

Abstract
RT-PCR and northern blots have long been used to study RNA isoforms usage for single genes. Recent advancements in long-read sequencing have yielded unprecedented information about the usage and abundance of these RNA isoforms. However, visualization of long-read sequencing data remains challenging due to the high information density. To alleviate these issues, we have developed NanoBlot, an open-source R-package that generates northern blot and RT-PCR-like images from long-read sequencing data. NanoBlot requires aligned, positionally sorted and indexed BAM files. Plotting is based around ggplot2 and is easily customizable. Advantages of NanoBlot include a robust system for designing probes to visualize isoforms including excluding reads based on the presence or absence of a specified region, an elegant solution to representing isoforms with continuous variations in length, and the ability to overlay multiple genes in the same plot using different colors. We present examples of nanoblots compared to actual northern blot data. In addition to traditional gel-like images, the NanoBlot package can also output other visualizations such as violin plots and 3′-RACE-like plots focused on 3′-end isoforms visualization. The use of the NanoBlot package should provide a simple answer to some of the challenges of visualizing long-read RNA-sequencing data.
Keywords: nanopore sequencing, long-read sequencing, data visualization, isoform visualization, alternative splicing, alternative polyadenylation
INTRODUCTION
mRNA isoforms usage is biologically important and dynamic. Alternative transcription start site, alternative splicing, and alternative polyadenylation site selection all result in the production of different mRNA isoforms from the same transcription unit. These diverse isoforms increase proteome complexity, and the presence of regulatory elements in each isoform can impact steady-state expression levels or RNA localization. RNA isoforms usage has long been studied on the scale of individual genes, first predominantly using northern blots and more recently by RT-PCR. While easily performed, RT-PCR and northern blots have limitations. RT-PCR cannot distinguish between isoforms with differences outside of the region targeted for PCR. Northern blots detect different isoforms which hybridize to the same probe, but they can only detect one or two gene products at a time. In addition, northern blots cannot easily visualize isoforms that differ by only a few nucleotides if the target RNA is long. The development of long-read sequencing technologies is an exciting innovation which allows for high-throughput studies of isoform usage. However, due to the information density, visualization of long-read sequencing data remains challenging and tools for visualization are scarce.
One common approach to visualization is using images from genome browsers such as the Integrative Genome Viewer (Robinson et al. 2011) or UCSC Genome Browser (http://genome.ucsc.edu) (Kent et al. 2002). This has the advantage of showing all the reads obtained in a specific genomic region. However, for genes with many isoforms or long introns relative to coding regions, compact visualization remains challenging. Tracks can be edited to shorten long introns and reads can be downsampled to increase information density. Tools such as ScisorWiz (Stein et al. 2022) help by automating much of this process. However, each additional sample requires a new track to be added. This takes up considerable space and makes the representation of more than a few samples in a single image non-feasible.
Another common approach is assigning each transcript to a discrete isoform. Tools such as IsoTV (Annaldasula et al. 2021) and Swan toolkit (Reese and Mortazavi 2021) automate this process and help create intuitive visualizations of isoforms. However, this requires detailed annotation of the host genome and assigns reads to distinct groups. This means that if the model organism being investigated is not well studied, or if there are continuous variations in isoform lengths, then counting is not possible.
A possible solution to these limitations is to represent each read as a data point representing the length of that read. The result of this is a plot which looks similar to an RT-PCR gel or a northern blot (Guilcher et al. 2021). To explore this idea, we have developed NanoBlot, an R-package which allows for the visualization of long-read sequencing data in an intuitive form reminiscent of northern blots or RT-PCR data. The most recent version of NanoBlot can be found on GitHub (https://github.com/SamDeMario-lab/NanoBlot).
RESULTS AND DISCUSSION
Main features of NanoBlot
NanoBlot was originally developed for visualization of RNA isoforms detected using Oxford Nanopore Technology (ONT) long-read sequencing data sets. However, NanoBlot should be able to handle data obtained with other long-read sequencing techniques such as PacBio. NanoBlot takes aligned, positionally sorted, and indexed BAM files as input. The location of the input data is provided as a BamFileList() created via Rsamtools. Each data set must include a unique name. The sequencing data used does not need to be normalized prior to running NanoBlot, as normalization is included. NanoBlot requires a series of target genomic regions referred to as “probes” (Fig. 1A). Probes are supplied as a standard six-column BED file, in which each entry must have a unique name in the fourth column. The subsetNanoblot() command is used to subset each of the input BAM files to only the sequencing reads which overlap with the specified probe region (Quinlan and Hall 2010). Multiple probes can be specified as a character vector in which case each read must overlap will all specified regions as in an “and” statement. If antiprobes are specified, reads which map to the antiprobe region(s) are excluded; this is an optional function which can be used to exclude specific reads to simplify plots. Next, the bamFileListToNanoblotData() command is used to extract the readID and length of each read from the subsetted BAM files and returns an R dataframe. Finally, the user can plot the lengths using either their own code or the included makeNanoblot() function (Fig. 1B). The makeNanoblot() function takes the output of bamFileListToNanoblotData() and can generate three different types of plots: (i) a plot where each read is represented as a series of horizontal points corresponding to its length. This type of plot is the most reminiscent of an actual northern blot or RT-PCR gel (Fig. 1C); (ii) a violin plot which is useful for representing samples where the isoforms of interest have significantly different sizes (Fig. 1D); or (iii) a ridge plot where density plots for each sample are slightly overlapped, making it useful for showing subtle differences in length (Fig. 1E).
