

Abstract
The vacuolar-type ATPase (V-ATPase) is a multisubunit protein composed of the cytosolic adenosine triphosphate (ATP) hydrolysis catalyzing V1 complex, and the integral membrane complex, Vo, responsible for proton translocation. The largest subunit of the Vo complex, subunit a, enables proton translocation upon ATP hydrolysis, mediated by the cytosolic V1 complex. Four known subunit a isoforms (a1–a4) are expressed in different cellular locations. Subunit a1 (also known as Voa1), the neural isoform, is strongly expressed in neurons and is encoded by the ATP6V0A1 gene. Global knockout of this gene in mice causes embryonic lethality, whereas pyramidal neuron-specific knockout resulted in neuronal cell death with impaired spatial and learning memory. Recently reported, de novo and biallelic mutations of the human ATP6V0A1 impair autophagic and lysosomal activities, contributing to neuronal cell death in developmental and epileptic encephalopathies (DEE) and early onset progressive myoclonus epilepsy (PME). The de novo heterozygous R740Q mutation is the most recurrent variant reported in cases of DEE. Homology studies suggest R740 deprotonates protons from specific glutamic acid residues in subunit c, highlighting its importance to the overall V-ATPase function. In this paper, we discuss the structure and mechanism of the V-ATPase, emphasizing how mutations in subunit a1 can lead to lysosomal and autophagic dysfunction in neurodevelopmental disorders, and how mutations to the non-neural isoforms, a2–a4, can also lead to various genetic diseases. Given the growing discovery of disease-causing variants of V-ATPase subunit a and its function as a pump-based regulator of intracellular organelle pH, this multiprotein complex warrants further investigation.
Keywords: V-ATPase, subunit a1, R740, neurodevelopmental disorders, developmental and epileptic encephalopahties, progressive myoclonus epilepsy
1. Introduction
The vacuolar H+-ATPase (V-ATPase) is an adenosine triphosphate (ATP)-dependent proton pump that regulates electrochemical gradients between membranes. These pumps are critical for pH homeostasis in the extracellular milieu and intracellular organelles, such as lysosomes, the Golgi network, and the endoplasmic reticulum (Wang L. et al., 2020). Membrane V-ATPases are crucial for various biological processes as they acidify specialized organelles involved in bone resorption (Toyomura et al., 2003), sperm maturation (Pietrement et al., 2006), and proton secretion by intercalated kidney cells (Wagner et al., 2004). V-ATPases are also involved in various signaling cascades, including Wnt, Notch, and mTOR, important for regulating cell proliferation and differentiation (Zoncu et al., 2011; Sun-Wada and Wada, 2015). Organellar V-ATPases establish and maintain the pH of endosomes and lysosomes for intracellular membrane trafficking and protein degradation (Forgac, 2007). V-ATPases are also suggested to play an important role in regulating membrane fusion and neurotransmitter release, such as glutamate and norepinephrine (Hinton et al., 2009). Furthermore, increased expression of V-ATPase on the plasma membrane facilitates cancer progression by maintaining an acidic tumor microenvironment (Pamarthy et al., 2018).
The V-ATPase is a large multisubunit rotary machine composed of two dissociable sectors: the peripheral V1 sector responsible for ATP hydrolysis and the membrane integral Vo sector governing proton translocation. Human V-ATPases consist of eight V1 subunits (A3, B3, C, D, E3, F, G3, H) and eight Vo subunits (a, c9, c″, d, e, Ac45, RNaseK and ATP6AP2), some of which are present in multiple copies as denoted by the subscript numbers. These subunits congregate and collaborate to hydrolyze ATP and subsequently conduct proton translocation across membranes (Wang L. et al., 2020). Subunit a is the largest of the Vo complex (~110 kDa) and mediates the entry and exit pathway of protons from the cytoplasmic to the luminal side, coupled with c-ring rotation powered by ATP hydrolysis (Kawasaki-Nishi et al., 2001b; Wang L. et al., 2020).
The V-ATPase pumps protons into the lumen of lysosomes, thereby decreasing the pH to activate autophagic enzymes. Disruption of neuronal lysosomes and their autophagic function cause aberrant accumulation of intracellular proteins and the formation of inclusion bodies, leading to neurodegeneration (Hara et al., 2006; Komatsu et al., 2006). Likewise, studies reported that dysfunction of the a1 subunit is implicated in neuronal impairment. For example, global knockout of the subunit a1 gene, ATP6V0A1 (located on chromosome 17q21.2), leads to embryonic lethality (Dickinson et al., 2016). Furthermore, impeded V-ATPase assembly and function due to impaired subunit a1 glycosylation and stability upon mutations to presenilin-1 (PS-1) was observed in the Alzheimer’s disease (AD) mouse model (Lee et al., 2015). Analysis of variant frequencies in AD reported that PS-1 mutations are the most likely pathogenic variant, followed by mutations to the amyloid precursor protein (APP) and presenilin-2 (PS-2) respectively, highlighting the indirect relationship between subunit a1 and AD (Xiao et al., 2021). Moreover, misrouted a1 is the main cause of lysosomal dysregulation in the mouse model of infantile neuronal ceroid lipofuscinoses, also known as Batten disease (Bagh et al., 2017). a1 conditional knockout in the forebrain pyramidal neurons of mice also resulted in general brain atrophy of hippocampal CA1 and displayed impaired learning and memory (Ma et al., 2019). Recently, studies have reported that various de novo and biallelic mutations to ATP6V0A1 are a major cause of DEE (developmental and epileptic encephalopathies) (Aoto et al., 2021; Bott et al., 2021). The R741Q mutation (R740Q based on the updated accession number NM_001130021.3) leads to impaired proton translocation and is a recurring DEE-causing variant. Additionally, a1 compound heterozygous variants, E149Kfs18 and R495W, are identified in early-onset PME (progressive myoclonus epilepsy) with ataxia (Bott et al., 2021). These findings demonstrate the crucial role of the a1 subunit in a functional V-ATPase for neuronal lysosome acidification and autophagosomes. It also highlights V-ATPases pathophysiology in neurodevelopmental and neurodegenerative disorders.
In this review, we will first cover the structure and mechanism of the V-ATPase, focusing on subunit a1. We will then discuss how various mutations to key a1 residues impair lysosomal and autophagosomal processes involved in neurodegenerative disease. Additionally, we will consider disease-associated variants of the other subunit a isoforms: a2, a3, and a4. Our review highlights the importance of understanding the pathophysiology of ATP6V0A1 mutations. Further studies will improve the knowledge surrounding V-ATPase’s role in brain development and neurodevelopmental disorders with lysosomal and autophagic dysfunction.
2. Structure and function of the V-ATPase and subunit a1
Characterizing the structure of the V-ATPase is critical to understanding its function. The yeast V-ATPase, the most thoroughly characterized V-ATPase, has been a useful model for studying its mammalian counterpart (Toei et al., 2010). It is composed of 31 polypeptides that are organized into two distinct structural sectors: a cytoplasmic V1 ATPase and a membrane-embedded Vo proton channel (Figure 1A; Zhao et al., 2015). The yeast V1 region contains subunits A3, B3, C, D, E3, F, G3, and H, while the Vo proton channel contains subunits a, c8, c′, c″, d, e, f, and Voa1p (note: subscript number indicates number of repeating subunits) (Mazhab-Jafari et al., 2016; Roh et al., 2018). The human V-ATPase V1 subunit is similar to its yeast counterpart in terms of subunit type and number (Wang L. et al., 2020). However, they differ in Vo composition, containing the following subunits: a, c9, c″, d, e, Ac45 (also known as ATP6AP1 and is equivalent to yeast Voa1p), RNaseK (equivalent to yeast f), and ATP6AP2 (Wang L. et al., 2020). Comparing the yeast and human subunit a isoforms reveals 30–50% (up to 85%) sequence similarity (Roh et al., 2018). This high similarity increases confidence when translating yeast research to the human subunit a of interest.
Figure 1.

Structure and mechanism of the human V-ATPase. (A) V-ATPase structure. The cytoplasmic V1 sector is responsible for ATP hydrolysis and contains the following subunits: A3, B3, C, D, E3, F, G3, and H. The membrane-embedded Vo sector mediates proton translocation and is composed of the following subunits: a1 (neuronal isoform), c9, c″, d, e, Ac45 (also known as ATP6AP1), RNaseK, ATP6AP2. The top of V1 includes the A3B3 hexamer head, the site of ATP binding and hydrolysis. ATP hydrolysis induces the rotation of the A3B3 hexamer which subsequently powers the rotation of the c-ring (composed of subunits c9 and c″) via linkage through subunits D, F, and d (the central stalk). The peripheral stalks, composed of subunits E and G, act as stators and connect the V1 sector with Vo. Protons enter and exit subunit a1 via the hemichannels and interact with key residues in a1 and the c-ring in the process of proton translocation. (B) ATP hydrolysis cycle. ATP hydrolysis occurs between the interface of the A and B subunits in the V1 complex. The C-terminal domains of A and B undergo conformational changes depending on their bound state to ATP and ADP. First, ATP is tightly bound between the A and B subunits and is subsequently hydrolyzed into ADP and inorganic phosphate (Pi). The Pi is released and the AB pair changes conformations to a less tight ADP-bound form. This triggers the adjacent AB pair to change conformations from a semi-open (nucleotide-free) conformation to a high ATP-affinity open confirmation. Next, ATP binds tightly to the open AB pair, changing the ADP-bound dwell into a semi-open conformation, thus releasing ADP. The conformational changes associated with the ATP hydrolysis cycle induce the tilting and oscillation of the A3B3 hexamer. (C) Proton translocation pathway. A proton enters the cytoplasmic hemichannel and protonates an essential glutamic acid residue on the c-ring (E139 subunit c or E98 subunit c”). Next, the c-ring, powered by ATP hydrolysis in V1, rotates clockwise through the lipid bilayer to bring the protonated c-ring glutamates close to an essential a1 residue, R740. A salt bridge forms between the protonated glutamate residue and R740, causing the release of the proton into the luminal hemichannel. Finally, the proton exits the membrane and enters the lumen by following a network of polar and negatively charged residues lining the luminal hemichannel (H748, E794, and R804), resulting in luminal acidification. For every three ATP molecules hydrolyzed, 10 protons are translocated. Amino acid positions are based on the updated accession number NM_001130021.3. The figure was created with BioRender.com.
2.1. The A3B3 hexamer
The top of the V1 complex is composed of an A3B3 hexamer head containing three pairs of the AB heterodimer alternatively arranged around a pseudo-6-fold symmetry axis (Figure 1A; Wang L. et al., 2020). ATP binding and hydrolysis occur between the interface of the A and B subunits (Liu et al., 1996, 1997). Initially, subunit A was believed to contain catalytic nucleotide binding sites, whereas subunit B had non-catalytic nucleotide-binding sites (Manolson et al., 1985; Feng and Forgac, 1992; Zhang et al., 1995; Vasilyeva and Forgac, 1996). The non-catalytic sites were hypothesized to be necessary to achieve maximum enzymatic activity and regulate activity in general (MacLeod et al., 1998). Contrastingly, recent evidence suggests that the mammalian V1 subunit does not have non-catalytic nucleotide binding sites (Abbas et al., 2020). Moreover, the A subunit in bacterial V-ATPase is thought to contain most of the catalytic residues but not all (Ueno et al., 2018). Although the specifics of the nucleotide binding site remain obscure, the ATP hydrolysis cycle within the A3B3 hexamer is well-reported. During the ATP hydrolysis cycle, the C-terminal domains of A and B undergo conformational changes depending on their bound state to ATP and ADP (Suzuki et al., 2016; Wang L. et al., 2020). First, ATP is tightly bound between the A and B subunits and is subsequently hydrolyzed into ADP and inorganic phosphate (Pi) (Figure 1B; Suzuki et al., 2016). The Pi is released and the AB pair changes conformations to a less tight ADP-bound form (Suzuki et al., 2016). This triggers the adjacent AB pair to change conformations from a semi-open (nucleotide-free) conformation to a high ATP-affinity open confirmation (Suzuki et al., 2016; Wang L. et al., 2020). Next, ATP binds tightly to the open AB pair, changing the ADP-bound dwell into a semi-open conformation, eventually releasing ADP (Suzuki et al., 2016). The conformational changes associated with the ATP hydrolysis cycle induce the tilting and oscillation of the A3B3 hexamer (Suzuki et al., 2016; Wang L. et al., 2020). These movements ultimately drive the 120o rotation of the central stalk (subunits D and F), which connects the middle hole of the A3B3 hexamer to the Vo complex, with a torque of ∼25 pNnm (Suzuki et al., 2016; Ueno et al., 2018; Wang L. et al., 2020). The rotation of the central stalk eventually leads to the transportation of a proton across the membrane (Nishi and Forgac, 2002). Subunit A is also involved in regulating V-ATPase disassembly. The non-homologous region of subunit A, a 90-amino acid domain not present in the β-subunit of the F-ATPase, interacts with the Vo complex to regulate glucose-dependent V-ATPase dissociation (Shao et al., 2003; Shao and Forgac, 2004).
2.2. Peripheral stalks
The V1 and Vo sectors are connected by three heterodimers of E and G subunits, known as the peripheral stalks (Figure 1A; Colina-Tenorio et al., 2018). The static peripheral stalks act as the stator of a motor to counteract the torque of the central stalk (Colina-Tenorio et al., 2018). Structurally, the EG heterodimer forms a long (150 Å) right-handed coiled-coil in the middle and is stabilized at their N and C termini by forming globular domains and interacting with other subunits (Oot et al., 2012). All three peripheral stalks (EG1-EG3) share the same sequence but slightly differ in their conformations, as their N-terminal domains (NTDs), contributed by both E and G NTDs, associate with different partners in the complex (Colina-Tenorio et al., 2018; Wang L. et al., 2020). EG1 interacts with the “foot” domain of a-NTD and subunit H; EG2 links with the “head” domain of a-NTD and the “foot” domain of subunit C; and EG3 only connects to the “head” domain of subunit C (Benlekbir et al., 2012; Zhao et al., 2015; Wang L. et al., 2020). Meanwhile, the C-terminal domains (CTDs) of the peripheral stalks, contributed by both E and G CTDs, interact with the N-termini of the B subunits of the A3B3 hexamer (Benlekbir et al., 2012; Zhao et al., 2015; Wang L. et al., 2020). Along with their stator role, the peripheral stalks are thought to be crucial in the disassembly and reassembly of V-ATPases in response to extracellular stimuli (Oot et al., 2017). The regulation of V-ATPase activity through its assembly and dissociation is known as reversible dissociation. Although the exact mechanism of the peripheral stalk role in reversible dissociation remains obscure, they are permissive to the movement of the complex and the rearrangement of other subunits, which ultimately drives V-ATPase reversible dissociation (Oot et al., 2012, 2017).
