

Abstract
Group 3 innate lymphoid cells (ILC3s) are innate immune effectors that contribute to host defense. Whether ILC3 functions are stably modified after pathogen encounter is unknown. Here, we assess the impact of a time-restricted enterobacterial challenge to long-term ILC3 activation in mice. We found that intestinal ILC3s persist for months in an activated state after exposure to Citrobacter rodentium. Upon rechallenge, these “trained” ILC3s proliferate, display enhanced interleukin-22 (IL-22) responses, and have a superior capacity to control infection compared with naïve ILC3s. Metabolic changes occur in C. rodentium–exposed ILC3s, but only trained ILC3s have an enhanced proliferative capacity that contributes to increased IL-22 production. Accordingly, a limited encounter with a pathogen can promote durable phenotypic and functional changes in intestinal ILC3s that contribute to long-term mucosal defense.
Specialized immune cells promote barrier function and maintain microbial tolerance at mucosal surfaces (1); these include effector and memory T cells that provide long-term immune-surveillance and recall responses as well as diverse innate lymphoid cells (ILCs) (2, 3). Group 3 ILCs (ILC3s) are highly enriched in the gut where they orchestrate lymphoid tissue development and mucosal defense (3, 4). Largely devoid of pattern recognition receptors (5), ILC3s are indirectly activated after infection by soluble factors [interleukin-1β (IL-1β) and IL-23] derived from epithelial and hematopoietic sentinel cells (4) and, in turn, secrete IL-17 and IL-22 to protect the host (6–9). ILC3s are active during fetal lymphoid tissue organogenesis (10, 11), and initial encounters with microbial flora in early postnatal life modifies ILC3 subset distribution and cytokine production (6, 12). Whether ILC3s exhibit any degree of long-term adaptation to a commensal or pathogen encounter that results in heightened immunological function remains to be demonstrated.
To study whether persistent functional changes occur in intestinal ILC3s after microbial encounter, we used Citrobacter rodentium, a mouse pathogen that provokes an enterocolitis with disease similarities to human enteropathogenic Escherichia coli infection (13). C. rodentium attaches and effaces the distal small intestine and colon, provoking innate dendritic cell (DC)–induced ILC3 activation as well as generation of adaptive antigen-specific B and T cells (6, 9, 14). To focus on intestinal ILC3 responses to C. rodentium that are independent of adaptive immune priming (14) and infection-associated dysbiosis (13, 15), we limited the infection window using antibiotics (Abx) (15) (fig. S1, A to E). Indeed, a short course of ciprofloxacin was sufficient to restrict adaptive T helper type 22 (TH22) responses, whereas innate ILC3-dependent IL-22 responses were activated normally (fig. S1, F to H). Although Abx treatment alone transiently modified intestinal bacterial communities (fig. S2, A to C), no long-term impact on ILC3 function (fig. S2, D to F) or on microbial diversity (fig. S3) was observed.
We characterized innate ILC3 and adaptive TH22 immune responses in this modified C. rodentium infection model using RorcGFP and Il22TdT reporter mice (16, 17) (Fig. 1, A and B; and fig. S4) (GFP, green fluorescent protein; TdT, TdTomato). T cell and ILC3 populations were generally stable during the initial infection, but C. rodentium reinfection 1 month after Abx treatment resulted in a rapid increase in intestinal NKp46+ and CCR6+ ILC3 subsets with little effect on T cells (Fig. 1, B and C). Absolute numbers and frequencies of IL-22–expressing ILC3s increased in a similar fashion, whereas IL-22–expressing T cells were largely unchanged (Fig. 1, D to F; and fig. S5). C. rodentium challenge and rechallenge experiments in mice not treated with ciprofloxacin showed similar ILC3 responses, indicating the innocuous effects of the Abx treatment (fig. S6, A to C). CD4+ T cell numbers and frequencies of IL-17A−, IL-22−, and interferon-γ (IFN-γ)–producing T cells were not affected (fig. S6, B to E). Thus, the homeostasis and function of intestinal IL-22–producing ILC3s can be selectively modified after an Abx-controlled subclinical C. rodentium infection.
Fig. 1. Tr-ILC3s efficiently control pathogenic bacteria rechallenge.