FIGURE 1.
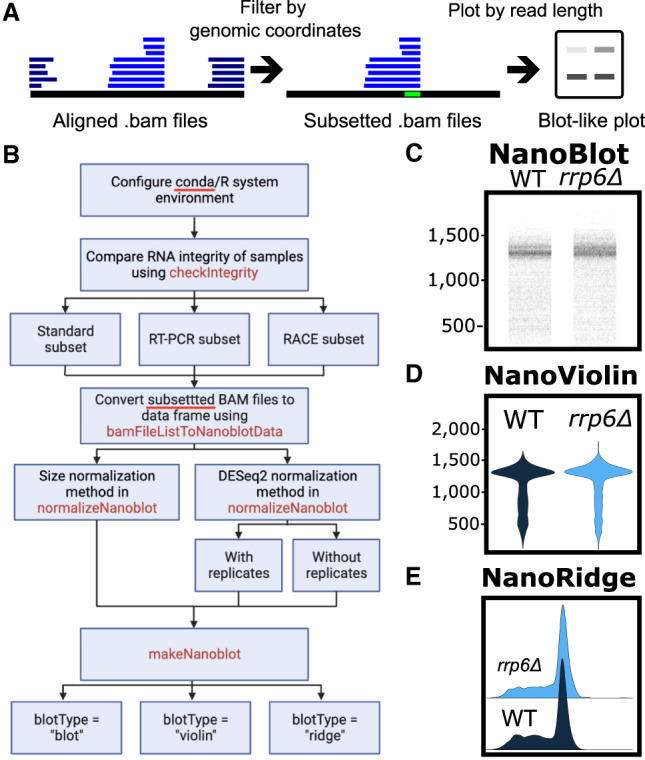
Overview of NanoBlot and example plots. (A) General overview of NanoBlot. NanoBlot's first step is the subsetting of BAM files based on a genomic region, shown in bright green. Reads are then represented as bands based on the length of the reads. Multiple samples and probes can be shown on the same plot. (B) Detailed workflow for using NanoBlot. (C–E) Example plots of the same data shown as a nanoblot (C), violin plot (D), and ridge plot (E).
Checking sequencing library and reads’ integrity
Oxford Nanopore sequencing data quality can vary between samples. This can be due to degradation inherent to the samples, incomplete DNA synthesis during library preparation or incomplete feeding of the RNA through the nanopore (Fig. 2A). Because NanoBlot is intended to be used for isoform usage comparisons between samples, it is critical that the data sets obtained from different samples have similar sequencing integrity. To facilitate these comparisons, we have included functionality to generate cumulative distribution plots showing the percentage of full-length reads in each sample compared to a gene annotation file. This function requires a series of user supplied annotations which specify genomic regions in which no 5′-ends are expected to be found. In the example provided below, an annotation file of all coding regions from the Saccharomyces cerevisiae genome was used. Because of the relative lack of alternative 5′ transcriptional start sites resulting in major changes in 5′ UTR lengths and because of the low number of introns in the S. cerevisiae genome this is a reasonable representation. For organisms with greater genomic complexity, a manually curated list of annotations may be required. However, the annotations do not need to be highly accurate as the metric is intended to be relative. If the same annotation file is used for multiple libraries the comparison is still meaningful. The calculateIntegrity() function counts the number of 5′-ends occurring within each of the genomic regions and returns a count table listing the number of reads overlapping with each annotation as well as the number of 5′-ends occurring in each. Finally, it also prints a cumulative distribution plot comparing the supplied BAM files.
FIGURE 2.