2.3. Subunits C and H
The single subunits of the V1 complex, C and H, play an important role in V-ATPase reversible dissociation and overall enzymatic activity (Liu et al., 2005; Diab et al., 2009; Pérez-Sayáns et al., 2012; Oot et al., 2016; Sharma et al., 2018). Subunits C and H lie between the V1Vo interface and stabilize the peripheral stalk NTDs as aforementioned (Figure 1; Colina-Tenorio et al., 2018; Wang L. et al., 2020). Structurally, subunit C consists of three different domains: A globular ‘head’ domain made of four antiparallel β-sheets and two α-helices; a long three-helix bundle ‘neck’ domain connected by salt bridges; and a globular ‘foot’ domain composed of similar structures to the ‘head’ domain and hosts both the N and C termini (Drory et al., 2004; Wang R. et al., 2020). First identified in yeast, glucose starvation results in the dissociation of the V1 structure from the Vo sector, and reassembly is initiated upon restoration of glucose in the growth medium (Kane, 1995). Following V-ATPase dissociation, subunit C separates from both sectors and is released into the cytosol, which may be mechanistically achieved through the disruption of the interaction between subunit C and EG3 (Sumner et al., 1995; Parra and Kane, 1998; Tabke et al., 2014; Oot et al., 2017). Meanwhile, with the help of subunit H, the dissociated sectors are functionally silent as V1 ATPase activity is inhibited and Vo proton translocation is stopped (Parra et al., 2000; Couoh-Cardel et al., 2016; Oot et al., 2016). Subunit H is structurally separated into two distinct domains, connected by a flexible four-residue loop (Sagermann et al., 2001; Liu et al., 2005; Wang L. et al., 2020). The NTD, the larger of the two domains, is characterized by its repetitive arrangement of 17 right-handed α-helices (Sagermann et al., 2001). The smaller CTD is composed of eight α-helices that form two turns of a right-handed superhelix (Sagermann et al., 2001). Following the dissociation of V1Vo in yeast, the H subunit silences the enzymatic activity of V1, preventing erroneous ATP hydrolysis (Parra et al., 2000; Oot et al., 2016). This inhibition is initiated when the separation of the sectors causes the CTD of subunit H to move and rotate away from its binding site on a-NTD to newly bind the central rotor subunit D and the catalytic site of subunit B, trapping ADP at the catalytic site and preventing ATP hydrolysis (Parra et al., 2000; Oot et al., 2016). Contrarily, the Regulator of the H+-ATPase of Vacuoles and Endosomes (RAVE) complex in yeast helps in V-ATPase reassembly upon glucose readdition (Smardon et al., 2002; Smardon and Kane, 2007). The RAVE complex first associates with sector V1 and then binds to available subunit C (Jaskolka et al., 2021). Next, RAVE directs V1 and C to Vo and is hypothesized to accelerate their association and assembly (Jaskolka et al., 2021). In mammalian cells, rabconnectin-3α, homologous to one of the RAVE protein complexes Rav1p, is also involved in V-ATPase assembly (Einhorn et al., 2012).
2.4. The proteolipid c-ring
As aforementioned, the membrane-embedded Vo complex contains subunits a, c9, c″, d, e, Ac45 (also known as ATP6AP1), RNaseK, and ATP6AP2 (Wang L. et al., 2020). While the V1 complex is responsible for ATP hydrolysis, the Vo complex canonically functions as a proton channel (Figure 1; Zhao et al., 2015). A key structure of the human Vo complex is the c-ring, containing two proteolipid variants, c and c″, in a 9:1 ratio (Figure 1A; Abbas et al., 2020; Wang L. et al., 2020). The c and c″ subunits are highly hydrophobic and are composed of four and five transmembrane helices, respectively (Flannery et al., 2004). Functionally, the c-ring is primarily involved in proton translocation, which is mediated by a key glutamic acid residue buried in each c/c″ subunit, undergoing reversible protonation (Figures 1A,B; Hirata et al., 1997). The conserved glutamic acid residues are E139 on subunit c and E98 on subunit c″, according to UniProt (P27449, Q99437) (Wang L. et al., 2020). To achieve proton translocation, the c-ring must rotate clockwise relative to the adjacent stator subunit a (Forgac, 2007). This movement is powered by ATP hydrolysis in V1, rotating the central stalk, which is linked to the c-ring via subunit d (Figure 1B; Iwata et al., 2004; Wang L. et al., 2020). The cone-shaped subunit d sits on top of the c-ring and attaches to the central stalk on its concave surface in a shape-complementary manner (Wang L. et al., 2020). The convex or N-terminus side of subunit d interacts with the c-ring, specifically at the cytosolic N-terminus loop of c″ (Roh et al., 2018; Wang L. et al., 2020). The d-to-c-ring interaction is reinforced by residues forming hydrogen bonds, hydrophobic contacts, and a salt bridge (Roh et al., 2018; Wang L. et al., 2020). Secondarily to proton transportation, the c-ring has a role in membrane fusion and neurotransmission, acting as a large-conductance transmembrane protein pore (Couoh-Cardel et al., 2016). This coincides with the knowledge that the Vo complex is implicated in exocytosis (Maxson and Grinstein, 2014; Morel and Poëa-Guyon, 2015).
2.5. V-ATPase subunit a
Subunit a of the Vo complex plays a crucial role in coupling ATP hydrolysis and proton translocation. Subunit a consists of a cytosolic N-terminal domain and a membrane-integrated C-terminal domain (Figure 2A) with eight transmembrane 𝛼-helices (Figure 2B). The highly tilted 𝛼7 and 𝛼8 are in contact with the c-ring (Figure 2B), forming the two offset hemichannels responsible for proton translocations (Mazhab-Jafari et al., 2016; Abbas et al., 2020). In yeast, there are two known isoforms of subunit a: the Vph1p and Stv1p, while there are four in fruit flies, worms, mice, and humans: a1, a2, a3, and a4 (Manolson et al., 1994; Nishi and Forgac, 2000; Toyomura et al., 2000; Oka et al., 2001a,b; Wagner et al., 2004). Each isoform is enriched in a specific subcellular location, making subunit a the determining factor for V-ATPase proper targeting. A chimeric study showed that the targeting information is controlled by the cytosolic N-terminal domain of subunit a in yeast and that Vph1p is localized at the vacuole, while Stv1p is at the Golgi (Kawasaki-Nishi et al., 2001b). Stv1p’s sorting signal on W83KY residues was found through a random mutagenesis study (Finnigan et al., 2012). However, unlike yeast, the specific targeting signal of mammalian subunit a remains elusive.
Figure 2.
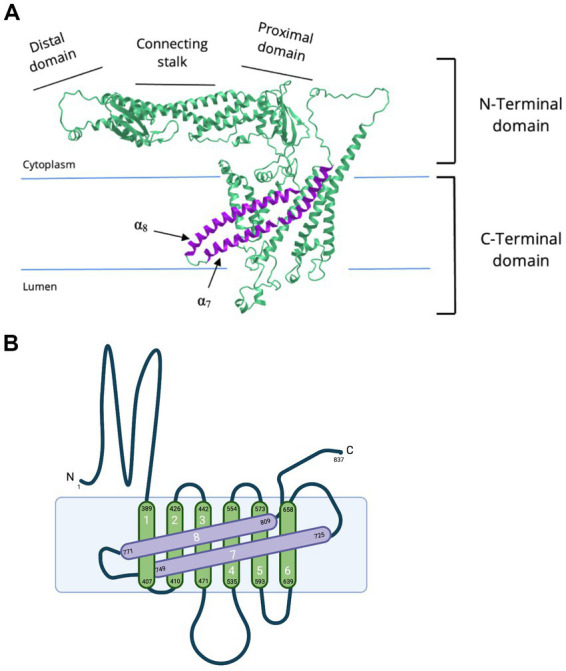
Cryo-EM of NTD and CTD of subunit a1. (A) Cryo-EM derived structure of the human V-ATPase subunit a (RCSB PDB: 6WLW, source: iCN3D) (Wang J. et al., 2020; Wang L. et al., 2020) and (B) a simplified topology representation of V-ATPase subunit a1. Subunit a1 is made up of two domains, a cytoplasmic NTD and membrane-embedded CTD. The a1-NTD is composed of a distal domain, connecting stalk, and proximal domain. a1-CTD consists of eight transmembrane α-helices connected by short linker loops on both cytoplasmic and luminal sides. a1-CTD is important for proton translocation. Transmembrane helices α7-α8 (in purple) are tilted. These transmembrane helices compose part of the hemichannels and hold important residues for proton translocation (R740 at α7 and R804 at α8). (B) was created with BioRender.com.
Subunit a1 is the neural isoform. In fruit flies, it is located in synaptic vesicles and membranes of presynaptic nerve terminals, where it interacts with Ca2+-Calmodulin in regulating SNARE assembly, which is important for Ca2+-dependent spontaneous neurotransmitter release (Zhang et al., 2008; Wang et al., 2014). Meanwhile, isoforms a2 and a3 are ubiquitously expressed with a2 located in the Golgi and a3 targeted to endosomes and lysosomes. Isoforms a3 and a4 are also localized to plasma membranes, with a3 found in developing osteoclasts and a4 in ion-transporting epithelia of the kidneys, ocular ciliary bodies, human ears, and epididymal cells (Oka et al., 2001a; Stover et al., 2002; Kawamura et al., 2010).
2.5.1. CTD of subunit a1
Subunit a1 is organized into two distinct domains, the cytoplasmic N-terminal domain (residues 1–388) and the membrane-embedded C-terminal domain (residues 389–837), each consisting of ~400 residues (Figures 1A, 2; Roh et al., 2018). The CTD of a1 is involved in proton translocation and includes eight transmembrane α-helices, with additional short linker-loops at the cytoplasmic and luminal sides (Figure 2; Roh et al., 2018; Abbas et al., 2020). Overall, the membrane-embedded a1 CTD is arranged beginning with the short, cytoplasmic-facing helices α1 and α2 [CTD α-helix numbering according to Toei et al. (2011)] (Mazhab-Jafari et al., 2016; Abbas et al., 2020). These two α-helices partially cross the lipid bilayer and are slightly exposed to the cytoplasm (Mazhab-Jafari et al., 2016; Abbas et al., 2020). Transmembrane helices α3-α6 form a central layer and are subsequently followed by the long and heavily tilted α7 and α8 helices (Figure 2A), terminating the CTD and forming most of the interface between subunit a1 and the c-ring, along with transmembrane helices of c1 and c″ (Mazhab-Jafari et al., 2016; Roh et al., 2018; Abbas et al., 2020). Moreover, the a1-CTD forms two aqueous cavities that extend only part way through the membrane and interface the c-ring, known as hemichannels (Mazhab-Jafari et al., 2016; Roh et al., 2018; Abbas et al., 2020). One cavity faces the cytoplasmic side, whereas the other faces the lumen, providing access to proton entry and exit for (from) the conserved glutamic acid residues on the c-ring (Figures 1A, C; Kawasaki-Nishi et al., 2001b; Mazhab-Jafari et al., 2016; Roh et al., 2018; Abbas et al., 2020). The cytoplasmic cavity is made from the cytoplasmic ends of α-helices 4, 5, 7, and 8, lined with charged and polar residues (Roh et al., 2018). The luminal-facing cavity is composed of the loops connecting α-helices 3, 4, 7, and 8, lined with several polar residues (Roh et al., 2018). In yeast, the key polar residues in the cytoplasmic cavity are E721, N725, and H729, which one is conserved in humans as E726 according to UniProt sequence alignment (P32563, Q93050) (Roh et al., 2018; Vasanthakumar and Rubinstein, 2020). The important yeast residues in the luminal cavity, D425, D481, and H743 correspond as D409, D466, and H748 in humans, according to UniProt sequence alignment (P32563, Q93050) (Roh et al., 2018; Vasanthakumar and Rubinstein, 2020). Non-conservative mutations to these residues lead to a significant loss in proton translocation activity, highlighting their importance (Toei et al., 2011). Furthermore, a single glycosylation at residue N488 of the first a1 luminal loop is required for protein stability and the incorporation of subunit a1 into the V-ATPase itself (Esmail et al., 2018b).
2.5.2. Subunit a1 and the proton translocation pathway
Mammalian proton transport requires collaboration between subunit a1 and the c-ring. Although there is little information regarding the proton transport system for mammalian V-ATPase, researchers have uncovered key residues and interactions within the proton transport pathway in yeast (Kawasaki-Nishi et al., 2003; Toei et al., 2011; Wang et al., 2014; Mazhab-Jafari et al., 2016; Roh et al., 2018; Abbas et al., 2020). Proton transport begins with the protonation of the essential glutamic acid residues on the c-ring (E139 subunit c and E98 subunit c” in humans; E137 subunit c, E145 subunit c’, and E108 subunit c” in yeast) (Figures 1A, C; Kawasaki-Nishi et al., 2001b; Roh et al., 2018; Abbas et al., 2020). Protonation of the c-ring glutamic acid residues occurs in the cytoplasmic cavity of subunit a, mediated by α7 residues S728 and E721 (conserved as S733 and E726 in human subunit a1) (Roh et al., 2018). The now neutrally charged c-ring glutamate residues are stabilized by hydrogen bonds formed with neighboring c-ring tyrosines, enabling their stable entry into the lipid bilayer (Roh et al., 2018; Abbas et al., 2020). Next, the c-ring, powered by ATP hydrolysis in V1, rotates clockwise through the lipid bilayer to bring the protonated c-ring glutamates close to an essential residue, R735, located on α7 (conserved as R740 in human subunit a1) (Wang et al., 2004; Abbas et al., 2020). A salt bridge forms between the protonated glutamate residue and R735/R740, causing the release of the proton into the luminal cavity (Roh et al., 2018; Abbas et al., 2020). Finally, the proton exits the membrane and enters the lumen by following a network of polar and negatively charged α7-8 residues lining the luminal cavity (Abbas et al., 2020). These charges residues include H743, E789, and R799 in yeast, which are conserved as H748, E794, and R804 in the human subunit a1, according to UniProt sequence alignment (P32563, Q93050) (Forgac, 2007; Abbas et al., 2020; Vasanthakumar and Rubinstein, 2020). For every three ATP molecules hydrolyzed, 10 protons are translocated (Abbas et al., 2020).
Alternatively, Roh et al. (2018) hypothesize that the deprotonation of the c-ring glutamates occurs at E789 in yeast via hydrogen bonding to neighboring proton binding site tyrosine residues. E789 then transfers the proton into the lumen using a network of amino acids, including D425, D481, and H743 (Roh et al., 2018). The deprotonated glutamate residues then interact with S792 and H796 before forming a salt bridge R735, acting as a fail-safety mechanism to ensure the deprotonation of the glutamates near the luminal cavity.
