

Abstract
Background and Objectives
Spinal muscular atrophy (SMA) is mainly caused by homozygous SMN1 gene deletions on 5q13. Non-5q SMA patients’ series are lacking, and the diagnostic yield of next-generation sequencing (NGS) is largely unknown. The aim of this study was to describe the clinical and genetic landscape of non-5q SMA and evaluate the performance of neuropathy gene panels in these disorders.
Methods
Description of patients with non-5q SMA followed in the different neuromuscular reference centers in France as well as in London, United Kingdom. Patients without a genetic diagnosis had undergone at least a neuropathy or large neuromuscular gene panel.
Results
Seventy-one patients from 65 different families were included, mostly sporadic cases (60.6%). At presentation, 21 patients (29.6%) showed exclusive proximal weakness (P-SMA), 35 (49.3%) showed associated distal weakness (PD-SMA), and 15 (21.1%) a scapuloperoneal phenotype (SP-SMA). Thirty-two patients (45.1%) had a genetic diagnosis: BICD2 (n = 9), DYNC1H1 (n = 7), TRPV4 (n = 4), VCP, HSBP1, AR (n = 2), VRK1, DNAJB2, MORC2, ASAH1, HEXB, and unexpectedly, COL6A3 (n = 1). The genetic diagnostic yield was lowest in P-SMA (6/21, 28.6%) compared with PD-SMA (16/35, 45.7%) and SP-SMA (10/15, 66.7%). An earlier disease onset and a family history of the disease or consanguinity were independent predictors of a positive genetic diagnosis. Neuropathy gene panels were performed in 59 patients with a 32.2% diagnostic yield (19/59). In 13 additional patients, a genetic diagnosis was achieved through individual gene sequencing or an alternative neuromuscular NGS.
Discussion
Non-5q SMA is genetically heterogeneous, and neuropathy gene panels achieve a molecular diagnosis in one-third of the patients. The diagnostic yield can be increased by sequencing of other neuromuscular and neurometabolic genes. Nevertheless, there is an unmet need to cluster these patients to aid in the identification of new genes.
Hereditary motor neuropathies (HMN) are a heterogeneous group of diseases characterized by peripheral motor neuron weakness with minimal or absent clinical and neurophysiologic sensory impairment, which differentiates them from Charcot-Marie-Tooth (CMT) disease and the hereditary sensory neuropathies.1
Distal HMN (dHMN) represent the most common clinical presentation of HMN.2,3 The term spinal muscular atrophy (SMA) is mainly used when referring to HMNs with proximal weakness at onset, either isolated (P-SMA) or concomitant with distal (PD-SMA) or scapuloperoneal (SP-SMA) motor weakness.4 A homozygous exon 7 and 8 deletion in the SMN1 gene represents the main cause of SMA,5 ranging from 50-94% of patients depending on the series.6,7 In the absence of this deletion (non-5q SMA), several genes have been described, such as TRPV4, DYNC1H1, BICD2, GLE1, UBA1, ASAH1, VRK1, EXOSC3, SLC52A3 and SLC52A2, VAPB, HEXB, SETX, LMNA, and TFG with different clinical phenotypes and ages at onset of disease.4,6,8-12 These and other genes may overlap with other nosological entities such as dHMN, CMT type 2, amyotrophic lateral sclerosis (ALS), hereditary spastic paraplegia (HSP), or even mitochondrial disease and myopathy.4,13 Nevertheless, around 70% or more of non-5q SMA patients do not have a definitive molecular diagnosis.6
Unfortunately, although some reviews of non-5q SMA patients have been published,4,14 clinical series of patients are lacking and the value of routine neuropathy next-generation sequencing (NGS) gene panels is largely unknown. The aim of this study was to describe the clinical and genetic landscape of a large multicentric series of patients with non-5q SMA and evaluate the performance of neuropathy gene panels in these patients.
Methods
Inclusion and Exclusion Criteria
Between April and May 2021, we retrospectively reassessed the clinical records of adult patients (18 years or older) with an HMN and predominant proximal weakness (SMA) from 10 neuromuscular reference centers in France. Sixty-one patients were included based on the following criteria: (1) Patients had a motor neuropathy or motor neuronopathy on electroneuromyography (ENMG), defined by the presence of large-size (>2 mV in amplitude) simple-shaped motor unit potentials and reduced recruitment. In addition, reductions of compound motor action potential (CMAP) amplitudes and less than 30% reduction of normal sensory nerve action potential (SNAP) amplitudes (reference values for adult patients: >10 µV in lower limbs, >8 µV for ulnar, and >15 µV for median and radial nerves) were accepted. (2) Patients were negative for the SMN1 deletion and (3) had a confirmed genetic diagnosis or at least undergone a neuropathy NGS gene panel. Patients with clinical or neurophysiologic disease progression at 6 months were excluded.