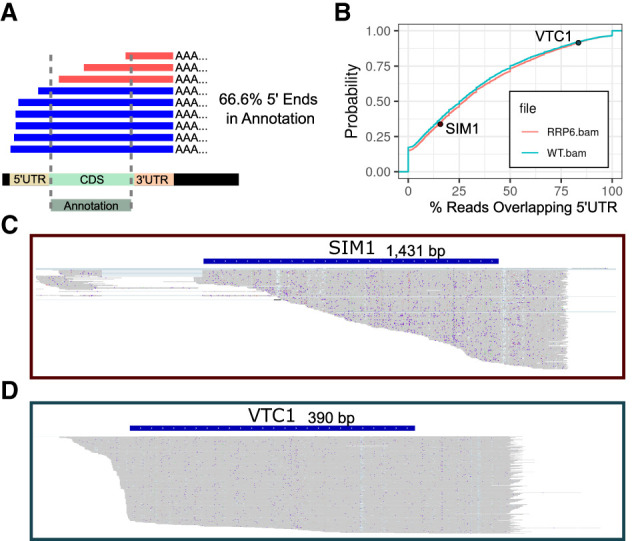
Integrity metrics and examples. (A) Illustration of partially sequenced reads aligned to a reference genome. An example annotation is shown which covers the entirety of the coding region. Note 33.3% of the read's 5′-ends are in the coding region, indicating partial sequencing. (B) Integrity plot comparing the integrity of two S. cerevisiae sequencing data sets from wild-type (WT) and rrp6Δ strains. Two representative examples are shown. (C) IGV screen shot of the reads obtained for SIM1 in the rrp6Δ data set. Most of the reads end within the coding region, resulting in a low integrity score. (D) Same as above for VTC1. Most of the reads do not end within the coding region, resulting in a high integrity score.
As an example, sequencing libraries from WT and rrp6Δ knockout strains from S. cerevisiae were compared (Fig. 2B). The overlap between the two distributions indicates that WT has slightly higher sequencing integrity. However, the difference is minor indicating that a direct comparison between isoform lengths in these two libraries is reasonable. We also provide two examples of sequencing integrity data for specific genes for the rrp6Δ data set, which highlight that the SIM1 gene exhibits a low percentage of full-length reads (Fig. 2C), while the VTC1 gene shows a much higher fraction of full-length reads (Fig. 2D). Because of the wide variety of samples analyzed by long-read sequencing, each user should decide on a specific minimum similarity for sequencing integrity required to make a meaningful comparison between samples.
Control of plotting and features for northern blot-like figures
Typical blot-like pictures were generated using NanoBlot and compared to actual northern blot data for RNAs extracted from S. cerevisiae WT and rrp6Δ mutant (Fig. 3A,B). The pictures generated by NanoBlot were strikingly similar to actual northern blot data, even for low abundance products such as an RNA cleavage product detected for RPL18A in the rrp6Δ mutant (Fig. 3A), or for low abundance isoforms of the ADI1 transcripts (Fig. 3B). In a nanoblot, the scale of the y-axis representing the size of the RNA isoforms can be changed allowing for more precise control over the distribution of bands. By default, nanoblots are scaled to include the full range of lengths. However, this occasionally results in plots that appear squished. Setting a custom length range can also increase plot legibility. If isoforms with large size differences are to be represented on the same blot, a logarithmic scale can be used for the y-axis (Fig. 3B).
FIGURE 3.
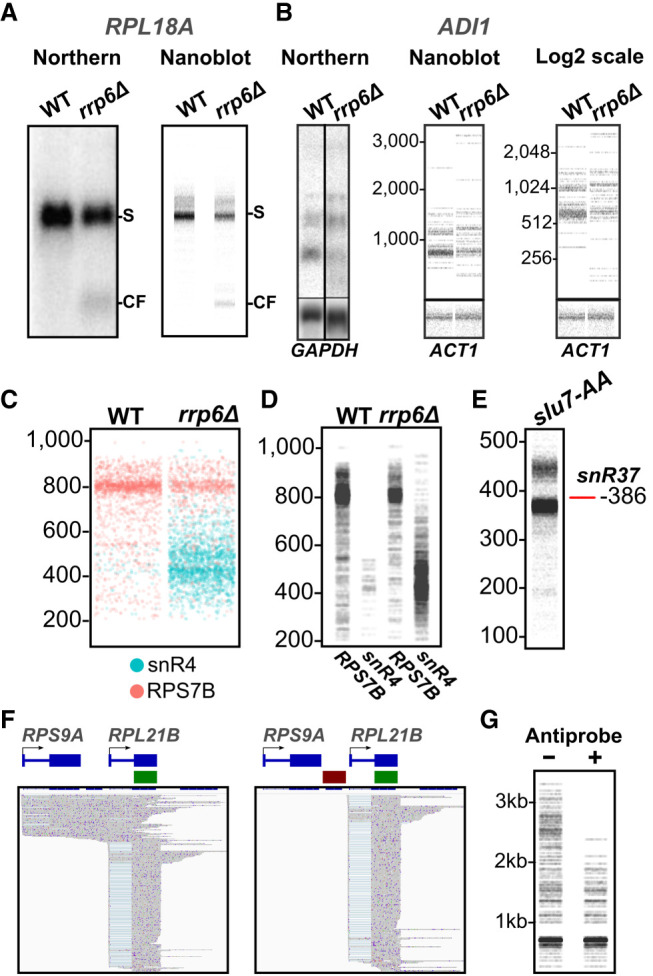
Examples of nanoblots, comparison to northern blots, overlaying, size estimation, and conditional probes. (A) Comparison between 32P northern Blot and nanoBlot targeting the 5′-exon of RPL18A. Notice the accumulation of the small molecular weight species in the rrp6Δ samples. S: Spliced RPL18A mRNA, CF: 5′ cleavage fragment. (B) Example of nanoblot on the low abundance gene, ADI1. Also shown is a nanoblot with the size axis shown on a log2 scale. (C) Overlaid nanoblot showing sequencing reads obtained from two genes probed with different colors: RPS7B = orange; snR4 = cyan. (D) The same data as in C but with each probe + samples combination separated into its own lane. (E) Nanoblot showing size misrepresentation of snR37. The red line indicates the true size of the mature snoRNA. slu7-AA: Direct RNA sequencing of Slu7-FRB rapamycin-treated RNAs preprocessed with Terminator exonuclease, rSAP, and in vitro polyadenylated. (F) IGV screenshots showing the reads selected by the use of a single probe (shown in green) (left) or a combination of one probe and an antiprobe (shown in red) (right). (G) Nanoblot produced for the data shown in F using the combination of probe only (−) or probe and antiprobe (+).