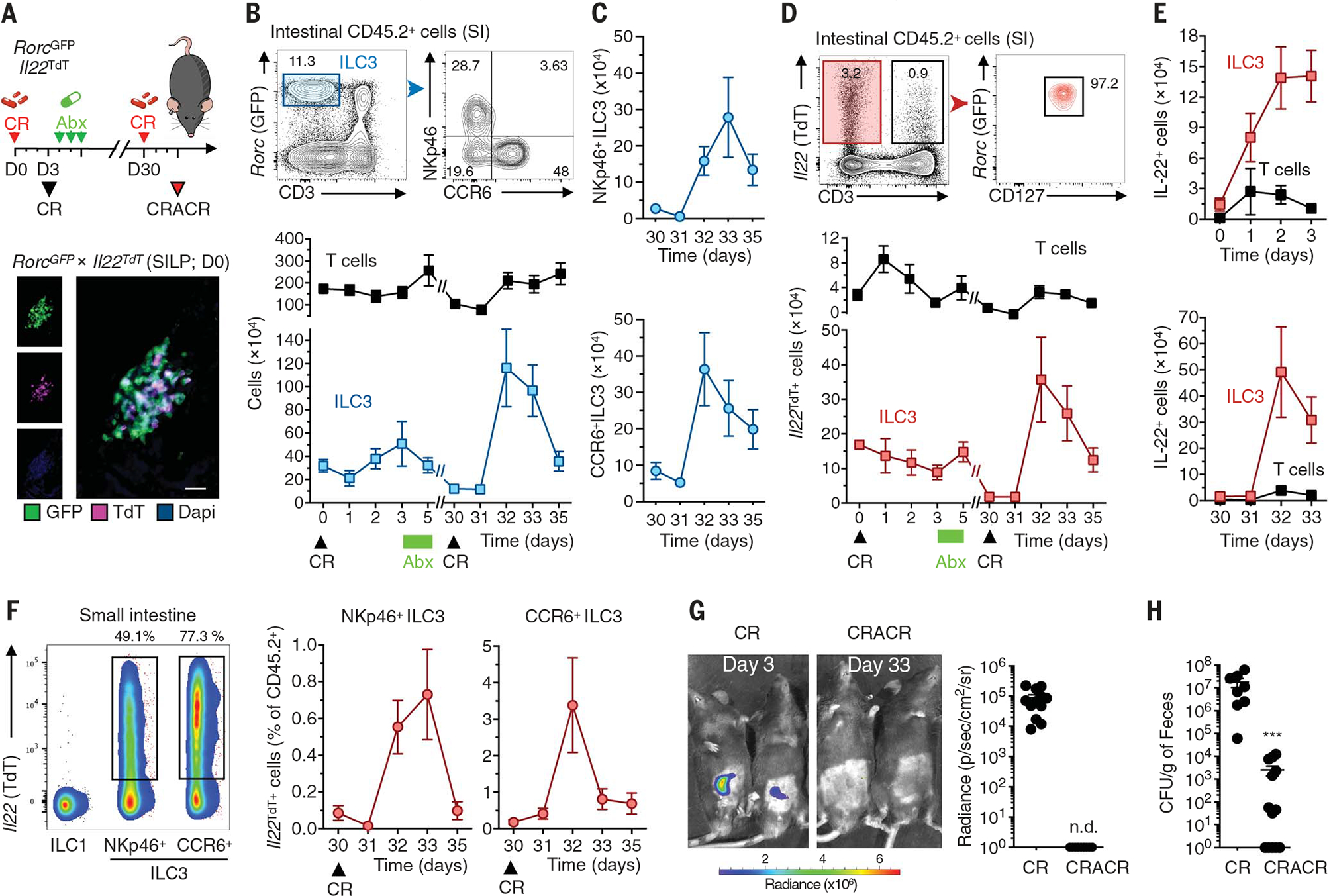
(A) The experimental design for (B) to (H) is shown at the top. RorcGFPIl22TdT mice received ciprofloxacin (Abx; 100 mg kg−1 day−1) after C. rodentium infection (CR). One month later, the mice were reinfected with C. rodentium (CRACR). A representative steady-state immunofluorescence analysis of RORγt+ (green) and Il22TdT+ (magenta) cells in the small intestine is shown at the bottom. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue) (scale bar, 20 μm). SILP, small intestine lamina propria; D0, day 0. (B) Intestinal NKp46+ and CCR6+ ILC3s were analyzed by flow cytometry (top). Absolute numbers of ILC3s and T cells in the small intestine lamina propria (n = 4 to 12 for each time point) are shown at the bottom. (C) Absolute numbers of intestinal NKp46+ (top) and CCR6+ (bottom) ILC3s determined with representative data from three independent analyses (n = 3 to 7 for each time point). (D) Small intestinal Il22TdT+ ILC3 (CD45+CD3−RorcGFP+) and T cell (CD45+CD3+) frequencies at day 0 (top). Absolute numbers of Il22TdT+ cells were determined with representative data from six independent analyses (n = 4 to 12 for each time point) (bottom). (E) Absolute numbers of IL-22+ (protein) cells were analyzed after ex vivo IL-23 (top) and IL-1β (bottom) stimulation (n = 4 to 9 for each time point). (F) Il22TdT expression in ILC1s (CD3−NKp46+NK1.1+), NKp46+ ILC3s, and CCR6+ ILC3s (left). Representative data are from three independent analyses. The frequencies of Il22TdT+NKp46+ and NKp46− ILC3 subsets in the small intestine lamina propria after reinfection are shown on the right (CRACR; n = 3 to 7 for each time point). (G) C. rodentium growth monitored by IVIS imaging (CR, n = 12; CRACR, n = 12). Representative pseudocolor images are shown (color scale in photons s−1 cm−2 sr−1). (H) Fecal C. rodentium counts (CR, n = 8; CRACR, n = 13). Data are representative of three independent experiments. Each graph corresponds to the mean ± SEM of the values obtained. n.d. indicates not detected and ***P < 0.001 using two-tailed Mann-Whitney test.