Furthermore, clustered data from 10 adult patients followed in the Centre for Neuromuscular Diseases, UCL Queen Square Institute of Neurology, London (United Kingdom) were obtained following the same inclusion and exclusion criteria.
Based on the clinical presentation, patients were classified in 3 groups: (1) Patients with P-SMA had pure proximal motor weakness at first visit, (2) patients with PD-SMA had proximal and distal weakness but pure proximal involvement at disease onset or proximal-predominant weakness at first visit, and (3) patients with SP-SMA had scapuloperoneal weakness. Demographic (date of birth, sex) and clinical information (age at onset, family history of neuropathy, consanguinity, neurologic, orthopedic, and respiratory manifestations) were collected. Additional data such as ENMG findings, creatine kinase (CK) levels, the need of noninvasive ventilation (NIV), and molecular biology results were collected when available, as well as loss of ambulation as functional outcome.
Molecular Biology Investigations
The molecular biology investigations performed in each patient are shown in eTable 1 (links.lww.com/NXG/A614).
Neuropathy Gene Panels
Fifty-nine neuropathy gene panels were performed in total. Forty-eight were performed at the molecular genetic laboratory of Hospices Civils de Lyon. DNA was extracted from blood samples using Nucleospin Blood L (Macherey-Nagel) on automat Microlab STARlet (Hamilton). Capture was performed with Roche KAPA Hypercap v3.0. Sequencing was performed using Illumina NextSeq 500. Copy number variations were detected using DeCovA_1.6.0. Sequencing depth at 30x was over 99,95%. One hundred two genes are included in this panel (eAppendix 1, links.lww.com/NXG/A613).
Eleven additional neuropathy gene panels were sequenced by the National Hospital for Neurology and Neurosurgery (NHNN) in London (n = 4, 15–32 genes) and the molecular genetic laboratories from Kremlin-Bicêtre (n = 4, 127 genes), Limoges (n = 2, 124 genes), and Angers (n = 1, 148 genes).
Alternative Neuromuscular Gene Panels and Individual Gene Sequencing
Eight patients underwent an alternative large neuromuscular gene panel: whole genome sequencing (WGS) with a ∼180 gene virtual panel performed by the NHNN in London (n = 3), a whole exome sequencing (WES) focused on known neuromuscular genes (n = 1), and large neuromuscular gene panels in Paris (n = 2), Strasbourg (n = 1), and Nancy (n = 1).
In 8 patients, a genetic diagnosis was confirmed through individual sequencing of other clinically suspected neuromuscular or neurometabolic genes.
Whole Exome Sequencing
Furthermore, in 20 patients without a molecular diagnosis after a neuropathy NGS panel, WES was performed at Hospices Civils de Lyon. Exome enrichment was performed with the SeqCap EZ MedExome Target Enrichment Kit (Roche Diagnostics). Sequencing was performed on a NextSeq 500 (Illumina) platform, aligned using Borrows-Weeler Aligner to a hg19 reference genome; variants were called using Genome Analysis Tool Kit Unified genotyper software, and filtering of data were performed based on an in-house pipeline: 1000 genomes frequency <0.01, maximum ExAC occurrences heterozygotes 250, maximum ExaC occurrences homozygotes 2, minimal read depth >7, mosaïcism >0.15, coding exons ± 10.
Variants were classified according to the 2015 American College of Medical Genetics Standards and Guidelines for the interpretation of sequence variants,15 and patients with pathogenic or likely pathogenic variants were considered as genetically confirmed.
Data Analysis and Statistics
Data analysis was performed using JASP version 0.16.2. Mean and median values, SD, and range of quantitative variables and absolute or relative frequencies of categorical variables were reported. Association was studied through the Student t test (continuous dependent variables) and χ2 test (categorical dependent variables). A multivariate logistic regression was performed to assess the association of relevant clinical variables with a definitive molecular diagnosis. Normality was verified through the Shapiro-Wilk test. Statistical significance was established at p ≤ 0.05.
Ethics Approval
In accordance with French legislation, the study was registered and is accessible to the French data protection authority (CNIL, Commission nationale de l'informatique et des libertés). This study was approved by the ethics committee of Île-de-France VI on 01/04/2021.
Data Availability
Anonymized data from this study will be shared by request from any qualified investigator.
Results
Clinical Description of Patients and Ancillary Testing
Seventy-one patients from 65 different families were included. Thirty-four patients (47.9%) were female, and most patients had no family history of neuromuscular disease or consanguinity and were therefore classified as sporadic (60.6%). Onset of the disease ranged from 0 to 58 years, and 36 patients (50.7%) had an early onset (younger than 18 years). At presentation (38.2 ± 14.9 years), 21 patients (29.6%) showed exclusive proximal weakness (P-SMA), 35 (49.3%) associated distal weakness (PD-SMA), and 15 patients (21.1%) a scapuloperoneal phenotype (SP-SMA). Clinical and ancillary data are shown in Table 1. Differences in clinical variables between the P-SMA, PD-SMA, and SP-SMA groups are shown in eTable 2 (links.lww.com/NXG/A615).