Traditional northern blots typically rely on the use of a single radioactive or fluorescent probe which detects RNAs hybridizing to the probe. Membrane stripping and subsequent hybridization can be performed to detect different RNAs (e.g., a loading control). In contrast, nanoblots can be overlapped and represented in multiple colors making overlaying multiple samples possible. To illustrate this feature, we used the data from WT and rrp6Δ mutant to generate blot-like pictures representing the relative abundances of the RPS7B and snR4 RNAs in these strains (Fig. 3C). Nanoblots can theoretically be overlayed infinitely. However, overplotting quickly becomes a concern. This can be partially alleviated by separating the probes into adjacent lanes, as shown for RPS7B and snR4 in Figure 3D.
Bands in nanoblots are not at risk of being masked by nearby highly abundant isoforms allowing for visualization of isoforms with similar sizes. However, it is worth noting that nanoblots generated from ONT sequencing data do not give single nucleotide resolution accurate size estimates. For instance, the mature form of the snR37 snoRNA is 386 nt long; however, a nanoblot shows a band slightly below the expected size (Fig. 3E). Limitations in base calling and mapping efficiency inherent to long-read sequencing result in small discrepancies in transcript length and NanoBlot includes insertions, deletions and soft-clipped regions on the 3′- and 5′-ends in the length counts. Because the frequency and lengths of insertions and deletions in ONT sequencing data are sequence-dependent, we cannot reasonably estimate this size misestimation. PacBio long-read sequencing may give a more accurate exact size due to the lower deletion rate and higher fidelity at the 5′-ends of sequences (Dohm et al. 2020).
Conditional probes and antiprobes
A useful feature of the NanoBlot package is that nanoprobes can be designed to select for more specific targets than what is possible with northern blots or RT-PCR. Nanoprobes can be designed to target reads in a conditional manner and to exclude reads mapping to a specific genomic region (hereby called antiprobe). To illustrate this feature, we used data obtained after the nuclear depletion of S. cerevisiae Nab2p. Nuclear depletion of Nab2p results in pervasive transcriptional readthrough which was demonstrated using ONT sequencing (Alpert et al. 2020). As shown in Figure 3F, the two genes RPS9A and RPL21B are located close to each other, and readthrough transcripts generated from RPS9A detected after nuclear depletion of Nab2p frequently run through the downstream RPL21B gene, convoluting the resulting plot (Fig. 3F,G) and making it difficult to identify transcripts originating from RPL21B. By using an antiprobe targeting the region between RPL21B and RPS9A, RPS9A readthrough transcripts can be excluded from the plots, which provide a clearer representation of transcripts that originate only from the RPL21B promoter (Fig. 3F,G).
NanoRT-PCR and NanoRACE
Some RNA isoforms have size differences which would be challenging to resolve on a nanoblot. This is the case for alternatively spliced species which contain alternative exons or use splice sites that result in only small size differences, or in the case of alternative transcription start sites that differ by only a few nucleotides. In order to visualize small size differences that focus on specific regions of transcripts, NanoBlot can also produce RT-PCR-like plots.
