

Abstract
Whether left ventricular noncompaction (LVNC) is a distinct cardiomyopathy or a morphologic trait shared by different cardiomyopathies remains controversial. Current guidelines from professional organizations recommend different strategies for diagnosing and treating patients with LVNC. This state-of-the-art review discusses new insights into the basic mechanisms leading to LVNC, its clinical manifestations, and treatment modalities, anatomy and pathology, embryology, genetics, epidemiology, and imaging. Three markers currently define LVNC: prominent left ventricular trabeculae; deep intertrabecular recesses; and a thin compacted layer. While new genetic data from mice and humans supports LVNC as a distinct cardiomyopathy, evidence for LVNC as a shared morphological trait is not ruled out. Criteria supporting LVNC as a shared morphological trait may depend on consensus guidelines from the multiple professional organizations. Enhanced imaging and increased use of genetics are both predicted to significantly impact our overall understanding of the basic mechanisms causing LVNC, and its optimal management.
Keywords: compacted, epidemiology, genetics, imaging, pathology, trabeculae
Introduction
Left ventricular noncompaction (LVNC) is defined by 3 markers: prominent left ventricular (LV) trabeculae; deep intertrabecular recesses; and the thin compacted layer (1). The spectrum of morphologic variability is extreme, ranging from hearts with a nearly absent compacted layer and an almost exclusively trabecular component in the LV apex, to hearts with prominent trabeculae and deep alternating recesses, but a well-represented compacted layer. Whether LVNC is a distinct cardiomyopathy or a morphologic trait shared by different types of cardiomyopathies is still debated. LVNC can be isolated or associated with cardiomyopathies, congenital heart diseases, and complex syndromes involving the heart. The American Heart Association classifies LVNC as a genetic cardiomyopathy (2) while the European Society of Cardiology classifies LVNC as an unclassified cardiomyopathy (3). The World Heart Organization’s International Classification of Diseases also reports LVNC as an unclassified cardiomyopathy. This state-of-the-art review includes data from embryology, genetic studies, epidemiology, and imaging studies (as outlined in a recent editorial on the evolution of translational medicine (4)), and provides an up-to-date view of current findings on the controversial topic of LVNC. Each of the authors selected published reports for this review on the basis of the most up-to-date criteria.
The LVNC trait may be familial (inherited) or nonfamilial (sporadic). Nonfamilial forms are diagnosed when LVNC is proven absent in relatives (5). Sporadic, LVNC can be acquired, as in highly trained athletes (6), sickle cell anemia patients (7), and in pregnancy (8). In the pregnancy study by Gati, 73% of affected women demonstrated complete resolution of the trabeculation during post-partum follow-up (8). In some cases, the trabeculation phenotype may occur in response to a mechanical load, and may disappear as the mechanical load dissipates. It is not known if there is a genetic underpinning to the disease in these cases (6) (7) (8). Cases in children suggest that 75% have ECG abnormalities, and most have depressed systolic function (9). Some children have transient recovery, followed by later deterioration, suggesting that these cases in children are genetic in nature. The genetic bases of familial LVNC are still a matter of research. Most familial cases identified to date are associated with mutations in the same genes that cause other types of cardiomyopathies (10,11). Whether these disease genes cause the cardiomyopathy or the LVNC phenotype remains to be clarified (12). A limitation of many (but not all) of these genetic studies is that most only screened genes associated with other cardiomyopathies, such as sarcomeric genes, which are also associated with hypertrophic, restrictive, and dilated cardiomyopathies.
Although there is no current gold standard for LVNC diagnosis, cardiac imaging is the best tool currently available. Pathoanatomic investigation in autopsy hearts or in hearts excised at transplantation provides data for pathoimaging correlations and assessment of imaging-based diagnoses (13). The most commonly used imaging modalities include echocardiography and cardiac magnetic resonance (CMR). Echocardiography provides the basic tool for diagnosis (14), while CMR adds anatomic details, and functional information on kinesis of the noncompacted versus compacted segments and fibrosis (15). The limitations of imaging will be discussed. Clinical management is based on the functional phenotype and related complications. The management of atrial and ventricular arrhythmias, device implantation, resynchronization, ablation procedures, and even LV surgical remodeling has been a matter of specific attention, raising the question of whether LVNC deserves specific medical strategies (4,16).
Embryogenic and nonembryogenic hypotheses
There can be multiple etiologic bases of LVNC: it may occur as an isolated trait or disease (I-LVNC); or in association with genetic diseases and congenital defects; or be sporadic and acquired in physiological (6) or pathologic conditions (7); or be permanent or transient (8). Therefore, LVNC can originate during embryonic development or be acquired later in life.
Nonembryogenic hypothesis.
Emerging evidence supports the hypothesis that the pathogenetic mechanisms leading to noncompaction or increased trabeculation may occur in adult life, leading to acquired LVNC. In young athletes, increased LV trabeculation may represent the effect of cardiac remodeling (6); in this case, trabeculation becomes more prominent, but the compacted layer is well represented. It has been suggested that “following ECG and echocardiography, 0.9% of highly trained athletes demonstrate concomitant T-wave inversion and reduced baseline indices of systolic function that may be considered diagnostic of LVNC” (6). The de novo LV trabeculations observed in a significant proportion (>25%) of pregnant women suggest that LV trabeculations may occur in response to increased LV loading conditions or other physiological adaptation mechanisms related to pregnancy (8). The increased trabeculation observed in individuals with sickle cell anemia may represent an exaggerated myocardial response to the increased cardiac preload (7). In sum, this evidence supports the hypothesis that particular phenotypic characteristics of LVNC are identified in cases including pregnancy, sickle cell anemia and athletes.