2.5.3. NTD of subunit a1
The NTD of subunit a1 forms a hairpin-like structure consisting of two globular segments, the proximal and distal domain, connected by a long coiled-coil known as the connecting stalk (Figure 2; Srinivasan et al., 2011; Abbas et al., 2020; Wang L. et al., 2020). In yeast, different subunit a isoforms have preferential organelle targeting, which is mediated by the a-NTD (Kawasaki-Nishi et al., 2001b). The N-terminus of the Vph1 isoform directly interacts with the phosphatidylinositol phosphate lipid PI(3,5)P2 for its recruitment to vacuoles (Kawasaki-Nishi et al., 2001b; Banerjee et al., 2019). Likewise, the Stv1 isoform is localized to the Golgi via its interaction with PI(4)P (Kawasaki-Nishi et al., 2001b; Banerjee and Kane, 2017). However, the specific cell-targeting information of the mammalian homologue remains unclear. As aforementioned, EG1 interacts with the “foot” (distal) domain of subunit a1, serving as a stator to anchor EG1 (Figure 1A; Qi and Forgac, 2008; Colina-Tenorio et al., 2018). This interaction occurs through the NTDs of subunits E and G that compose EG1, along with location-specific residues of the proximal a-NTD portion (residues 347–369 in yeast, 331–385 in human a1) (Qi and Forgac, 2008). Conformational changes to this interaction are also hypothesized to trigger the dissociation of the Vo and V1 sectors (Qi and Forgac, 2008). Specifically, this interaction, along with contact from the C-terminal helices of subunit H, may create an environment that keeps subunit a1 in a conformation favorable for proton translocation (Wang R. et al., 2020). Furthermore, both the proximal and distal domains of a1-NTD bind to subunit d when Vo is separated from V1 (Qi and Forgac, 2008; Roh et al., 2018). This interaction locks the rotary subunit, preventing rotation and proton translocation during Vo dissociation (Nishi and Forgac, 2000; Roh et al., 2018).
3. Subunit a1-associated neurological disorders
The V-ATPase plays a crucial role in maintaining the homeostatic pH of intracellular vesicles and mutations that impair its function are implicated in various diseases. As the Vo complex couples ATP hydrolysis with proton translocation, many variants to genes encoding subunit a1 have debilitating effects on vesicle acidification, which affects lysosomal activities and neurotransmission.
3.1. Variants of ATP6V0A1 in developmental and epileptic encephalopathies (DEE) and progressive myoclonus epilepsy (PME)
Recently, two groups of researchers identified de novo and biallelic variants of ATP6V0A1, encoding the a1 subunit, in individuals with rare neurological disorders: DEE and early onset PME (Aoto et al., 2021; Bott et al., 2021). DEE is a condition consisting of both severe epilepsies and significant cognitive developmental delay or loss (encephalopathies), which manifest early in life. Increasing evidence has attributed DEE to various genetic causes, which are believed to be responsible for epilepsy and adverse encephalopathy independently (Berg et al., 2021). PME is a group of rare disorders with symptoms including myoclonus (sudden muscle jerking), epilepsy, and progressive neurological deterioration. While also uncommon, the onset of this disease is more variable than DEE, ranging from early childhood to adolescence and adulthood (Orsini et al., 2019).
The first group, Aoto et al. (2021) studied individuals with DEE and identified two unrelated individuals with the same de novo heterozygous missense mutation, R741Q (corresponds to R740Q (c.2219G > A), based on the updated accession number NM_001130021.3) (Figures 3A, 4; Table 1). Additionally, they identified two inherited missense mutations, A512P and N534D, each in compound heterozygosity with a 50-kb deletion (c.del(17)(q21.2)) and a splice site mutation (c.196 + 1G > A), respectively. According to the updated accession NM_001130021.3, the aforementioned mutations are A505P (c.1513C > G) and N527D (c.1579A > G) (Figures 3A, 4; Table 1). Using an in vitro culture, authors demonstrated that when homozygous, all missense mutations mentioned above resulted in a loss of function and higher lysosomal pH. However, upon further investigation using a mouse model, Aoto et al. (2021) showed that they do so at different degrees. They generated mice harboring human A505P and R740Q variants, corresponding to A506P and R741Q in mice (note: mice lines are named based on human residue variants). Atp6v0a1R740Q/R740Q embryonic lethality suggests this mutation leads to a complete V-ATPase loss-of-function which impairs neuronal development. On the other hand, Atp6v0a1A505P/A505P survived for 2 weeks and displayed impaired motor function and ataxia. Nonetheless, these pups had smaller brains and fewer neurons, suggesting that the A505P mutation affects neurodevelopment and synaptic formation. This retardation in cell growth is due to A505P effect on mTORC1, a signaling pathway involved in the regulation of cell proliferation and differentiation. Immunoblot analysis showed a decrease in the phosphorylated S6 level, which is a downstream target of mTORC1. Additionally, Atp6v0a1A505P/A505P affects the expression of other V-ATPase subunits, as shown by a decrease in the A subunit of the V1 complex in the cerebellum (encoded by ATP6V1A).
Figure 3.
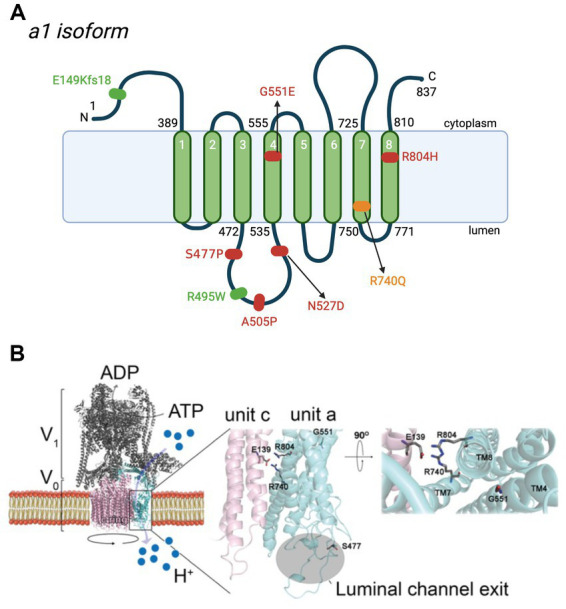
De novo and biallelic mutations to ATP6V0A1 impair V-ATPase functions in cases of DEE and PME. (A) Schematic representation of the location of mutations to subunit a1 in DEE (red), early onset PME (green), and both diseases (yellow) (Aoto et al., 2021; Bott et al., 2021). The most common mutation is R740Q, which is de novo, heterozygous, and follows a dominant inheritance pattern. Other de novo heterozygote variants are S477P, G551E, and R804H. Biallelic mutations, E149Kfs18 and R495W, are reported in 5 individuals with PME, 4 of whom are from the same family. In DEE individuals, A505P and N527D are in compound heterozygosity, each with a 50-kb deletion [del(17)(q21.2)] and a splice site mutation (c.196C + 1G > A), respectively. Positions are based on the updated accession number NM_001130021.3. For the purpose of annotation, transmembrane α7-α8 are not tilted. Amino acid sequences are based on Q93050 in UniProt. (A) was created with BioRender.com. (B) Homology model of human V-ATPase and locations of residues reported in de novo mutations on subunit a1 (Bott et al., 2021). Residues S477, G551, R740, and R804 are represented as stick structures relative to the key glutamic acid residue, E139 of subunit c. S477, at the second short linker loop (connecting transmembrane α3-α4) of luminal hemichannel.
Figure 4.

ATP6V0A1 variants implicated in DEE and PME cause lysosomal and autophagic dysfunction. Normal ATP6V0A1 encodes for a functional subunit a1 of the V-ATPase, which couples ATP hydrolysis and proton translocation through the c-ring. Protons are pumped into the lumen of lysosomes to achieve a pH of ~5.5 for optimal enzymatic activities in autophagosome clearance. On the other hand, variants of ATP6V0A1 compromise V-ATPase function and interfere with proper lysosomal acidification, leading to the accumulation of autophagosome and lysosomal bodies, as reported in cases of DEE and PME. R740Q mutant was reported in DEE and PME patients, while R804 and A505P were found in DEE patients (Aoto et al., 2021; Bott et al., 2021). Based on studies in animal models by Aoto et al. (2021) and Bott et al. (2021), R740Q and R804H in the 7th and 8th transmembrane region, respectively, are key residues in the proton translocation, adversely affected by such substitutions. A505P variant on the 2nd luminal linker region is also implicated in DEE, affecting autophagic function (Aoto et al., 2021). It is still unclear how A505P impairs V-ATPase function in autophagic dysfunction. The figure was created with BioRender.com.
Table 1.
List of ATP6V0A1, ATP6V0A2, ATP6V0A3, and ATP6V0A4 variants.
| Gene | Nucleotide change | Amino acid change | Mutation type | Zygosity | Impact on V-ATPase | Associated disease | Variant identifier | Reference |
|---|---|---|---|---|---|---|---|---|
| ATP6V0A1 | c.T1429C | p.S477P | Missense | Heterozygous | Alters conformation of luminal channel, hindering proton translocation | DEE | VAR_087489 | Bott et al. (2021) |
| ATP6V0A1 | c.C1483T | p.R495W | Missense | Compound heterozygous with frameshift mutation: c.445delG, p.E149Kfs18 | Loss of positively charged residue near the luminal channel exit hinders proton translocation | PME and ataxia | rs781278654 | Bott et al. (2021) |
| ATP6V0A1 | c.C1513G | p.A505P | Missense | Compound heterozygous with deletion: c.del(17)(q21.2) | Undetermined | DEE | VAR_087491 | Aoto et al. (2021) |
| ATP6V0A1 | c.A1579G | p.N527D | Missense | Compound heterozygous with splice site mutation: c.196+1G>A | Undetermined | DEE | rs766856192 | Aoto et al. (2021) |
| ATP6V0A1 | c.G1652A | p.G551E | Missense | Heterozygous | Deforms environment mediating proton exchange | DEE | VAR_087493 | Bott et al. (2021) |
| ATP6V0A1 | c.G2219A | p.R740Q | Missense | Heterozygous | Impedes glutamate protonation thereby disrupting proton translocation | DEE, PME, autism, microcephaly | rs1567871600 | Aoto et al. (2021); Bott et al. (2021) |
| ATP6V0A1 | c.G2411A | p.R804H | Missense | Heterozygous | Impedes glutamate protonation thereby disrupting proton translocation | DEE | VAR_087495 | Bott et al. (2021) |
| ATP6V0A2 | c.187T | p.R63X | Nonsense | Homozygous | Truncated protein, non-functional subunit a2 | ARCL II | rs80356750 | Kornak et al. (2008) |
| ATP6V0A2 | c.C1214T | p.P405L | Missense | Homozygous | Compromises subunit a2 stability, complex assembly and Golgi trafficking | ARCL II | rs750788949 | Hucthagowder et al. (2009); Esmail et al. (2018a) |
| ATP6V0A2 | c.C2293T | p.Q765X | Nonsense | Homozygous | Truncated protein, non-functional subunit a2 | ARCL II | rs80356758 | Kornak et al. (2008) |
| ATP6V0A2 | c.C2432C | p.L811P | Missense | Compound heterozygous with splicing mutation: c.2055+2dupT | Changes last α-helix conformation, hindering subunit interactions and proton translocation | ARCL II | rs778642108 | Fischer et al. (2012) |
| ATP6V0A3 | c.IVS2+4A→T | p.V26-D39del | Deletion | Homozygous | Unable to assemble with V1 subunit | MIOP | N/A | Kornak et al. (2000) |
| ATP6V0A3 | c.G1024T | p.E342X | Stop | Heterozygous | Truncated protein, non-functional subunit a3 | MIOP | N/A | Sobacchi et al. (2001); Susani et al. (2004) |
| ATP6V0A3 | c.G1213A | p.G405R | Missense | Homozygous or compound heterozygous with R444L | Steric hindrance of 1st cytoplasmic loop | MIOP | VAR_019569 | Sobacchi et al. (2001); Susani et al. (2004) |
| ATP6V0A3 | c.G1331T | p.R444L | Missense | Homozygous or compound heterozygous with G405R | Protein instability due to hydrophobic Arginine (R) | MIOP | rs137853151 | Sobacchi et al. (2001); Susani et al. (2004) |
| ATP6V0A3 | c.G2412A | p.W805X (updated NM_006019.4) | Stop | Heterozygous | Truncated protein, non-functional subunit a3 | MIOP | rs1555000308 | Kornak et al. (2000) |
| 1. ATP6V0A4 2. ATP6V1B1 | 1.c.C419T 2.c.A437G | 1. p.T140M 2. p.D146G | Missense | Digenic heterozygous | 1. Slight conformational change hindering subunit trafficking and assembly 2. Alteration of subunit surface charge leading to the disruption of ATP binding sites | dRTA | 1. rs144802156 2. rs782681801 | Nagara et al. (2018) |
| ATP6V0A4 | c.C1631T | p.S544L | Missense | Heterozygous | Unable to bind to B1 subunit, assembly issues, weakened ATPase activity | Complete and incomplete dRTA | rs1026435 | Imai et al. (2016); Chen et al. (2020) |
| ATP6V0A4 | c.G2420A | p.R807Q | Missense | Homozygous | Reduction of subunit a4 production leading to V-ATPase assembly and activity deficits | dRTA, Sensorineural hearing loss | rs28939081 | Stover et al. (2002) |
| ATP6V0A4 | c.G2458C | p.G820R | Missense | Homozygous | Loss of a4 binding to phosphofructokinase-1, formation of salt bridge disrupting proton translocation | dRTA | rs267606671 | Esmail et al. (2018a) |
ARCL II, autosomal recessive cutis laxa type II; DEE, developmental and epileptic encephalopathies; dRTA, distal renal tubular acidosis; MIOP, malignant infantile osteopetrosis; PME, progressive myoclonus epilepsy.
Staining of Atp6v0a1A505P/A505P brain sections revealed lower levels of mature Cathepsin D, abnormal distribution of lysosomes, and an increase in neuronal cell death (Aoto et al., 2021). Cathepsin D is an aspartic protease whose trafficking and maturation in the endolysosomal compartment depends on optimum acidic pH (pH 3–5) (Steinfeld et al., 2006). This suggests that Atp6v0a1A505P/A505P impairs lysosomal activity, leading to an abnormal cellular distribution of lysosomes and consequently, cell death. Accordingly, the authors attributed the decrease in mTORC1 signaling and increased neuronal cell death induced by lysosomal dysfunction to the Atp6v0a1A505P/A505P pup’s smaller brain size. Furthermore, lysosomal dysfunction leads to failure in autophagosome clearance, as shown by the accumulation of autophagosomes and lysosomes in Atp6v0a1A505P/A505P. Additionally, Atp6v0a1A505P/A505P electrophysiology analysis reported lower amplitude and frequency of miniature excitatory postsynaptic current (mEPSC) and miniature inhibitory postsynaptic current (mIPSC) relative to the wildtype group. Thus, it suggests a reduction in neurotransmitter content inside synaptic vesicles (reduced neurotransmitter loading) and a decrease in vesicle fusion events for release. Therefore, the V-ATPase plays a crucial role in determining synaptic vesicle content and release.