Mucosal IL-22 production activates epithelial responses during pathogen infection and is required for resistance to C. rodentium (18, 19). Bacterial growth after first infection (denoted “CR”) was not observed after reinfection of Abx-cleared infected mice (denoted CR-AbxCR or “CRACR”) (Fig. 1, G and H), suggesting that enhanced ILC3 function in CRACR mice might play a role in the resistance to bacterial rechallenge. DC activation was noted during rechallenge, whereas monocytes, macrophages, and granulocytes were largely unchanged (fig. S7). Taken together, a limited initial exposure of intestinal ILC3s to pathogenic bacteria can generate a highly functional and persistent ILC3 subset, which we refer to hereafter as “trained ILC3s” (Tr-ILC3s).
Enhanced Tr-ILC3 responses persisted 4 months after C. rodentium reinfection of Abx-cleared mice (Fig. 2A), with reduced bacterial loads upon reinfection (Fig. 2B). Absolute numbers of total ILC3s (Fig. 2C) and IL-22+ ILC3s (Fig. 2, D and E) were significantly increased in CRACR mice that were reinfected 4 months later with diverse ILC3 subsets as dominant IL-22 producers (Fig. 2, F and G). Because the DNA-binding protein inhibitor ID2 is highly expressed in differentiated ILCs (20, 21), we used Id2-regulated inducible fate-mapping (22) to track ILCs under steady state and after infection (Fig. 2H). All ILCs were red fluorescent protein (RFP)–labeled, with a somewhat higher percentage of RFP+ ILC3s after C. rodentium infection (CR and CRA conditions; Fig. 2I). By contrast, most RFP+ ILC1s and ILC2s were lost in CRACR mice, whereas labeled RFP+ ILC3 subsets were maintained (Fig. 2I). These results indicate that diverse Tr-ILC3s can persist after a limited exposure to C. rodentium and may preferentially expand after pathogen reencounter.
Fig. 2. Tr-ILC3s show long-term persistence.
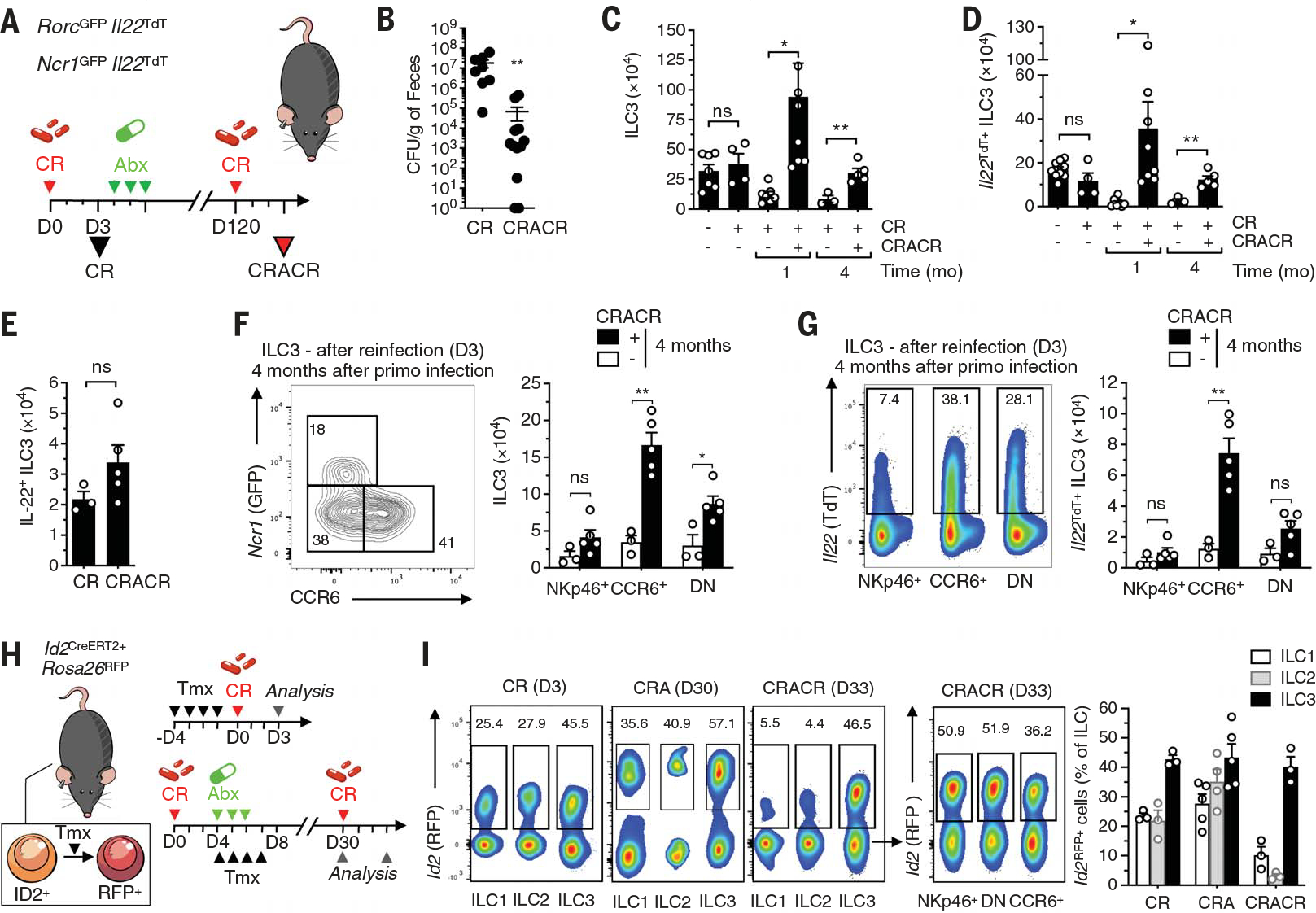