Embryogenic hypothesis.
Most data supporting the embryogenic hypothesis of LVNC come from experimental studies. Fetal echocardiographic studies may contribute to elucidation of the embryogenic mechanisms of LVNC and its association with other cardiac diseases. In a very elegant study by Arunamata et al., 22 of 24 fetuses with LVNC had congenital heart disease, and 15 had complete heart block (17). Studies in identical twins may further expand the routes of investigation, especially when identical phenotypes are expected, but not observed (18). Studies in experimental models suggest that the process of cardiac trabeculation begins after the cardiac looping stage. Trabeculae formation begins with the emergence of myocytes through delamination (migration) from the compacted myocardium (19). Emerging evidence suggests that the myocytes forming the trabeculae arise from a different clonal origin in the heart wall (19,20). Myocytes project radially into the cavity and are covered by the endocardial layer. This array guarantees the best perfusion of the myocytes by increasing the contact surface between the left ventricular cavity and the myocytes, while the coronary tree is not yet developed. Intertrabecular spaces are transformed into capillary vessels. Failure at this stage corresponds to the formation of thin elongated trabecular projections separated by deep recesses. The compact layers of myocytes proliferate and the epicardium enters the myocardial wall and forms the coronary vasculature (21,22). Recent studies in zebrafish and mice suggest that cardiac trabeculation is mediated by endocardial neuregulin 1 through the ErbB4 and ErbB2 receptor complex (19,23–27). Deletion or mutation of the homologs of Drosophila Mindbomb 1, Notch1, Neuregulin 1, Erbb4, or Errb2 in zebrafish or mice results in the absence of trabecular formation. ErbB3 activation by Neuregulin 1 phosphorylates focal adhesion kinase (FAK). Systemic deletion of FAK in mouse also results in a phenotype similar to LVNC (28,29). Thus, Neuregulin 1 signaling through ErbB4 and ErbB2 leading to FAK phosphorylation appears integral to cardiac trabecular formation. Moreover, Notch signaling in the endocardium is also critical for cardiac trabecular formation (30,31).
As cardiac development progresses, myocytes compact by organizing into ordered bundles that progressively generate the compacted myocardial walls (more prominent in the left than in the right ventricle). The trabecular portion of the myocardial wall is tiny and thinner in the LV than in the right, and the compacted wall is more prominent in the thicker LV wall. Two embryologic morphogenetic hypotheses were formulated as potential explanations of LVNC pathogenesis. Hypothesis 1 states that arrested or abnormal myocardial morphogenesis leading to LVNC occurs during heart development, when myocyte organization fails to evolve from the embryonic spongiform condition to the compacted, mature state. Although both ventricles may be involved, the LV is generally affected (32). Hypothesis 2 states that LVNC occurs as a result of inhibiting the regression of embryonic structures (33). Sponginess would result from the looseness of cells or of cell bundles. LVNC describes a macroscopic mismatch between the noncompacted trabeculae and the compacted myocyte layers. Myocytes in the trabeculae do not show histologic differences from those forming the compacted layer, explaining why LVNC histology (i.e., endomyocardial biopsy) does not specifically contribute to the diagnosis. The diagnostic hallmark of LVNC is the macroscopic appearance that correlates with imaging findings.
Anatomy and Pathology
In hearts excised at transplantation or at autopsy, LVNC diagnosis is on the basis of the prominent appearance of LV trabeculae and the ratio between the compacted and noncompacted LV wall (13). Sectioning of formalin-fixed hearts provides the best way of measuring compacted and noncompacted layers (34). Prominent trabeculae and thin, compacted myocardial layers can be described either as suggestive for LVNC or as increased trabeculation when the noncompacted/compacted ratio does not meet that commonly used (2.3) in imaging diagnosis (35). The pathologic diagnosis should not be forced in imaging criteria, but should provide data for pathology-imaging correlation (Figure 1). Imaging is especially useful for identifying mural thrombi wedged within the intertrabecular recesses (especially common in hypokinetic LVs)(Figure 2).
Figure 1. Two Hearts Depicting the Variability In Both Extension and Depth Of Trabeculae and Recesses.