Table 1.
Clinical Features and Ancillary Examinations of Non-5q Spinal Muscular Atrophy (SMA) Patients
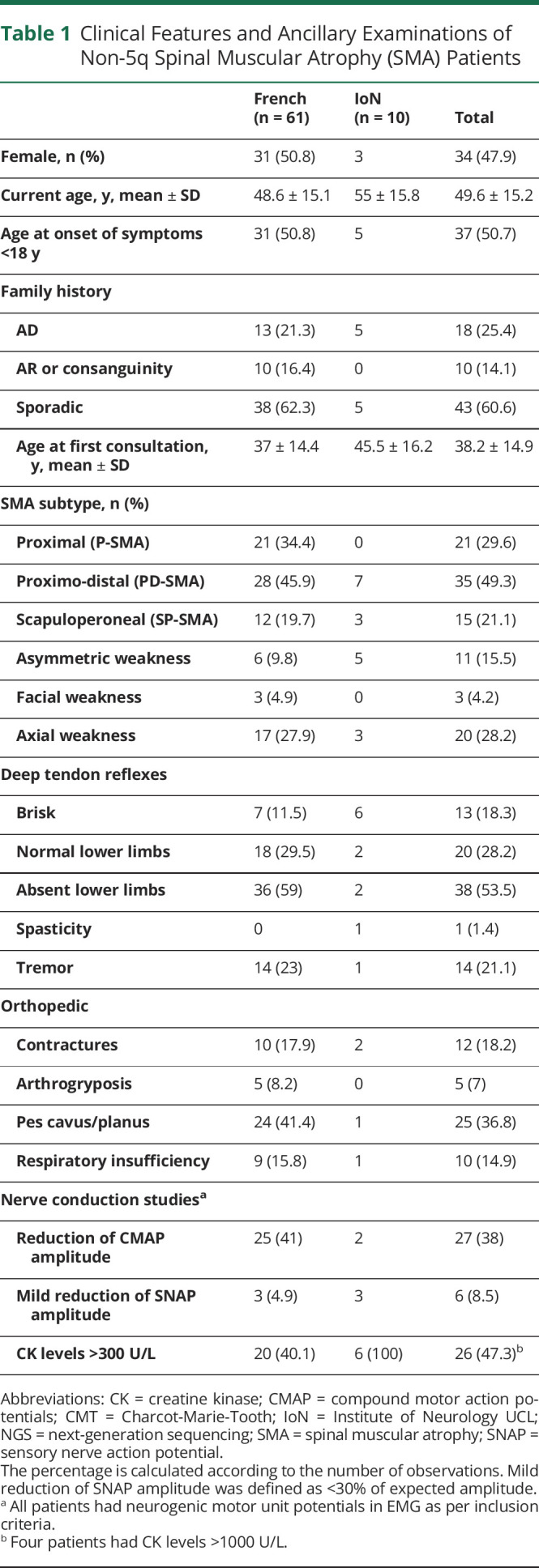
On ENMG, all patients had neurogenic motor unit potentials as per inclusion criteria, with 27 (38%) showing reduction of CMAP amplitude and 6 (8.5%) a minimal reduction of SNAPs associated with the peripheral motor neuron involvement.
For 60 patients, follow-up data were available and 5 patients (8.3%) had lost ambulation at a median of 5 years (range 4–54 years) from symptom's onset.
Genetic Diagnosis and Predictive Variables
In 32 patients (45.1%), a genetic diagnosis was achieved. The molecular diagnostic yield was lowest in the P-SMA (6/21, 28.6%) vs the PD-SMA (16/35, 45.7%) and SP-SMA (10/15, 66.7%) clinical groups. The different genes found in each non-5q SMA clinical group can be seen in Figure 1.
Figure 1. Distribution of Patients With a Positive Genetic Diagnosis According to the Spinal Muscular Atrophy (SMA) Subtype.

Thirty-two patients with a genetic diagnosis achieved through neuropathy gene panel (n = 19: DYNC1H1 x6, BICD2 x4, TRPV4 x4, HSPB1, VCP, VRK1, DNAJB2, MORC2), individual gene sequencing (n = 8: BICD2 x3, AR x2, HSPB1, HEXB, and COL6A3), and other neuromuscular NGS (n = 5: BICD2 x2, DYNC1H1, VCP, and ASAH1). Neg: negative; NGS: next-generation sequencing; P-SMA: proximal SMA; PD-SMA; proximo-distal SMA; SP-SMA: scapuloperoneal SMA.