The Aoto et al. (2021) Atp6v0a1A505P/A505P mouse model allows for a deeper investigation of ATP6V0A1 missense mutations and their impact on a1 subunit synthesis, protein complex assembly, V-ATPase loss of function, along with their effects on lysosome and autophagosome function, and mTORC-mediated neuronal development. Additionally, this study showed the importance of the a1 linker loop in proton translocation. Thus, the mouse model warrants further investigation on ATP6V0A1 variants with mutations on linker loops (not only on the transmembrane α-helices), such as S477P, G551E, and R495W, and how such mutations lead to impaired proton translocation.
The second group, Bott et al. (2021) identified 12 individuals with de novo missense variants to ATP6V0A1 presenting phenotypes of DEE and five individuals with biallelic variants showing early onset PME with ataxia. R740Q is the most common de novo missense variant, found in 8 out of 12 individuals with DEE. Identification of this mutation hotspot in multiple unrelated DEE individuals provides stronger evidence of the causal relation between this candidate de novo mutation with DEE. They also identified three other de novo heterozygous variants: S477P (c.1429 T > C), G551E (c.1652G > A), and R804H (c.2411G > A) on the third linker loop domain (between transmembrane α-helix 3 and 4), α4, and α8, respectively, (Figure 3A). In five patients with early onset PME with ataxia (four are from the same family), they found a novel compound heterozygous ATP6V0A1 variant: E149Kfs18 (c.445delG) and R495W (c.1483C > T) (Figure 3A; Table 1).
To circumvent the embryonic lethality in the mouse model reported by Aoto et al. (2021), Bott et al. (2021) conducted a R740Q in vitro cell culture study, which revealed impaired protonation, abolished level of Cathepsin D, and accumulation of LC3-II, a standard marker for autophagosomes. These results indicate impaired lysosome activity and autophagosome turnover. Next, Bott et al. (2021) observed Caenorhabditis elegans harboring the unc-32 mutation, an ATP6V0A1 ortholog. They found that knockdown of unc-32 increases endogenous LGG-1::mCherry fluorescence (nematode ortholog of LC3) without increasing its expression level, suggesting impaired protein clearance. unc-32 knockdown reduced the expression of autophagic machinery components and lysosomal hydrolytic enzymes while increasing sqst-1 expression, a stress-responsive autophagic receptor. Moreover, C. elegans with the corresponding R740Q homozygous mutation, unc-32(rm20), showed developmental arrest and the accumulation of proteins in the nerve ring.
Homology models in yeast and fly demonstrate the importance of residue R740 for proton translocation. A yeast study showed the importance of residue R735 of Vph1p for proton translocation (R740 in human ATP6V0A1) (Kawasaki-Nishi et al., 2001a). R735K leads to a complete loss in proton transport while retaining approximately 20% of the wild-type ATP hydrolysis function. On the other hand, R735 substitution with Asn, Glu, or Gln results in the complete impairment of ATP hydrolysis and proton transport function. This suggests that any substitution, including conserved substitutions (ex. R735K), cannot rescue enzymatic function, highlighting the importance of this residue. Moreover, Drosophila melanogaster harbouring a homologous mutation (R755A in VHA100-1) showed lysosomal abnormality and autophagosome accumulation (Williamson et al., 2010).
By forming a salt bridge, R740 removes a proton bound to a glutamic acid residue of the proteolipid ring subunit c (E139), allowing its translocation into the lumen (Figures 1C, 3B; Aoto et al., 2021; Bott et al., 2021). The missense mutations, R740Q and R804H, impair this crucial deprotonation process (Figures 3B, 4). A more extensive understanding into the role of R740 in proton translocation requires conducting studies using a conditional or inducible knockout mice model. These models could potentially circumvent embryonic lethality and will help explain the pathophysiology of R740Q variant-related diseases in the mammalian system. Additionally, future in vitro and in vivo analysis of the R804H is important to complete our understanding of proton translocation throughout the luminal hemichannel and how R804 mutations translate clinically. Moreover, the G551A variant on α4 alters the structure of the protein region crucial for proton exchange (Figure 3). The luminal domain variant, S477P, modifies the loop contour of the luminal hemichannel, impairing proton translocation (Figure 3B). Altogether, these findings emphasized the importance of the a1 subunit for proton translocation, especially residue R740, identified in most cases of DEE and PME with ATP6V0A1 mutations.
Independent of its role in acidification, the Vo complex is suggested to be required for membrane fusion (Peters et al., 2001; Hiesinger et al., 2005; Peri and Nüsslein-Volhard, 2008; Saw et al., 2011; Wang et al., 2014). The direct interaction between subunit a1/V100 (subunit a1 Drosophila ortholog) and soluble N-ethylmaleimide-sensitive factor activating protein receptors (SNAREs) has also been well documented (Perin et al., 1991; Galli et al., 1996). SNARE proteins mediate vesicle fusion by tethering vesicles to their target membrane (Söllner et al., 1993). A V100 loss-of-function study showed that V100 regulates synaptic vesicle fusion downstream of SNARE-dependent vesicle priming (Hiesinger et al., 2005). Moreover, in mouse hippocampal neurons, subunit a1 is not directly involved in vesicle fusion but instead modulates neurotransmitter release upstream of docking, favoring the fusion of acidified and loaded synaptic vesicles (Bodzęta et al., 2017). Wang et al. (2014) also proposed an acidification-independent mechanism whereby a1/V100 interacts with SNAREs to mediate SNARE assembly and spontaneous release, in a Ca2+–Calmodulin -dependent manner. Without Ca2+–Calmodulin, V100 competitively binds to the target-membrane SNAREs synaptobrevin and syntaxin, disrupting SNARE complex formation and hindering spontaneous release. However, the addition of Ca2+–Calmodulin rescues SNARE assembly. This process is mediated by the NTD of V100, as it hosts key SNARE and Calmodulin binding residues (Hiesinger et al., 2005; Zhang et al., 2008; Wang et al., 2014). Furthermore, this study provides evidence that subunit a1 is involved in the regulation of exocytosis, but does not contribute to the fusion pore, unlike the c-ring and other Vo subunits (Couoh-Cardel et al., 2016).
Additional findings on the reduction in spontaneous neurotransmitter release (mEPSC and mIPSC) in the A505P variant, reported by Aoto et al. (2021), highlights the critical role of Voa1-containing V-ATPase in driving neurotransmitter uptake and overall neurotransmission. Voa1 and Voa2 are the two isoforms found on secretory vesicles and contain critical and overlapping roles in acidification and neurotransmitter uptake (Kawasaki-Nishi et al., 2001b). Loading neurotransmitters into synaptic vesicles requires the collaborative work of a V-ATPase and a vesicular neurotransmitter transporter. The V-ATPase creates an electrochemical gradient which is utilized by the vesicular neurotransmitter transporter to pump neurotransmitters into the vesicular lumen. Recently reported, ATP concentration regulates V-ATPase pumping probability, while the electrochemical proton gradient controls the pumping rate (Kosmidis et al., 2022). Investigations on Voa1-containing V-ATPase role in neurotransmitter loading have been difficult since knockout models of this key subunit are lethal (Dickinson et al., 2016; Ma et al., 2019). Thus, the Atp6v0a1A505P/A505P mouse model creates an opportunity to further examine the effects of Voa1 mutations on V-ATPase proton pumping kinetics and neurotransmitter loading, to better comprehend its role in secretory vesicles and related pathophysiology, including DEE and PME-related seizures resulting from excitatory and inhibitory imbalance.
3.2. Impaired subunit a1 trafficking due to mutations in presenilin-1 and Alzheimer’s disease (AD)
In addition to impaired V-ATPase function due to mutations within the a1 subunit itself, studies have reported variants in Voa1-interacting proteins affecting the a1 activity. Namely, mutations in presenilin-1 (PS-1) which is implicated in AD. AD is the most common cause of dementia and is characterized by impaired memory and cognitive abilities. AD neuropathology has been associated with two types of protein depositions: initiation by extracellular Amyloid-β (Aβ) accumulation, followed by hyperphosphorylation of intracellular tau protein forming the neurofibrillary tangle. Although studies have also reported tau pathology progression independently of Aβ (van der Kant et al., 2020). Aβ originates from the cleavage of an Amyloid Precursor Protein (APP) by extracellular β- and intracellular 𝛾-secretases. PS-1 is a component of the 𝛾-secretase responsible for the intracellular cleavage and processing of APP. Most familial forms of AD (FAD) are associated with mutations to PS-1, resulting in an increased formation of Aβ aggregates (Lee et al., 2010). Besides promoting the accumulation of Aβ plaque, loss-of-function of PS-1 affects the proper targeting of Voa1-containing V-ATPases to lysosomes, worsening clearance of the aberrant protein aggregate (van der Kant et al., 2020). Normally, PS-1 binds to unglycosylated Voa1, priming its N-glycosylation by the oligosaccharyltransferase. N-glycosylation is required to efficiently deliver Voa1 from the ER to the lysosome, which acidifies and activates various enzymes important for autophagosomes and the clearance of protein aggregates (Lee et al., 2010). Thus, PS-1 is a common mechanism linking the dual pathogenic processes in AD; it increases Aβ formation and decreases Aβ aggregate clearance. It also highlights the importance of proper localization of Voa1 in the context of AD. Although the PS-1 variant is reported to be the most pathogenic variant in AD (Xiao et al., 2021), further studies are required to clarify the exact interaction between PS-1 and Voa1 of V-ATPase. One study reported impaired Voa1 N-glycosylation of PS-1 and lysosomal acidification in 5xFAD mice (Avrahami et al., 2013). However, another study found a contradictory result, with no change to Voa1 N-glycosylation in mouse embryonic fibroblast nor alteration in lysosomal and autophagic function (Zhang et al., 2012).
4. Diseases related to non-neural subunit a isoforms
4.1. Diseases related to ATP6V0A2 mutations
Early mice studies reported a near-ubiquitous expression of subunit a2 mRNA (excluding skeletal muscle), with the strongest expression in the kidney and liver (Nishi and Forgac, 2000). In humans, a2 is encoded by ATP6V0A2 on chromosome 12q.24.3, consisting of 21 exons (Morava et al., 2009). Mutations leading to the loss of ATP6V0A2 gene function and impaired a2 production are responsible for autosomal recessive cutis laxa type II (ARCL II), which is a distinct type of the congenital disorders of glycosylation (CDG) (Kornak et al., 2008; Morava et al., 2009). ARCL is a connective tissue disorder divided into three types, with clinical features including loose and inelastic skin shared among all three. Specifically, the ARCL II phenotype includes developmental delays, skeletal abnormalities, a variable severity of cutis laxa (loose skin), and neurological abnormalities such as epilepsy, cognitive delay, and mental deterioration (Morava et al., 2009). The phenotypic spectrum of cutis laxa ranges from mild wrinkly skin syndrome (WSS) to the more severe Debré-type cutis laxa (Kornak et al., 2008). Individuals with Debré-type cutis laxa displayed more neurological disorders relative to WSS patients (Maldergem et al., 2023). Patients with ARCL II arising from ATP6V0A2 also have defects in N- and O-glycosylation and sialylation, giving the disease its CDG classification (Kornak et al., 2008; Morava et al., 2008).
Glycosylation occurs at the endoplasmic reticulum and the Golgi to modify protein structure, function, and stability (Reily et al., 2019). This process is highly dependent on compartment-specific enzyme activity. The a2 subunit displays Golgi-localization in the cell type responsible for ARCL II manifestations: the human dermal fibroblast (Fischer et al., 2012). The pathophysiology of ARCL II arising from ATP6V0A2 mutations comes from V-ATPase pumps’ failure to maintain proper pH in the Golgi for optimum glycosyltransferase activity, thus impairing glycosylation and organelle transport (Kornak et al., 2008; Morava et al., 2008; Rivinoja et al., 2009). This leads to abnormal elastin synthesis as tropoelastin accumulates inside the cell and prevents mature elastin transport to the extracellular space, creating the deleterious cutaneous phenotype observed in ARCL II (Hucthagowder et al., 2009; Shafagh Shishavan and Morovvati, 2022). The neurological abnormalities associated with ARCL II are caused by alterations to neurometabolic homeostasis initiated by aberrant glycosylation (Hucthagowder et al., 2009; Callewaert and Urban, 2022). As a result, the processing and secretion of many brain proteins is compromised.
There are various ATP6V0A2-associated mutations that manifest as ARCL II, including the homozygous R63X and Q765X nonsense mutations (Kornak et al., 2008). These mutations are unique as they have been found in several patients who differ geographically and genetically (Hucthagowder et al., 2009; Bahena-Bahena et al., 2014). The two mutations also have lower mRNA expression and a truncated protein product, compromising V-ATPase function and leading to glycosylation and neurological defects (Kornak et al., 2008). Moreover, the heterozygous L811P missense mutation is located on the last α-helix and changes the helix conformation, thereby disturbing proton translocation and subunit a2 interactions with other subunits (Fischer et al., 2012). However, the heterozygous L811P mutation alone likely does not cause ARCL II and forms a somewhat stable protein. Nevertheless, L811P exists as a compound heterozygous mutation along with the splicing mutation c.2055 + 2dupT, which caused the associated disease (Fischer et al., 2012). Lastly, the homozygous P405L missense mutation is located in the first α-helix and is a well-studied ARCL II-causing mutation (Hucthagowder et al., 2009; Esmail et al., 2018a). Interestingly, the P405L subunit a4 is N-glycosylated considering ARCL II is characterized by defective N-glycosylation (Esmail et al., 2018a). Despite this, the P405L variant experiences high degradation rates, so the authors proposed that P405 is required for complex assembly, stability, and Golgi trafficking (Esmail et al., 2018a).
Some other early reported ATP6V0A2-causing ARCL II variants include the frameshift mutations K117fsX144 and E442fsX506, along with the nonsense mutation E442X (Figure 5A; Kornak et al., 2008). Kornak et al. (2008) also reported ATP6V0A2 frameshift mutations present in individuals with WSS: V66fsX107 and T643fsX683. Furthermore, the missense mutations R510I and P792R, the heterozygous frameshift variant E432fsX444, and the compound heterozygous mutation: H763Y and c.1326 + 1G > A are ATP6V0A2-causing ARCL II variants (Hucthagowder et al., 2009; Fischer et al., 2012).
Figure 5.