To produce an RT-PCR plot, a viewing window must be specified in the subsetNanoblot() command. NanoBlot then checks each read to ensure that they overlap with the beginning and end of the viewing window. Any read which does not is excluded from the output. The reads are then hard clipped to the bounds of the viewing window. To illustrate this feature, we generated nanoblot and an RT-PCR-like plot to visualize the presence of an isoform containing an alternative poison exon inclusion event for the BAG1 mRNA. The poison exon contains a premature translation termination codon, and isoforms that contain the poison exon can only be detected upon siRNA knockdowns of human nonsense-mediated mRNA decay (NMD) factors (Karousis et al. 2021), particularly when the NMD factors SMG6 and SMG7 are codepleted. Full-length BAG1 transcripts range from 1 to 4 kb long and the alternative poison exon is only 94 bp long, which makes it difficult to discriminate the poison exon containing mRNAs from the other BAG1 species on a standard nanoblot (Fig. 4A), even though it is clear that the total amount of BAG1 RNAs increase after knockdown of NMD factors. In contrast, generating a NanoRT-PCR plot highlights the stabilization of the RNAs containing the poison exon cassette upon NMD factors knockdown (Fig. 4B), providing an example for the use of Nano RT-PCR.
FIGURE 4.

NanoRT-PCR, NanoRACE, and NanoRidge. (A) Nanoblot plot showing an alternative exon inclusion event (SCR1) Scramble 1, (SCR2) Scramble 2, (UPF1) UPF1 Knockdown, (SMG6) SMG6 Knockdown, (SMG7) SMG7 Knockdown, (SMG6 + 7) SMG6 and SMG7 Double Knockdown. (B) NanoRT-PCR plot with a viewing window spanning the alternative exon. Labels same as in A. (C) IGV screenshots of WT sequencing data and NanoRACE plots. RPB2 shows alternative polyadenylation site usage while DIS3 shows a single poly(A) site. Note the abundance of short reads making representation via a nanoblot challenging. (D) NanoRACE of reads mapping to the Arabidopsis thaliana PRPL34 gene obtained from the VIR control or vir1-1 mutant samples shown as a blot. (E) NanoRACE representation of PRPL34 data shown as a ridge plot. The vertical black lines indicate the median read length.
A standard nanoblot visualization has limitations when focusing on transcripts 3′-ends or to highlight differences in polyadenylation sites which do not result in major differences in the size of transcripts. To directly visualize alternative 3′-ends or polyadenylation sites of specific transcripts, we have included the “RACE” parameter in subsetNanoblot(). When “RACE” is set to TRUE, reads are not clipped downstream from the 3′-end of the viewing window. This makes NanoBlot a useful tool for the visualization of differences in 3′-ends or polyadenylation sites. The use of the “RACE” parameter circumvents the issue of partial coverage of the sequencing reads, as many transcripts exhibit a bias of sequencing toward the 3′-end. To illustrate this feature, we used sequencing reads for the S. cerevisiae RPB2 and DIS3 transcripts. Many of the reads for these two genes do not cover the entire gene (Fig. 4C), so they could not be used for accurate size estimation. However, these reads still provide useful information regarding the poly(A) sites used, as the reads coverage is biased toward the 3′-end of these transcripts. Accurate density plots can be produced by setting the ViewingWindow parameter of subsetNanoblot() to cover the poly(A) sites. The NanoRace plots of Figure 4C show that the RPB2 mRNA exhibits two major poly(A) sites used at approximately equal rates, while the DIS3 mRNA shows only a single poly(A) site.
Quantification of isoforms with small length changes using ridge plots
Representation of isoforms with minor length changes can be challenging using nanoblots. For instance, the inactivation of the VIR gene in A. thaliana causes increased usage of proximal poly(A) sites (Parker et al. 2020). However, because the distance between poly(A) sites can be relatively small (25 nt) visualizing the size change in a nanoblot, even when using nanoRACE, is not easy (Fig. 4D). Using the ridge plot type and adding a line indicating the median read length helps to illustrate this subtle change (Fig. 4E).
Advantages and limitations of nanoblots
Issues and limitations linked mapping and repeated or homologous sequences
ONT sequencing has some limitations. Lower average read quality leads to significantly more reads being mismapped or rendered unmappable compared to standard short reads sequencing. Of particular concern are genes with multiple copies or paralogs. As an example, the S. cerevisiae TDH1 gene has two closely related paralogs, TDH2 and TDH3. Long-read aligners (e.g., Minimap2) have a particularly difficult time uniquely mapping these reads and therefore they are either not represented in the results BAM files or flagged as secondary alignments. As a result, NanoProbes targeting TDH1 can produce blank plots or plots with extra unintended reads depending on how the mapping was handled. To check for this the NanoBlot plotting function scans for potentially multimapped reads. By default, if more than 10% of the reads in a subsetted BAM file are flagged as secondary alignments then a warning is issued. The specific percentage of secondary alignments required to trigger a warning can be changed by the end user. Because NanoBlot performs this check it is recommended that users initially run aligners allowing for secondary alignments. However, due to the nature of ONT sequencing excessively short or homopolymer-rich exons can be skipped during alignment. This generally results in a lower alignment score, which is reported as a secondary alignment. In these cases, it is recommended that the alignment be rerun without allowing for secondary alignments to prevent duplication of the data.