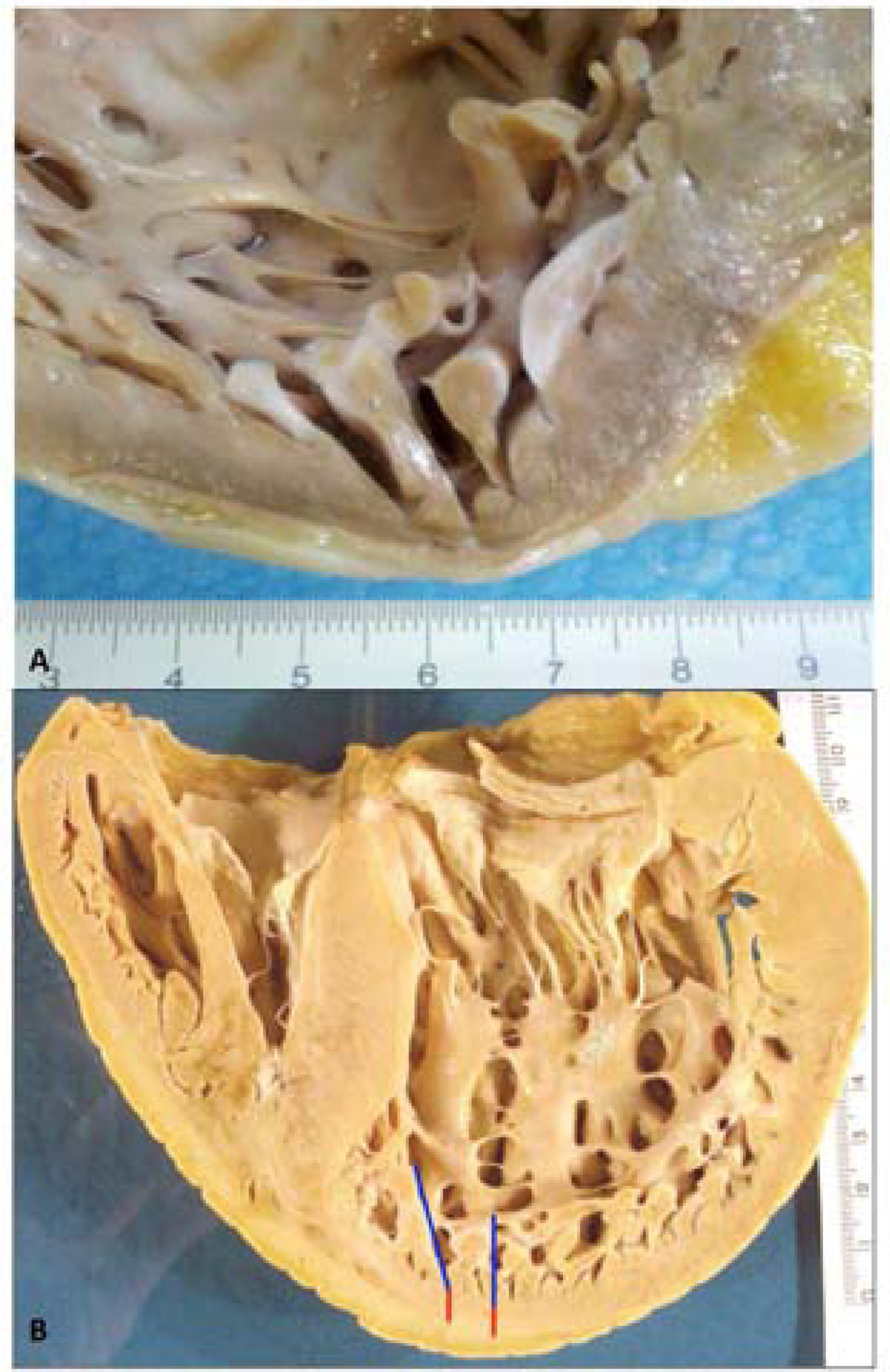
A) In this high magnification view of the apical wall of the heart, the noncompacted area is limited to a few apical trabeculae. The patient harbored mutations p.(Arg495Trp) in Myosin Binding Protein Cardiac 3 (MYBPC3) and p.(Asp117Asn) in Lim domain binding protein 3 (LDB3) genes [MH+D OH GAD EG-MYBPC3[p.Arg495Trp]+LDB3 [p.Asp117Asn]SC-IV]. Although LBD3 is a candidate gene for LVNC, in this family, the disease segregated with the mutation in MYBPC3.
B) In this heart, the prominent trabeculations (blue line) and deep recesses (red line) involve the entire LV apex. LV = left ventricle.
Figure 2. High Magnification View of Intertrabecular (BLUE →) and Endocardial (GREEN→) Thrombotic Stratification.
