained for the inner nuclear membrane protein emerin to monitor for the presence of micronuclei. Scale bar, 25 μm.
(B) Percentage of LOX or LOXARF6-Q67L cells containing micronuclei were counted. The data represent 450 cells across 4 independent biological replicates and are presented as means ± SDs. n = 4. The p value was obtained by an unpaired 2-tailed t test.
(C) Whole-cell lysate and TMV lysates were generated from LOX and LOXARF6-Q67L cells. Equal amounts of protein lysates from whole cells and TMVs were then separated by SDS-PAGE and the emerin content was examined by western blotting. The relative TMV emerin content was quantified and graphed. The data are presented as means ± SDs. n = 4. The p value was obtained by an unpaired 2-tailed t test.
(D) Cytosolic DNA was isolated from LOX and LOXARF6-Q67L cells. Isolated DNA was then quantified by Quant-iT high-sensitivity dsDNA assay according to the manufacturer’s instructions. The data are presented as means ± SDs. n = 4. The p value was obtained by an unpaired 2-tailed t test.
(E) DNA isolated from TMVs shed from LOX or LOXARF6-Q67L cells was analyzed by qRT-PCR, and the quantity of DNA per TMV determined by absolute quantitation using control human genomic DNA. The data are presented as means ± SDs. n = 4. The p value was obtained by an unpaired 2-tailed t test.
(F) EGFP cDNA was transfected into LOX or LOXARF6-Q67L melanoma cells actively shedding TMVs. Following incubation with EV-free media for the times indicated, TMVs were isolated as described in Method details, and EGFP DNA within whole cells or TMVs was quantified by qRT-PCR. The data are presented as means ± SDs. n = 3. The p value was obtained by an unpaired 2-tailed t test with Bonferonni’s correction for multiple comparisons.
(G) Cytosolic DNA was isolated from LOX, LOXARF6-Q67L, and LOXARF6-T27N cells. Isolated DNA was then quantified using the Quant-iT high-sensitivity dsDNA kit. The data are presented as means ± SDs. n = 3. The p value was obtained by ANOVA with Sidak’s correction for multiple comparisons.
(H) Total dsDNA content was measured in media conditioned by LOX, LOXARF6-Q67L, and LOXARF6-T27N cells, following 24-h incubation in EV-free media. DNA quantification was determined by the Quant-iT high-sensitivity dsDNA kit. The data are presented as means ± SDs. n = 6. The p value was obtained by ANOVA with Sidak’s correction for multiple comparisons.
(I–K) Total dsDNA content was measured in media conditioned by LOX, LOXARF6-Q67L, and LOXARF6-T27N cells, following 24-h incubation in EV-free media. BeforeDNA quantification, media was subjected to 0.2 μm filtration alone or following 60-min incubation with 0.1% Triton X-100. DNA was then quantified by the Quant-iT high-sensitivity dsDNA kit and relative amounts of dsDNA were graphed. For each panel, the data are presented as means ± SDs. n = 3. The p value was obtained by ANOVA with Sidak’s correction for multiple comparisons.
Consistent with the above findings, biochemical analysis confirms increased levels of cytosolic DNA in ARF6-T27N cells (Figure 4G), indicating that the efflux of dsDNA is decreased, when ARF6 activation is blocked by the expression of the T27N mutant. Next, we examined the amount of dsDNA in conditioned media from parental, ARF6-Q67L, and ARF6-T27N cells following low-speed centrifugation to remove detached cells and other particulates, and 0.2 μm filtration to remove TMVs. The expression of ARF6-T27N led to a significant decrease in extracellular dsDNA, while ARF6-Q67L expression only partially reduced the detectable dsDNA (Figure 4H). Since ARF6-T27N cells do not shed meaningful numbers of TMVs (Muralidharan-Chari et al., 2009), we reasoned that the differences between the active and inhibitory mutants were likely due in part to dsDNA inclusion within TMVs, which are removed during filtration. Pre-treatment of conditioned media with Triton X-100 revealed that when compared to the parental cell line, in ARF6-Q67L cells, a higher proportion of effluxed dsDNA was contained within TMV-sized particles and only detected following detergent disruption of EVs (Figures 4I and 4J). In ARF6-T27N cells, only a few, if any, TMVs are released and lost during filtration (Figure 4K). Taken together, these data support the contention that ARF6 activation facilitates the efflux DNA via TMVs.
ARF6 activation regulates intracellular cGAS levels
As ARF6 is not known to directly bind nucleic acids, we hypothesized that it was instead facilitating the efflux of DNA-protein complexes. To investigate how ARF6 activity could affect dsDNA shuttling to the cell surface for inclusion into budding TMVs, we turned to the literature to examine which, if any, known DNA binding proteins have also been identified as ARF6 interactors. Apurinic/apyrimidinic endodeoxyribonuclease 1 (APEX1), the multifunctional DNA repair enzyme, is one such protein that was identified as an ARF6 binding partner through a large-scale mapping of protein-protein interactions (Ewing et al., 2007). Western blotting of TMV lysates revealed that although APEX1 is included within shed TMVs, there was no enrichment relative to the total cell. Instead, we observed a decrease in APEX1 in the presence of constitutively active ARF6, suggesting that its interaction with the GTPase would be unlikely in TMVs, as ARF6-GTP is highly enriched in shed TMVs (Muralidharan-Chari et al., 2009) (Figure S5A). It is possible, however, that DNA-APEX complexes efflux via TMVs.
With the strong cytoplasmic signal from dsDNA immunofluorescence (Figure 3C), we next examined possible roles for the cytosolic dsDNA sensor/receptor cGAS. Human cGAS is activated by the direct binding of dsDNA, with a preference for longer fragments that led to the formation of more stable, ladder-like oligomers of cGAS (Motwani et al., 2019; Zhou et al., 2018; Li et al., 2013). Western blot analysis revealed the consistent enrichment of cGAS within shed TMVs and that cGAS TMV levels were even further increased by the activation of ARF6 (Figure 5A). These findings prompted the further investigation of cGAS recruitment to TMVs. cGAS was recently reported to undergo liquid phase condensation, resulting in the formation of liquid-like droplets upon dsDNA binding, droplets with the potential to pellet along with TMVs following serial centrifugation (Du and Chen, 2018). To assess any potential contribution from phase droplets, we conducted demulsification experiments using media conditioned by cells expressing cytosolic GFP. Combined large EV pellets obtained following centrifugation of conditioned media at 2,000 × g and 10,000 × g were then subjected to demulsification by repeated cycles of freezing at −80°C and heating to 37°C before re-isolation by serial centrifugation. Demulsification results in the loss of soluble GFP from the 2,000 × g pellet without altering the associated cGAS content, suggesting that larger structures, which appear to lyse during freeze-thaw cycles, contain little cGAS cargo content (Figure 5B). TMVs, which are re-isolated by centrifugation at 10,000 × g, saw no appreciable loss of GFP or cGAS (Figure 5B). Together, these results indicate that cGAS is contained within TMVs, while phase droplets appear to pellet with other particulates such as apoptotic bodies.
Figure 5. ARF6 and cGAS coordinate DNA delivery to TMVs.

(A) TMVs were isolated from LOX and LOXARF6-Q67L cells and lysed. Equal amounts of TMV protein were separated by SDS-PAGE and probed for cGAS content by western blotting. Relative TMV cGAS levels were quantified and graphed. The data are presented as means ± SDs. n = 4. The p value was obtained by an unpaired 2-tailed t test.
(B) Equal numbers of large EVs were divided, with half being subjected to demulsification to monitor for the presence of cGAS liquid phase droplets. Following repeated cycles of freezing and heating, vesicle pellets were re-isolated and both cGAS and cytosolic GFP levels were examined by western blotting. The data are presented as means ± SDs. n = 3. The p value was obtained by an unpaired 2-tailed t test.
(C) cGAS preferentially binds active, GTP-bound ARF6 in vitro. Recombinant GST-WT-ARF6 conjugated beads were incubated with melanoma cell lysate in the presence of 100 μM GTP-γ-S, 1 mM GDP-β-S, or vehicle control, for 60 min at 37° C. Bound proteins were then precipitated, separated by SDS-PAGE for western blotting, and relative amounts of co-precipitating cGAS were quantified. The data are presented as means ± SDs. n = 4. The p value was obtained by ANOVA with Sidak’s correction for multiple comparisons.
(D) Immunofluorescence was used to examine the localization of endogenous cGAS in LOX, LOXARF6-Q67L, and LOXARF6-T27N cells. In parental and LOXARF6-Q67L cells, cGAS can be found at the cell periphery, where it is incorporated into shedding TMVs. Scale bars, 15 μm.
(E) Equal amounts of whole-cell lysate from LOX, LOXARF6-Q67L, and LOXARF6-T27N cells were separated by SDS-PAGE, and relative amounts cGAS were quantified by western blotting. The data are presented as means ± SDs. n = 4. The p value was obtained by ANOVA with Sidak’s correction for multiple comparisons.
(F) Whole-cell lysates from LOX, LOXARF6-Q67L, and LOXARF6-T27N cells treated for 6 h with ML-7, U0126, or vehicle control were separated by SDS-PAGE, and relative amounts of cGAS were quantified by western blotting. The data are presented as means ± SDs. n = 3. The p value was obtained by ANOVA with Sidak’s correction for multiple comparisons.
(G) Endogenous cGAS and dsDNA were examined by immunofluorescence. Dominant inhibition of ARF6 by ARF6(T27N) expression results in the accumulation of large, intracellular puncta containing both dsDNA and cGAS. Imaging parameters result in the overexposure of intracellular aggregates in LOXARF6-T27N cells. See Figure S5D for alternate exposure. Scale bars, 15 μm.
(H) Total dsDNA content was measured in TMVs isolated from LOXLCV–NT or LOXcGAS–KD cells (left), or from cytosol of LOX, LOXLCV–NT, or LOXcGAS–KD cells (left). DNA quantification was determined by the Quant-iT high-sensitivity dsDNA kit. The data are presented as means ± SEMs. n = 5 (TMVs) or 3 (cytosol). The p value was obtained by t test (TMVs) or ANOVA with Sidak’s correction for multiple comparisons (cytosol).
(I) TMVs were isolated from EdU-labeled LOXLCV–NT or LOXcGAS–KD cells and fixed to poly-l-lysine-coated coverslips as described in Method details. TMVs were fixed, stained, and imaged by confocal microscopy. Scale bar, 10 μm.