(A) Experimental design for (B) to (H). RorcGFP×Il22TdT and Ncr1GFP×Il22TdT mice received ciprofloxacin (Abx; 100 mg kg−1 day−1) after C. rodentium infection. Four months later, the mice were reinfected with C. rodentium (CRACR). (B) Fecal C. rodentium counts (CR, n = 8; CRACR, n = 12). CFU, colony forming units. (C and D) Absolute numbers of total (C) (n = 3 to 8) and Il22TdT+ (D) (n = 3 to 9) ILC3s in the small intestine lamina propria. (E) Absolute numbers of intestinal IL-22+ ILC3s (protein; n = 3 to 5) after ex vivo IL-23 and IL-1β stimulation 3 days after infection or reinfection. (F and G) Frequency and absolute numbers of ILC3s (F) and Il22TdT+ ILC3 (G) subsets in CRACR mice were determined (n = 3 to 5). DN, double negative. (H) Fate-mapping protocol. Id2CreERT2+×Rosa26RFP mice received ciprofloxacin (Abx; 100 mg kg−1 day−1) 3 days after C. rodentium infection. Mice received tamoxifen (Tmx) by intraperitoneal injection and were analyzed as shown. (I) Analysis of Id2RFP+ cells in ILC1 (CD3− NKp46+NK1.1+), ILC2 (CD3−CD127+ CD90.2+KLRG1+), and ILC3 subsets (left) for the protocol shown in (H). Percentages of Id2RFP+ cells of the total ILCs in CR, CRA, and CRACR mice were determined (n = 3 to 5) (right). Each graph corresponds to the mean ± SEM of the values obtained. ns indicates not significant, *P < 0.05, and **P < 0.01 using two-tailed Mann-Whitney test.
We determined if Tr-ILC3s could be generated in the absence of adaptive immunity (Fig. 3A). Absolute numbers of IL-22+ ILC3s were significantly increased in CRACR Rag2−/− mice (Fig. 3B), which had reduced bacterial loads compared with the CR-infected Rag2−/− mice (Fig. 3C). IL-22 production was required for Tr-ILC3–mediated protection against C. rodentium (Fig. 3A) because Rag2−/− Il22−/− CRACR mice did not survive reinfection (Fig. 3, D and E). We next compared the protective capacity of adoptively transferred naïve versus Tr-ILC3s in vivo (Fig. 3F). CR-infected Il22−/− mice that received naïve ILC3s exhibited body weight loss and succumbed about 2 weeks after infection (Fig. 3G). By contrast, CR-infected Il22−/− mice that received Tr-ILC3s recovered and survived (Fig. 3G). Thus, the enhanced functional capacity of Tr-ILC3s can be adoptively transferred and can operate in a T cell–independent fashion during pathogen rechallenge.
Fig. 3. Tr-ILC3s show enhanced IL-22–mediated protection.