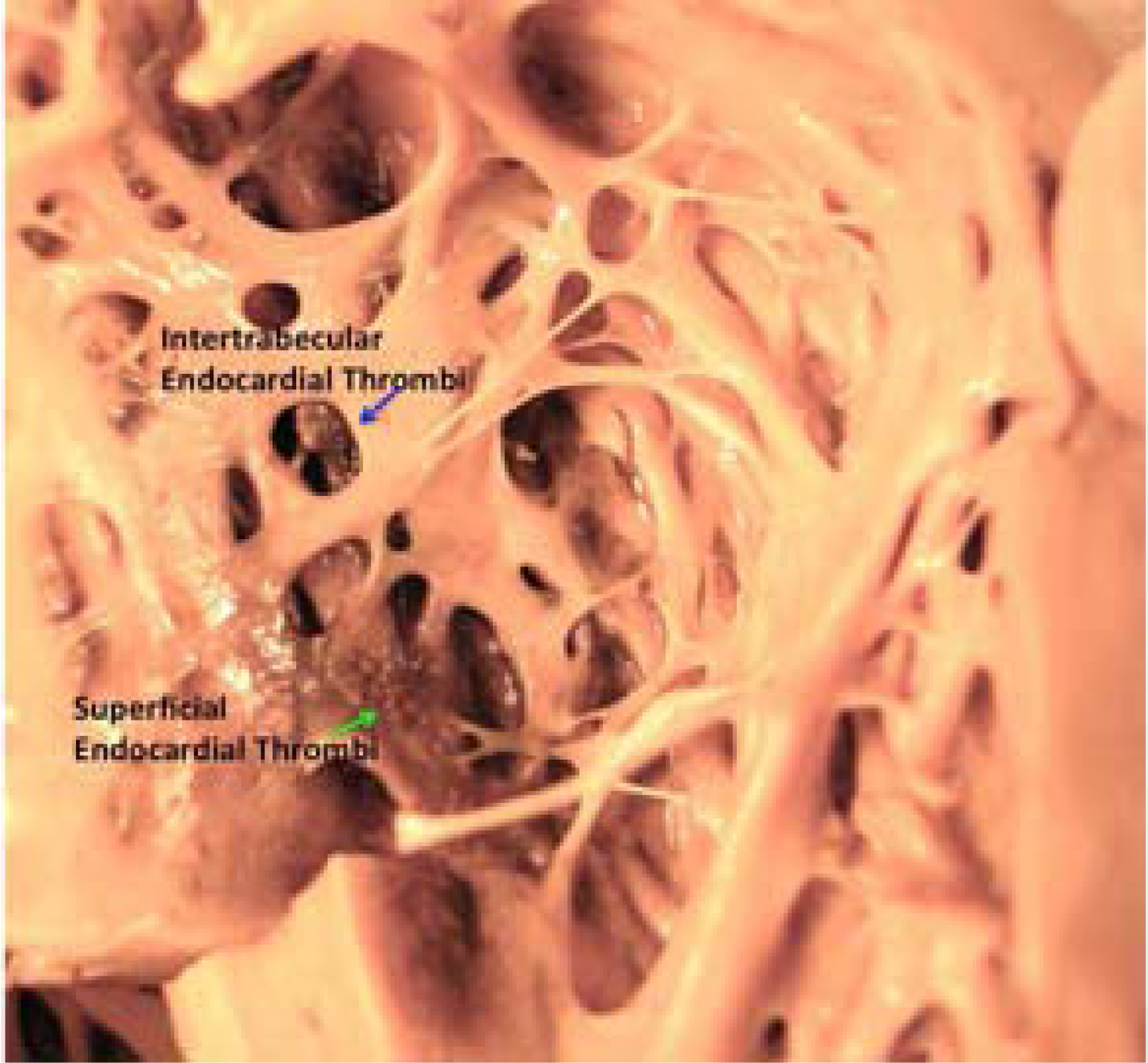
LV dilation and LV hypertrophy can be present or absent and do not influence LVNC diagnosis. Given the common localization of the noncompacted areas in the apex and the common localization on LV hypertrophy at the septum, the 2 diagnoses, HCM and LVNC can coexist. The topographic distribution of LVNC does not typically extend to the interventricular septum, although the septum may be involved in rare cases. LVNC has also been described in association with restrictive cardiomyopathy (RCM). In pure RCM, the enlarged atria and the small nonhypertrophic ventricles support the pathologic diagnosis. The “restriction” is a functional diagnostic clue that can be inferred in pathologic studies by the atrial/ventricular size mismatch in the absence of significant LV hypertrophy. LVNC may also coexist with arrhythmogenic right ventricular cardiomyopathy (ARVC). In classical ARVC without involvement of the LV, the presence of LVNC is independent of the right side cardiomyopathy. In this context, the causes of ARVC and LVNC may not coincide. In biventricular and predominantly left arrhythmogenic cardiomyopathies, the presence of LVNC may be either considered as an independent trait or as part of the arrhythmogenic cardiomyopathy involving the LV. The pathology study should contribute to characterization of LVNC as an isolated finding or as a trait present in cardiomyopathy in autopsied hearts and in hearts excised at transplantation. Finally, fibrous endocardial thickening can be present; it may reflect the effect of volume overload in LVNC in dilated cardiomyopathy (DCM), or the organization pattern of mural thrombi. Overall, LVNC can be observed in all types of cardiomyopathies.
Epidemiology
LVNC occurs in infants (0.81 per 100,000 infants/year), children (0.12 cases per 100,000 children (21) and adults (prevalence 0.014%)(33). It can occur as an isolated myocardial trait or associated with cardiomyopathies (hypertrophic, restrictive, dilated and arrhythmogenic), congenital heart diseases (36) and complex syndromes affecting multiple organs and tissues including mitochondrial diseases caused by mutations in both nuclear (23) and mitochondrial genes (24). In isolated LVNC, the intertrabecular recesses communicate with the LV cavity. LVNC was first described in 1984, in Engberding and Bender’s description of the first echocardiographic diagnosis of persistence of isolated myocardial sinusoids (37). In patients with LVNC associated with other congenital heart diseases, the deep intertrabecular recesses communicate with both the LV cavity and the coronary circulation (33). LVNC was first described by Bellet and Gouley in 1932, when they observed abnormally “spongy” myocardial walls associated with aortic atresia and coronary-ventricular fistula in an autopsy of a newborn with congenital heart disease (38).