The increase in TMV cGAS content upon ARF6 activation suggested the possibility that ARF6 was directing the trafficking of cGAS into nascent TMVs, potentially through a direct interaction. To confirm this result, we precipitated ARF6 binding proteins from total cell lysates using recombinant wild-type ARF6 (WT-ARF6) conjugated to magnetic Sepharose beads, in which we were able to co-precipitate cGAS only with WT-ARF6 and not beads alone (Figure 5C). Furthermore, the amount of co-precipitating cGAS was increased in the presence of the non-hydrolyzable GTP-γ-S, and decreased in the presence of guanosine diphosphate (GDP)-β-S (Figure 5C). Immunofluorescence analysis of melanoma cells expressing constitutively active (ARF6-Q67L/ARF6-GTP) or dominant inhibitory (ARF6-T27N/ARF6-GDP) revealed subtle differences in cGAS localization. In the parental and Q67L cells, cGAS could be found at the cell periphery, where it can be seen within forming TMVs (Figure 5D, insets). Further investigation revealed differences in cGAS protein levels with alterations in ARF6 nucleotide cycling. Interestingly, expression of either ARF6-Q67L or ARF6-T27N reduced cGAS protein levels within the cell (Figure 5E). The reduction of cGAS in both instances led us to hypothesize that ARF6 is at least partially responsible for regulating intracellular cGAS trafficking, with a portion of cGAS being trafficked to TMVs for release, while inactivation of the GTPase blocks this trafficking and likely leads to some amount of protein degradation.
Since proteasomal and lysosomal degradation have been reported for cGAS (Han et al., 2020; Seo et al., 2018), we investigated the effects of inhibiting these pathways on cGAS levels in cells expressing ARF6-activated or dominant inhibitory mutants. Treatment of LOX, LOXARF6-Q67L, or LOXARF6-T27N melanoma cells with the proteasome inhibitor MG132 led to a slight and consistent increase in cGAS levels in all three cell lines, together with the accumulation of higher-molecular-weight, likely ubiquitinated cGAS (Seo et al., 2018) (Figure S5B, arrow). Treatment with chloroquine to inhibit lysosomal acidification rescued levels of cGAS in LOXARF6-T27N cells, but only partially restored levels in LOXARF6-Q67L cells in the same time frame (Figure S5C). Similarly, inhibition of the previously characterized release of TMVs downstream of ARF6 by small-molecule inhibitors against ERK and MLC activation, U0126 or ML-7, respectively (Muralidharan-Chari et al., 2009), led to increased levels of cGAS in parental and LOXARF6-Q67L cells but had little effect on the minimally shedding LOXARF6-T27N cell line (Figure 5F). Modulating ARF6 activation resulted in stark differences in dsDNA localization, particularly with the expression of ARF6-T27N, in which dsDNA can be seen accumulating in large peripheral aggregates, some of which are decorated with endogenous cGAS (Figures 5G, S5D, and S5E). Taken together, these results indicate that multiple trafficking pathways, including ARF6-regulated release of TMVs, govern intracellular cGAS levels.
cGAS regulates DNA abundance in TMVs
To confirm the role of cGAS in dsDNA trafficking to TMVs we used cGAS (LOXcGA–KD) or non-targeting (LOXLCV2–NT) control CRISPR-Cas9 techniques to knock down endogenous cGAS (Figure S5F). In line with previous reports, the loss of cGAS resulted in a decrease in cells expressing strong focal staining for phospho-H2A.X at Ser139 (Figure S5G) and an increase in the percentage of cells containing abnormal nuclear morphology (Figure S5H), micronuclei (Figure S5I), and/or intracellular dsDNA aggregates (Figure S5J) (Mohr et al., 2021; Liu et al., 2018; Mackenzie et al., 2017). Furthermore, cGAS knockdown led to a significant decrease in dsDNA included within shed TMVs, together with an increase in cytosolic dsDNA (Figure 5H), while having no effect on the amount of ARF6 trafficked as TMV cargo (Figure S5F). These results, together with the decrease in p-H2A.X levels, suggest that it is not simply an accumulation of damaged DNA resulting in TMV DNA cargo, rather that DNA trafficking is an active process. To investigate the incorporation of recently synthesized DNA, LOXcGAS–KD or LOXLCV2–NT cells were labeled with the thymidine analog 5-ethynyl-2′-deoxyuridine (EdU), which is incorporated during DNA synthesis. Newly synthesized EdU-labeled cytosolic DNA was then colocalized with cGAS (Figure S5K). Furthermore, EdU-labeled DNA was readily detected in TMVs isolated from control cells 12 h post-treatment, while labeled TMVs were virtually absent from LOXcGAS–KD cells (Figure 5I). Notably, the interrogation of publicly available The Cancer Genome Atlas (TCGA) data reveals that cGAS levels are increased in multiple tumor types (An et al., 2019). Furthermore, ARF6, the ARF6 exchange factor PSD4/EFA6B, and cGAS transcript levels are increased in melanoma metastasis relative to primary tumors (Figure S5L). While no strong correlation exists between ARF6 and cGAS transcript levels, this is not surprising given the myriad functions of the GTPase, in which it is activity, rather than bulk levels, that are critical for downstream signaling. However, it is worth noting that PSD4/EFA6B levels show a positive correlation with cGAS expression, further suggesting potential regulatory links underlying dsDNA sensing and endomembrane trafficking (Figure S5L).
TMV DNA can be transferred to recipient cells leading to increased anchorage-independent growth
EVs, including TMVs, have previously been shown to transfer protein and RNA cargo to recipient cells, altering the content and behavior of those cells (Clancy et al., 2019a, 2019b; Peinado et al., 2012; Skog et al., 2008). To determine whether TMVs were capable of transferring genomic DNA cargo to recipient cells, we made use of the SRY gene, encoding the sex-determining region Y protein together with our well-characterized model for TMV shedding, the melanoma cell line LOX, which originates from a male patient (Fodstad et al., 1988). DNA encoding SRY could be detected by qRT-PCR within TMVs susceptible to detergent disruption (Figure 6A). Publicly available TCGA data reports varying SRY expression levels in tumors of distinct tissue types, including higher expression in patients with prostate cancer and lower expression in melanoma patients. In line with these reports, using prostate carcinoma cell lines LNCaP or DU145 as controls, we were unable to detect SRY mRNA (Figure 6B) or protein (Figure 6C) within shed LOX TMVs, despite low levels of protein expression in the shedding cells. Moreover, we observed that the amount of TMV SRY DNA cargo is reduced in vesicles released from LOXcGAS–KD cells (Figure 6D). The addition of isolated TMVs to cultures of mammary cells (MCF-10A, MDA-MB-231) leads to the transfer of DNA containing the SRY gene, which could then be detected within recipient cells of female origin, revealing the TMV-mediated intercellular transfer of endogenous dsDNA (Figure 6E). TMV-mediated transfer of dsDNA to recipient mammary cells leads to the expression of SRY protein, detected by western blotting (Figure 6F). We have previously demonstrated that the transfer of TMV cargo leads to alterations in recipient cell protein expression and invasive capacity (Clancy et al., 2019a, 2019b). To investigate the possible contributions of dsDNA cargo, we examined the invasive capacity of recipient melanoma cells following incubation with control or cGAS knockdown TMVs. Similar to prior reports, incubation with LOX TMVs resulted in an increase in cells invading through Matrigel-coated Boyden chambers, and this increased invasion was reduced when invading cells were incubated with LOXcGAS–KD TMVs (Figure S6A). Similarly, treatment of the non-transformed mammary epithelial cell line MCF-10A with LOX TMVs led to an increase in growth rate, which, like invasive capacity, was mitigated when cells were incubated with TMVs from LOXcGAS–KD cells (Figures 6G and S6B). Of note, SRY expression has been linked to the survival of hepatocellular carcinoma cells in anchorage-independent conditions (Murakami et al., 2014), conditions which are believed to enrich for tumor initiating cells in breast cancer models (Dontu, et al., 2003). Treatment of MCF-10A cells with male TMVs leads to an increase in sphere-forming efficiency, with a concomitant expression of SRY protein (Figure 6H). SRY expression can be reduced, with a subsequent reduction in mammosphere-forming efficiency using small hairpin RNA (shRNA)-targeting SRY (Figure 6I). These results indicate that TMVs can serve as a vehicle for the intercellular transfer of cGAS-regulated cargo such as dsDNA, and that the transferred cargo can lead to alterations in recipient cells.
Figure 6. TMVs transfer dsDNA to recipient cells, leading to protein expression and behavioral changes.

(A) Using absolute quantification by qRT-PCR, SRY genomic DNA was identified and measured within TMVs treated with benzonase or co-treated with Triton X-100. The data are presented as means, with whiskers indicating maximum and minimum values. n = 9. The p value was obtained by an unpaired 2-tailed t test.
(B) Total RNA was isolated from 3 benzonase-treated independent pools of LOX, TMVs, or LNCaP cells. SRY mRNA was then examined by RT-PCR.
(C) SRY protein was examined by western blot in lysates from whole cells or isolated TMVs from the male cell lines DU145 or LOX.
(D) SRY genomic DNA is reduced when measured by qRT-PCR on equal numbers of benzonase-treated TMVs from LOX or LOXcGAS–KD cells. The data are presented as means, with whiskers indicating maximum and minimum values. n = 6. The p value was obtained by an unpaired 2-tailed t test.
(E) SRY genomic DNA can be detected by qRT-PCR in female mammary cell lines, MDA-MB-231, or MCF-10A, following incubation with male (LOX) TMVs. Maximal threshold cycle (Ct) count for each qRT-PCR run was set to 40, and SRY was undetected in untreated MDA-MB-231 or MCF-10A cells. The data are presented as means ± SDs. n = 5. The p value was obtained by an unpaired 2-tailed t test.
(F) SRY protein can be detected by western blotting in female cell lines, MDA-MB-231, or MCF-10A, when incubated with LOX TMVs.
(G) Incubation with LOX TMVs increases growth rate in MCF-10A cells, while loss of cGAS mitigates this increase. The data are presented as means ± SDs for each time point. n = 8. The p value was obtained by the Wilcoxon rank-sum test.
(H) Treatment with LOX TMVs leads to SRY protein expression and increases anchorage-independent growth as measured by mammosphere sphere-forming efficiency. The data are presented as means, with whiskers indicating maximum and minimum values. n = 12. The p value was obtained by an unpaired 2-tailed t test. Scale bar, 75 μm.
(I) SRY shRNA reduces TMV-mediated changes to anchorage-independent growth as measured by mammosphere sphere-forming efficiency and blocks expression of SRY protein in mammosphere cultures. The data are presented as means ± SDs. n = 12 (sphere-forming efficiency), n = 5 (protein expression). The p value was obtained by ANOVA with Sidak’s correction for multiple comparisons (mammospheres) or 2-tailed t test (protein levels).