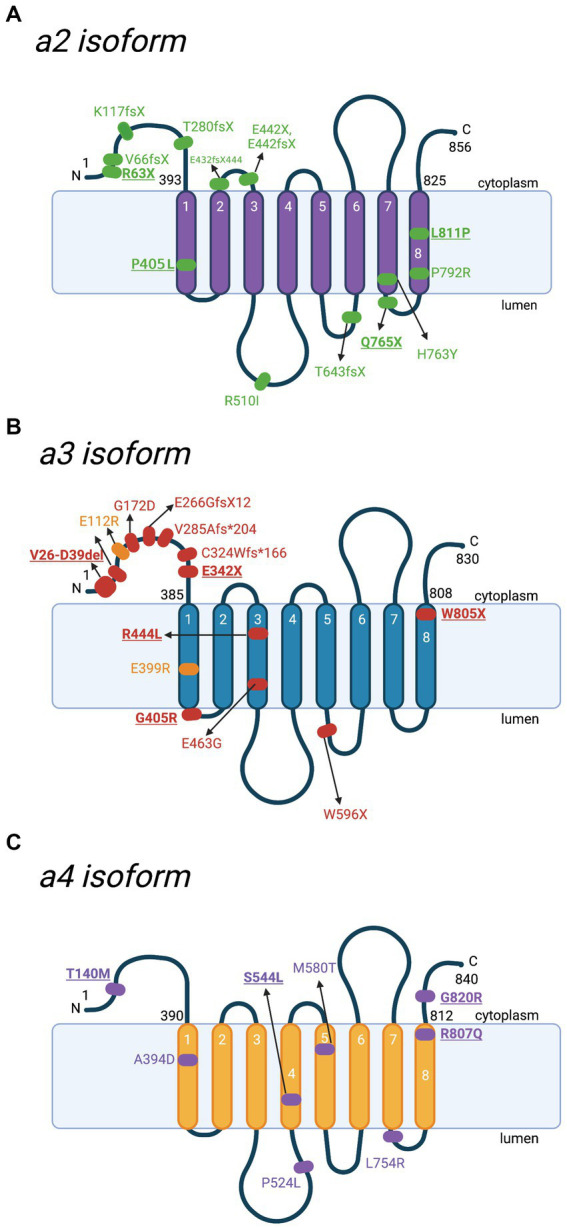
Schematic representation of a2-a4 subunits and location of mutations with the corresponding diseases. Besides a1, there are 3 other isoforms of V-ATPase subunit a. Various mutations have been reported in different diseases. Key mutations that are heavily discussed in the text are bolded and underlined. (A) Locations of various mutations to the a2 subunit, identified in cases of autosomal recessive cutis laxa type II (ARCL II). Amino acid sequences are based on Q9Y487 in UniProt. (B) Locations of identified mutations to the a3 subunit contributing to malignant infantile osteopetrosis (MIOP) (red) and its milder form (yellow). Amino acid sequences are based on Q13488 in UniProt. (C) Locations of mutations to the a4 subunit involved in distal renal tubular acidosis (dRTA). Amino acid sequences are based on Q9HBG4 in UniProt. For the purpose of annotation, transmembrane helices α7-α8 are not tilted. The figure was created with BioRender.com.
4.2. Diseases related to ATP6V0A3 mutations
The ATP6V0A3 gene on chromosome 11q13 encodes the human a3 subunit (gene is also known as TCIRG1, Atp6i, or OC116) (Heaney et al., 1998; Heinemann et al., 1999). The gene transcript encodes both OC116 in the osteoclast and T-cell Immune Response cDNA 7 (TIRC7) protein ubiquitously. In mice, the ATP6V0A3 transcript is strongly expressed in the liver and heart (Nishi and Forgac, 2000). Additionally, osteoclast differentiation induces a3 expression on the plasma membrane (Toyomura et al., 2000). Osteoclasts are cells responsible for bone degradation through the acidification of a sealed extracellular interface, ultimately regulated by the V-ATPase (Kornak et al., 2000). V-ATPases are found in the ruffled membrane borders, pumping protons to maintain a low pH, which is crucial for dissolving inorganic bone material and promoting the activity of proteases that further degrades the bone matrix (Blair, 1998).
Mutations to the ATP6V0A3 gene are the most common genetic cause of the rare autosomal recessive disease, malignant infantile osteopetrosis (MIOP), which manifests early in life and is fatal if left untreated (Kornak et al., 2000; Michigami et al., 2002). In this condition, osteoclasts fail to resorb bone and clinical symptoms include osteosclerosis, thrombocytopenia, anemia, hepatosplenomegaly, and in some cases, visual impairment (Kornak et al., 2000). Alteration to normal bone resorption and remodeling results in an abundance of osteoid, which reduces bone marrow space, a region where hematopoiesis occurs, leading to the gradual reduction of blood production (Vomero et al., 2019). In severe cases, patients show neurological complications, including cranial neuropathies, due to progressive compression of the cranial nerves, spinal cords, and blood vessels (Steward, 2003).
There are various mutations reported in MIOP patients, which lead to V-ATPase loss of function (Figure 5B; Table 1). The earliest study by Kornak et al. (2000) identified a biallelic splice site mutation of the OC116 gene, V26-D39del, of the cytoplasmic Vo N-terminus. The N-terminus is crucial for forming the V1 complex and V-ATPase assembly in-vivo. Thus, mutations to this region often result in V-ATPase dysfunction (Liberman et al., 2013). Furthermore, protein truncation, due to premature stop codons at linker regions after the 3rd or 5th transmembrane domains (A480fsX or W596X), results in total loss of a3 function (Kornak et al., 2000). Two heterozygous stop mutations were also identified: E342 on the N-terminus and W804 on the C-terminus (W805 based on the updated accession number NM_006019.4), which is suspected to be in compound heterozygosity with other mutations yet to be discovered (Kornak et al., 2000). Substitution by an arginine in the G405R variant introduced a sterical hindrance for the 1st vacuolar loop bending between the 1st and 2nd 𝛼-helices (Figure 5B; Table 1; Sobacchi et al., 2001; Susani et al., 2004). Substitution of the R444 residue in the 3rd transmembrane 𝛼-helix with a hydrophobic leucine causes instability to the basic residue (Figure 5B; Table 1; Sobacchi et al., 2001).
Additionally, mutations on the splice donor site, such as a T-to-C transition on intron 19, a common region to OC116 and TIRC7, potentially resulted in abnormal splicing of both transcripts (Michigami et al., 2002). Therefore, MIOP patients commonly presented immune anomalies and a high incidence of infections (Vomero et al., 2019). Some other reported variants in the ATP6V0A3 are compound heterozygous mutations of E266GfsX12 with E463G (Vomero et al., 2019), E399RTer with E112R in a milder version of the disease (Luong et al., 2022), G172D, V285Afs*204, and c.C324Wfs*X166 (Figure 5B; Liu et al., 2021).
4.3. Disease related to ATP6V0A4 mutations
The human a4 isoform is encoded by the ATP6V0A4 gene located on chromosome 7q34 and is expressed in α-intercalated and β-intercalated cells of the kidneys, ocular ciliary bodies, pigmented epithelial cells of the retina, the human inner ear, and the epididymis (Oka et al., 2001a; Stover et al., 2002; Pietrement et al., 2006; Kawamura et al., 2010). ATP6V0A4 was identified as an early causative gene of distal renal tubular acidosis (dRTA) along with the gene encoding subunit B1, ATP6V1B1 (Karet et al., 1999; Smith et al., 2000; Stover et al., 2002). ATP6V0A4 and ATP6V1B1-related dRTA account for ~50–60% of primary dRTA cases and display autosomal recessive inheritance (Wagner et al., 2023). Distal renal tubular acidosis is characterized by the inability to secrete protons in α-intercalated cells of the cortical and outer medullary collecting ducts (Wagner et al., 2023). This reduces urinary acidification and ammonium excretion and leads to acidosis. Clinically, dRTA patients develop a range of phenotypes arising directly from cellular defects caused by dRTA-related genes and the indirect effect of acidosis (Wagner et al., 2023). Some acidosis-related effects include rickets, hypokalaemia, nephrocalcinosis, and hypercalciuria (Caldas et al., 1992; Alexander et al., 2016; Vallés and Batlle, 2018). Hypokalaemia arises due to the altered balance between proton, potassium, and sodium transport (Sebastian et al., 1971; Batlle et al., 2006). Meanwhile, the increased acid in the blood causes bone to release calcium as a means of buffering. This, along with decreased calcium absorption due to acidosis, contributes to nephrocalcinosis and hypercalciuria (Alexander et al., 2016). Mutations in subunit a4 of the V-ATPase can lead to impaired V-ATPase trafficking, assembly, activity, or its interaction with other proteins, thereby affecting urine acidification and causing dRTA (Ochotny et al., 2006).
Inspired by two naturally occurring homozygous missense mutations in subunit a4, R807Q, and G820R, that cause dRTA, Su et al. (2008) recreated these mutations in yeast (Vph1p isoform) to explore them further (Figure 5C; Table 1). The two aforementioned residues lie within the phosphofructokinase-1-binding domain of a4, which is a key regulator of glycolysis (Su et al., 2008). The study revealed that the G820R mutant led to a complete loss of binding for phosphofructokinase-1, followed by the severe deterioration of proton translocation and the mild destruction of ATPase activity. The mild interruption of ATPase activity (36% loss) combined with the severe decline of proton translocation (78% loss) suggests that the a4-phosphofructokinase-1 interaction functionally couples the ATPase activity to proton transport (Su et al., 2008; Ghazi et al., 2020). Nevertheless, more recent 3D molecular modeling displayed that the G820R mutation forms a putative salt bridge with the negatively charged a4 Glu-729 residue, which is important in the proton translocation pathway (Toei et al., 2011; Esmail et al., 2018a). The formation of this salt bridge is believed to interfere with the proton channel structure and thus hinder proton translocation (Esmail et al., 2018a). The conformational change induced by the salt bridge may also contribute to the damaged a4-phosphofructokinase-1 interaction (Esmail et al., 2018a). Moreover, the R807Q mutation reduced subunit a4 production, leading to the severe loss of enzymatic function, but did not affect phosphofructokinase-1 binding (Su et al., 2008). R807 in a4 is a paralog to a1 R804, meaning it serves as a key residue to transport protons into the lumen (Su et al., 2008; Abbas et al., 2020). The significance of this conserved residue accounts for the severe R807Q mutation phenotype observed.
The first known heterozygous mutation in the a4 subunit (S554L), causing incomplete distal renal tubular acidosis, was identified in a 40-year-old man (Imai et al., 2016). Clinical features included urinary proton excretion dysfunction causing excess loss of potassium instead of proton, along with hypokalaemia and nephrocalcinosis. However, metabolic acidosis was absent. The same heterozygous missense mutation was later observed in four related patients who exhibited complete dRTA (Chen et al., 2020). Transfected HEK293T cells carrying the S544L mutation also exhibited hindered ATPase activity and the inability to bind subunit B1, which may affect V-ATPase assembly (Chen et al., 2020). This finding is interesting and warrants further investigation, as this is the only missense mutation from a pool of over 40 a4 mutations where dRTA is inherited in a dominant manner (Su et al., 2008; Pereira et al., 2015; Gómez et al., 2016; Imai et al., 2016; Chen et al., 2020). Another notable ATP6V0A4-related dRTA mutation includes a patient with a digenic inheritance of dRTA, being heterozygous for the ATP6V1B1 (D146G) and ATP6V0A4 (T140M) missense mutations (Nagara et al., 2018). This is the first observed digenic inheritance of dRTA and helps increase our understanding of disease inheritance and genetic diagnostics (Nagara et al., 2018). Some other ATP6V0A4-associated dRTA mutations include the homozygous missense variants A394D, P524L, M580T, and L754R (Figure 5C; Smith et al., 2000; Gao et al., 2014; Park et al., 2018).
Fortunately, dRTA is a treatable disease, with the main line of treatment consisting of alkali supplementation (Lopez-Garcia et al., 2019). The alkali agents reduce acidosis to maintain acid–base homeostasis, which leads to the reduction of various symptoms. However, sensorineural hearing impairments, which are commonly associated with ATP6V0A4-associated dRTA, cannot be treated by alkali supplementation (Wagner et al., 2023). ATP6V0A4 is highly expressed in the marginal cells in the stria vascularis, which is needed for regulating endolymph pH in the cochlea of the inner ear (Stanković et al., 1997). ATP6V0A4 mutations affecting V-ATPase function may alkalinize the cochlear endolymph, leading to sensorineural deafness (Stover et al., 2002; Lopez-Garcia et al., 2019). Therefore, treating ATP6V0A4-associated dRTA with alkali manages the symptoms of acidosis, not the genetic manifestation of hearing loss.
5. Conclusion
The V-ATPase is an ATP-dependent proton pump that holds a prominent housekeeping role in maintaining characteristic acidic pH for homeostatic purposes and, eventually, organism survival. It is present in both plasma and intracellular organelle membranes and is a multisubunit protein complex that operates by a rotary mechanism, coupling ATP hydrolysis at the cytosolic domain (V1) with proton translocation at the membrane-embedded domain (Vo). Recently, the structures of human and other mammalian V-ATPases have been uncovered via cryo-electron microscopy (Abbas et al., 2020; Wang L. et al., 2020; Wang R. et al., 2020). Elucidating these structures is imperative to understand the functional and structural consequences of various V-ATPase subunit mutations for disease pathophysiology and treatment. Moreover, as a multisubunit structure, the V-ATPase exhibits cell-specific subunit isoforms regulating its localization and specialized function. Aberrant mutations to any subunits have been associated with various human diseases. Mutations to subunit a1 are involved in neurodevelopmental disorders, namely PME and DEE. Mutations to isoform a2 result in glycosylation impairments, leading to the connective tissue disorder ARCL II and a3 variants are the leading cause of MIOP. Finally, isoform a4 mutations often cause dRTA and sensorineural hearing loss.
So far, studies have evaluated the effect of Voa1 mutations on lysosomal acidification and autophagic function in neurodevelopment (Aoto et al., 2021; Bott et al., 2021). However, their animal models did not display epileptic events, which are the common symptoms of DEE and PME. Conversely, a previous study reported that a conditional knockout of Voa1 in the mouse hippocampus resulted in hyperexcitability of the CA3 network, which manifested as nonconvulsive electrographic seizures events in the electroencephalogram (Ma et al., 2019). Thus, further investigations looking specifically at how such mutations resulted in a seizure phenotype would help understand the impact of Voa1 mutations on neuronal electrical firing diseases, propelling the advancement in treatment options, including precision therapy. It is also possible that the resulting symptoms in DEE and PME come from a complex interaction of multiple mutated genes. Consequently, it is imperative to inspect the interactions of multiple genes in DEE and PME pathophysiology for potential polygenic inheritance mode. Examining relations between epilepsy and developmental impairment in ATP6V0A1 variants is also instrumental in the treatment plan, whether seizure management would help dampen neurodevelopmental impairment or if the two are separated.