Sensitivity
Nanoblots are subject to the limitations of Nanopore sequencing. One of the major issues is that short RNA fragments or isoforms are not efficiently mapped. As a result, smaller fragments cannot be efficiently represented in a nanoblot. Sensitivity is also a concern. It is challenging to quantify how sensitive Nanopore sequencing is compared to traditional northern blots. As an example, ADI1, a low abundance RNA transcript, is shown as either a 32P northern blot, or a nanoblot (Fig. 3B). As with any sequencing approach that includes a library preparation, potential biases could be introduced by the ONT sequencing kits. As new strategies are developed to prevent biases, they can be seamlessly incorporated into NanoBlot.
Issues linked to the presence of degradation products
RNA degradation and incomplete transcript sequencing are a problem for accurate transcript size estimation. Transcripts partially degraded from the 5′-end but with an intact poly(A) tail will still be sequenced via ONT sequencing resulting in shortened read sequences. In nanoblots, this results in a low molecular weight smear which can make representation difficult especially for longer transcripts. One possible solution is to specify two regions as probes, one at the 5′-end of the transcript and another at the 3′-end. This will result in the visualization of only near full-length transcripts. It is worth noting that if isoforms of substantially different lengths are being investigated this type of selection could result in the blot being biased toward shorter transcripts.
Detection and visualization of polyadenylated and nonpolyadenylated species and poly(A) tail length
Standard Nanopore RNA sequencing is poly(A)-based meaning that any transcript lacking a poly(A) tail will not be sequenced. There do exist ways to prepare RNAs for sequencing non-poly(A) RNAs. RNAs can be tailed or in vitro polyadenylated prior to library synthesis (Tudek et al. 2021; Liu et al. 2022) and several approaches have been developed to sequence specific types of RNAs (Drexler et al. 2021; Ibrahim et al. 2021; Vo et al. 2021). However, some specific RNAs lack terminal 3′ hydroxyls (e.g., the U6 snRNA; Lund and Dahlberg 1992) or have modifications at their 3′-ends which prevent their polyadenylation in vitro (e.g., aminoacylated tRNAs). While specific ad hoc enzymatic treatments such as deacylation of tRNAs in vitro (see, for example, Czech 2020) can help to promote in vitro polyadenylation of certain RNA, it is challenging to develop a general strategy which would allow the detection of all classes of non-polyadenylated RNAs using Nanopore RNA sequencing, so ad hoc strategies similar to the ones described in the studies cited above should be used.
Finally, poly(A) tail length is not included in the bands shown on nanoblots. The poly(A) tail length could theoretically be included if a direct RNA sequencing strategy is used, because poly(A) tail length can be estimated and added to the length although the initial release of NanoBlot does not have this functionality (Krause et al. 2019).
MATERIALS AND METHODS
Yeast growth
WT and rrp6Δ knockout yeast strains are from the BY4741 genetic background. Yeast cultures were grown in YPD (1% w/v yeast extract, 2% w/v peptone, and 2% w/v dextrose). Briefly, 50 mL cultures were grown at the standard 30°C to an OD600 of ∼0.4. Cells were then pelleted and flash frozen in liquid nitrogen for RNA isolation. RNA isolation was performed as described (Wang et al. 2020).
RNA sequencing libraries preparation
Total RNAs were treated with DNase I (Invitrogen, catalog #: 18-068-015) following the manufacturer's protocol. Sequencing libraries were prepared using the Direct RNA Sequencing Kit from Oxford Nanopore (ONT, catalog #: SQK-RNA002) according to the manufacturer's instructions. Sequencing was performed using R9.4 flow cells on a MinION Mk1B device and sequenced for 48 h.
BAM file preprocessing
Base-calling was performed using Guppy Basecaller (version 6.1.1+1f6bfa7f8). Reads were then mapped to the S. cerevisiae genome: (S288C_reference_sequence_R64-3-1) using Minimap 2 (version 2.17-r941). Mapped reads were sorted and indexed using IGVtools (Robinson et al. 2011).
Normalization
Because NanoBlots are generated from full RNA-sequencing data sets, many different normalization techniques are viable. Nanoblot includes the normalizeNanoblotData() function which can perform normalization using DESeq2 or a simple library size normalization. By default, NanoBlot accepts an annotation file as input and normalization is done via DESeq2 (Love et al. 2014). First, NanoBlot counts read using Rsubread (Liao et al. 2019). Next, NanoBlot runs the estimateSizeFactors() command from DESeq2 to generate size factors. Finally, the user supplies the size factors to the makeNanoblot() function. NanoBlot generates plots with increased sampling for smaller libraries. Unnormalized data are used to produce density plots. Multiple replicates can be used in the DESeq normalization.
If an annotation file is not available, NanoBlot can also generate normalization factors based solely on library depth. Other normalization techniques can be used depending on the specific data sets used. NanoBlot can accept either normalized or unnormalized reads.