DISCUSSION
The understanding of EV cargo is constantly evolving, which is in line with the rapid expansion of the field of EV biology as a whole. The question of nucleic acid cargo in particular is one that has garnered an increasing amount of attention, given its seemingly limitless potential as a target for therapy and liquid biopsy (Yu et al., 2021; Clancy and D’Souza-Schorey, 2018). As such, nucleic acid localization and recruitment to various EV subtypes is highly relevant as these characteristics have major ramifications with respect to their clinical utility. Here, we have shown that quantifiable amounts of genomic DNA are included as cargo within shed tumor cell-derived MVs released from the cell surface. The data further demonstrate that this cargo does not traffic with components of the amphisome or micronuclei. Our experiments identify a role for the small signaling GTPase ARF6 in altering the intracellular trafficking of the cytosolic DNA receptor cGAS and dsDNA, in which constitutive activation leads to the rapid release of endogenous or exogenous cytosolic DNA within TMVs, and dominant inhibition results in the buildup of cGAS and dsDNA, blocking efflux. We also present data demonstrating that TMVs can serve as a platform for the intercellular transfer of genomic DNA, highlighting their potential as packets of signaling information that facilitate the development and progression of disease states. The results described here indicate that the inclusion of dsDNA within shed TMVs is an active process facilitated by the interactions of cGAS with the ARF6-regulated endocytic machinery. They also provide insights to a more detailed understanding of EV subpopulations, and in particular, further our knowledge of TMV cargo and cargo trafficking, both of which stand to have profound impacts on human health and disease.
During tumorigenesis, the nascent tumor experiences some form of oncogenic stress, including, for example, DNA damage, which elicits a counter response and an attempt to eliminate the damaged cell. It was recently reported that ARF6 is one of multiple proteins regulating vesicular trafficking that are upregulated in response to the accumulation of UV-induced DNA damage in a Caenorhabditis elegans model (Edifizi et al., 2017). Our data suggest that the DNA damage-associated increase in ARF6 levels could represent one mechanism through which a cell attempts to remove unrepaired lesions in the genome. If, however, those cells are capable of evading the cell death-inducing and proliferation-limiting mechanisms that contain normal somatic cell expansion, they may well be primed to further facilitate tumor development and metastases, as indicated by examination of TCGA data.
In addition to the well-documented role of the cGAS-STING pathway in innate immunity, emerging research points to its roles during tumor development, where cGAS-STING activity is crucial to cancer immunity (Andzinski et al., 2016). Thus, the enrichment of cGAS in TMVs, as well as cGAS-mediated inclusion of DNA within shed TMVs, highlights a critical intersection between intracellular trafficking and intercellular communication. This is a particularly intriguing intersection with our data, demonstrating the translation and expression of transferred DNA, the lack of robust histone trafficking, and prior reports highlighting that nucleosome binding is a mechanism to prevent cGAS activation (Kujirai et al., 2020). Furthermore, research has, for example, indicated that initial engagement of the cGAS-STING pathway but subsequent inhibition during the latter stages both favored tumor growth (Yang et al., 2017). One could hypothesize, then, that tumorigenic changes result in increased levels of cGAS and ARF6 within the cell, while increased efflux in TMVs serves as a means to reduce their cellular levels. However, if the oncogenic insults overcome these mechanisms, then those same cells would be primed to use ARF6 to facilitate TMV-mediated invasion and metastasis. Furthermore, the inclusion of nucleic acid cargo opens the potential of TMVs to deliver driver mutations to neighboring cells or cargo components, which prime a metastatic niche or transform neighboring cells. Our results demonstrating abundant shedding of cGAS+ TMVs in response to elevated ARF6 activation, together with reports of elevated levels of cytosolic DNA found in many malignant cells (Wu et al., 2019; Bakhoum et al., 2018), raises the possibility that targeted inhibition of ARF6 may constitute a viable therapeutic avenue. In total, these results highlight the importance of a more detailed understanding of EV subpopulations, and in particular, a thorough knowledge of TMV cargo and mechanisms of cargo transfer, as they stand to have profound effects on human health and disease.
Limitations of the study
We recognize that there are limitations to the present study, most of which stem from the increasingly understood heterogeneity that exists within EV populations. While our results support the hypothesis that some pool of dsDNA is contained within TMVs, current techniques lack the sensitivity to accurately determine how that DNA is broadly distributed within individual vesicles that make up a TMV pellet. As is the case with protein components, it is unlikely that dsDNA is evenly distributed among TMVs, but more likely that some pool of TMVs contains a large percentage of the protected dsDNA. Furthermore, while our analysis did identify some genes consistently shed within TMVs, it was by no means exhaustive. Although beyond the scope of the present study, large-scale sequencing of multiple, highly purified TMV pellets from multiple cell types will help us better understand the distribution, trafficking, and consistency of dsDNA cargo. This work will also likely point to new trafficking pathways, and likely disease-specific cargo patterns as the sensitivity is greater than PCR-based approaches.
Accurate assessment of EV contents necessitates the precise separation of distinct vesicle and particle subclasses to appropriately attribute cargo and pathophysiological responses. Here, we have used a high-resolution Optiprep gradient to better purify the TMV samples used for downstream analysis. However, our results demonstrate the inclusion of smaller particles following gradient-based enrichment for TMVs, leaving the results and interpretation to some extent influenced by the carrying over of additional EV populations. Future refinements may warrant revisiting the precise constituents of a TMV pellet that contains dsDNA. This is a common challenge in EV biology and one that highlights the needs for better and more consistent isolation and purification methods.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources or reagents should be directed to and will be fulfilled by the lead contact, Crislyn D’Souza-Schorey (cdsouzas@nd.edu).
Materials availability
Plasmids generated in this study are available from the lead contact upon request.
Data and code availability
This paper analyzes existing, publicly available (The Cancer Genome Atlas) data. The accession numbers for the datasets used are listed in the key resources table.
Additional data reported in this paper will be shared by the lead author upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| β-Actin | Cell Signaling Technologies | Cat#4970, RRID:AB_2223172 |
| HA tag | Cell Signaling Technologies | Cat#3724, RRID:AB_1549585 |
| DsDNA | Abcam | Cat#ab27156, RRID:AB_470907 |
| Histone H3 (tri-methyl K9) | Abcam | Cat#ab8898, RRID:AB_306848 |
| β1-integrin | MilliporeSigma | Cat#AB1952, RRID:AB_91150 |
| α-tubulin | MilliporeSigma | Cat#T9026, RRID:AB_477593 |
| CD63 | System Biosciences International | Cat#EXOAB-CD63A-1, RRID:AB_2561274 |
| VAMP3 | ProteinTech | Cat#107021-AP, RRID:AB_2212628 |
| ARF6 | ProteinTech | Cat#20225-1-AP, RRID:AB_10646481 |
| Emerin | ProteinTech | Cat#10351-1-AP, RRID:AB_2100056 |
| APEX1 | ProteinTech | Cat#10203-1-AP, RRID:AB_2057927 |
| Annexin-A1 | ProteinTech | Cat#21990-1-AP, RRID:AB_11182596 |
| Flotillin | ProteinTech | Cat#15571-1-AP, RRID:AB_2106746 |
| Alix | ProteinTech | Cat#12422-1-AP, RRID:AB_2162467 |
| CD81 | ProteinTech | Cat#66866-1-Ig, RRID:AB_2882203 |
| TSG-101 | ProteinTech | Cat#28283-1-AP, RRID:AB_2881104 |
| Histone-H3 | ProteinTech | Cat#17168-1-AP, RRID:AB_2716755 |
| CGAS | ProteinTech | Cat#26416-1-AP, RRID:AB_2880507 |
| CD63 | Developmental Studies Hybridoma Bank | Cat#h5c6, RRID:AB_528158 |
| SRY | ProteinTech | Cat#67349-1-lg, RRID:AB_2882607 |
| GFP | ThermoFisher | Cat#33-2600, RRID:AB_2533111 |
| Donkey anti-mouse Alexa Fluor Plus 488 | ThermoFisher | Cat#A32766, RRID:AB_2762823 |
| Donkey anti-rabbit Alexa Fluor Plus 555 | ThermoFisher | Cat#A32794, RRID:AB_2762834 |
| Donkey anti-mouse Alexa Fluor Plus 680 | ThermoFisher | Cat#A32788, RRID:AB_2762831 |
| Donkey anti-rabbit Alexa Fluor Plus 800 | ThermoFisher | Cat# A32808, RRID:AB_2762837 |
| Bacterial and virus strains | ||
| NEB Stable | New England Biolabs | Cat#C3040I |
| Biological samples | ||
| Control human genomic DNA | ThermoFisher | Cat#360486 |
| Chemicals, peptides, and recombinant proteins | ||
| Alexa Fluor 555 Plus phalloidin | ThermoFisher | Cat#A30106 |
| Alexa Fluor 647 Plus phalloidin | ThermoFisher | Cat#A30107 |
| To-pro-3 iodide | ThermoFisher | Cat#T3605 |
| DRAQ-5 | ThermoFisher | Cat#62254 |
| Protease Inhibitor Cocktail | MilliporeSigma | Cat#P8340-5ML |
| Paraformaldehyde | Electron Microscopy Sciences | Cat#15713-5 |
| Poly-L-lysine | MilliporeSigma | Cat#P4832-50ML |
| Poly-HEMA | MilliporeSigma | Cat#P3932-10G; CAS#25249-16-5 |
| Matrigel | Corning | Cat#354230 |
| Horse Serum | ThermoFisher | Cat#16050122 |
| EGF | Peprotech | Cat#AF10015 |
| Hydrocortisone | MilliporeSigma | Cat#H0888-1G |