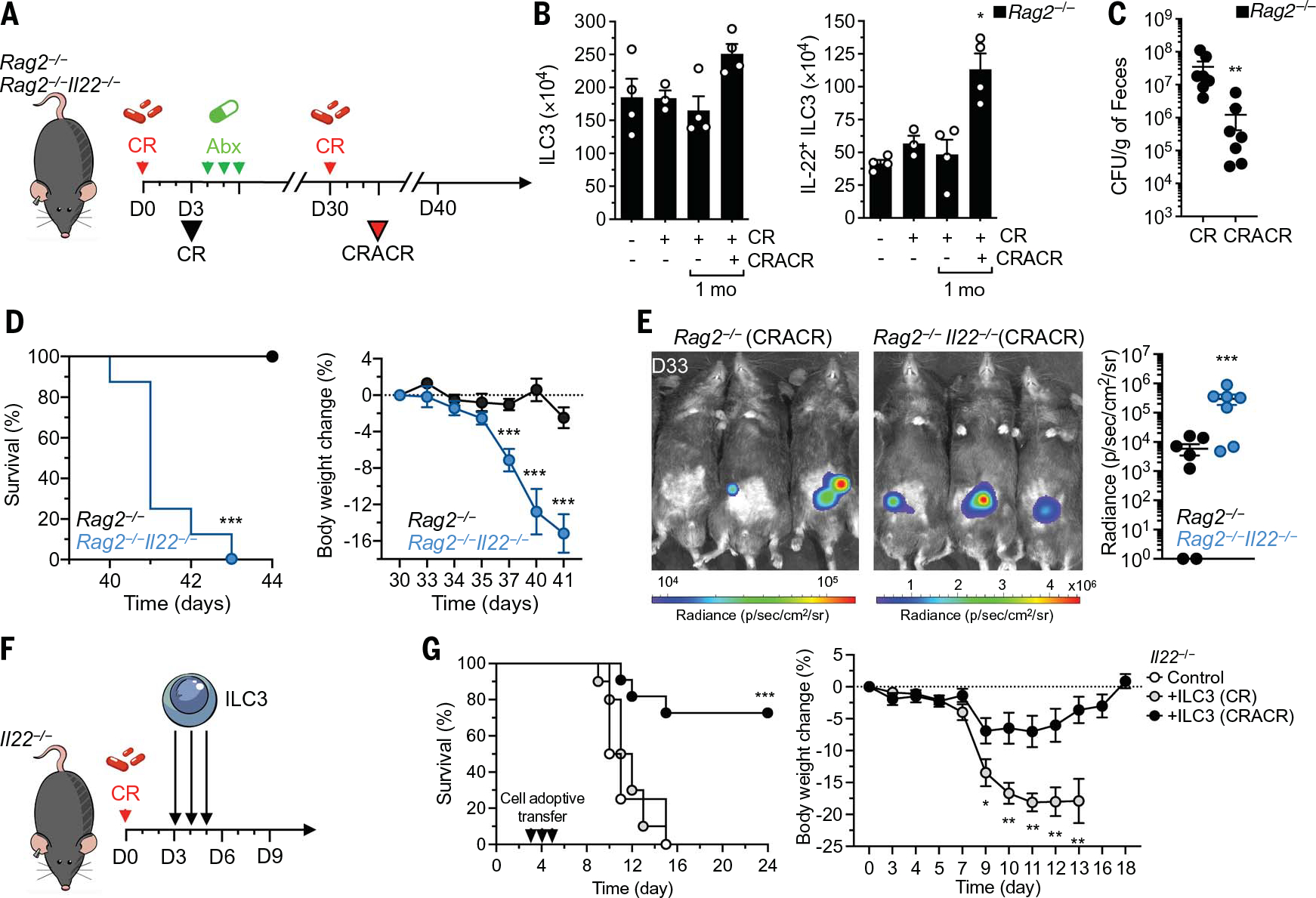
(A) Experimental design for (B) to (E). Rag2−/− and Rag2−/− Il22−/− mice received ciprofloxacin (Abx; 100 mg kg−1 day−1) after C. rodentium infection. One month later, the mice were reinfected with CR (CRACR). (B) Analysis of ILC3s (left) and IL-22+ ILC3s (right) in Rag2−/− mice (n = 3 to 4). (C) Fecal C. rodentium counts (n = 7). (D) Survival (left) and body weight (right) in reinfected Rag2−/− and Rag2−/− IL22−/− mice (n = 7). (E) C. rodentium growth monitored by IVIS imaging 3 days after reinfection (left). Relative C. rodentium growth was determined in Rag2−/− and Rag2−/− IL22−/− mice (n = 7) (right). (F) Experimental design for (G). ILC3s were purified from infected (CR) or reinfected (CRACR) mice 3 days after infection and transferred into C. rodentium–infected Il22−/− mice. (G) Survival and body weight were assessed at the indicated times after infection [n = 4 for Il22−/− and ILC3 (CR); n = 10 for Il22−/− and ILC3 (CRACR); n = 11 for Il22−/− per group, pool of two independent experiments]. Each graph corresponds to the mean ± SEM of the values obtained. *P < 0.05, **P < 0.01, and ***P < 0.001 using Krustal-Wallis test (B), two-tailed Mann-Whitney test [(C) to (G)], or log-rank (Mantel-Cox) test [(D) and (G)].
Although antigen specificity has long been considered as a cardinal feature of adaptive B and T cell memory (23), innate populations such as myeloid cells (24), natural killer (NK) cells (25, 26), and ILC2s (27) have been shown to have “memory-like” properties. To investigate pathogen-specific properties of Tr-ILC3s, we analyzed whether C. rodentium–activated Tr-ILC3s could protect against Listeria monocytogenes. L. monocytogenes infection (LM) elicits IL-23 release that can trigger ILC3 activation and IL-22 production, which is involved in the intestinal epithelial response (28). We found enhanced ILC3 responses after rechallenge with L. monocytogenes (CRALM compared with LM) (fig. S9, A and B). Thus, Tr-ILC3 responses are not restricted to C. rodentium and can potentially extend protection to unrelated pathogens. We also assessed whether Tr-ILC3s can be generated by cytokines alone. Although repeated IL-23 and IL-1β injection triggered higher IL-22 production in intestinal ILC3s (fig. S9, C and D), we failed to detect increased ILC3 numbers in this noninfectious context (fig. S9E). Tr-ILC3s therefore lack antigen specificity and require tissue-dependent signals beyond IL-1β and IL-23 for their generation.