By itself, “LVNC” does not necessarily describe a disease; it describes an anatomic variant of LV structure (39,40). There is wide variability in the ratio between trabeculated and compacted layers of the LV. At one extreme, severe forms of LV apex noncompaction and inferior/lateral walls are typically seen in children with Barth syndrome. In these patients, LVNC is associated with LV dilation and dysfunction (41). At the other extreme, hypertrabeculation with prominent (but less pronounced) trabeculations and intertrabecular recesses associated with a preserved, compacted layer is more common. Ethnic differences in the amount of trabeculation have been observed; Gati et al. suggested prominent LV trabeculation was more prevalent in African American subjects (6).
Genetics
This paper’s title posed the question of whether LVNC is a, “distinct cardiomyopathy or a trait shared by different cardiac diseases.” Human genetic studies suggest that several genes are associated with LVNC (see Table 1).
Table 1.
Genes Associated With LVNC.
| Phenotype | Gene/Locus Additional | Mode of Inheritance | References | ||||
|---|---|---|---|---|---|---|---|
| Location Phenotype | MIM number | Gene/Locus | MIM number | Phenotypes | |||
| 1p36.32 |
Left ventricular
noncompaction 8 |
615373 | PRDM16 | 605557 | Dilated cardiomyopathy | Autosomal Dominant |
(77) |
| 1q32.1 |
Left ventricular
noncompaction 6 |
601494 | TNNT2 | 191045 | Dilated cardiomyopathy Dominant | Autosomal Dominant |
(78) |
| 10q23.2 | Left ventricular noncompaction 3 |
601493 | LDB3 | 605906 | Dilated cardiomyopathy | Autosomal Dominant |
(79) |
| 11p15 |
Left ventricular
noncompaction 2 |
609470 | None | 609470 | ------ | Autosomal Dominant |
(80) |
| 11p11.2 |
Left ventricular
noncompaction 10 |
615396 | MYBPC3 | 600958 | Dilated cardiomyopathy |
Autosomal Dominant |
(81) |
| 14q11.2 |
Left ventricular
noncompaction 5 |
613426 | MYH7 | 160760 | Dilated cardiomyopathy | Autosomal Dominant |
(80,82) |
| 15q14 |
Left ventricular
noncompaction 4 |
613424 | ACTC1 | 102540 | Dilated cardiomyopathy | Autosomal Dominant |
(83) |
| 15q22.2 |
Left ventricular
noncompaction 9 |
611878 | TPM1 |
191010
|
Dilated cardiomyopathy | ----- | (81) |
| 18q11.2 |
Left ventricular
noncompaction 7 |
615092 | MIB1 | 608677 | ----- |
Autosomal Dominant |
(30) |
| 18q12.1 | Left ventricular noncompaction 1 |
604169 | DTNA | 601239 | With or without congenital heart defects |
Autosomal Dominant |
(84) |
| Xq28 | Barth syndrome | 302060 | G4.5, TAZ | 300394 | Failure to thrive Failure to grow |
X-linked recessive |
(71) |
This table was adapted from Online Mendelian Inheritance in Man (OMIM). Additional information is available at the OMIM website (85). From left to right, the table provides the location for each locus, the phenotype (LVNC), with a number that refers to phenotypes associated with the particular genes or loci), the phenotype MIM number, gene/locus, the gene/locus MIM number, additional phenotypes, mode of inheritance, and references. MIM number refers to a numerical assignment for genes and functional segments of DNA, as well as to inherited diseases. ACTC1 = actin, alpha, cardiac muscle; DTNA = dystrobrevin alpha; LDB3 = Lim domain-binding 3; LVNC = left ventricular noncompaction;MIB1 = homolog of Drosophila mindbomb; MYBPC3 = myosin-binding protein C, cardiac; MYH7 = myosin heavy chain 7, cardiac muscle, beta; PRDM16 = PR domain–containing protein 16; TAZ = tafazzin; TNNT2 = troponin T2; TPM1 = tropomyosin1. X-linked recessive (both matching genes must be abnormal to cause disease).
Nearly all of the genes associated with LVNC are associated with additional phenotypes, like cardiomyopathies or congenital heart defects. However mutations in 1 gene, the homolog of Drosophila Mindbomb 1 (MIB1), segregated with autosomal dominant LVNC in 2 Spanish families and a conditional loss of function allele in a mouse also led to LVNC (30). The hypertrabeculation and noncompaction seen in the Mib1 mouse was mimicked in a mouse with inactivation of Jagged1 in the myocardium or Notch1 in the endocardium, suggesting that the Notch1 signaling pathway was, indeed, involved (30). Chen and colleagues recently reported an important role in trabeculation for endocardial expression of a Notch ligand, Fkbp1a (31). These findings firmly support the hypothesis that in some circumstances, LVNC is a cardiomyopathy and dysregulated Notch signaling in the endocardium leads to disrupted trabeculation. Barth syndrome is an X-linked recessive disorder diagnosed either prenatally or in infants that is characterized by failure to thrive, growth retardation, and cardiovascular abnormalities including LVNC (5,42). It is probably 1 of the few cardiomyopathies that can be prenatally recognized with imaging (42), and typically includes hypokinetic dilated cardiomyopathy with LVNC that can cause death in early infancy. Barth syndrome is associated with the gene, G4.5, encoding tafazzin (TAZ), a mitochondrial protein critical for remodeling of the phospholipid, cardiolipin. TAZ knockdown mice die embryonically with cardiomyopathy characterized by hypertrabeculation and noncompaction (43). The mouse model and together with human TAZ and Barth syndrome data provide additional evidence that genetic pathways can lead directly to hypertrabeculation and noncompaction, suggesting that in some instances, LVNC is a cardiomyopathy (30,44).