Data set generation
Bedtool's intersect function is used to subset the input files to only reads which overlap with the region of interest. An arbitrary number of probes can be specified as a character vector and will be treated as AND probes, requiring each read to overlap with all specified probes. Probes to be used for NOT statements are specified as a character vector in the “targetAntiProbe” option. OR logical statements must be done as two separate selections and be merged by the end user.
NanoBlot can also produce RT-PCR or RACE-like plots where the length of each read is counted as the number of mapped bases in a viewing window. The “viewingWindow” option is used to specify the RT-PCR function. See README for additional information.
R packages and plotting
All required R packages are installed via Bioconductor (Huber et al. 2015). The subsetted BAMs are read into R using scanBAM() from the Rsamtools package (Morgan et al. 2022). The length of each read is calculated from the CIGAR string and listed in the qwidth column. Data are reformatted for plotting with the dplyr (Wickham et al. 2022) package. Plot generation is done via ggplot2 (Wickham 2016; Wilke 2021).
DATA DEPOSITION
Data for Saccharomyces cerevisiae SLU7 Anchor-Away were downloaded from the SRA, accession number PRJNA827814. Data for Saccharomyces cerevisiae Nab2 Anchor-Away were downloaded from the GEO, accession number GSE156133. Data for Arabidopsis thaliana VIR and vir-1 mutant were downloaded from the ENA, accession number PRJEB32782. Data for human NMD factors knockdown were downloaded from the ArrayExpress database at EMBL-EBI (www.ebi.ac.uk/arrayexpress) under accession number: E-MTAB-10452. Sequencing data generated for this study are available at the SRA, accession number: PRJNA891745.
All custom R script used to generate figures in this publication are available in the NanoBlot GitHub (https://github.com/SamDeMario-lab/NanoBlot). NanoBlot is available under the MIT license. DOI: 10.5281/zenodo.7213547.
ACKNOWLEDGMENTS
We thank members of the Chanfreau laboratory for helpful discussions and feedback on the manuscript figures. This work was supported by grant GM130370 from the National Institute of General Medical Sciences (NIGMS) to G.F.C.
Author contributions: S.D. and G.F.C. conceived the study and wrote the manuscript. S.D. and K.X. authored the code. K.H. generated sequencing libraries and performed beta testing. S.D. created figures. All authors reviewed the final manuscript.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079505.122.
Freely available online through the RNA Open Access option.
MEET THE FIRST AUTHOR
Samuel DeMario.

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Samuel DeMario is the first author of this paper, “NanoBlot: An R-package for visualization of RNA isoforms from long-read RNA-sequencing data.” Samuel is a PhD candidate in the Biochemistry, Molecular and Structural Biology program at UCLA. He joined the department in 2018 and works in the laboratory of Guillaume Chanfreau. His research focuses on RNA degradation pathways and computational methods in long-read sequencing.
What are the major results described in your paper and how do they impact this branch of the field?
Here, we present NanoBlot our R-package for the creation of northern blot-like images from long-read sequencing data. As sequencing technologies have advanced the amount of information we get about specific RNA isoforms increases dramatically, necessitating new ways to represent data. We hope NanoBlot becomes a useful part of the data visualization toolbelt.
What led you to study RNA or this aspect of RNA science?
During the 2021 RNA Society meeting, one of the speakers showed a northern blot and said they “were keeping the art of the northern blot alive.” Later, I was talking to my lab mate about that comment, and he mentioned that northern blots are satisfying to look at. I tend to agree with him. For many biochemists, agarose gels are among their first experiences with raw data. Because of this, interpretation of blots or blot-like data is intuitive. This realization led us to develop NanoBlot.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
The first nanoblot we made was incredibly surprising to us. I had the idea for NanoBlot around 1 P.M. and by 5 P.M. the first NanoBlot had been made. The next morning, I invited Guillaume to look at the plot I had made. I pulled up the nanoblot and explained to him what I did. There was a moment of awkward silence where Guillaume waited for me to pull up the nanoblot, followed by laughter when he realized he was already looking at it. Since then, we have changed the default way that nanoblots are displayed to make them look less like real northern blots.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
I wish I remembered where I first heard this, but it was pointed out to me that Einstein's E = mc2 equation did not come out of a perfectly designed experiment or incredibly complex derivation. Rather, it's a reassembling of previously characterized equalization into a form with interesting implications. What I took away from this is that the way you represent your results is just as important as the results themselves.
What are your subsequent near- or long-term career plans?
In the future, I'd like to work on personal genomics for early medical intervention.