RNA sequencing (RNA-seq) analysis on intestinal ILC3 subsets from naïve (C), C. rodentium–infected (CR), C. rodentium–infected and ciprofloxacin-treated (CRA), and CRACR mice allowed us to validate previous ILC3 datasets (5) and to identify more than 1000 genes in CCR6+ ILC3s and 500 genes in CD49a+ ILC3s modified by C. rodentium infection (Fig. 4, A and B; fig. S10A; and table S1). By contrast, comparison of CR and CRACR ILC3 profiles revealed only 152 differentially expressed genes in CCR6+ ILC3s and 138 genes in CD49a+ ILC3s, suggesting that altered transcriptomic profiles stably persist in ILC3s after C. rodentium infection (fig. S10, A and B). We documented increased effector functions in ILC3 subsets in C. rodentium–exposed mice compared with control naïve mice (Fig. 4C), with Tr-ILC3s strongly expressing transcripts for Il22, Il17f, Gzmb, and Gzmc (fig. S10C).
Fig. 4. Gene expression analysis and metabolic profiles of Tr-ILC3s.
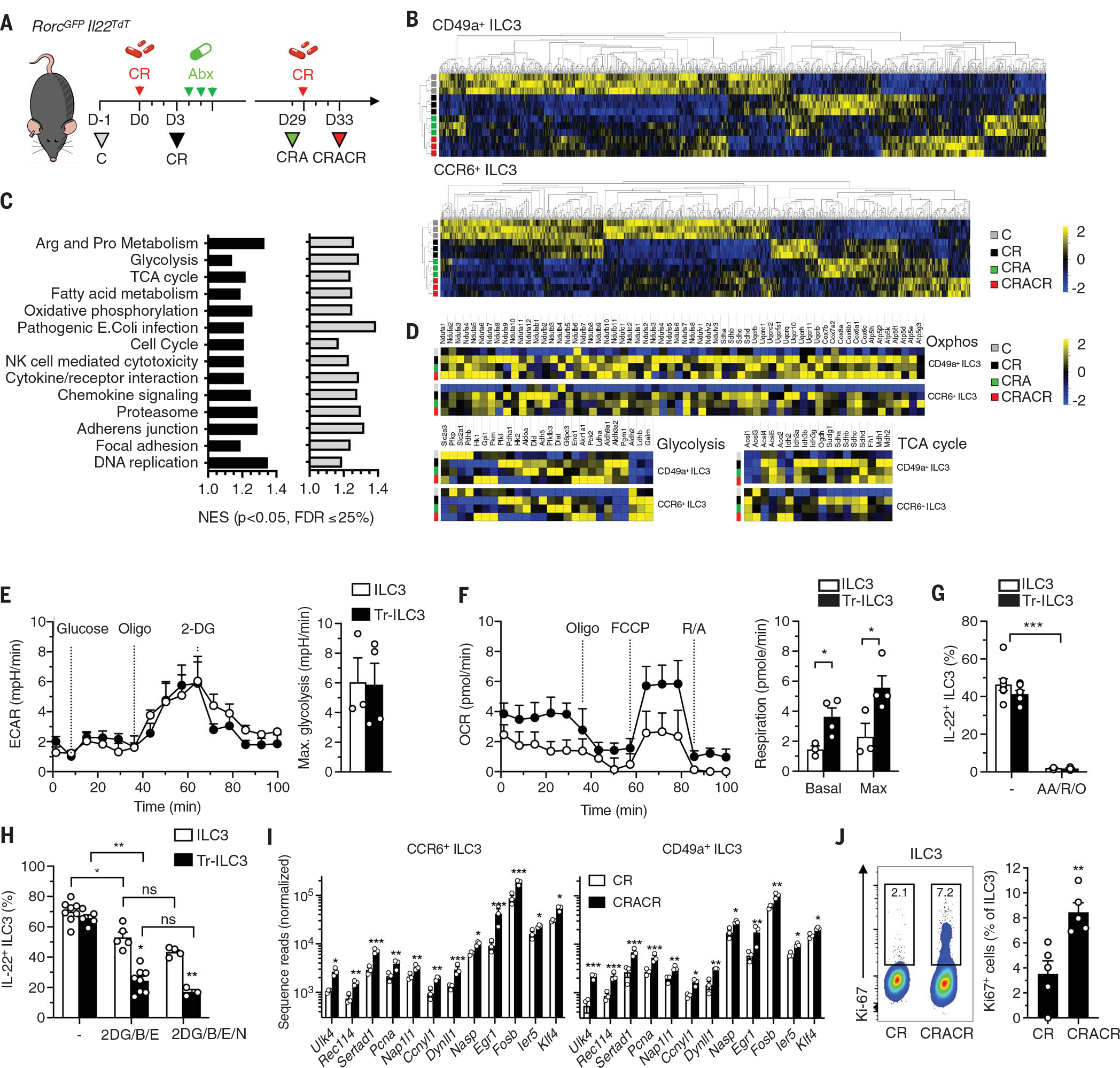
(A) CCR6+ and CD49a+ ILC3 from naïve (C) and infected mice (CR, CRA, and CRACR) were sorted for RNA-seq analysis. (B and C) A heatmap showing the relative expression levels of differentially expressed genes is shown in (B). In (C), pathway analysis was performed and gene pathways were organized into clusters to compare CR, CRA, and CRACR conditions to C (n = 3). FDR, false discovery rate; NES, normalized enrichment score. (D) Heatmap with clustering of differentially expressed metabolism-associated genes (n = 3). (E and F) Seahorse analysis of freshly sorted ILC3s (C) and Tr-ILC3s (CRACR) in response to a mitochondrial uncoupler [fluoro-carbonylcyanide phenylhydrazone (FCCP)], oligomycin (oligo), rotenone and antimycin (R/A), and 2-DG. Representative metabolic profiles (OCR and ECAR), glycolysis, and respiration were determined (three independent experiments; n = 3 to 4). (G) Effect of OXPHOS inhibition (18 hours) on IL-22 production by intestinal ILC3s (n = 5). AA, antimycin; R, rotenone; O, oligomycin. (H) Effect of metabolic pathway inhibition (2 hours) on IL-22 production by intestinal ILC3s (two independent experiments; n = 3 to 8). B, BPTES; E, etomoxir; N, Nor-NOHA. (I) Differentially expressed genes in CR and CRACR ILC3s (n = 3). (J) Frequency of Ki67+ ILC3 from infected (CR) and reinfected (CRACR) mice (two independent experiments; n = 5). Each graph corresponds to the mean ± SEM of the values obtained. ns indicates not significant, *P < 0.05, **P < 0.01, and ***P < 0.001 using two-tailed unpaired Student’s t test [(E) and (F)], two-tailed Mann-Whitney test [(G) and (J)], or Krustal-Wallis test (H).