Overall, recent findings on ATP6V0A1 mutations in neurodevelopmental disorders emphasize the paramount role of V-ATPases in the development and maintenance of proper lysosomal and autophagosomal activity. In light of these growing discoveries, further structural and functional investigation of human subunit a1 mutations and other isoforms will be beneficial to help elucidate their pathophysiology for disease treatment.
Author contributions
SS conceived and designed the manuscript. KI and PA drafted and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Natural Sciences and Engineering Research Council of Canada (RGPIN 2020 07139) and the Canadian Institute of Health Research (CIHR PJT 165917).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
- Abbas Y. M., Wu D., Bueler S. A., Robinson C. V., Rubinstein J. L. (2020). Structure of V-ATPase from mammalian brain. Science 367, 1240–1246. doi: 10.1126/science.aaz2924, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander R. T., Cordat E., Chambrey R., Dimke H., Eladari D. (2016). Acidosis and urinary calcium excretion: insights from genetic disorders. J. Am. Soc. Nephrol. 27, 3511–3520. doi: 10.1681/ASN.2016030305, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto K., Kato M., Akita T., Nakashima M., Mutoh H., Akasaka N., et al. (2021). ATP6V0A1 encoding the a1-subunit of the V0 domain of vacuolar H+-ATPases is essential for brain development in humans and mice. Nat. Commun. 12:2107. doi: 10.1038/s41467-021-22389-5, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avrahami L., Farfara D., Shaham-Kol M., Vassar R., Frenkel D., Eldar-Finkelman H. (2013). Inhibition of glycogen synthase Kinase-3 ameliorates β-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: IN VIVO AND IN VITRO STUDIES *. J. Biol. Chem. 288, 1295–1306. doi: 10.1074/jbc.M112.409250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagh M. B., Peng S., Chandra G., Zhang Z., Singh S. P., Pattabiraman N., et al. (2017). Misrouting of v-ATPase subunit V0a1 dysregulates lysosomal acidification in a neurodegenerative lysosomal storage disease model. Nat. Commun. 8:14612. doi: 10.1038/ncomms14612, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahena-Bahena D., López-Valdez J., Raymond K., Salinas-Marín R., Ortega-García A., Ng B. G., et al. (2014). ATP6V0A2 mutations present in two Mexican mestizo children with an autosomal recessive cutis laxa syndrome type IIA. Mol Genet Metab Rep 1, 203–212. doi: 10.1016/j.ymgmr.2014.04.003, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S., Clapp K., Tarsio M., Kane P. M. (2019). Interaction of the late endo-lysosomal lipid PI(3,5)P2 with the Vph1 isoform of yeast V-ATPase increases its activity and cellular stress tolerance. J. Biol. Chem. 294, 9161–9171. doi: 10.1074/jbc.RA119.008552, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S., Kane P. M. (2017). Direct interaction of the Golgi V-ATPase a-subunit isoform with PI(4)P drives localization of Golgi V-ATPases in yeast. Mol. Biol. Cell 28, 2518–2530. doi: 10.1091/mbc.E17-05-0316, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batlle D., Moorthi K. M. L. S. T., Schlueter W., Kurtzman N. (2006). Distal renal tubular acidosis and the potassium enigma. Semin. Nephrol. 26, 471–478. doi: 10.1016/j.semnephrol.2006.12.001, PMID: [DOI] [PubMed] [Google Scholar]
- Benlekbir S., Bueler S. A., Rubinstein J. L. (2012). Structure of the vacuolar-type ATPase from Saccharomyces cerevisiae at 11-Å resolution. Nat. Struct. Mol. Biol. 19, 1356–1362. doi: 10.1038/nsmb.2422, PMID: [DOI] [PubMed] [Google Scholar]
- Berg A. T., Mahida S., Poduri A. (2021). KCNQ2-DEE: developmental or epileptic encephalopathy? Ann. Clin. Transl. Neurol. 8, 666–676. doi: 10.1002/acn3.51316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair H. C. (1998). How the osteoclast degrades bone. BioEssays 20, 837–846. doi: [DOI] [PubMed] [Google Scholar]
- Bodzęta A., Kahms M., Klingauf J. (2017). The presynaptic v-ATPase reversibly disassembles and thereby modulates exocytosis but is not part of the fusion machinery. Cell Rep. 20, 1348–1359. doi: 10.1016/j.celrep.2017.07.040, PMID: [DOI] [PubMed] [Google Scholar]
- Bott L. C., Forouhan M., Lieto M., Sala A. J., Ellerington R., Johnson J. O., et al. (2021). Variants in ATP6V0A1 cause progressive myoclonus epilepsy and developmental and epileptic encephalopathy. Brain Commun 3:fcab245. doi: 10.1093/braincomms/fcab245, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldas A., Broyer M., Dechaux M., Kleinknecht C. (1992). Primary distal tubular acidosis in childhood: clinical study and long-term follow-up of 28 patients. J. Pediatr. 121, 233–241. doi: 10.1016/s0022-3476(05)81194-1, PMID: [DOI] [PubMed] [Google Scholar]
- Callewaert B. L., Urban Z. (2022). “ELN-related cutis Laxa” in GeneReviews®. eds. Adam M. P., Mirzaa G. M., Pagon R. A., Wallace S. E., Bean L. J., Gripp K. W., et al. (Seattle (WA): University of Washington, Seattle; ) [Google Scholar]
- Chen L., Wang H.-L., Zhu Y.-B., Jin Z., Huang J.-B., Lin X.-F., et al. (2020). Screening and function discussion of a hereditary renal tubular acidosis family pathogenic gene. Cell Death Dis. 11:159. doi: 10.1038/s41419-020-2354-y, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colina-Tenorio L., Dautant A., Miranda-Astudillo H., Giraud M.-F., González-Halphen D. (2018). The peripheral stalk of rotary ATPases. Front. Physiol. 9:1243. doi: 10.3389/fphys.2018.01243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couoh-Cardel S., Hsueh Y.-C., Wilkens S., Movileanu L. (2016). Yeast V-ATPase proteolipid ring acts as a large-conductance transmembrane protein pore. Sci. Rep. 6:24774. doi: 10.1038/srep24774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diab H., Ohira M., Liu M., Cobb E., Kane P. M. (2009). Subunit interactions and requirements for inhibition of the yeast V1-ATPase*. J. Biol. Chem. 284, 13316–13325. doi: 10.1074/jbc.M900475200, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson M. E., Flenniken A. M., Ji X., Teboul L., Wong M. D., White J. K., et al. (2016). High-throughput discovery of novel developmental phenotypes. Nature 537, 508–514. doi: 10.1038/nature19356, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drory O., Frolow F., Nelson N. (2004). Crystal structure of yeast V-ATPase subunit C reveals its stator function. EMBO Rep. 5, 1148–1152. doi: 10.1038/sj.embor.7400294, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einhorn Z., Trapani J. G., Liu Q., Nicolson T. (2012). Rabconnectin3α promotes stable activity of the H+ pump on synaptic vesicles in hair cells. J. Neurosci. 32, 11144–11156. doi: 10.1523/JNEUROSCI.1705-12.2012, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esmail S., Kartner N., Yao Y., Kim J. W., Reithmeier R. A. F., Manolson M. F. (2018a). Molecular mechanisms of cutis laxa– and distal renal tubular acidosis–causing mutations in V-ATPase a subunits, ATP6V0A2 and ATP6V0A4. J. Biol. Chem. 293, 2787–2800. doi: 10.1074/jbc.M117.818872, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esmail S., Kartner N., Yao Y., Kim J. W., Reithmeier R. A. F., Manolson M. F. (2018b). N-linked glycosylation of a subunit isoforms is critical for vertebrate vacuolar H+ -ATPase (V-ATPase) biosynthesis. J. Cell. Biochem. 119, 861–875. doi: 10.1002/jcb.26250 [DOI] [PubMed] [Google Scholar]
- Feng Y., Forgac M. (1992). Cysteine 254 of the 73-kDa A subunit is responsible for inhibition of the coated vesicle (H+)-ATPase upon modification by sulfhydryl reagents. J. Biol. Chem. 267, 5817–5822. doi: 10.1016/S0021-9258(18)42626-9, PMID: [DOI] [PubMed] [Google Scholar]
- Finnigan G. C., Cronan G. E., Park H. J., Srinivasan S., Quiocho F. A., Stevens T. H. (2012). Sorting of the yeast vacuolar-type, proton-translocating ATPase enzyme complex (V-ATPase): identification of a necessary and sufficient Golgi/endosomal retention signal in Stv1p*. J. Biol. Chem. 287, 19487–19500. doi: 10.1074/jbc.M112.343814, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer B., Dimopoulou A., Egerer J., Gardeitchik T., Kidd A., Jost D., et al. (2012). Further characterization of ATP6V0A2-related autosomal recessive cutis laxa. Hum. Genet. 131, 1761–1773. doi: 10.1007/s00439-012-1197-8, PMID: [DOI] [PubMed] [Google Scholar]
- Flannery A. R., Graham L. A., Stevens T. H. (2004). Topological characterization of the c, c, and c subunits of the vacuolar ATPase from the yeast Saccharomyces cerevisiae. J. Biol. Chem. 279, 39856–39862. doi: 10.1074/jbc.M406767200, PMID: [DOI] [PubMed] [Google Scholar]
- Forgac M. (2007). Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 8, 917–929. doi: 10.1038/nrm2272, PMID: [DOI] [PubMed] [Google Scholar]
- Galli T., McPherson P. S., De Camilli P. (1996). The V0 sector of the V-ATPase, synaptobrevin, and synaptophysin are associated on synaptic vesicles in a Triton X-100-resistant, freeze-thawing sensitive, complex. J. Biol. Chem. 271, 2193–2198. doi: 10.1074/jbc.271.4.2193 [DOI] [PubMed] [Google Scholar]
- Gao Y., Xu Y., Li Q., Lang Y., Dong Q., Shao L. (2014). Mutation analysis and audiologic assessment in six Chinese children with primary distal renal tubular acidosis. Ren. Fail. 36, 1226–1232. doi: 10.3109/0886022X.2014.930332 [DOI] [PubMed] [Google Scholar]
- Ghazi S., Bourgeois S., Gomariz A., Bugarski M., Haenni D., Martins J. R., et al. (2020). Multiparametric imaging reveals that mitochondria-rich intercalated cells in the kidney collecting duct have a very high glycolytic capacity. FASEB J. 34, 8510–8525. doi: 10.1096/fj.202000273R, PMID: [DOI] [PubMed] [Google Scholar]
- Gómez J., Gil-Peña H., Santos F., Coto E., Arango A., Hernandez O., et al. (2016). Primary distal renal tubular acidosis: novel findings in patients studied by next-generation sequencing. Pediatr. Res. 79, 496–501. doi: 10.1038/pr.2015.243, PMID: [DOI] [PubMed] [Google Scholar]
- Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., et al. (2006). Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889. doi: 10.1038/nature04724, PMID: [DOI] [PubMed] [Google Scholar]
- Heaney C., Shalev H., Elbedour K., Carmi R., Staack J. B., Sheffield V. C., et al. (1998). Human autosomal recessive Osteopetrosis maps to 11q13, a position predicted by comparative mapping of the murine Osteosclerosis (oc) mutation. Hum. Mol. Genet. 7, 1407–1410. doi: 10.1093/hmg/7.9.1407, PMID: [DOI] [PubMed] [Google Scholar]
- Heinemann T., Bulwin G. C., Randall J., Schnieders B., Sandhoff K., Volk H. D., et al. (1999). Genomic organization of the gene coding for TIRC7, a novel membrane protein essential for T cell activation. Genomics 57, 398–406. doi: 10.1006/geno.1999.5751, PMID: [DOI] [PubMed] [Google Scholar]
- Hiesinger P. R., Fayyazuddin A., Mehta S. Q., Rosenmund T., Schulze K. L., Zhai R. G., et al. (2005). The v-ATPase V0 subunit a1 is required for a late step in synaptic vesicle exocytosis in Drosophila. Cells 121, 607–620. doi: 10.1016/j.cell.2005.03.012, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton A., Bond S., Forgac M. (2009). V-ATPase functions in normal and disease processes. Pflugers Arch. Eur. J. Physiol. 457, 589–598. doi: 10.1007/s00424-007-0382-4 [DOI] [PubMed] [Google Scholar]
- Hirata R., Graham L. A., Takatsuki A., Stevens T. H., Anraku Y. (1997). VMA11 and VMA16 encode second and third proteolipid subunits of the Saccharomyces cerevisiae vacuolar membrane H+-ATPase. J. Biol. Chem. 272, 4795–4803. doi: 10.1074/jbc.272.8.4795 [DOI] [PubMed] [Google Scholar]
- Hucthagowder V., Morava E., Kornak U., Lefeber D. J., Fischer B., Dimopoulou A., et al. (2009). Loss-of-function mutations in ATP6V0A2 impair vesicular trafficking, tropoelastin secretion and cell survival. Hum. Mol. Genet. 18, 2149–2165. doi: 10.1093/hmg/ddp148, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai E., Kaneko S., Mori T., Okado T., Uchida S., Tsukamoto Y. (2016). A novel heterozygous mutation in the ATP6V0A4 gene encoding the V-ATPase a4 subunit in an adult patient with incomplete distal renal tubular acidosis. Clin. Kidney J. 9, 424–428. doi: 10.1093/ckj/sfw008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata M., Imamura H., Stambouli E., Ikeda C., Tamakoshi M., Nagata K., et al. (2004). Crystal structure of a central stalk subunit C and reversible association/dissociation of vacuole-type ATPase. Proc. Natl. Acad. Sci. U. S. A. 101, 59–64. doi: 10.1073/pnas.0305165101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaskolka M. C., Tarsio M., Smardon A. M., Khan M. M., Kane P. M. (2021). Defining steps in RAVE-catalyzed V-ATPase assembly using purified RAVE and V-ATPase subcomplexes. J. Biol. Chem. 296:100703. doi: 10.1016/j.jbc.2021.100703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane P. M. (1995). Disassembly and reassembly of the yeast vacuolar H(+)-ATPase in vivo. J. Biol. Chem. 270, 17025–17032. doi: 10.1016/S0021-9258(17)46944-4, PMID: [DOI] [PubMed] [Google Scholar]