Fifty-nine neuropathy NGS gene panels were performed, with a positive genetic diagnosis in 19/59 (32.2%). There was no significant difference in the performance of the neuropathy gene panel from Hospice Civils de Lyon (n = 48) vs other neuropathy gene panels in this series (n = 11) (eTable 3, links.lww.com/NXG/A616). In 8 additional patients, a genetic diagnosis was achieved through individual gene sequencing: BICD2 (3 patients from a previously reported family with 6 affected individuals),16 AR (n = 2), HSPB1, HEXB, and COL6A3. Reasons for this were genetic testing before routine gene panel sequencing in family members were an index case was found to carry a variant in the gene through exome sequencing (BICD2),16 genetic testing before routine gene panel sequencing and a plausible clinical picture (HSPB1), a particular or noncorresponding pattern on muscle MRI (HEXB and COL6A3), gynecomastia and elevated CK levels (patient with Kennedy disease), and a negative neuropathy gene panel and elevated CK levels (second patient with Kennedy disease). Finally, 5 patients were diagnosed through an alternative neuromuscular NGS: BICD2 (n = 2), DYNC1H1, VCP, and ASAH1. Furthermore, 20 patients underwent WES, which did not confirm any additional pathogenic or likely pathogenic variants in a known neurologic disease-causing gene.
Patients who received a definitive genetic diagnosis were more likely to have an earlier age at onset, a family history of the disease or consanguinity, and contractures and/or arthrogryposis. In a multivariate logistic regression, an earlier onset and a family history of the disease or consanguinity retained statistical significance (Table 2).
Table 2.
Predictors of a Positive Genetic Diagnosis

Clinical Features of the Most Common Non-5q SMA–Associated Genes
The most common genes in non-5q SMA patients were BICD2 (n = 9) and DYNC1H1 (n = 7), associated with SMA with lower extremity predominance (SMA-LED), and TRPV4 (n = 4), implicated in SP-SMA. These 3 genes are dominantly inherited; however, only 13/20 patients had a clear AD family history of the disease, the rest were therefore categorized as sporadic cases. We summarize below the features of these 3 most common genes.
BICD2 (9 patients from 7 different families). Variants: c.1502G>C (n = 3) from the previously reported family,16 c.2042C>T (n = 2), c.2108C>T, c.2080C>T, c.1617C>G, and c.380A>G. The latter 2 had never been reported: The c.1617C>G variant segregated correctly within the family and predicted as likely pathogenic in silico; the c.380A>G variant was predicted as a likely pathogenic in silico (because it is absent from population databases, well conserved, and situated in a mutational hotspot, with a clinical and muscle MRI phenotype compatible with BICD2 SMA-LED).17 However, given that SMA-LED is not a disease with a single genetic etiology and that segregation was not performed, we retain this variant as a variant of unknown significance (VUS) for now. Globally, although all types of phenotypical presentations were possible between and within families [PD-SMA (n = 5), SP-SMA (n = 3) or P-SMA (n = 1)], BICD2 patients frequently showed some degree of distal weakness on presentation (7/9) and a disease onset younger than 10 years (8/9). Normal or brisk deep tendon reflexes (DTR) (6/9) with 1 patient presenting some degree of spasticity, scapula alata (4/9), and axial weakness (3/9) were relatively frequent. Two patients had mild respiratory insufficiency, 1 needing overnight NIV. No patient had lost ambulation at follow-up. All patients had a motor neuronopathy on ENMG, one with mild sensory involvement.
DYNC1H1 (7 patients from 7 families). Variants: c.2327C>T (n = 2), c.752G> A, c.1792C>T, c.752G>T, c.1427T>C, c.596A>C. The latter 2 variants have never been reported and were predicted as likely pathogenic in silico. Segregation for the c.596A>C confirmed a de novo origin; however, segregation for the c.1427T>C variant could not be performed because the patient was adopted. Clinically, DYNC1H1 patients presented most frequently a PD-SMA (5/7). Disease onset was always in younger than 5 years, 3/7 patients had neonatal arthrogryposis, and 3/7 learning difficulties since childhood. DTRs were mostly abolished (6/7). Only 1 patient had moderate respiratory insufficiency in the context of obesity. Of the 4 patients who were followed up, none had lost ambulation. All patients had a pure motor neuronopathy on ENMG.
TRPV4 (4 patients from 3 families). Variants: c.557G>A (n = 2) and c.694C>T (n = 2), all previously reported. These patients showed predominantly a SP-SMA (3/4), although 1 patient presented with a PD-SMA without scapula alata. Although onset of the first progressive symptom was in adult age (18–53 years), 2 patients had a personal history of arthrogryposis and one of scoliosis and pes cavus. DTRs were always abolished. Three patients had some degree of respiratory insufficiency, none requiring ventilation. Three of these patients also had vocal cord paralysis with hypophonia. One patient showed a minimal reduction of normal SNAP on ENMG. All but 1 patient remain ambulant, the latter losing ambulation at age 5 years (before the onset of weakness) because of arthrogryposis.