| Cholera Toxin | MilliporeSigma | Cat#C8052-2MG |
| Insulin | MilliporeSigma | Cat#I1822-100MG |
| B27 | ThermoFisher | Cat#17504044 |
| Glutaraldehyde (electron microscopy grade) | Electron Microscopy Sciences | Cat#16120 |
| GTP-γ-S | MilliporeSigma | Cat#G8634-1MG; CAS# 94825-44-2 |
| GDP-β-S | MilliporeSigma | Cat# G7637-5MG; CAS# 97952-36-8 |
| MG132 | MilliporeSigma | Cat#M8699-1MG; CAS# 1211877-36-9 |
| Chloroquine phosphate | Cayman Chemical | Cat#14194; CAS# 50-63-5 |
| U0126 | Cayman Chemical | Cat#70970; CAS# 109511-58-2 |
| Sulforhodamine B | MilliporeSigma | Cat#230162-5G; CAS# 3520-42-1 |
| Trichloroacetic Acid | MilliporeSigma | Cat#T6399-250G; CAS# 76-03-9 |
| Triton-X100 | Fisher Scientific | Cat#BP151-100 |
| Critical commercial assays | ||
| DNeasy Blood and Tissue Kit | Qiagen | Cat#69504 |
| BCA Protein Assay Kit | ThermoFisher | Cat#23227 |
| Ready Probes Cell Viability Imaging Kit | ThermoFisher | Cat#R37609 |
| Quant-iT High Sensitivity dsDNA Assay Kit | ThermoFisher | Cat#2129422 |
| SuperScript IV First-Strand Synthesis System with ezDNAse | ThermoFisher | Cat#18091150 |
| Monarch Total RNA Miniprep Kit | New England Biolabs | Cat#T2010S |
| Click-iT EdU Alexa Fluor 488 Cell Proliferation Kit | ThermoFisher | Cat#C10086 |
| Deposited data | ||
| The Cancer Genome Atlas | http://cancergenome.nih.gov/ | RRID:SCR_003193 |
| Experimental models: Cell lines | ||
| LOX | Fodstad et al. 1988 | N/A |
| H460 | ATCC | Cat#HTB-177, RRID:CVCL_0459 |
| MDA-MB-231 | ATCC | Cat#CRL-12532, RRID:CVCL_0062 |
| MDA-MB-468 | ATCC | Cat#HTB-132, RRID:CVCL_0419 |
| T-47D | ATCC | Cat#HTB-133, RRID:CVCL_0553 |
| PC-3 | ATCC | Cat#CRL-1435, RRID:CVCL_0035 |
| A375-P | ATCC | Cat#CRL-3224, RRID:CVCL_6233 |
| A375-MA2 | ATCC | Cat#CRL-3223, RRID:CVCL_X495 |
| 293FT | ThermoFisher | Cat#R70007 |
| DU145 | ATCC | Cat#HTB-81, RRID:CVCL_0105 |
| LNCaP | ATCC | Cat#CRL-1740, RRID:CVCL_1379 |
| MCF-10A | Joan Brugge | N/A |
| SW480 | ATCC | Cat#CCL-228, RRID:CVCL_0546 |
| Oligonucleotides | ||
| For complete list of oligonucleotides please see Table S1 | N/A | |
| Recombinant DNA | ||
| pEGFP-C1 | Clontech | Cat#6084-1 |
| LentiCRISPR v2 | Sanjana et al. 2014 | Addgene plasmid #52961; RRID:Addgene_52961 |
| psPAX2 | Didier Trono | Addgene plasmid #12260; RRID:Addgene_12260 |
| pMD2.G | Didier Trono | Addgene plasmid #12259; RRID:Addgene_12259 |
| pLKO.1-TRC | Moffat et al., 2006 | Addgene plasmid #10878; RRID:Addgene_10878 |
| pGEX-3X GST-wt-ARF6 | Schweitzer and D’Souza-Schorey 2005 | N/A |
| Software and algorithms | ||
| FIJI | www.imagej.net | Version 2.0.0-rc-69/1.52p |
| Graphpad Prism | www.graphpad.com | Version 9.2.0 |
| Microsoft Excel | https://www.microsoft.com/en-us/microsoft-365/excel | Version 16.16.27 |
| Histogram | Apogee Flow Systems; www.apogeeflow.com | N/A |
| UALCAN | Chandrashekar et al. 2017 | RRID:SCR_015827 |
| UCSC Xena | Goldman et al., 2020 | RRID:SCR_018938 |
| Other | ||
| Glutathione high capacity magnetic beads | MilliporeSigma | Cat#G0924-1ML |
| Exosome depleted FBS | ThermoFisher | Cat#A27208-01 |
| EquaFetal Serum | Atlas Biologicals | Cat#EF-0500-A |
| Hyclone FBS | Cytiva Life Sciences | Cat#SH30070.03 |
| Benzonase | MilliporeSigma | Cat#71206 |
| Immobilon PVDF-FL | MilliporeSigma | Cat#IPFL85R |
| Prolong diamond antifade mounting media | ThermoFisher | Cat#P36970 |
| Lipofectamine2000 Transfection Reagent | ThermoFisher | Cat#11668-027 |
| Micro flow cytometry calibration beads (Apogee Mix) | Apogee Flow Systems | Cat#1493 |
| Optiprep Density Gradient Medium | MilliporeSigma | Cat#D1555-250ML |
| ddPCR Supermix for probes (no dUTP) | BioRad | Cat#1863024 |
| TaqMan Fast Advance Master Mix | ThermoFisher | Cat#4444554 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
The LOX melanoma cell line was generated via the subcutaneous xenograft of cells originating from the lymph-node metastasis of a male patient with metastatic melanaoma and kindly provided by Prof. Oystein Fodstad, Oslo University (Fodstad, et al., 1988). H460 (male, RRID: CVCL_0459); MDA-MB-231 (female, RRID: CVCL_0062); MDA-MB-468 (female, RRID: CVCL_0419); SW480 (male, RRID: CVCL_0546); T-47D (female, RRID: CVCL_0553); PC-3 (male, RRID: CVCL_0035); DU145 (male, RRID: CVCL_0105); LNCaP (male, RRID: CVCL_1379), A375-P (female, RRID: CVCL_6233); and A375-MA2 (female, RRID: CVCL_X495) cell lines were originally purchased from ATCC. 293FT (female) cells were obtained from ThermoFisher Scientific. MCF-10A cells were kindly provided by Dr. Joan Brugge, Harvard University School of Medicine. LOX, T-47D, LNCaP, and H460 cells were cultured in RPMI supplemented with 10% (v/v) EquaFetal Serum, 2 mM L-glutamine and 100 U/mL penicillin-streptomycin, LNCaP with the addition of 1mM sodium pyruvate. PC-3 cells were grown in F-12K with 10% EquaFetal Serum and 100 U/mL penicillin-streptomycin. MDA-MB-231, MDA-MB-468, SW480, A375P, and A375-MA2 cells were grown in DMEM supplemented with 1 mM sodium pyruvate, 10% EquaFetal Serum, 2 mM L-glutamine, and 100 U/mL penicillin-streptomycin. 293FT cells were cultured according to manufacturer’s specifications in DMEM supplemented with 10% fetal bovine serum, 0.1 mM non-essential amino acids, 6 mM L-glutamine, 1 mM sodium pyruvate, 100 U/mL penicillin-streptomycin, and 500 μg/mL G418. DU145 cells were grown in EMEM supplemented with 1 mM sodium pyruvate, 10% EquaFetal Serum, 2 mM L-glutamine, and 100 U/mL penicillin-streptomycin.All cell lines were maintained in humidified incubators at 37°C with 5% CO2. MCF-10A cells were maintained in DMEM/F12 supplemented with 5% horse serum, 2 mM L-glutamine, 20 ng/mL EGF, 500 ng/mL hydrocortisone, 100 ng/mL cholera toxin, 10 μg/mL insulin, and 100 U/mL penicillin-streptomycin. Where indicated, cells were treated with MG132 (10 μM), chloroquine (50 μM), ML-7 (10 μM), or U0126 (30 μM) for 6 hours in complete culture media.
Antibodies and reagents
Antibodies to β-actin and HA tag were purchased from Cell Signaling Technologies. Antibodies against double stranded DNA and Histone-H3 (tri-methyl K9) were purchased from Abcam. β1-integrin and α-tubulin antibodies were purchased from MilliporeSigma. CD63 antibody used for immunoblotting was obtained from System Biosciences International. VAMP3, ARF6, Emerin, APEX1, Annexin-A1, Flotillin, Alix, CD81, TSG-101, SRY, and Histone H-3 antibodies, along with rabbit polyclonal antibody against cGAS, were purchased from Proteintech. Mouse monoclonal antibody against human CD63 (H5C6) used for immunofluorescence was purchased from the Developmental Studies Hybridoma Bank. Antibody against GFP was purchased from Life Technologies. Fluorophore conjugated secondary antibodies (donkey anti-mouse Alexa Fluor Plus 488, donkey anti-rabbit Alexa Fluor Plus 555, donkey anti-mouse Alexa Fluor Plus 680, and donkey anti-rabbit Alexa Fluor Plus 800), rhodamine phalloidin, Alexa Fluor 647-phalloidin, To-Pro-3 iodide, Nuc Green stain, and DRAQ-5 were purchased from Life Technologies. High capacity magnetic sepharose beads were purchased from MilliporeSigma. Unless noted, all other chemicals were purchased from V.W.R.
METHOD DETAILS
TMV isolation
Cells for TMV isolation were plated to reach ~80% confluency at the time of conditioned media collection. Prior to TMV isolation, cells were washed twice with sterile filtered PBS, and media replaced with complete growth media made with EV-free serum (Life Technologies). Conditioned media was collected from cells, and the culture vessel rinsed 1× with sterile PBS which was subsequently added to the collected media. The mixture was then centrifuged for 15 minutes at 300 g to remove cells and large debris. The supernatant was then subjected to centrifugation for 20 minutes at 2,000 g to pellet apoptotic bodies. TMVs were then isolated by centrifugation at 10,000 g for 30 minutes. The resulting pellet was washed in sterile PBS 2 times before a final isolation prior to downstream use. 10,000 g supernatants were centrifuged for an additional 60 minutes at 10,000 g, before being centrifuged at 100,000 g for 60 minutes to isolate exosomes.
dsDNA and RNA isolation
TMVs, isolated as described above, were resuspended in 100 μL of 0.1 μm filtered PBS. Where indicated, TMVs were treated with 50 U/mL of Benzonase alone, or with 0.1% Triton X-100 and mixed by gentle vortexing. The mixtures were then incubated at 37°C for 2 hours. Following incubation, the treated TMVs were cooled on ice, diluted 10-fold with additional filtered PBS, and vesicles re-isolated by centrifugation at 10,000 g for 30 min at 4°C. When examining the effect of Triton-X100 alone on DNA, dsDNA was isolated directly from the reaction mix without re-isolation. DNA was isolated using the DNeasy Blood and Tissue kit (Qiagen) and eluted into 100 μL according to manufacturer’s protocol. Total RNA was isolated from Benzonase treated TMVs or total cells using the Monarch Total RNA Miniprep Kit according to manufacturer’s protocol, including DNAse digest step.
Iodixanol (optiprep) gradient centrifugation
TMVs were isolated as described above, resuspended in ice-cold 0.1 μm filtered PBS and mixed with Optiprep (MilliporeSigma) to a final concentration of 36% iodixanol. This mixture was then carefully added to the bottom of an ultracentrifugation tube. Additional solutions of descending concentrations of iodixanol in PBS were carefully layered to form a discontinuous gradient, with the lightest fraction being 12% iodixanol as previously published (Jeppesen, et al., 2019). Gradients were centrifuged at 100,000 g for 16 hours at 4°C. 11 individual fractions of 1mL were collected from the top of the gradient and added to new centrifuge tubes. Fractions were diluted to 10 mL with additional ice cold 0.1 μm filtered PBS before being centrifuged at 10,000 g and the resulting pellets used for downstream analysis.