Classical and innate immunological memory requires metabolic rewiring (29, 30) that orchestrates cell survival, proliferation, differentiation, and long-term persistence of these cells. ILC3 subsets demonstrated a profound metabolic shift from glycolysis and glutaminolysis to enhanced tricarboxylic acid (TCA) cycle, oxidative phosphorylation (OXPHOS), fatty acid synthesis, and oxidation-associated gene expression after C. rodentium infection, which was preserved in CRA and CRACR mice (Fig. 4, C and D; and fig. S10C). As such, intestinal ILC3s experience durable metabolic rewiring after pathogen encounter.
Bioenergetic profiles, including extracellular acidification rate (ECAR) and cellular oxygen-consumption rate (OCR), define mitochondrial respiration and aerobic glycolytic activity, respectively. Tr-ILC3s showed higher OCRs, indicating increased mitochondrial fitness compared with naïve ILC3s (Fig. 4, E and F), although both required OXPHOS for IL-22 production (Fig. 4G). Diverse nutrients (including glucose, glumatine, and fatty acids) fuel immune cell metabolism and mitochondrial bioenergetic pathways (31). In addition, urea cycle–associated genes [arginase-1 (Arg1)] can drive ILC2 and T cell proliferation as well as proinflammatory functions (32, 33). Inhibition of glycolysis [2-deoxy-D-glucose (2-DG)], glutamine conversion [bis-2-(5-phenylacetamido-1,3,4--thiadiazol-2-yl)ethyl sulfide (BPTES)], arginine metabolism [Nω-hydroxy-nor-L-arginine (Nor-NOHA)], or fatty acid β-oxidation (etomoxir) did not differentially affect IL-22 production from naïve versus Tr-ILC3s (fig. S10, D to F). However, combined treatment with 2-DG–BPTES and etomoxir significantly decreased IL-22 production from Tr-ILC3s, whereas Arg1 inhibition synergized with 2-DG, BPTES, and etomoxir to significantly reduce IL-22 production from naïve ILC3s (Fig. 4H). Therefore, L-arginine may act as a metabolic source for naïve ILC3s but not Tr-ILC3s.
We further focused our attention on transcripts that were selectively modified in CRACR ILC3s (table S1). From the 51 transcripts that were up-regulated in both CD49+ and CCR6+ ILC3s, 12 were associated with the regulation of cell proliferation (Fig. 4I). Higher Ki67 levels were detected in ILC3s from CRACR compared with CR mice (Fig. 4J), consistent with rapid Tr-ILC3 proliferation after activation. Our results suggest that pathogen encounter is accompanied by durable metabolic changes in intestinal ILC3s, generating TrILC3 subsets with enhanced proliferative capacity and contributing to long-term mucosal defense.