Other LVNC-associated genes in Table 1 are also associated with additional phenotypes, including cardiomyopathies (45). LVNC lacks genome-wide association studies (GWAS) for LVNC, which would be challenging, given that patients present with pleiotropic phenotypes (46). Additional limitations include that most studies reported to date are underpowered, limiting their strength. While whole-genome and whole-exome sequencing permit the discovery of a new complexity of genotypes (46), many studies reported to date do not report clinical whole-genome or whole-exome sequencing, but instead sequence candidate genes. Pairing new complex genotypes with complex phenotypes like LVNC requires criteria for phenotyping and quality control for genotyping. Thus, at this early stage, we can not rule out modifier genes or the potential for several genes to influence the LVNC phenotype. Epigenetic (i.e., DNA methylation) or environmental causes, such as increased mechanical load or stress that may induce the phenotype, are additional possibilities. A recent study using high-resolution episcopic microscopy and 3D reconstruction shows (with high sensitivity and quantification) hypertrabeculation in the Mib1 loss-of-function allele mouse, compared to a wild type mouse (47).
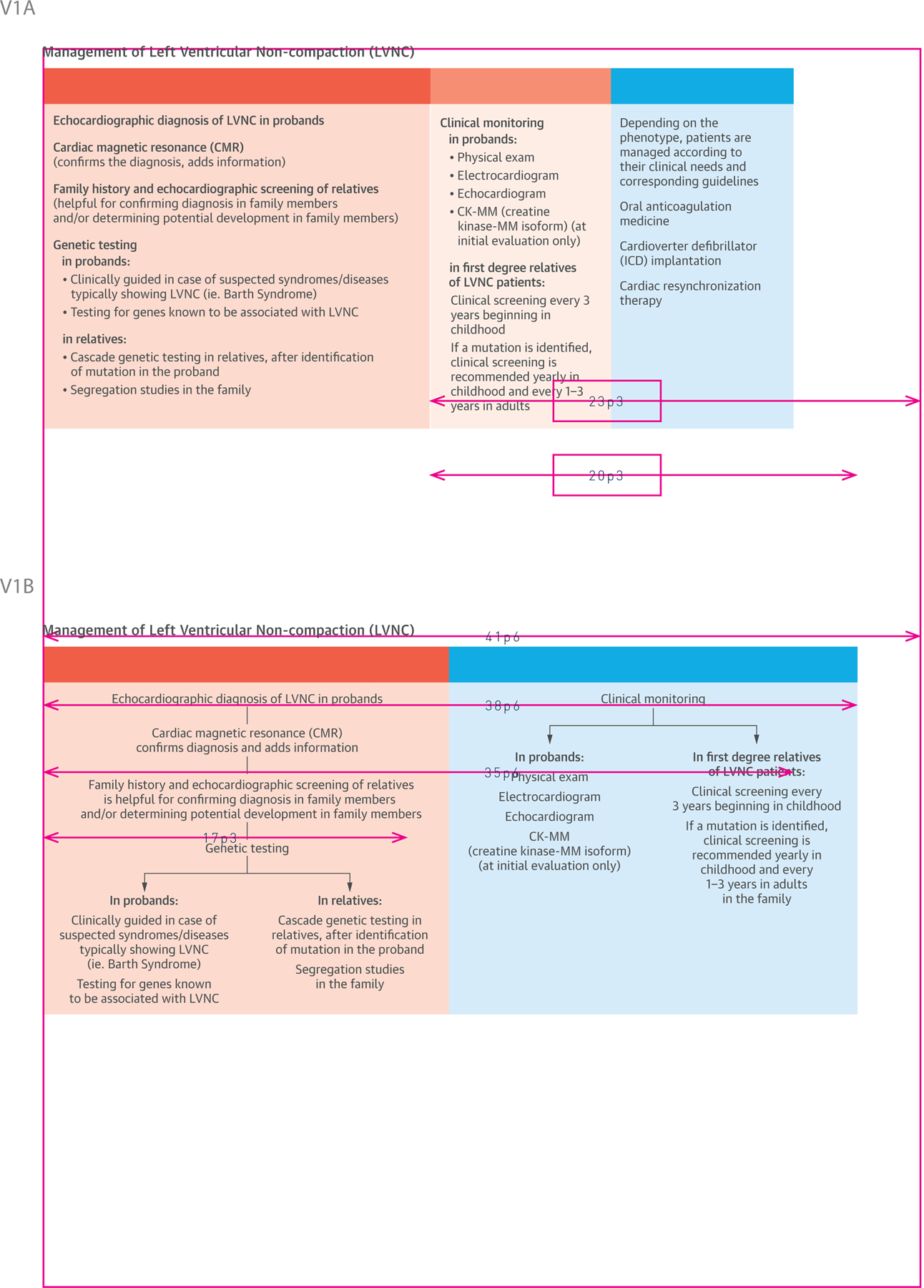
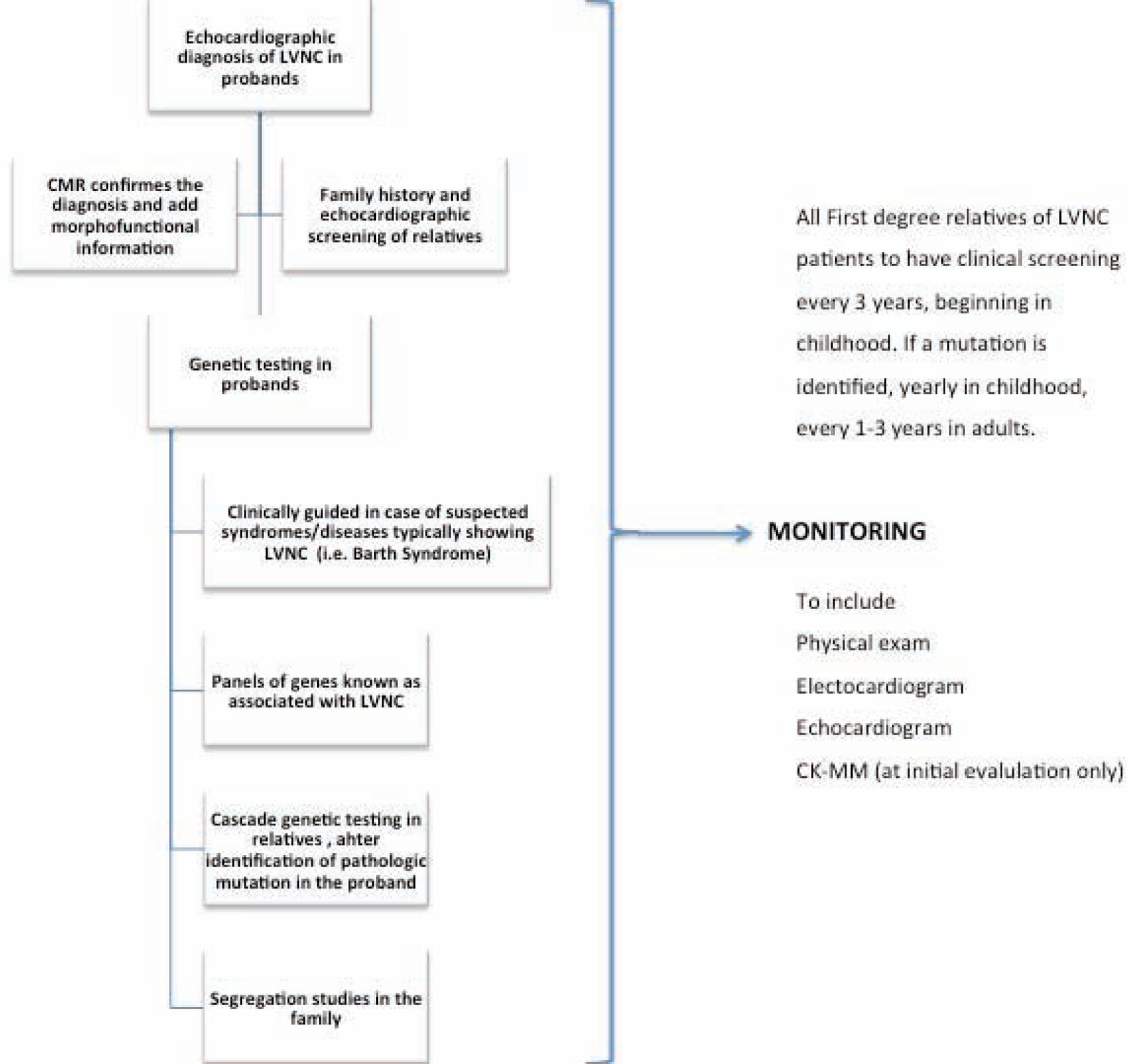
The Central Illustration outlines LVNC management. In the left-hand panel, under diagnosis, we suggest imaging for the initial diagnostic tool in the proband. To confirm diagnosis or determine potential involvement in family members, family history and echocardiographic screening are 2 potential options. In cases of suspected syndromes, such as Barth Syndrome (an X-linked recessive disorder) genetic testing in probands is suggested, given the relatively high fatality rate.. Genetic testing in relatives is an additional option identification options after the mutation in the proband is identified (Central Illustration).
Central Illustration. A Clinical Management Outline for Left Ventricular Noncompaction (LVNC).

Diagnosis and screening strategies for probands and relatives are listed in the left panel, clinical monitoring guides are listed in the middle panel, and treatment options are outlined in the right panel. Data for this table was selected from the Online Mendelian Inheritance in Man URL, established as a collaboration between the Institute of Genetic Medicine, Johns Hopkins Medicine, and the National Human Genome Research Institute.