Western blotting
For western blotting, cells were lysed in buffer containing 150 mM NaCl, 25 mM Tris-HCl pH 7.6, 1% sodium deoxycholate, 0.1% SDS, and 1% Triton X-100 (all purchased from VWR). Just prior to use, mammalian protease inhibitor cocktail (Millipore Sigma was added to complete the buffer. Triton insoluble fractions were removed by centrifugation at 15,000 g for 15 minutes. Cleared lysates were quantified by BCA assay (Pierce) and equal amounts of protein mixed with 4× SDS loading dye and incubated in a boiling water bath for 6 minutes. Lysates were then separated by SDS-PAGE. Separated proteins were then transferred to PDVF-FL membrane (Millipore Sigma). Non-specific binding was blocked by incubation in 5% nonfat milk in TBS for 2 hours at room temperature. Primary antibody incubation was carried out in 2% nonfat milk in TBS +0.2% Tween-20 overnight at 4°C with gentle rocking. Membranes were washed (4 × 5 min washes) in TBS +0.1% Tween 20 before incubation with Alexa Fluor Plus secondary antibodies in 2% nonfat milk in TBS +0.2% Tween-20 for 1 hour at room temperature. Membranes were again washed (4 × 5 min washes) in TBS +0.1% Tween 20 before being imaged using a LiCor Odyssey scanner.
PCR
For 2 step reverse transcription PCR, cDNA was first generated by assembling 15 μL reactions containing 100 ng of total RNA using the SuperScript IV First-Strand Synthesis System with ezDNAse (ThermoFisher). PCR was then carried out using TaqMan probe sets (key resources table and Table S1) containing 5 μL of cDNA reaction, and cycled according to manufacturer’s recommendations. PCR products were analyzed using 8% TBE polyacrylamide gels. Quantitative RT-PCR (qRT-PCR) was performed on a Step-One-Plus real-time PCR machine (Applied Biosystems) and analyzed by the comparative CT method, or absolute quantification as indicated in relevant Figures. Amplification was monitored using TaqMan probes listed in key resources table, and reactions were cycled according to manufacturer’s instructions. For absolute quantification, control human genomic DNA was obtained from Life Technologies and used to generate standard curves. For digital PCR, 22 μL reactions were assembled containing 10 ng of dsDNA, ddPCR Supermix for probes (no dUTP) (BioRad), and TaqMan probes (see key resources table and Table S1) according to manufacturer’s specifications. The reactions were partitioned into droplets using the QX200 droplet generator before being cycled in a C1000 Touch thermocycler (BioRad). Reaction conditions were 95°C for 10 minutes; [94°C for 30 seconds, 60°C for 60 seconds] x 45 cycles; 98°C for 10 minutes before being cooled to 4°C. Droplets were then read using a QX200 droplet reader (BioRad).
Immunofluorescence
Cells were plated on glass coverslips, TMVs plated on poly-L-lysine (MilliporeSigma) coated coverslips, and allowed to adhere for a minimum of 18 hours before being fixed in 2% PFA (Electron Microscopy Supply). Fixed cells were washed 3 times with filtered 1 × PBS; pre permeabilized with 1 × PBS +0.3% Triton X-100; blocked and permeabilized in 5% BSA, 0.2% Triton X-100, 0.05% Tween 20 in 1 × PBS; and incubated with primary antibodies diluted in blocking buffer as indicated in Figures. Cells were then incubated with fluorophore conjugated secondary antibodies together with rhodamine phalloidin Alexa Fluor 647 phalloidin, Nuc Green, or ToPro-3 iodide as indicated in Figures (Life Technologies). Coverslips were mounted using anti-fade gold mounting media (Life Technologies). To evaluate colocalization, background was subtracted from images, and nuclei segmented and removed by hand. The resulting images were then analyzed using the Coloc_2 function in ImageJ.
EGFP cDNA experiments
EGFP cDNA was PCR amplified from pEGFP-C2 plasmid DNA with the following primers: FWD 5′-CACGATGATGATATCGCCAC-3′; REV 5′-TATTGATCTAGAGTCGCGGC-3′. The resulting PCR product was gel purified and quantified using a Qubit fluorometer. 150,000 cells were transfected with 50 ng of EGFP PCR product using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s protocol. 16 hours post transfection, cells were rinsed extensively with sterile PBS, and EV-free growth media added to the wells. After 24 hours, TMVs were isolated, treated with nuclease, DNA extracted as described above, and examined by qPCR. For time course experiments, DNA was extracted from TMVs or whole cells, as described, at the indicated times post addition of EV-free growth media. Equal amounts of isolated DNA was used as input for qRT-PCR.
Isolation of cytosolic DNA fractions by sucrose density-gradient centrifugation
Cytosolic DNA was prepared by modifying a previously published method (Kawashima, et al., 2011). Cells were rinsed extensively in sterile filtered PBS, removed from the plate by scraping, and collected in a 1.5 mL microcentrifuge tube. Cells were centrifuged for 1 min in a microcentrifuge, the supernatant discarded, and the pelleted resuspended in 500 μL of 0.3 M sucrose, 2% Tween-40, 10 mM HEPES KOH (pH 7.9), 10 mM KCl, 1.5 mM MgCl2, and 0.1 mM EDTA and homogenized by pipetting 50 times using a P200 tip. The homogenate was overlaid onto 500 μL of 1.5 M sucrose, 10 mM HEPES KOH (pH 7.9), 10 mM KCl, 1.5 mM MgCl2, and 0.1 mM EDTA and centrifuged at 13,200 rpm for 10 min. The upper layer (~400 μL) was transferred to another tube, and cytosolic DNA was purified using the Qiagen kit described previously.
Sulforhodamine B (SRB) cell growth analysis
Active log-phase cultures of MCF-10A cells were trypsinzed, resuspended in growth media, and counted. 1,500 cells were plated per well in 96 well plates, in 200 μL of growth media. 3 wells were plated per condition per time point. Cells were allowed 6 hours to adhere before beginning the assay. At each time point, media was removed from the wells, and cells were fixed in 75 μL of 10% trichloroacetic acid for 1 hour at room temperature. The wells were then rinsed 5 times with 150 μL of distilled water and allowed to dry completely. Dried plates can be stored, covered, at room temperature until the file time point to allow for simultaneous staining. Once completely dry, cells were stained with 75 μL of 0.4% SRB in 1% acetic acid for 1 hour at room temperature. Wells were rinsed 5 times with 150 μL of 1% acetic acid and allowed to dry completely. SRB is solubilized by the addition of 100 μL of 10mM TRIS to each well. Plates are incubated for 20 minutes at room temperature, tapped gently to insure solubilization and 554 nm absorbance read using a plate reader.
In vitro ARF6-cGAS pulldown
GST-wt-ARF6 plasmid was described previously (Schweitzer and D’Souza-Schorey, 2005). Recombinant ARF6 protein was conjugated to glutathione high capacity magnetic agarose beads according to manufacturer’s instructions. LOX cells, grown to 80% confluency, were lysed in co-immunoprecipitation buffer (50 mM Tris-HCl pH 7.4, 500 mM NaCl, 10 mM MgCl2, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, with mammalian protease inhibitor added just prior to use) for 15 minutes at 4°C and detergent insoluble fractions removed by centrifugation at 15,000 g for 15 minutes. While lysing cells, 50 μL of 50% bead slurry per sample were blocked in lysis buffer with 5% BSA for 10 minutes at 4°C with gentle inversion. Following blocking, beads were rinsed twice with lysis buffer. 400 μg of total protein in pre-cleared lysates (diluted as needed to 1 μg/μL with additional lysis buffer) were added to blocked beads together with 100 μM GTP-γ-S, 1 mM GDP-β-S, or vehicle control for 60 min at 37°C. Following protein binding, beads were washed 3× with 1 mL of ice-cold lysis buffer, resuspended in 1X SDS sample buffer and boiled for 5 minutes, separated by SDS-PAGE, and probed by western blotting.
cGAS liquid-phase droplet analysis
Large EVs (2,000 g and 10,000 g pellets) were isolated from cells transfected with pEGFP-C1 to express unconjugated cytosolic GFP. The pellets were resuspended in 0.1 μm filtered PBS and combined to a total volume of 1mL. The sample was then divided into 2, 500 μL fractions. One fraction was subjected to demulsification by 5 cycles of freezing (−80 C) and heating (37 C) for 5 minutes. Both groups were then subjected to differential centrifugation and pellets isolated at 2,000 g, 10,000 g, and 20,000 g. Pellets were then lysed, separated by SDS-PAGE, and both cGAS and GFP levels examined by western blotting.
Scanning electron microscopy
Isolated TMVs or isolated exosomes were incubated overnight on poly-L-lysine (0.1% w/v) coated coverslips in naive EV-free culture media. Samples were fixed in 2% electron microscopy grade glutaraldehyde (Electron Microscopy Sciences) diluted in SEM buffer (0.1M sodium cacodylate, pH 7.5) for 60 minutes at room temperature. Samples were rinsed (3 × 10 minutes) in SEM buffer prior to fixation in 1% osmium tetroxide (in SEM buffer) for 60 minutes at room temperature. Following a second rinsing step (3 × 10 minutes) in fresh SEM buffer, samples were taken through a series of dehydrating rinses in 50%, 70%, 80%, 95%, and 100% EM grade ethanol (2 × 10-minute incubations each). Samples in 100% ethanol were subjected to critical point drying by replacing ethanol with 100% liquid CO2 and bringing the CO2 to its critical point. Dried coverslips were mounted to SEM stubs (Ted Pella Inc.) using carbon tape and conductive silver paste. Samples were sputter coated with iridium to a thickness of 4 nm and imaged using a Magellan 400 (FEI) scanning electron microscope.
TMV-mediated DNA transfer
TMVs were isolated as described above, resuspended in filtered PBS, and a small sample quantified using flow cytometry. Equal numbers of TMVs at a ratio of 50:1 TMVs:recipient cells, were pelleted and resuspended in naïve EV free cell culture media, and added to recipient cells in culture. Cells were incubated for 36 hours before being used for PCR or Western blot experiments. For growth curve analysis, cells were incubated with TMVs as described above, for 24 hours prior to plating in 96 well plates. Additional TMVs, resuspended in naïve media at a ratio of approximately 50:1 TMVs:cells, were added to the growth media at the time of plating. For mammosphere analysis, cells were incubated with TMVs using the conditions outlined above, for 48 hours in complete growth media prior to being plated in mammosphere suspensions. Additional TMVs, resuspended in naïve media at a ratio of approximately 50:1 TMVs:cells, were added to the growth media at the time of plating.
Flow cytometry analysis
Isolated TMVs were washed extensively and resuspended with 0.1 μm filtered PBS. Resuspended samples were then analyzed by flow cytometry using a Micro flow cytometer (Apogee Flow Systems). Calibration gates were determined using a mixture of silica beads at 180, 240, 300, 590, 880, and 1300 nm diameters. Positive fluorescent signal was measured as increase above background levels detected using unstained control samples. Data analysis was carried out using Histogram Software (Apogee Flow Systems).