Other Non-5q SMA–Associated Genes
Other variants in genes implicated in HMNs were found. In 2 SP-SMA patients with lower-limb distal involvement, scapula alata, and a disease onset in adult life, HSPB1 variants were found (AD transmission). Two patients (SP-SMA and P-SMA) carried variants in the VCP and one (SP-SMA) in the VRK1 gene (both AD), all 3 with an adult disease onset. A previously reported patient with an adult-onset PD-SMA carried a variant in the MORC2 gene.18 One patient with an infantile-onset PD-SMA carried a homozygous pathogenic variant in the DNAJB2 gene (AR). The specific genetic variants can be seen in eTable 1 (links.lww.com/NXG/A614).
It is important that 2 patients with a PD-SMA phenotype without clinical nor ENMG sensory involvement and an onset of symptoms in their late 20s carried CAG repeat expansions in the AR gene (AD transmission). None of these patients had bulbar symptoms, and only one had very high CK levels (2000U/L) and gynecomastia.
Furthermore, 2 patients carried biallelic variants (AR transmission) in genes implicated in lysosomal storage disorders (LSD). One patient carries 2 new compound heterozygous pathogenic variants in the HEXB gene (c.881A>G and c.1082+2T>C), compatible with a diagnosis of Sandhoff disease. This patient presented with a pure P-SMA, an onset of symptoms at 11 years, and no other neurologic manifestation of the disease (cerebellar, extrapyramidal, cognitive, or psychiatric); DTRs were normal, and there was no upper limb tremor. ENMG was compatible with a motor neuronopathy. Importantly, the patient was found to carry a VUS in the DYNC1H1 gene, but the muscle MRI pattern did not correspond to the one previously described in DYNC1H1-associated SMA (Figure 2),19 so a leukocyte hexosaminidase analysis was performed, finding deficient A and B enzymatic activities. A second patient which we have previously reported20 carries compound heterozygous variants in the ASAH1 gene (c.77C>G and c.125+1G>A) and has a deficient leukocyte ceramidase activity. This patient presented with a pure P-SMA, an onset of symptoms at 19 years and again no other neurologic manifestation of the disease (epilepsy). The patient had scoliosis and club feet, upper limb tremor, and retained DTRs. ENMG was compatible with a motor neuronopathy.
Figure 2. Differing MRI Muscle Involvement in Non-5q Spinal Muscular Atrophy (SMA) Patients.

T1-weighted MRI sequences of 5 patients with variants in the 3 most frequent non-5q SMA genes (DYNC1H1, BICD2, and TRPV4) as well as in Sandhoff disease (HEXB) and COL6A3. Muscles with yellow arrows had marked fat replacement; muscles with white arrows were completely or partially spared. (A) Quadriceps, semitendinosus, and adductor magnus muscle degeneration in a patient with Sandhoff disease presenting as P-SMA. (B) The previously described quadriceps muscle degeneration with adductor longus and semitendinosus sparing in a patient with DYNC1H1 PD-SMA.19 In this case, sartorius and gracillis muscle fat replacement is also observed. (C) Quadriceps and sartorious muscle degeneration with more important lower limb involvement in a patient with BICD2 PD-SMA. Notice some degree of paraspinal muscle atrophy and important scoliosis. (D) Tibialis anterior and gastrocnemius medialis involvement with sparing of muscles of the thigh and paraspinal and deltoid muscle degeneration in a patient with TRPV4 SP-SMA. (E) Finally, the characteristic muscle fat replacement in a “sandwich patter” of soleus and quadriceps muscles (yellow arrowheads) in a patient with COL6A3 mutations and a pure SMA phenotype. AL = adductor longus; AM = adductor magnus; De = deltoid; GM = gastrocnemius medialis; Gr = gracillis; PS = paraspinal; Quad = quadriceps; Sa = sartorious; ST = semitendinosus; TA = tibialis anterior.
Finally, in 1 patient with a PD-SMA, retained DTRs, contractures in all 4 limbs since early childhood, and a neurogenic ENMG and muscle biopsy with no evidence of myopathy, who had no genetic diagnosis (after a 102 gene neuropathy panel, SMN1 deletion, and hexosaminidase analysis); a characteristic “sandwich” sign on muscle MRI, typical of collagenopathies, was observed (Figure 2).21 Direct sequencing of the COL6A3 gene found a previously unreported likely pathogenic homozygous c.5867A>G variant as well as a previously described heterozygous missense variant (c.7447A>G), which probably acts as a modulator of the disease.22 A sibling, who presents a similar clinical phenotype and a predominant neurogenic ENMG and muscle biopsy, carried the same variants. Their mother was heterozygous for the c.5867A>G variant, and the father was deceased.
Discussion
We have described the clinical and genetic spectrum of non-5q SMA, whether P-SMA, PD-SMA or SP-SMA, in a large series of patients without the SMN1 deletion. Furthermore, this study provides evidence that the diagnostic yield of neuropathy NGS gene panels in non-5q SMA is still low (almost one-third of patients were diagnosed) but differs across the different clinical presentations.