- Karet F. E., Finberg K. E., Nelson R. D., Nayir A., Mocan H., Sanjad S. A., et al. (1999). Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat. Genet. 21, 84–90. doi: 10.1038/5022, PMID: [DOI] [PubMed] [Google Scholar]
- Kawamura N., Tabata H., Sun-Wada G.-H., Wada Y. (2010). Optic nerve compression and retinal degeneration in Tcirg1 mutant mice lacking the vacuolar-type H+-ATPase a3 subunit. PLoS One 5:e12086. doi: 10.1371/journal.pone.0012086, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki-Nishi S., Bowers K., Nishi T., Forgac M., Stevens T. H. (2001a). The amino-terminal domain of the vacuolar proton-translocating ATPase a subunit controls targeting and in vivo dissociation, and the carboxyl-terminal domain affects coupling of proton transport and ATP hydrolysis*. J. Biol. Chem. 276, 47411–47420. doi: 10.1074/jbc.M108310200 [DOI] [PubMed] [Google Scholar]
- Kawasaki-Nishi S., Nishi T., Forgac M. (2001b). Arg-735 of the 100-kDa subunit a of the yeast V-ATPase is essential for proton translocation. Proc. Natl. Acad. Sci. U. S. A. 98, 12397–12402. doi: 10.1073/pnas.221291798, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki-Nishi S., Nishi T., Forgac M. (2003). Interacting helical surfaces of the transmembrane segments of subunits a and c’ of the yeast V-ATPase defined by disulfide-mediated cross-linking. J. Biol. Chem. 278, 41908–41913. doi: 10.1074/jbc.M308026200, PMID: [DOI] [PubMed] [Google Scholar]
- Komatsu M., Waguri S., Chiba T., Murata S., Iwata J., Tanida I., et al. (2006). Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884. doi: 10.1038/nature04723 [DOI] [PubMed] [Google Scholar]
- Kornak U., Schulz A., Friedrich W., Uhlhaas S., Kremens B., Voit T., et al. (2000). Mutations in the a3 subunit of the vacuolar H(+)-ATPase cause infantile malignant osteopetrosis. Hum. Mol. Genet. 9, 2059–2063. doi: 10.1093/hmg/9.13.2059, PMID: [DOI] [PubMed] [Google Scholar]
- Kornak U., Reynders E., Dimopoulou A., van Reeuwijk J., Fischer B., Rajab A., et al. (2008). Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat. Genet. 40, 32–34. doi: 10.1038/ng.2007.45, PMID: [DOI] [PubMed] [Google Scholar]
- Kosmidis E., Shuttle C. G., Preobraschenski J., Ganzella M., Johnson P. J., Veshaguri S., et al. (2022). Regulation of the mammalian-brain V-ATPase through ultraslow mode-switching. Nature 611, 827–834. doi: 10.1038/s41586-022-05472-9, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.-H., McBrayer M. K., Wolfe D. M., Haslett L. J., Kumar A., Sato Y., et al. (2015). Presenilin 1 maintains lysosomal ca(2+) homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep. 12, 1430–1444. doi: 10.1016/j.celrep.2015.07.050, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.-H., Yu W. H., Kumar A., Lee S., Mohan P. S., Peterhoff C. M., et al. (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cells 141, 1146–1158. doi: 10.1016/j.cell.2010.05.008, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman R., Cotter K., Baleja J. D., Forgac M. (2013). Structural analysis of the N-terminal domain of subunit a of the yeast vacuolar ATPase (V-ATPase) using accessibility of single cysteine substitutions to chemical modification *. J. Biol. Chem. 288, 22798–22808. doi: 10.1074/jbc.M113.460295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Ajmal M., Akram Z., Ghafoor T., Farhan M., Shafique S., et al. (2021). Genetic analysis of osteopetrosis in Pakistani families identifies novel and known sequence variants. BMC Med. Genet. 14:264. doi: 10.1186/s12920-021-01117-4, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Kane P. M., Newman P. R., Forgac M. (1996). Site-directed mutagenesis of the yeast V-ATPase B subunit (Vma2p). J. Biol. Chem. 271, 2018–2022. doi: 10.1074/jbc.271.4.2018, PMID: [DOI] [PubMed] [Google Scholar]
- Liu Q., Leng X. H., Newman P. R., Vasilyeva E., Kane P. M., Forgac M. (1997). Site-directed mutagenesis of the yeast V-ATPase A subunit. J. Biol. Chem. 272, 11750–11756. doi: 10.1074/jbc.272.18.11750 [DOI] [PubMed] [Google Scholar]
- Liu M., Tarsio M., Charsky C. M. H., Kane P. M. (2005). Structural and functional separation of the N- and C-terminal domains of the yeast V-ATPase subunit H*. J. Biol. Chem. 280, 36978–36985. doi: 10.1074/jbc.M505296200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Garcia S. C., Emma F., Walsh S. B., Fila M., Hooman N., Zaniew M., et al. (2019). Treatment and long-term outcome in primary distal renal tubular acidosis. Nephrol. Dial. Transplant. 34, 981–991. doi: 10.1093/ndt/gfy409, PMID: [DOI] [PubMed] [Google Scholar]
- Luong L. H., Nguyen H. D., Trung T. N., Minh T. M. T., Khanh T. L., Son T. P., et al. (2022). Case report of mild TCIRG1-associated autosomal recessive osteopetrosis in Vietnam. Am. J. Med. Genet. A 188, 3096–3099. doi: 10.1002/ajmg.a.62897, PMID: [DOI] [PubMed] [Google Scholar]
- Ma K., Bin N.-R., Shi S., Harada H., Wada Y., Wada G.-H.-S., et al. (2019). Observations from a mouse model of forebrain Voa1 knockout: focus on hippocampal structure and function. Front. Cell. Neurosci. 13:484. doi: 10.3389/fncel.2019.00484, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod K. J., Vasilyeva E., Baleja J. D., Forgac M. (1998). Mutational analysis of the nucleotide binding sites of the yeast vacuolar proton-translocating ATPase. J. Biol. Chem. 273, 150–156. doi: 10.1074/jbc.273.1.150 [DOI] [PubMed] [Google Scholar]
- Maldergem L. V., Dobyns W., Kornak U. (2023). “ATP6V0A2-related cutis Laxa” in GeneReviews®. eds. Adam M. P., Mirzaa G. M., Pagon R. A. (Seattle: University of Washington; ) [Google Scholar]
- Manolson M. F., Rea P. A., Poole R. J. (1985). Identification of 3-O-(4-benzoyl)benzoyladenosine 5′-triphosphate- and N,N’-dicyclohexylcarbodiimide-binding subunits of a higher plant H+-translocating tonoplast ATPase. J. Biol. Chem. 260, 12273–12279. doi: 10.1016/S0021-9258(17)39021-X, PMID: [DOI] [PubMed] [Google Scholar]
- Manolson M. F., Wu B., Proteau D., Taillon B. E., Roberts B. T., Hoyt M. A., et al. (1994). STV1 gene encodes functional homologue of 95-kDa yeast vacuolar H(+)-ATPase subunit Vph1p. J. Biol. Chem. 269, 14064–14074. doi: 10.1016/S0021-9258(17)36755-8, PMID: [DOI] [PubMed] [Google Scholar]
- Maxson M. E., Grinstein S. (2014). The vacuolar-type H+-ATPase at a glance - more than a proton pump. J. Cell Sci. 127, 4987–4993. doi: 10.1242/jcs.158550 [DOI] [PubMed] [Google Scholar]
- Mazhab-Jafari M. T., Rohou A., Schmidt C., Bueler S. A., Benlekbir S., Robinson C. V., et al. (2016). Atomic model for the membrane-embedded VO motor of a eukaryotic V-ATPase. Nature 539, 118–122. doi: 10.1038/nature19828, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michigami T., Kageyama T., Satomura K., Shima M., Yamaoka K., Nakayama M., et al. (2002). Novel mutations in the a3 subunit of vacuolar H+-adenosine triphosphatase in a Japanese patient with infantile malignant osteopetrosis. Bone 30, 436–439. doi: 10.1016/S8756-3282(01)00684-6, PMID: [DOI] [PubMed] [Google Scholar]
- Morava E., Guillard M., Lefeber D. J., Wevers R. A. (2009). Autosomal recessive cutis laxa syndrome revisited. Eur. J. Hum. Genet. 17, 1099–1110. doi: 10.1038/ejhg.2009.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morava E., Lefeber D. J., Urban Z., de Meirleir L., Meinecke P., Gillessen Kaesbach G., et al. (2008). Defining the phenotype in an autosomal recessive cutis laxa syndrome with a combined congenital defect of glycosylation. Eur. J. Hum. Genet. 16, 28–35. doi: 10.1038/sj.ejhg.5201947, PMID: [DOI] [PubMed] [Google Scholar]
- Morel N., Poëa-Guyon S. (2015). The membrane domain of vacuolar H(+)ATPase: a crucial player in neurotransmitter exocytotic release. Cell. Mol. Life Sci. 72, 2561–2573. doi: 10.1007/s00018-015-1886-2, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagara M., Papagregoriou G., Ben Abdallah R., Landoulsi Z., Bouyacoub Y., Elouej S., et al. (2018). Distal renal tubular acidosis in a Libyan patient: evidence for digenic inheritance. Eur. J. Med. Genet. 61, 1–7. doi: 10.1016/j.ejmg.2017.10.002, PMID: [DOI] [PubMed] [Google Scholar]
- Nishi T., Forgac M. (2000). Molecular cloning and expression of three isoforms of the 100-kDa a subunit of the mouse vacuolar proton-translocating ATPase. J. Biol. Chem. 275, 6824–6830. doi: 10.1074/jbc.275.10.6824, PMID: [DOI] [PubMed] [Google Scholar]
- Nishi T., Forgac M. (2002). The vacuolar (H+)-ATPases--nature’s most versatile proton pumps. Nat. Rev. Mol. Cell Biol. 3, 94–103. doi: 10.1038/nrm729 [DOI] [PubMed] [Google Scholar]
- Ochotny N., van Vliet A., Chan N., Yao Y., Morel M., Kartner N., et al. (2006). Effects of human a3 and a4 mutations that result in Osteopetrosis and distal renal tubular acidosis on yeast V-ATPase expression and activity*. J. Biol. Chem. 281, 26102–26111. doi: 10.1074/jbc.M601118200, PMID: [DOI] [PubMed] [Google Scholar]
- Oka T., Murata Y., Namba M., Yoshimizu T., Toyomura T., Yamamoto A., et al. (2001a). a4, a unique kidney-specific isoform of mouse vacuolar H+-ATPase subunit a. J. Biol. Chem. 276, 40050–40054. doi: 10.1074/jbc.m106488200, PMID: [DOI] [PubMed] [Google Scholar]
- Oka T., Toyomura T., Honjo K., Wada Y., Futai M. (2001b). Four subunit a isoforms of Caenorhabditis elegans vacuolar H+-ATPase. Cell-specific expression during development. J. Biol. Chem. 276, 33079–33085. doi: 10.1074/jbc.M101652200 [DOI] [PubMed] [Google Scholar]
- Oot R. A., Couoh-Cardel S., Sharma S., Stam N. J., Wilkens S. (2017). Breaking up and making up: the secret life of the vacuolar H+ -ATPase. Protein Sci. 26, 896–909. doi: 10.1002/pro.3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oot R. A., Huang L.-S., Berry E. A., Wilkens S. (2012). Crystal structure of the yeast vacuolar ATPase heterotrimeric EGChead peripheral stalk complex. Structure 20, 1881–1892. doi: 10.1016/j.str.2012.08.020, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oot R. A., Kane P. M., Berry E. A., Wilkens S. (2016). Crystal structure of yeast V1-ATPase in the autoinhibited state. EMBO J. 35, 1694–1706. doi: 10.15252/embj.201593447, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsini A., Valetto A., Bertini V., Esposito M., Carli N., Minassian B. A., et al. (2019). The best evidence for progressive myoclonic epilepsy: A pathway to precision therapy. Seizure 71, 247–257. doi: 10.1016/j.seizure.2019.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamarthy S., Kulshrestha A., Katara G. K., Beaman K. D. (2018). The curious case of vacuolar ATPase: regulation of signaling pathways. Mol. Cancer 17:41. doi: 10.1186/s12943-018-0811-3, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E., Cho M. H., Hyun H. S., Shin J. I., Lee J. H., Park Y. S., et al. (2018). Genotype–phenotype analysis in pediatric patients with distal renal tubular acidosis. Kidney Blood Press. Res. 43, 513–521. doi: 10.1159/000488698, PMID: [DOI] [PubMed] [Google Scholar]
- Parra K. J., Kane P. M. (1998). Reversible association between the V1 and V0 domains of yeast vacuolar H+-ATPase is an unconventional glucose-induced effect. Mol. Cell. Biol. 18, 7064–7074. doi: 10.1128/MCB.18.12.7064, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra K. J., Keenan K. L., Kane P. M. (2000). The H subunit (Vma13p) of the yeast V-ATPase inhibits the ATPase activity of cytosolic V1 complexes. J. Biol. Chem. 275, 21761–21767. doi: 10.1074/jbc.M002305200, PMID: [DOI] [PubMed] [Google Scholar]
- Pereira P. C. B., Melo F. M., De Marco L. A. C., Oliveira E. A., Miranda D. M., Simões e Silva A. C. (2015). Whole-exome sequencing as a diagnostic tool for distal renal tubular acidosis. J. Pediatr. 91, 583–589. doi: 10.1016/j.jped.2015.02.002 [DOI] [PubMed] [Google Scholar]
- Pérez-Sayáns M., Suárez-Peñaranda J. M., Barros-Angueira F., Diz P. G., Gándara-Rey J. M., García-García A. (2012). An update in the structure, function, and regulation of V-ATPases: the role of the C subunit. Braz. J. Biol. 72, 189–198. doi: 10.1590/s1519-69842012000100023 [DOI] [PubMed] [Google Scholar]
- Peri F., Nüsslein-Volhard C. (2008). Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cells 133, 916–927. doi: 10.1016/j.cell.2008.04.037, PMID: [DOI] [PubMed] [Google Scholar]
- Perin M. S., Fried V. A., Stone D. K., Xie X. S., Südhof T. C. (1991). Structure of the 116-kDa polypeptide of the clathrin-coated vesicle/synaptic vesicle proton pump. J. Biol. Chem. 266, 3877–3881. doi: 10.1016/S0021-9258(19)67875-0, PMID: [DOI] [PubMed] [Google Scholar]
- Peters C., Bayer M. J., Bühler S., Andersen J. S., Mann M., Mayer A. (2001). Trans-complex formation by proteolipid channels in the terminal phase of membrane fusion. Nature 409, 581–588. doi: 10.1038/35054500, PMID: [DOI] [PubMed] [Google Scholar]
- Pietrement C., Sun-Wada G.-H., da Silva N., McKee M., Marshansky V., Brown D., et al. (2006). Distinct expression patterns of different subunit isoforms of the V-ATPase in the rat epididymis. Biol. Reprod. 74, 185–194. doi: 10.1095/biolreprod.105.043752, PMID: [DOI] [PubMed] [Google Scholar]