Cell permeable and impermeable staining of TMVs
TMVs were isolated as described above and resuspended in filtered PBS. TMVs were then stained with either NucBlue Live or NucGreen Dead reagents (Ready Probes Cell Viability Imaging Kit, Thermofisher) for 2 hours at room temperature with gentle inversion, according to manufacturer’s specifications. Stained TMVs were centrifuged, washed with additional filtered PBS, and analyzed by flow cytometry.
Quant-iT DNA analysis
TMVs were isolated from individual cell lines and DNA extracted from the resulting pellets as described above. DNA was quantified using the Quant-iT High Sensitivity dsDNA Assay kit (Thermofisher) in 96 well format. The assay was carried out according to manufacturer’s instructions. The assay is highly selective for double-stranded DNA over RNA, and in the range of 0.2–100 ng of DNA, the fluorescence signal is linear. Samples were measured in technical triplicates.
Click-iT EdU labeling
Click-iT EdU labeling was conducted using the Click-iT EdU Alexa Fluor 488 Cell Proliferation Kit for Imaging (ThermoFisher) according to manufacturer’s instructions. Following 60-minute incubation with EdU label, cells were washed extensively and the media replaced with EV-free complete cell culture media to allow for TMV shedding. TMVs were isolated as described above, and plated on poly-L-lysine coated coverslips for imaging. Cells and TMVs were fixed and subjected to Click-iT reaction according to manufacturer’s recommended protocol before staining with primary and secondary antibodies as described above.
Short hairpin RNA
shRNA against SRY was generated as described by Addgene, from whom the pLKO.1-TRC cloning vector was sourced. A short hairpin RNA targeting SRY mRNA was subcloned into the lentiviral expression plasmid pLKO.1-TRC using the primer sequences listed in key resources table. The oligo nucleotides were annealed by placing in a 1-liter water bath heated to 95°C, and allowing the bath to cool slowly to room temperature. The annealed oligos were then ligated between the AgeI and EcoRI sites of the pLKO.1-TRC-cloning vector. Positive clones were screened and validated by sequencing. Lentiviral particles were generated as described below, and the knockdown of SRY was validated using DU145 and LNCaP cell lines prior to use in MCF-10A cells. Following infection, MCF-10A cells expressing SRY shRNA were selected using 2μg/mL puromycin for 14 days, and maintained in culture at 1 μg/mL.
CRISPR/Cas9 knockdown
Guide RNA sequence targeting cGAS along with non-targeting control were selected from the GECKO library (sequences listed in key resources table) and cloned into the LentiCRISPR v2 plasmid, a gift from Feng Zhang (Addgene plasmid # 52,961; http://n2t.net/addgene:52961; RRID:Addgene_52961) as previously published (Sanjana, et al., 2014). LOX cells were infected as described below, and grown for 3 days before being selected with 400 ng/mL puromycin. Following selection, cGAS levels were monitored by western blotting.
Lentiviral production and infection
5 × 105 293FT cells were plated in 10 cm dishes in complete growth media without antibiotics and allowed to adhere for 24 hours. Transfection mix was prepared using 9 μg of psPAX2, 3 μg of pMD2.G, 3 μg of lentiviral transfer plasmid, and 36 μL of Lipofectamine 2000 (ThermoFisher) according to the manufacturer’s protocol and added dropwise to cells in 5 mL of complete growth media without antibiotics. Cells were incubated with transfection mix for 6 hours at which time media was replaced with 10 mL of complete media without antibiotics to allow viral shedding. Conditioned media harvested 48 hours post transfection. Viral supernatant was centrifuged at 3000 rpm for 15 minutes, filtered through 0.45 μm PVDF syringe filters, aliquoted, and stored at −80°C until use. To infect tumor cells, viral aliquots were thawed on ice, mixed 1:1 with tumor growth media without antibiotics, and added to cells for 24 hours. Polybrene was added at a final concentration of 8 μg/μL at the time of infection.
Mammosphere assay
To prevent cell adhesion and promote suspension culture, 12 well tissue culture plates were coated in poly(2-hydroxyethyl methacrylate) (poly-HEMA). 1.2% (w/v) poly-HEMA solution was generated by dissolving 0.6 g poly-HEMA in 95% ethanol and allowing to rock overnight at room temperature. 270 μL of poly-HEMA solution was added to each well of the plate and gently rocked to ensure coverage. The plates were allowed to dry uncovered in a tissue culture hood overnight before being coated again to ensure full coverage of the tissue culture plastic. Actively growing cultures were trypsinzed, resuspended in culture media and counted to allow plating of 3000 cells/well. Resuspended cells were then centrifuged at 500 g for 5 minutes and resuspended in mammosphere assay media (growth media, 2% B27, 20 ng/mL EGF) and passed through a 25G needle to ensure formation of a single cell suspension. Single cell suspension cells were then divded and added to wells containing 1.5 mL of assay media. Mammosphere plates were placed in an incubator and allowed to grow for 96 hours before being imaged and mammospheres (spheres greater than 50 μm in diameter) counted. Sphere forming efficiency is calculated as [(Mammosphere#/Plated cell#)*100].
Matrigel coated boyden chamber invasion assay
Transwell inserts (Corning) with 8 μm diameter pores were coated with Matrigel as recommended by the manufacturer. Briefly, for 24 well tissue culture plate inserts, Matrigel was diluted to a final concentration of 500 μg/mL in ice cold coating buffer (0.01M Tris (pH 8.0), 0.7% NaCl sterile filtered). Transwell inserts were placed into tissue culture plates, and 100 μL of coating Matrigel mix added to the upper chamber of the transwell. Plates were incubated at 37°C for 2 hours, before any remaining coating buffer was removed, leaving polymerized Matrigel on the upper surface of the transwell membrane. Trypsinze and count cells, and centrifuge appropriate volume to yield sufficient cells to plate 2,500 cells/well. Resuspend cells in serum free growth media (2,500 cells/200 μL). Add 200 μL of cell mid to each transwell. Add 750 μL of complete growth media to the lower chambers. Cap and place in the incubator. Following invasion, remove non-invading cells using a moistened cotton swab and gently rubbing the upper surface of the transwell membrane. Repeat with a fresh cotton swab. Fix and stain the transwells by adding appropriate solutions (see immunofluorescence section) to the upper and lower chambers. Remove the transwell, place on microscope slide invading cell side up, cover with mounting media, place coverglass, seal with nail polish, and image.
QUANTIFICATION AND STATISTICAL ANALYSIS
Relevant statistical details are included in individual figure legends. Statistical analyses were performed using GraphPad Prism 9 or Microsoft Excel version 16.12. Student’s t test was used when comparing two groups with data that appeared to be normally distributed with similar variances. When comparing multiple treatment groups to a single control and the data were normally distributed, Bonferonni’s correction for multiple comparisons was employed. When multiple groups were being analyzed and each group was compared to all other groups and the data appeared normally distributed, we performed a one-way ANOVA with Sidak’s correction for multiple comparisons. To analyze growth curves, data was evaluated using Wilcoxon’s rank sum analysis. No outliers were excluded from analysis and all experiments were performed on at least N = 3 biological replicates under similar conditions. For imaging experiments, representative images are shown, with conclusions or quantification drawn from at least n = 10 fields of view acquired from each of at least N = 3 biological replicates. Note: where indicated in figure legends n = individual cell number or other relevant metric from individual biological replicates. In order to present relative data, control sample means from biological replicates R1, R2,…RN were averaged to yield the normalization value Z. Biological replicate means from control (R) and experimental (S) conditions were then normalized to the value Z (eg. R1/Z, R2/Z,…RN/Z; and S1/Z, S2/Z,…SN/Z), which results in control values having a normalized average of 1, while maintaining the replicate variance and allowing for statistical comparisons.
Supplementary Material
Highlights.
Extracellular dsDNA is protected from nuclease degradation in TMV membrane fractions
DNA inclusion in TMVs occurs with cGAS, but not amphisome or micronuclei components
DNA efflux to TMVs is enhanced by ARF6 activation
TMV dsDNA transfer to recipient cells provokes cellular responses in recipient cells
ACKNOWLEDGMENTS
This work was supported in part by funds from the National Cancer Institute (UG3CA241684) and the Boler Foundation. A.C.B. is supported by a predoctoral fellowship from the Berthiaume Institute for Precision Health at the University of Notre Dame.