This study first confirms that variants in 3 genes, BICD2, DYNC1H1, and TRPV4, account for almost two-thirds of genetically confirmed non-5q SMA patients. Nevertheless, each gene shows a distinctive clinical presentation, and while both BICD2 and DYNC1H1 patients had an early disease onset and some degree of distal weakness, BICD2 patients had more frequently scapula alata and preserved or brisk DTRs, while DYNC1H1 patients had more frequently orthopedic manifestations and learning difficulties. TRPV4 patients present most frequently with SP-SMA and even though orthopedic manifestations since childhood are frequent, onset of motor weakness occurs in adult life.
Variants in 8 additional genes were found and account for a significant genetic heterogeneity in non-5q SMA, with multiple possible pathophysiologic mechanisms. These genes can overlap with other nosological entities such as CMT and dHMN (HSBP1, DNAJB2, MORC2),2,23 Kennedy disease (AR),24 myopathies (VCP, COL6A3), ALS (VCP),25,26 LSDs (ASAH1 and HEXB),27 and other developmental disorders (VRK1, MORC2).28-31
Non-5q SMA is also a clinically heterogenous disorder, with different ages at onset of symptoms, patterns of muscle weakness, and associated signs such as abnormal DTRs or orthopedic manifestations. It is important that these clinical features affect the performance of genetic studies. While an earlier onset and a family history of the disease or consanguinity are independent predictors of a positive genetic diagnosis, contractures and/or arthrogryposis as well as SP-SMA phenotype also seem to increase the likelihood of a definitive genetic diagnosis. A previous genetic-focused study testing the diagnostic yield of a panel of 62 non-5q SMA genes vs a broad 479 neuromuscular gene panel6 reported a diagnostic yield of 13% for the non-5q SMA panel vs a 33% for the broad neuromuscular panel; the latter comparable with the performance of neuropathy gene panels in our study. Therefore, neuropathy gene panels (coupled to in-depth phenotyping) seem an adequate strategy in the genetic diagnosis of non-5q SMA patients.
With the increase throughput capability of NGS technologies, comes an increase in the number of VUS, which require an important amount of time and expertise to interpret the variants and avoid misclassification. An example of this is the patient with Sandhoff disease, who was previously found to carry a VUS in the DYNC1H1 gene. In this case, careful analysis of the muscle MRI prompted the search for other alternative diagnosis. This further shows that ancillary testing, and in particular muscle MRI, is crucial to increase the proportion of correctly diagnosed non-5q SMA patients in the advent of NGS techniques. Muscle MRI can not only help discriminate between neuropathy and myopathy32 but also show particular patterns of muscle fat replacement described in DYNC1H1 and BICD2 patients17,19 or characteristic MRI signs, like the “sandwich” sign in collagenopathies.21
Nevertheless, a significant proportion of non-5q SMA patients do not have a definitive genetic diagnosis. Strategies that may increase the number of genetically diagnosed patients can be extracted from the results of this study. First, Kennedy disease should be more frequently searched for, even in the absence of other features such as gynecomastia or sensory involvement, which are not systematic.33 Second, the performance of NGS panels in non-5q SMA could be increased if other neurometabolic disease–associated genes were to be included. This is especially critical because some motor neuron mimics such as SLC52A2 and SLC52A3 genes show a dramatic clinical response to riboflavin34 and that therapeutic trials are ongoing in LSDs such as Sandhoff disease (NCT04221451). Third, the frontier between neuropathies and myopathies is increasingly porous. A well-known example of this is the VCP gene, with varied neurologic presentations (myopathy, neuropathy, or ALS) for most genetic variants.35 Furthermore, almost half of the patients showed moderate elevations of CK levels, a feature which is fairly common in diseases involving motor nerve damage.36 Unexpectedly, 1 patient with a pure SMA (based on repeated ENMG and muscle biopsy findings) but contractures and a muscle MRI compatible with a collagenopathy was found to carry likely pathogenic variants in the COL6A3 gene. Indeed, collagen VI animal models show alterations in myelination and axonal growth,37 and in humans, a previous COL6A3 patient with myopathic and neuropathic changes on ENMG has been described.22 However, this is a report of a patient with pure SMA probably because of mutations in the COL6A3 gene. Finally, WGS, and particularly long-read WGS, may be a more suitable approach than WES in genetically unconfirmed non-5q SMA to search for intronic variants as well as structural variants (SV) because these may account for significant part of the missing heritability in non-5q SMA, as is the case in other neuromuscular diseases.38-40 In Figure 3, the reader can find our recommended diagnostic workflow for an adult patient with a non-5q SMA.
Figure 3. Flow Diagram With the Recommended Diagnostic Approach in Non-5q Spinal Muscular Atrophy (SMA) Patients.

All 3 different sections (genetic, clinical, and ancillary findings) are critical for a correct final diagnosis. Findings in each section should be confronted to findings in the other 2 sections before establishing a definitive diagnosis. Genes underscored show an autosomal dominant (AD) transmission while the rest show an autosomal recessive (AR) transmission, with the exception of the COL6 genes (COL6A1, COL6A2, COL6A3), which may be both AD and AR. The Figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license. CK = creatine kinase; NCS = nerve conduction studies; NGS = next-generation sequencing; SNAP = sensory nerve action potential.