REFERENCES
- Alpert T, Straube K, Carrillo Oesterreich F, Herzel L, Neugebauer KM. 2020. Widespread transcriptional readthrough caused by Nab2 depletion leads to chimeric transcripts with retained introns. Cell Rep 33: 108324. 10.1016/j.celrep.2020.108324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annaldasula S, Gajos M, Mayer A. 2021. IsoTV: processing and visualizing functional features of translated transcript isoforms. Bioinformatics 37: 3070–3072. 10.1093/bioinformatics/btab103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech A. 2020. Deep sequencing of tRNA's 3′-termini sheds light on CCA-tail integrity and maturation. RNA 26: 199–208. 10.1261/rna.072330.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohm JC, Peters P, Stralis-Pavese N, Himmelbauer H. 2020. Benchmarking of long-read correction methods. NAR Genom Bioinform 2: lqaa037. 10.1093/nargab/lqaa037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler HL, Choquet K, Merens HE, Tang PS, Simpson JT, Churchman LS. 2021. Revealing nascent RNA processing dynamics with nano-COP. Nat Protoc 16: 1343–1375. 10.1038/s41596-020-00469-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilcher M, Liehrmann A, Seyman C, Blein T, Rigaill G, Castandet B, Delannoy E. 2021. Full length transcriptome highlights the coordination of plastid transcript processing. Int J Mol Sci 22: 11297. 10.3390/ijms222011297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. 2015. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods 12: 115–121. 10.1038/nmeth.3252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim F, Oppelt J, Maragkakis M, Mourelatos Z. 2021. TERA-Seq: true end-to-end sequencing of native RNA molecules for transcriptome characterization. Nucleic Acids Res 49: e115. 10.1093/nar/gkab713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karousis ED, Gypas F, Zavolan M, Mühlemann O. 2021. Nanopore sequencing reveals endogenous NMD-targeted isoforms in human cells. Genome Biol 22: 223. 10.1186/s13059-021-02439-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. 2002. The human genome browser at UCSC. Genome Res 12: 996–1006. 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M, Niazi AM, Labun K, Torres Cleuren YN, Müller FS, Valen E. 2019. tailfindr: alignment-free poly(A) length measurement for Oxford Nanopore RNA and DNA sequencing. RNA 25: 1229–1241. 10.1261/rna.071332.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2019. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res 47: e47. 10.1093/nar/gkz114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, DeMario S, He K, Gibbs MR, Barr KW, Chanfreau GF. 2022. Splicing inactivation generates hybrid mRNA-snoRNA transcripts targeted by cytoplasmic RNA decay. Proc Natl Acad Sci 119: e2202473119. 10.1073/pnas.2202473119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund E, Dahlberg JE. 1992. Cyclic 2′,3′-phosphates and nontemplated nucleotides at the 3′ end of spliceosomal U6 small nuclear RNA's. Science 255: 327–330. 10.1126/science.1549778 [DOI] [PubMed] [Google Scholar]
- Morgan M, Pagès H, Obenchain V, Hayden N. 2022. Rsamtools: Binary alignment (BAM), FASTA, variant call (BCF), and tabix file import. https://bioconductor.org/packages/Rsamtools. [Google Scholar]
- Parker MT, Knop K, Sherwood AV, Schurch NJ, Mackinnon K, Gould PD, Hall AJ, Barton GJ, Simpson GG. 2020. Nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and m6A modification. Elife 9: e49658. 10.7554/eLife.49658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese F, Mortazavi A. 2021. Swan: a library for the analysis and visualization of long-read transcriptomes. Bioinformatics 37: 1322–1323. 10.1093/bioinformatics/btaa836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein AN, Joglekar A, Poon C-L, Tilgner HU. 2022. ScisorWiz: visualizing differential isoform expression in single-cell long-read data. Bioinformatics 38: 3474–3476. 10.1093/bioinformatics/btac340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudek A, Krawczyk PS, Mroczek S, Tomecki R, Turtola M, Matylla-Kulińska K, Jensen TH, Dziembowski A. 2021. Global view on the metabolism of RNA poly(A) tails in yeast Saccharomyces cerevisiae. Nat Commun 12: 4951. 10.1038/s41467-021-25251-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo JM, Mulroney L, Quick-Cleveland J, Jain M, Akeson M, Ares MJ. 2021. Synthesis of modified nucleotide polymers by the poly(U) polymerase Cid1: application to direct RNA sequencing on nanopores. RNA 27: 1497–1511. 10.1261/rna.078898.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Liu Y, DeMario SM, Mandric I, Gonzalez-Figueroa C, Chanfreau GF. 2020. Rrp6 moonlights in an RNA exosome-independent manner to promote cell survival and gene expression during stress. Cell Rep 31: 107754. 10.1016/j.celrep.2020.107754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. 2016. ggplot2: elegant graphics for data analysis. Springer-Verlag, New York. [Google Scholar]
- Wickham H, François R, Henry L, Müller K. 2022. dplyr: a grammar of data manipulation. https://CRAN.R-project.org/package=dplyr. [Google Scholar]
- Wilke CO. 2021. ggridges: Ridgeline Plots in “ggplot2”. https://CRAN.R-project.org/package=ggridges. [Google Scholar]