INCLUSION AND DIVERSITY
We worked to ensure diversity in experimental samples through the selection of the cell lines. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110443.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abels ER, and Breakefield XO (2016). Introduction to extracellular vesicles: biogenesis, RNA cargo selection, content, release, and uptake. Cell Mol. Neurobiol 36, 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An X, Zhu Y, Zheng T, Wang G, Zhang M, Li J, Ji H, Li S, Yang S, Xu D, et al. (2019). An analysis of the expression and association with immune cell infiltration of the cGAS/STING pathway in pan-cancer. Mol. Ther. Nucleic Acids 14, 80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andzinski L, Spanier J, Kasnitz N, Kroger A, Jin L, Brinkmann MM, Kalinke U, Weiss S, Jablonska J, and Lienenklaus S (2016). Growing tumors induce a local STING dependent type I IFN response in dendritic cells. Int. J. Cancer 139, 1350–1357. [DOI] [PubMed] [Google Scholar]
- Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et al. (2018). Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaj L, Lessard R, Dai L, Cho YJ, Pomeroy SL, Breakefield XO, and Skog J (2011). Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun 2, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, and Zhao K (2007). High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837. [DOI] [PubMed] [Google Scholar]
- Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, and Lyden D (2016). Extracellular vesicles in cancer: cell-to-cell mediators of metastasis. Cancer Cell 30, 836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolukbasi MF, Mizrak A, Ozdener GB, Madlener S, Strobel T, Erkan EP, Fan JB, Breakefield XO, and Saydam O (2012). miR-1289 and "Zipcode"-like sequence enrich mRNAs in microvesicles. Mol. Ther. Nucleic Acids 1, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi B, and Varambally S (2017). UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 19, 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy J, and D’Souza-Schorey C (2018). Extracellular vesicles in cancer: purpose and promise. Cancer J. 24, 65–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy JW, Schmidtmann M, and D’Souza-Schorey C (2021). The ins and outs of microvesicles. FASEB Bioadv. 3, 399–06. 10.1096/fba.2020-00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy JW, Sedgwick A, Rosse C, Muralidharan-Chari V, Raposo G, Method M, Chavrier P, and D’Souza-Schorey C (2015a). Regulated delivery of molecular cargo to invasive tumour-derived microvesicles. Nat. Commun 6, 6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy JW, Tricarico CJ, and D’Souza-Schorey C (2015b). Tumor-derived microvesicles in the tumor microenvironment: how vesicle heterogeneity can shape the future of a rapidly expanding field. Bioessays 37, 1309–1316. [DOI] [PubMed] [Google Scholar]
- Clancy JW, Tricarico CJ, Marous DR, and D’Souza-Schorey C (2019a). Coordinated regulation of intracellular fascin distribution governs tumor microvesicle release and invasive cell capacity. Mol. Cell Biol 39, e00264–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy JW, Zhang Y, Sheehan C, and D’Souza-Schorey C (2019b). An ARF6-Exportin-5 axis delivers pre-miRNA cargo to tumour microvesicles. Nat. Cell Biol 21, 856–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza-Schorey C, and Schorey JS (2018). Regulation and mechanisms of extracellular vesicle biogenesis and secretion. Essays Biochem. 62, 125–133. [DOI] [PubMed] [Google Scholar]
- Diamond JM, Vanpouille-Box C, Spada S, Rudqvist NP, Chapman JR, Ueberheide BM, Pilones KA, Sarfraz Y, Formenti SC, and Demaria S (2018). Exosomes shuttle TREX1-sensitive IFN-stimulatory dsDNA from irradiated cancer cells to DCs. Cancer Immunol. Res 6, 910–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, and Wicha MS (2003). In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 17, 1253–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror S, Sander L, Schwartz H, Sheinboim D, Barzilai A, Dishon Y, Apcher S, Golan T, Greenberger S, Barshack I, et al. (2016). Melanoma miRNA trafficking controls tumour primary niche formation. Nat. Cell Biol 18, 1006–1017. [DOI] [PubMed] [Google Scholar]
- Du M, and Chen ZJ (2018). DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361, 704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edifizi D, Nolte H, Babu V, Castells-Roca L, Mueller MM, Brodesser S, Kruger M, and Schumacher B (2017). Multilayered reprogramming in response to persistent DNA damage in C. elegans. Cell Rep. 20, 2026–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing RM, Chu P, Elisma F, Li H, Taylor P, Climie S, McBroom-Cerajewski L, Robinson MD, O’Connor L, Li M, et al. (2007). Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol. Syst. Biol 3, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodstad O, Aamdal S, McMenamin M, Nesland JM, and Pihl A (1988). A new experimental metastasis model in athymic nude mice, the human malignant melanoma LOX. Int. J. Cancer 41, 442–449. [DOI] [PubMed] [Google Scholar]
- Goldman MJ, Craft B, Hastie M, Repecka K, McDade F, Kamath A, Banerjee A, Luo Y, Rogers D, Brooks AN, et al. (2020). Visualizing and interpreting cancer genomics data via the xena platform. Nat. Biotechnol 38, 675–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Chen H, Gong H, Tang X, Huang N, Xu W, Tai H, Zhang G, Zhao T, Gong C, et al. (2020). Autolysosomal degradation of cytosolic chromatin fragments antagonizes oxidative stress-induced senescence. J. Biol. Chem 295, 4451–4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch EM, Fischer AH, Deerinck TJ, and Hetzer MW (2013). Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 154, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun K, Jeon J, Park K, and Kim J (2017). Writing, erasing and reading histone lysine methylations. Exp. Mol. Med 49, e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen DK, Fenix AM, Franklin JL, Higginbotham JN, Zhang Q, Zimmerman LJ, Liebler DC, Ping J, Liu Q, Evans R, et al. (2019). Reassessment of exosome composition. Cell 177, 428–445 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlert C, Melo SA, Protopopov A, Tang J, Seth S, Koch M, Zhang J, Weitz J, Chin L, Futreal A, et al. (2014). Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J. Biol. Chem 289, 3869–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, and LeBleu VS (2016). Discovery of double-stranded genomic DNA in circulating exosomes. Cold Spring Harb. Symp. Quant. Biol 81, 275–280. [DOI] [PubMed] [Google Scholar]
- Kalluri R, and LeBleu VS (2020). The biology, function, and biomedical applications of exosomes. Science 367, eaau6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima A, Tanigawa K, Akama T, Wu H, Sue M, Yoshihara A, Ishido Y, Kobiyama K, Takeshita F, Ishii KJ, et al. (2011). Fragments of genomic DNA released by injured cells activate innate immunity and suppress endocrine function in the thyroid. Endocrinology 152, 1702–1712. [DOI] [PubMed] [Google Scholar]
- Kujirai T, Zierhut C, Takizawa Y, Kim R, Negishi L, Uruma N, Hirai S, Funabiki H, and Kurumizaka H (2020). Structural basis for the inhibition of cGAS by nucleosomes. Science 370, 455–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latifkar A, Hur YH, Sanchez JC, Cerione RA, and Antonyak MA (2019). New insights into extracellular vesicle biogenesis and function. J. Cell Sci 132, jcs222406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaro-Ibanez E, Sanz-Garcia A, Visakorpi T, Escobedo-Lucea C, Siljander P, Ayuso-Sacido A, and Yliperttula M (2014). Different gDNA content in the subpopulations of prostate cancer extracellular vesicles: apoptotic bodies, microvesicles, and exosomes. Prostate 74, 1379–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Li C, Zhang Y, Zhang D, Otterbein LE, and Jin Y (2019). Caveolin-1 selectively regulates microRNA sorting into microvesicles after noxious stimuli. J. Exp. Med 216, 2202–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TH, Chennakrishnaiah S, Audemard E, Montermini L, Meehan B, and Rak J (2014). Oncogenic ras-driven cancer cell vesiculation leads to emission of double-stranded DNA capable of interacting with target cells. Biochem. Biophys. Res. Commun 451, 295–301. [DOI] [PubMed] [Google Scholar]
- Li X, Shu C, Yi G, Chaton CT, Shelton CL, Diao J, Zuo X, Kao CC, Herr AB, and Li P (2013). Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 39, 1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, Jiang Y, Fei Y, Zhu C, Tan R, et al. (2018). Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 563, 131–136. [DOI] [PubMed] [Google Scholar]
- Lu A, Wawro P, Morgens DW, Portela F, Bassik MC, and Pfeffer SR (2018). Genome-wide interrogation of extracellular vesicle biology using barcoded miRNAs. Elife 7, e41460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. (2017). cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie AJ, Hoshino D, Hong NH, Cha DJ, Franklin JL, Coffey RJ, Patton JG, and Weaver AM (2016). KRAS-MEK signaling controls Ago2 sorting into exosomes. Cell Rep. 15, 978–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo SA, Sugimoto H, O’Connell JT, Kato N, Villanueva A, Vidal A, Qiu L, Vitkin E, Perelman LT, Melo CA, et al. (2014). Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 26, 707–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. (2006). A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298. [DOI] [PubMed] [Google Scholar]
- Mohr L, Toufektchan E, von Morgen P, Chu K, Kapoor A, and Maciejowski J (2021). ER-directed TREX1 limits cGAS activation at micronuclei. Mol. Cell 81, 724–738 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motwani M, Pesiridis S, and Fitzgerald KA (2019). DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet 20, 657–674. [DOI] [PubMed] [Google Scholar]
- Murakami S, Chishima S, Uemoto H, Sakamoto E, Sato T, Kurabe N, Kawasaki Y, Shibata T, Akiyama H, and Tashiro F (2014). The male-specific factor sry harbors an oncogenic function. Oncogene 33, 2978–2986. [DOI] [PubMed] [Google Scholar]
- Muralidharan-Chari V, Clancy J, Plou C, Romao M, Chavrier P, Raposo G, and D’souza-Schorey C (2009). ARF6-Regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr. Biol 19, 1875–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, Garcia-Santos G, Ghajar C, et al. (2012). Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med 18, 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phinney DG, Di Giuseppe M, Njah J, Sala E, Shiva S, St Croix CM, Stolz DB, Watkins SC, Di YP, Leikauf GD, et al. (2015). Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat. Commun 6, 8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raposo G, and Stahl PD (2019). Extracellular vesicles: a new communication paradigm? Nat. Rev. Mol. Cell Biol 20, 509–510. [DOI] [PubMed] [Google Scholar]
- Reis-Sobreiro M, Chen JF, Novitskaya T, You S, Morley S, Steadman K, Gill NK, Eskaros A, Rotinen M, Chu CY, et al. (2018). Emerin deregulation links nuclear shape instability to metastatic potential. Cancer Res. 78, 6086–6097. [DOI] [PubMed] [Google Scholar]
- Sakha S, Muramatsu T, Ueda K, and Inazawa J (2016). Exosomal microRNA miR-1246 induces cell motility and invasion through the regulation of DENND2D in oral squamous cell carcinoma. Sci. Rep 6, 38750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer JK, and D’Souza-Schorey C (2005). A requirement for ARF6 during the completion of cytokinesis. Exp. Cell Res 311, 74–83. [DOI] [PubMed] [Google Scholar]
- Sedgwick AE, Clancy JW, Olivia Balmert M, and D’Souza-Schorey C (2015). Extracellular microvesicles and invadopodia mediate non-overlapping modes of tumor cell invasion. Sci. Rep 5, 14748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedgwick AE, and D’Souza-Schorey C (2018). The biology of extracellular microvesicles. Traffic 19, 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo GJ, Kim C, Shin WJ, Sklan EH, Eoh H, and Jung JU (2018). TRIM56-mediated monoubiquitination of cGAS for cytosolic DNA sensing. Nat. Commun 9, 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan C, and D’Souza-Schorey C (2019). Tumor-derived extracellular vesicles: molecular parcels that enable regulation of the immune response in cancer. J. Cell Sci 132, jcs235085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shurtleff MJ, Temoche-Diaz MM, Karfilis KV, Ri S, and Schekman R (2016). Y-box protein 1 is required to sort microRNAs into exosomes in cells and in a cell-free reaction. Elife 5, e19276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skog J, Würdinger T, van Rijn S, Meijer D, Gainche L, Curry W, Carter B, Krichevsky A, and Breakefield X (2008). Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol 10, 1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi A, Okada R, Nagao K, Kawamata Y, Hanyu A, Yoshimoto S, Takasugi M, Watanabe S, Kanemaki MT, Obuse C, et al. (2017). Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun 8, 15287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur BK, Zhang H, Becker A, Matei I, Huang Y, Costa-Silva B, Zheng Y, Hoshino A, Brazier H, Xiang J, et al. (2014). Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 24, 766–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torralba D, Baixauli F, Villarroya-Beltri C, Fernandez-Delgado I, Latorre-Pellicer A, Acin-Perez R, Martin-Cofreces NB, Jaso-Tamame AL, Iborra S, Jorge I, et al. (2018). Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through antigen-driven contacts. Nat. Commun 9, 2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagner T, Spinelli C, Minciacchi VR, Balaj L, Zandian M, Conley A, Zijlstra A, Freeman MR, Demichelis F, De S, et al. (2018). Large extracellular vesicles carry most of the tumour DNA circulating in prostate cancer patient plasma. J. Extracell. Vesicles 7, 1505403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Niel G, D’Angelo G, and Raposo G (2018). Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol 19, 213–228. [DOI] [PubMed] [Google Scholar]
- Welsh JA, Holloway JA, Wilkinson JS, and Englyst NA (2017). Extracellular vesicle flow cytometry analysis and standardization. Front. Cell Dev. Biol 5, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Turner KM, Nguyen N, Raviram R, Erb M, Santini J, Luebeck J, Rajkumar U, Diao Y, Li B, et al. (2019). Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 575, 699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CA, Huang HY, Chang YS, Lin CL, Lai IL, and Chang JG (2017). DNA-sensing and nuclease gene expressions as markers for colorectal cancer progression. Oncology 92, 115–124. [DOI] [PubMed] [Google Scholar]
- Yu W, Hurley J, Roberts D, Chakrabortty SK, Enderle D, Noerholm M, Breakefield XO, and Skog JK (2021). Exosome-based liquid biopsies in cancer: opportunities and challenges. Ann. Oncol 32, 466–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F, Sun L, Yang N, Zheng W, Shen P, Huang Y, and Lu Y (2020). Increased release of microvesicles containing mitochondria is associated with the myeloid differentiation of AML-M5 leukaemia cells. Exp. Cell Res 395, 112213. [DOI] [PubMed] [Google Scholar]
- Zhou W, Whiteley AT, de Oliveira Mann CC, Morehouse BR, Nowak RP, Fischer ES, Gray NS, Mekalanos JJ, and Kranzusch PJ (2018). Structure of the human cGAS-DNA complex reveals enhanced control of immune surveillance. Cell 174, 300–311.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publicly available (The Cancer Genome Atlas) data. The accession numbers for the datasets used are listed in the key resources table.