Adaptive immune responses are classically associated with the development of immunological memory. The innate immune system can likewise adapt to environmental inflammatory signals that generate NK and myeloid cells with new long-lived phenotypes (24, 26). Here, we describe intestinal “trained” ILC3s that emerge and persist after initial pathogen encounter. Upon reinfection, Tr-ILC3s proliferate and robustly produce IL-22, thus promoting mucosal defense. The distinct metabolic activity in memory T cells (34, 35) and trained monocytes and macrophages (24) is associated with epigenetic reprogramming. The precise mechanisms that underlie the distinct functional features of Tr-ILC3s and the signals that promote them remain to be defined. Tr-ILC3 targeting may provide an avenue for prevention or treatment of disease caused by inflammation or pathogens that invade barrier surfaces.
Supplementary Material
ACKNOWLEDGMENTS
We thank all the members of the Di Santo laboratory for critical review of the manuscript, helpful discussions, and support. We thank G. Eberl (Institut Pasteur, France) and O. Mandelboim (Hebrew University, Israel) for providing RorcGFP and Ncr1GFP mice, respectively. We are indebted to S. Novault (Technology Core of the Center for Translational Science at Institut Pasteur, France) for cell sorting and M. Berard, R. Chennouf (Institut Pasteur, France, animal facilities), and M. Lecuit (Institut Pasteur, France) for support. We thank P. Bousso (Institut Pasteur, France) for critical comments on the manuscript.
Funding:
This work is supported by grants from the Institut National de la Santé et de la Recherche Médicale (INSERM), the Institut Pasteur, the Agence National pour le Recherche (ANR - ILC_MEMORY), and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (695467 – ILC_REACTIVITY). A.J. is supported by the French Ministry of Higher Education, Research, and Innovation and by the Fondation pour la recherche médicale.
Footnotes
Competing interests: The authors declare no competing financial interests.
Data and materials availability:
All data are available in the main text, supplementary materials, or public data repositories [16S rRNA at the NCBI Sequence Read Archive (SUB10763153/BioProject ID: PRJNA786093; RNA-seq datasets: GSE191167)].
REFERENCES AND NOTES
- 1.Hooper LV, Macpherson AJ, Nat. Rev. Immunol. 10, 159–169 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Spits H et al. , Nat. Rev. Immunol. 13, 145–149 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Sonnenberg GF, Artis D, Nat. Med. 21, 698–708 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klose CSN, Artis D, Nat. Immunol. 17, 765–774 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Robinette ML et al. , Nat. Immunol. 16, 306–317 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Satoh-Takayama N et al. , Immunity 29, 958–970 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D, Immunity 34, 122–134 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buonocore S et al. , Nature 464, 1371–1375 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cella M et al. , Nature 457, 722–725 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshida H et al. , Int. Immunol. 11, 643–655 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Mebius RE, Rennert P, Weissman IL, Immunity 7, 493–504 (1997). [DOI] [PubMed] [Google Scholar]
- 12.Sawa S et al. , Nat. Immunol. 12, 320–326 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Collins JW et al. , Nat. Rev. Microbiol. 12, 612–623 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Basu R et al. , Immunity 37, 1061–1075 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mullineaux-Sanders C et al. , Cell Rep. 21, 3381–3389 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eberl G et al. , Nat. Immunol. 5, 64–73 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Shen W, Hixon JA, McLean MH, Li WQ, Durum SK, Front. Immunol. 6, 662 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolk K et al. , Immunity 21, 241–254 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Zheng Y et al. , Nat. Med. 14, 282–289 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Cherrier M, Sawa S, Eberl G, J. Exp. Med. 209, 729–740 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu W et al. , Immunity 50, 1054–1068.e3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rawlins EL, Clark CP, Xue Y, Hogan BL, Development 136, 3741–3745 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ordovas-Montanes J, Beyaz S, Rakoff-Nahoum S, Shalek AK, Nat. Rev. Immunol. 20, 308–320 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saeed S et al. , Science 345, 1251086 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cooper MA et al. , Proc. Natl. Acad. Sci. U.S.A. 106, 1915–1919 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun JC, Beilke JN, Lanier LL, Nature 457, 557–561 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez-Gonzalez I et al. , Immunity 45, 198–208 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Disson O et al. , J. Exp. Med. 215, 2936–2954 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arts RJW, Joosten LAB, Netea MG, Semin. Immunol. 28, 425–430 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Bantug GR, Galluzzi L, Kroemer G, Hess C, Nat. Rev. Immunol. 18, 19–34 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Muri J, Kopf M, Nat. Rev. Immunol. 21, 363–381 (2021). [DOI] [PubMed] [Google Scholar]
- 32.Geiger R et al. , Cell 167, 829–842.e13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monticelli LA et al. , Nat. Immunol. 17, 656–665 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caputa G, Castoldi A, Pearce EJ, Nat. Immunol. 20, 793–801 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Buck MD, O’Sullivan D, Pearce EL, J. Exp. Med. 212, 1345–1360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text, supplementary materials, or public data repositories [16S rRNA at the NCBI Sequence Read Archive (SUB10763153/BioProject ID: PRJNA786093; RNA-seq datasets: GSE191167)].