- Qi J., Forgac M. (2008). Function and subunit interactions of the N-terminal domain of subunit a (Vph1p) of the yeast V-ATPase. J. Biol. Chem. 283, 19274–19282. doi: 10.1074/jbc.M802442200, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reily C., Stewart T. J., Renfrow M. B., Novak J. (2019). Glycosylation in health and disease. Nat. Rev. Nephrol. 15, 346–366. doi: 10.1038/s41581-019-0129-4, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivinoja A., Hassinen A., Kokkonen N., Kauppila A., Kellokumpu S. (2009). Elevated Golgi pH impairs terminal N-glycosylation by inducing mislocalization of Golgi glycosyltransferases. J. Cell. Physiol. 220, 144–154. doi: 10.1002/jcp.21744, PMID: [DOI] [PubMed] [Google Scholar]
- Roh S.-H., Stam N. J., Hryc C. F., Couoh-Cardel S., Pintilie G., Chiu W., et al. (2018). The 3.5-Å CryoEM structure of Nanodisc-reconstituted yeast vacuolar ATPase Vo Proton Channel. Mol. Cell 69, 993–1004.e3. doi: 10.1016/j.molcel.2018.02.006, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagermann M., Stevens T. H., Matthews B. W. (2001). Crystal structure of the regulatory subunit H of the V-type ATPase of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 98, 7134–7139. doi: 10.1073/pnas.131192798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saw N. M. N., Kang S.-Y. A., Parsaud L., Han G. A., Jiang T., Grzegorczyk K., et al. (2011). Vacuolar H+-ATPase subunits Voa1 and Voa2 cooperatively regulate secretory vesicle acidification, transmitter uptake, and storage. Mol. Biol. Cell 22, 3394–3409. doi: 10.1091/mbc.E11-02-0155, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian A., McSherry E., Morris R. C. (1971). On the mechanism of renal potassium wasting in renal tubular acidosis associated with the Fanconi syndrome (type 2 RTA). J. Clin. Invest. 50, 231–243. doi: 10.1172/JCI106479, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafagh Shishavan N., Morovvati S. (2022). A novel deletion mutation in the ATP6V0A2 gene in an Iranian patient affected by autosomal recessive cutis laxa. Ir. J. Med. Sci. doi: 10.1007/s11845-022-03246-w [DOI] [PubMed] [Google Scholar]
- Shao E., Forgac M. (2004). Involvement of the nonhomologous region of subunit A of the yeast V-ATPase in coupling and in vivo dissociation. J. Biol. Chem. 279, 48663–48670. doi: 10.1074/jbc.M408278200, PMID: [DOI] [PubMed] [Google Scholar]
- Shao E., Nishi T., Kawasaki-Nishi S., Forgac M. (2003). Mutational analysis of the non-homologous region of subunit A of the yeast V-ATPase. J. Biol. Chem. 278, 12985–12991. doi: 10.1074/jbc.M212096200, PMID: [DOI] [PubMed] [Google Scholar]
- Sharma S., Oot R. A., Wilkens S. (2018). MgATP hydrolysis destabilizes the interaction between subunit H and yeast V1-ATPase, highlighting H’s role in V-ATPase regulation by reversible disassembly. J. Biol. Chem. 293, 10718–10730. doi: 10.1074/jbc.RA118.002951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smardon A. M., Kane P. M. (2007). RAVE is essential for the efficient assembly of the C subunit with the vacuolar H(+)-ATPase. J. Biol. Chem. 282, 26185–26194. doi: 10.1074/jbc.M703627200 [DOI] [PubMed] [Google Scholar]
- Smardon A. M., Tarsio M., Kane P. M. (2002). The RAVE complex is essential for stable assembly of the yeast V-ATPase. J. Biol. Chem. 277, 13831–13839. doi: 10.1074/jbc.M200682200 [DOI] [PubMed] [Google Scholar]
- Smith A. N., Skaug J., Choate K. A., Nayir A., Bakkaloglu A., Ozen S., et al. (2000). Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat. Genet. 26, 71–75. doi: 10.1038/79208, PMID: [DOI] [PubMed] [Google Scholar]
- Sobacchi C., Frattini A., Orchard P., Porras O., Tezcan I., Andolina M., et al. (2001). The mutational spectrum of human malignant autosomal recessive osteopetrosis. Hum. Mol. Genet. 10, 1767–1773. doi: 10.1093/hmg/10.17.1767, PMID: [DOI] [PubMed] [Google Scholar]
- Söllner T., Whiteheart S. W., Brunner M., Erdjument-Bromage H., Geromanos S., Tempst P., et al. (1993). SNAP receptors implicated in vesicle targeting and fusion. Nature 362, 318–324. doi: 10.1038/362318a0 [DOI] [PubMed] [Google Scholar]
- Srinivasan S., Vyas N. K., Baker M. L., Quiocho F. A. (2011). Crystal structure of the cytoplasmic N-terminal domain of subunit I, a homolog of subunit a, of V-ATPase. J. Mol. Biol. 412, 14–21. doi: 10.1016/j.jmb.2011.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanković K. M., Brown D., Alper S. L., Adams J. C. (1997). Localization of pH regulating proteins H+ATPase and cl-/HCO3- exchanger in the guinea pig inner ear. Hear. Res. 114, 21–34. doi: 10.1016/s0378-5955(97)00072-5, PMID: [DOI] [PubMed] [Google Scholar]
- Steinfeld R., Reinhardt K., Schreiber K., Hillebrand M., Kraetzner R., Brück W., et al. (2006). Cathepsin D deficiency is associated with a human neurodegenerative disorder. Am. J. Hum. Genet. 78, 988–998. doi: 10.1086/504159, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward C. G. (2003). Neurological aspects of osteopetrosis. Neuropathol. Appl. Neurobiol. 29, 87–97. doi: 10.1046/j.1365-2990.2003.00474.x, PMID: [DOI] [PubMed] [Google Scholar]
- Stover E. H., Borthwick K. J., Bavalia C., Eady N., Fritz D. M., Rungroj N., et al. (2002). Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J. Med. Genet. 39, 796–803. doi: 10.1136/jmg.39.11.796, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y., Blake-Palmer K. G., Sorrell S., Javid B., Bowers K., Zhou A., et al. (2008). Human H+ATPase a4 subunit mutations causing renal tubular acidosis reveal a role for interaction with phosphofructokinase-1. Am. J. Physiol. Renal Physiol. 295, F950–F958. doi: 10.1152/ajprenal.90258.2008, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumner J. P., Dow J. A., Earley F. G., Klein U., Jäger D., Wieczorek H. (1995). Regulation of plasma membrane V-ATPase activity by dissociation of peripheral subunits. J. Biol. Chem. 270, 5649–5653. doi: 10.1074/jbc.270.10.5649, PMID: [DOI] [PubMed] [Google Scholar]
- Sun-Wada G.-H., Wada Y. (2015). Role of vacuolar-type proton ATPase in signal transduction. Biochimica et Biophysica Acta (BBA). Bioenergetics 1847, 1166–1172. doi: 10.1016/j.bbabio.2015.06.010, PMID: [DOI] [PubMed] [Google Scholar]
- Susani L., Pangrazio A., Sobacchi C., Taranta A., Mortier G., Savarirayan R., et al. (2004). TCIRG1-dependent recessive osteopetrosis: mutation analysis, functional identification of the splicing defects, and in vitro rescue by U1 snRNA. Hum. Mutat. 24, 225–235. doi: 10.1002/humu.20076, PMID: [DOI] [PubMed] [Google Scholar]
- Suzuki K., Mizutani K., Maruyama S., Shimono K., Imai F. L., Muneyuki E., et al. (2016). Crystal structures of the ATP-binding and ADP-release dwells of the V1 rotary motor. Nat. Commun. 7:13235. doi: 10.1038/ncomms13235, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabke K., Albertmelcher A., Vitavska O., Huss M., Schmitz H.-P., Wieczorek H. (2014). Reversible disassembly of the yeast V-ATPase revisited under in vivo conditions. Biochem. J. 462, 185–197. doi: 10.1042/BJ20131293, PMID: [DOI] [PubMed] [Google Scholar]
- Toei M., Saum R., Forgac M. (2010). Regulation and isoform function of the V-ATPases. Biochemistry 49, 4715–4723. doi: 10.1021/bi100397s [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toei M., Toei S., Forgac M. (2011). Definition of membrane topology and identification of residues important for transport in subunit a of the vacuolar ATPase. J. Biol. Chem. 286, 35176–35186. doi: 10.1074/jbc.M111.273409, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyomura T., Murata Y., Yamamoto A., Oka T., Sun-Wada G.-H., Wada Y., et al. (2003). From lysosomes to the plasma membrane: localization of vacuolar-type H+ -ATPase with the a3 isoform during osteoclast differentiation. J. Biol. Chem. 278, 22023–22030. doi: 10.1074/jbc.M302436200, PMID: [DOI] [PubMed] [Google Scholar]
- Toyomura T., Oka T., Yamaguchi C., Wada Y., Futai M. (2000). Three subunit a isoforms of mouse vacuolar H(+)-ATPase. Preferential expression of the a3 isoform during osteoclast differentiation. J. Biol. Chem. 275, 8760–8765. doi: 10.1074/jbc.275.12.8760 [DOI] [PubMed] [Google Scholar]
- Ueno H., Suzuki K., Murata T. (2018). Structure and dynamics of rotary V1 motor. Cell. Mol. Life Sci. 75, 1789–1802. doi: 10.1007/s00018-018-2758-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallés P. G., Batlle D. (2018). Hypokalemic Distal Renal Tubular Acidosis. Adv. Chronic Kidney Dis. 25, 303–320. doi: 10.1053/j.ackd.2018.05.003 [DOI] [PubMed] [Google Scholar]
- van der Kant R., Goldstein L. S. B., Ossenkoppele R. (2020). Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 21, 21–35. doi: 10.1038/s41583-019-0240-3 [DOI] [PubMed] [Google Scholar]
- Vasanthakumar T., Rubinstein J. L. (2020). Structure and roles of V-type ATPases. Trends Biochem. Sci. 45, 295–307. doi: 10.1016/j.tibs.2019.12.007 [DOI] [PubMed] [Google Scholar]
- Vasilyeva E., Forgac M. (1996). 3’-O-(4-benzoyl)benzoyladenosine 5′-triphosphate inhibits activity of the vacuolar (H+)-ATPase from bovine brain clathrin-coated vesicles by modification of a rapidly exchangeable, noncatalytic nucleotide binding site on the B subunit. J. Biol. Chem. 271, 12775–12782. doi: 10.1074/jbc.271.22.12775, PMID: [DOI] [PubMed] [Google Scholar]
- Vomero A., Tapie A., Arroyo C., Ragggio V., Peluffo G., Dufort G., et al. (2019). Malignant Infantile osteopetrosis. Rev. Chil. Pediatr. 90, 443–447. doi: 10.32641/rchped.v90i4.987, PMID: [DOI] [PubMed] [Google Scholar]
- Wagner C. A., Finberg K. E., Breton S., Marshansky V., Brown D., Geibel J. P. (2004). Renal vacuolar H+-ATPase. Physiol. Rev. 84, 1263–1314. doi: 10.1152/physrev.00045.2003 [DOI] [PubMed] [Google Scholar]
- Wagner C. A., Unwin R., Lopez-Garcia S. C., Kleta R., Bockenhauer D., Walsh S. (2023). The pathophysiology of distal renal tubular acidosis. Nat. Rev. Nephrol. 19, 384–400. doi: 10.1038/s41581-023-00699-9 [DOI] [PubMed] [Google Scholar]
- Wang D., Epstein D., Khalaf O., Srinivasan S., Williamson W. R., Fayyazuddin A., et al. (2014). Ca2+–calmodulin regulates SNARE assembly and spontaneous neurotransmitter release via v-ATPase subunit V0a1. J. Cell Biol. 205, 21–31. doi: 10.1083/jcb.201312109, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Inoue T., Forgac M. (2004). TM2 but not TM4 of subunit c″ interacts with TM7 of subunit a of the yeast V-ATPase as defined by disulfide-mediated cross-linking *. J. Biol. Chem. 279, 44628–44638. doi: 10.1074/jbc.M407345200, PMID: [DOI] [PubMed] [Google Scholar]
- Wang R., Long T., Hassan A., Wang J., Sun Y., Xie X.-S., et al. (2020). Cryo-EM structures of intact V-ATPase from bovine brain. Nat. Commun. 11:3921. doi: 10.1038/s41467-020-17762-9, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Wu D., Robinson C. V., Wu H., Fu T.-M. (2020). Structures of a complete human V-ATPase reveal mechanisms of its assembly. Mol. Cell 80, 501–511.e3. doi: 10.1016/j.molcel.2020.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Youkharibache P., Zhang D., Lanczycki C. J., Geer R. C., Madej T., et al. (2020). iCn3D, a web-based 3D viewer for sharing 1D/2D/3D representations of biomolecular structures. Bioinformatics 36, 131–135. doi: 10.1093/bioinformatics/btz502, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson W. R., Wang D., Haberman A. S., Hiesinger P. R. (2010). A dual function of V0-ATPase a1 provides an endolysosomal degradation mechanism in Drosophila melanogaster photoreceptors. J. Cell Biol. 189, 885–899. doi: 10.1083/jcb.201003062, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X., Liu H., Liu X., Zhang W., Zhang S., Jiao B. (2021). APP, PSEN1, and PSEN2 variants in Alzheimer’s disease: systematic re-evaluation according to ACMG guidelines. Front. Aging Neurosci. 13:695808. doi: 10.3389/fnagi.2021.695808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Garbett K., Veeraraghavalu K., Wilburn B., Gilmore R., Mirnics K., et al. (2012). A role for presenilins in autophagy revisited: normal acidification of lysosomes in cells lacking PSEN1 and PSEN2. J. Neurosci. 32, 8633–8648. doi: 10.1523/JNEUROSCI.0556-12.2012, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Vasilyeva E., Feng Y., Forgac M. (1995). Inhibition and labeling of the coated vesicle V-ATPase by 2-azido-[32P]ATP. J. Biol. Chem. 270, 15494–15500. doi: 10.1074/jbc.270.26.15494 [DOI] [PubMed] [Google Scholar]
- Zhang W., Wang D., Volk E., Bellen H. J., Hiesinger P. R., Quiocho F. A. (2008). V-ATPase V0 sector subunit a1 in neurons is a target of calmodulin. J. Biol. Chem. 283, 294–300. doi: 10.1074/jbc.M708058200, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Benlekbir S., Rubinstein J. L. (2015). Electron cryomicroscopy observation of rotational states in a eukaryotic V-ATPase. Nature 521, 241–245. doi: 10.1038/nature14365, PMID: [DOI] [PubMed] [Google Scholar]
- Zoncu R., Bar-Peled L., Efeyan A., Wang S., Sancak Y., Sabatini D. M. (2011). mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science 334, 678–683. doi: 10.1126/science.1207056 [DOI] [PMC free article] [PubMed] [Google Scholar]