Additional data reported in this paper will be shared by the lead author upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| β-Actin | Cell Signaling Technologies | Cat#4970, RRID:AB_2223172 |
| HA tag | Cell Signaling Technologies | Cat#3724, RRID:AB_1549585 |
| DsDNA | Abcam | Cat#ab27156, RRID:AB_470907 |
| Histone H3 (tri-methyl K9) | Abcam | Cat#ab8898, RRID:AB_306848 |
| β1-integrin | MilliporeSigma | Cat#AB1952, RRID:AB_91150 |
| α-tubulin | MilliporeSigma | Cat#T9026, RRID:AB_477593 |
| CD63 | System Biosciences International | Cat#EXOAB-CD63A-1, RRID:AB_2561274 |
| VAMP3 | ProteinTech | Cat#107021-AP, RRID:AB_2212628 |
| ARF6 | ProteinTech | Cat#20225-1-AP, RRID:AB_10646481 |
| Emerin | ProteinTech | Cat#10351-1-AP, RRID:AB_2100056 |
| APEX1 | ProteinTech | Cat#10203-1-AP, RRID:AB_2057927 |
| Annexin-A1 | ProteinTech | Cat#21990-1-AP, RRID:AB_11182596 |
| Flotillin | ProteinTech | Cat#15571-1-AP, RRID:AB_2106746 |
| Alix | ProteinTech | Cat#12422-1-AP, RRID:AB_2162467 |
| CD81 | ProteinTech | Cat#66866-1-Ig, RRID:AB_2882203 |
| TSG-101 | ProteinTech | Cat#28283-1-AP, RRID:AB_2881104 |
| Histone-H3 | ProteinTech | Cat#17168-1-AP, RRID:AB_2716755 |
| CGAS | ProteinTech | Cat#26416-1-AP, RRID:AB_2880507 |
| CD63 | Developmental Studies Hybridoma Bank | Cat#h5c6, RRID:AB_528158 |
| SRY | ProteinTech | Cat#67349-1-lg, RRID:AB_2882607 |
| GFP | ThermoFisher | Cat#33-2600, RRID:AB_2533111 |
| Donkey anti-mouse Alexa Fluor Plus 488 | ThermoFisher | Cat#A32766, RRID:AB_2762823 |
| Donkey anti-rabbit Alexa Fluor Plus 555 | ThermoFisher | Cat#A32794, RRID:AB_2762834 |
| Donkey anti-mouse Alexa Fluor Plus 680 | ThermoFisher | Cat#A32788, RRID:AB_2762831 |
| Donkey anti-rabbit Alexa Fluor Plus 800 | ThermoFisher | Cat# A32808, RRID:AB_2762837 |
| Bacterial and virus strains | ||
| NEB Stable | New England Biolabs | Cat#C3040I |
| Biological samples | ||
| Control human genomic DNA | ThermoFisher | Cat#360486 |
| Chemicals, peptides, and recombinant proteins | ||
| Alexa Fluor 555 Plus phalloidin | ThermoFisher | Cat#A30106 |
| Alexa Fluor 647 Plus phalloidin | ThermoFisher | Cat#A30107 |
| To-pro-3 iodide | ThermoFisher | Cat#T3605 |
| DRAQ-5 | ThermoFisher | Cat#62254 |
| Protease Inhibitor Cocktail | MilliporeSigma | Cat#P8340-5ML |
| Paraformaldehyde | Electron Microscopy Sciences | Cat#15713-5 |
| Poly-L-lysine | MilliporeSigma | Cat#P4832-50ML |
| Poly-HEMA | MilliporeSigma | Cat#P3932-10G; CAS#25249-16-5 |
| Matrigel | Corning | Cat#354230 |
| Horse Serum | ThermoFisher | Cat#16050122 |
| EGF | Peprotech | Cat#AF10015 |
| Hydrocortisone | MilliporeSigma | Cat#H0888-1G |
| Cholera Toxin | MilliporeSigma | Cat#C8052-2MG |
| Insulin | MilliporeSigma | Cat#I1822-100MG |
| B27 | ThermoFisher | Cat#17504044 |
| Glutaraldehyde (electron microscopy grade) | Electron Microscopy Sciences | Cat#16120 |
| GTP-γ-S | MilliporeSigma | Cat#G8634-1MG; CAS# 94825-44-2 |
| GDP-β-S | MilliporeSigma | Cat# G7637-5MG; CAS# 97952-36-8 |
| MG132 | MilliporeSigma | Cat#M8699-1MG; CAS# 1211877-36-9 |
| Chloroquine phosphate | Cayman Chemical | Cat#14194; CAS# 50-63-5 |
| U0126 | Cayman Chemical | Cat#70970; CAS# 109511-58-2 |
| Sulforhodamine B | MilliporeSigma | Cat#230162-5G; CAS# 3520-42-1 |
| Trichloroacetic Acid | MilliporeSigma | Cat#T6399-250G; CAS# 76-03-9 |
| Triton-X100 | Fisher Scientific | Cat#BP151-100 |
| Critical commercial assays | ||
| DNeasy Blood and Tissue Kit | Qiagen | Cat#69504 |
| BCA Protein Assay Kit | ThermoFisher | Cat#23227 |
| Ready Probes Cell Viability Imaging Kit | ThermoFisher | Cat#R37609 |
| Quant-iT High Sensitivity dsDNA Assay Kit | ThermoFisher | Cat#2129422 |
| SuperScript IV First-Strand Synthesis System with ezDNAse | ThermoFisher | Cat#18091150 |
| Monarch Total RNA Miniprep Kit | New England Biolabs | Cat#T2010S |
| Click-iT EdU Alexa Fluor 488 Cell Proliferation Kit | ThermoFisher | Cat#C10086 |
| Deposited data | ||
| The Cancer Genome Atlas | http://cancergenome.nih.gov/ | RRID:SCR_003193 |
| Experimental models: Cell lines | ||
| LOX | Fodstad et al. 1988 | N/A |
| H460 | ATCC | Cat#HTB-177, RRID:CVCL_0459 |
| MDA-MB-231 | ATCC | Cat#CRL-12532, RRID:CVCL_0062 |
| MDA-MB-468 | ATCC | Cat#HTB-132, RRID:CVCL_0419 |
| T-47D | ATCC | Cat#HTB-133, RRID:CVCL_0553 |
| PC-3 | ATCC | Cat#CRL-1435, RRID:CVCL_0035 |
| A375-P | ATCC | Cat#CRL-3224, RRID:CVCL_6233 |
| A375-MA2 | ATCC | Cat#CRL-3223, RRID:CVCL_X495 |
| 293FT | ThermoFisher | Cat#R70007 |
| DU145 | ATCC | Cat#HTB-81, RRID:CVCL_0105 |
| LNCaP | ATCC | Cat#CRL-1740, RRID:CVCL_1379 |
| MCF-10A | Joan Brugge | N/A |
| SW480 | ATCC | Cat#CCL-228, RRID:CVCL_0546 |
| Oligonucleotides | ||
| For complete list of oligonucleotides please see Table S1 | N/A | |
| Recombinant DNA | ||
| pEGFP-C1 | Clontech | Cat#6084-1 |
| LentiCRISPR v2 | Sanjana et al. 2014 | Addgene plasmid #52961; RRID:Addgene_52961 |
| psPAX2 | Didier Trono | Addgene plasmid #12260; RRID:Addgene_12260 |
| pMD2.G | Didier Trono | Addgene plasmid #12259; RRID:Addgene_12259 |
| pLKO.1-TRC | Moffat et al., 2006 | Addgene plasmid #10878; RRID:Addgene_10878 |
| pGEX-3X GST-wt-ARF6 | Schweitzer and D’Souza-Schorey 2005 | N/A |
| Software and algorithms | ||
| FIJI | www.imagej.net | Version 2.0.0-rc-69/1.52p |
| Graphpad Prism | www.graphpad.com | Version 9.2.0 |
| Microsoft Excel | https://www.microsoft.com/en-us/microsoft-365/excel | Version 16.16.27 |
| Histogram | Apogee Flow Systems; www.apogeeflow.com | N/A |
| UALCAN | Chandrashekar et al. 2017 | RRID:SCR_015827 |
| UCSC Xena | Goldman et al., 2020 | RRID:SCR_018938 |
| Other | ||
| Glutathione high capacity magnetic beads | MilliporeSigma | Cat#G0924-1ML |
| Exosome depleted FBS | ThermoFisher | Cat#A27208-01 |
| EquaFetal Serum | Atlas Biologicals | Cat#EF-0500-A |
| Hyclone FBS | Cytiva Life Sciences | Cat#SH30070.03 |
| Benzonase | MilliporeSigma | Cat#71206 |
| Immobilon PVDF-FL | MilliporeSigma | Cat#IPFL85R |
| Prolong diamond antifade mounting media | ThermoFisher | Cat#P36970 |
| Lipofectamine2000 Transfection Reagent | ThermoFisher | Cat#11668-027 |
| Micro flow cytometry calibration beads (Apogee Mix) | Apogee Flow Systems | Cat#1493 |
| Optiprep Density Gradient Medium | MilliporeSigma | Cat#D1555-250ML |
| ddPCR Supermix for probes (no dUTP) | BioRad | Cat#1863024 |
| TaqMan Fast Advance Master Mix | ThermoFisher | Cat#4444554 |