In summary, non-5q SMA is a clinically and genetically heterogenous disorder, and neuropathy NGS gene panels achieve a genetic diagnosis in about one-third of patients followed in specialized neuromuscular reference centers, with an earlier onset and a family history of the disease being an independent predictor of a positive genetic diagnosis. Two-thirds of these patients carry pathogenic variants in 3 genes (BICD2, DYNC1H1, and TRPV4); however, non-5q SMA may also be caused by other overlapping genes, whose identification is laborious if not included in the NGS gene panel. Unfortunately, more than half of the patients do not have a precise genetic diagnosis despite gene panels and WES. Thus, there is an unmet need to cluster these rare patients in view of the identification of new common genes through higher throughput NGS techniques.
Glossary
- ALS
amyotrophic lateral sclerosis
- CK
creatine kinase
- CMAP
compound motor action potentials
- CMT
Charcot-Marie-Tooth
- DTR
deep tendon reflexes
- ENMG
electroneuromyography
- HMN
hereditary motor neuropathies
- HSP
hereditary spastic paraplegia
- NGS
next-generation sequencing
- NHNN
National Hospital for Neurology and Neurosurgery
- NIV
noninvasive ventilation
- SMA
spinal muscular atrophy
- SNAP
sensory nerve action potentials
- VUS
variant of unknown significance
- WES
whole exome sequencing
- WGS
whole genome sequencing
Appendix. Authors
Study Funding
The authors report no targeted funding.
Disclosure
The authors report no relevant disclosures. Go to Neurology.org/NG for full disclosures.
References
- 1.Pipis M, Rossor AM, Laura M, Reilly MM. Next-generation sequencing in Charcot–Marie–Tooth disease: opportunities and challenges. Nat Rev Neurol. 2019;15(11):644-656. [DOI] [PubMed] [Google Scholar]
- 2.Rossor AM, Kalmar B, Greensmith L, Reilly MM. The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatry. 2012;83(1):6-14. [DOI] [PubMed] [Google Scholar]
- 3.Beijer D, Baets J. The expanding genetic landscape of hereditary motor neuropathies. Brain. 2020;143(12):3540-3563. [DOI] [PubMed] [Google Scholar]
- 4.Peeters K, Chamova T, Jordanova A. Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrophies. Brain. 2014;137(11):2879-2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brzustowicz LM, Lehner T, Castilla LH, et al. Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q1 1.2-13.3. Nature. 1990;344(6266):540-541. [DOI] [PubMed] [Google Scholar]
- 6.Karakaya M, Storbeck M, Strathmann EA, et al. Targeted sequencing with expanded gene profile enables high diagnostic yield in non-5q-spinal muscular atrophies. Hum Mutat. 2018;39(9):1284-1298. [DOI] [PubMed] [Google Scholar]
- 7.Wirth B, Herz M, Wetter A, et al. Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am J Hum Genet. 1999;64(5):1340-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farrar MA, Kiernan MC. The genetics of spinal muscular atrophy: progress and challenges. Neurotherapeutics. 2015;12(2):290-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zerres K, Rudnik-Schöneborn S. 93rd ENMC international workshop: non-5q-spinal muscular atrophies (SMA) – clinical picture (6-8 April 2001, Naarden, The Netherlands). Neuromuscul Disord. 2003;13(2):179-183. [DOI] [PubMed] [Google Scholar]
- 10.Wee CD, Kong L, Sumner CJ. The genetics of spinal muscular atrophies. Curr Opin Neurol. 2010;23(5):450-458. [DOI] [PubMed] [Google Scholar]
- 11.Darras BT. Non-5q spinal muscular atrophies: the alphanumeric soup thickens. Neurology. 2011;77(4):312-314. [DOI] [PubMed] [Google Scholar]
- 12.Echaniz-Laguna A, Dubourg O, Carlier P, et al. Phenotypic spectrum and incidence of TRPV4 mutations in patients with inherited axonal neuropathy. Neurology. 2014;82(21):1919-1926. [DOI] [PubMed] [Google Scholar]
- 13.Keller N, Paketci C, Altmueller J, et al. Genomic variants causing mitochondrial dysfunction are common in hereditary lower motor neuron disease. Hum Mutat. 2021;42(4):460-472. [DOI] [PubMed] [Google Scholar]
- 14.de Rezende Pinto WBV, de Souza PVS, Badia BML, et al. Adult-onset non-5q proximal spinal muscular atrophy: a comprehensive review. Arq Neuropsiquiatr. 2021;79(10):912-923. [DOI] [PubMed] [Google Scholar]
- 15.Richards S, Aziz N, Bale S, Bick D, Das S, et al. ; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17(5):405-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oates EC, Rossor AM, Hafezparast M, et al. Mutations in BICD2 cause dominant congenital spinal muscular atrophy and hereditary spastic paraplegia. Am J Hum Genet. 2013;92(6):965-973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rossor AM, Oates EC, Salter HK, et al. Phenotypic and molecular insights into spinal muscular atrophy due to mutations in BICD2. Brain. 2015;138(Pt 2):293-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacquier A, Ribault S, Mendes M, et al. Expanding the phenotypic variability of MORC2 gene mutations: from Charcot‐Marie‐Tooth disease to late ‐ onset pure motor neuropathy. Hum Mutat. 2022;43(12):1898-1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scoto M, Rossor AM, Harms MB, et al. Novel mutations expand the clinical spectrum of DYNC1H1-associated spinal muscular atrophy. Neurology. 2015;84(7):668-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.AME van der Beek N, Nelson I, Froissart R, et al. A new case of SMA phenotype without epilepsy due to biallelic variants in ASAH1. Eur J Hum Genet. 2019;27(3):337-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mercuri E, Lampe A, Allsop J, et al. Muscle MRI in Ullrich congenital muscular dystrophy and Bethlem myopathy. Neuromuscul Disord. 2005;15(4):303-310. [DOI] [PubMed] [Google Scholar]
- 22.Villar-Quiles RN, Donkervoort S, De Becdelièvre A, et al. Clinical and molecular spectrum associated with COL6A3 c.7447A>G p.(Lys2483Glu) variant: elucidating its role in collagen VI-related myopathies. J Neuromuscul Dis. 2021;8(4):633-645. [DOI] [PubMed] [Google Scholar]
- 23.Sevilla T, Lupo V, Martínez-Rubio D, et al. Mutations in the MORC2 gene cause axonal Charcot-Marie-Tooth disease. Brain. 2016;139(Pt 1):62-72. [DOI] [PubMed] [Google Scholar]
- 24.Spada ARL, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352(6330):77-79. [DOI] [PubMed] [Google Scholar]
- 25.Weihl CC, Pestronk A, Kimonis VE. Valosin-containing protein disease: inclusion body myopathy with Paget's disease of the bone and fronto-temporal dementia. Neuromuscul Disord. 2009;19(5):308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson JO, Mandrioli J, Benatar M, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68(5):857-864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Platt FM, d'Azzo A, Davidson BL, Neufeld EF, Tifft CJ. Lysosomal storage diseases. Nat Rev Dis Prim. 2018;4(1):27. [DOI] [PubMed] [Google Scholar]
- 28.Renbaum P, Kellerman E, Jaron R, et al. Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am J Hum Genet. 2009;85(2):281-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hyun YS, Hong YBin, Choi BO, Chung KW. Clinico-genetics in Korean Charcot-Marie-Tooth disease type 2Z with MORC2 mutations. Brain. 2016;139(7):e40. [DOI] [PubMed] [Google Scholar]
- 30.Stafki SA, Turner J, Littel HR, et al. The spectrum of MORC2-related disorders: a potential link to cockayne syndrome. Pediatr Neurol. 2023:141:79-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guillen Sacoto MJ, Tchasovnikarova IA, Torti E, et al. De novo variants in the ATPase module of MORC2 cause a neurodevelopmental disorder with growth retardation and variable craniofacial dysmorphism. Am J Hum Genet. 2020;107(2):352-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bugiardini E, Morrow JM, Shah S, et al. The diagnostic value of MRI pattern recognition in distal myopathies. Front Neurol. 2018;9:456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferrante MA, Wilbourn AJ. The characteristic electrodiagnostic features of Kennedy's disease. Muscle Nerve. 1997;20(3):323-329. [DOI] [PubMed] [Google Scholar]
- 34.Carreau C, Benoit C, Ahle G, et al. Late-onset riboflavin transporter deficiency: a treatable mimic of various motor neuropathy aetiologies. J Neurol Neurosurg Psychiatry. 2021;92(1):27-35. [DOI] [PubMed] [Google Scholar]
- 35.Schiava M, Ikenaga C, Villar-Quiles RN, et al. Genotype-phenotype correlations in valosin-containing protein disease: a retrospective muticentre study. J Neurol Neurosurg Psychiatry. 2022:1099-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silvestri NJ, Wolfe GI. Asymptomatic/pauci-symptomatic creatine kinase elevations (hyperckemia). Muscle Nerve. 2013;47(6):805-815. [DOI] [PubMed] [Google Scholar]
- 37.Gregorio I, Braghetta P, Bonaldo P, Cescon M. Collagen VI in healthy and diseased nervous system. DMM Dis Model Mech. 2018;11(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ishiura H, Shibata S, Yoshimura J, et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat Genet. 2019;51(8):1222-1232. [DOI] [PubMed] [Google Scholar]
- 39.Cortese A, Simone R, Sullivan R, et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet. 2019;51(4):649-658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data from this study will be shared by request from any qualified investigator.