Clinical monitoring options in probands (middle panel) include physical exam, electrocardiogram, echocardiogram, and creatine kinase MM isoform. For monitoring of first-degree relatives, options include clinical screening every 3 years beginning in childhood, and, if a mutation is identified, annual clinical screening in children and every 1 to 3 years in adults. Treatment and management options are listed in the far right panel and depend entirely on the patient’s phenotype and clinical needs, and the corresponding guidelines (see Management section). Three topics will be touched on: oral anticoagulation medicine, cardioverter defibrillator implantation, and cardiac resynchronization therapy (Central Illustration).
Imaging and diagnosis
Echocardiography.
Standard echocardiography is the first diagnostic tool for LVNC in both index patients and family members. 2D grayscale echocardiography is the most common and useful for LVNC diagnosis, showing both broad trabeculae and deep intertrabecular recesses in the LV myocardium, typically located in the LV apex and the midinferior and lateral walls. In contrast, the basal and midinterventricular septum scanned by an apical 4-chamber view is typically free of trabeculae (Figure 3). In most patients, it is necessary to image the LV, not only with standard defined imaging views, but also with atypical views to image the more apical segments of the LV and detect the prominent trabeculae (Figure 4). Several echocardiographic diagnostic criteria for isolated LVNC are available (48–51), but none can be considered the gold standard for LVNC diagnosis. Furthermore, the criteria are indirect, assessing morphological abnormalities. After careful evaluation of all criteria, the most important echocardiographic criterion remains the ratio of noncompacted/compacted >2.0 in end-systole (49,50). However, when using this ratio to diagnose LVNC, one must keep its quite high interobserver and intraobserver variability in mind as a limitation. Importantly, for LVNC diagnosis, the aforementioned imaging criteria may be considered together with family history and genetics.
Figure 3. Echocardiographic 4-chamber views distinguishing prominent trabeculation (A) vs. hypertrabeculation (B).
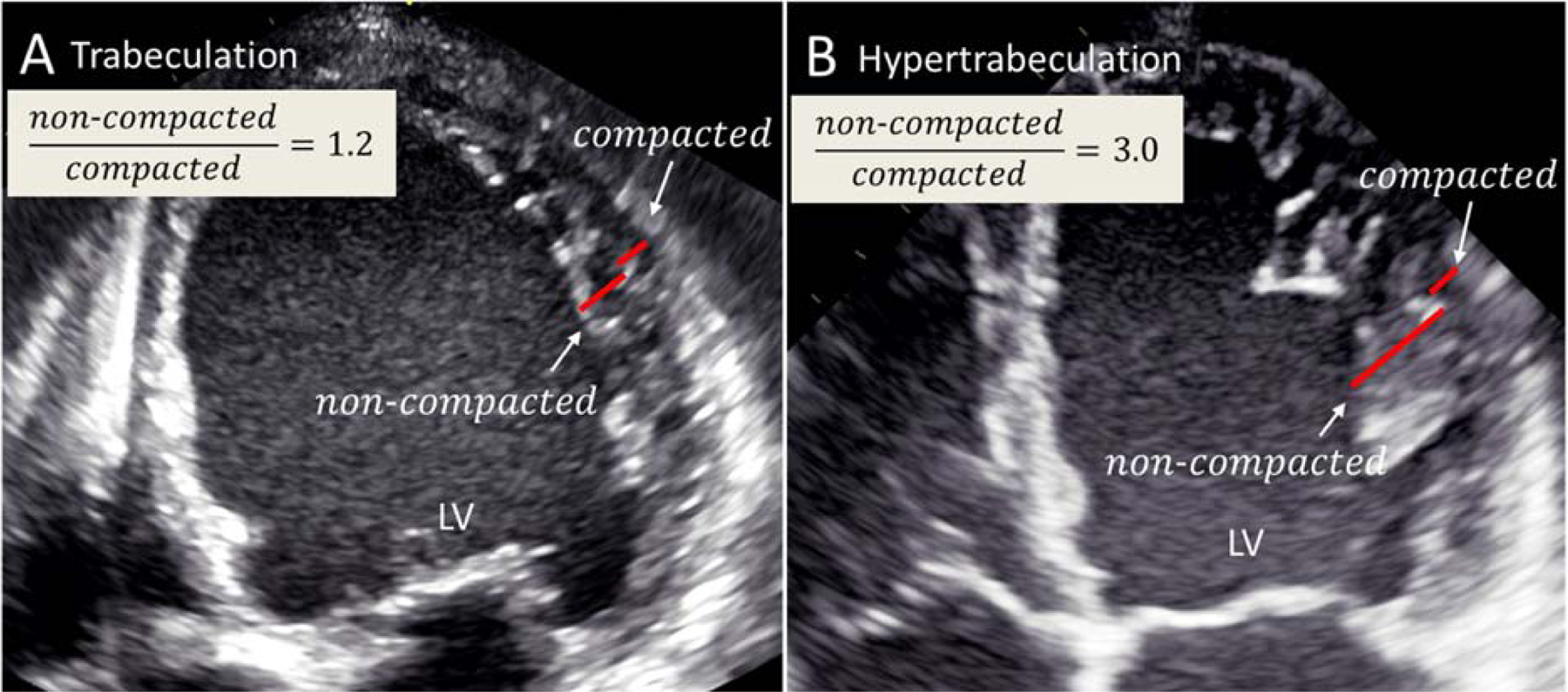
(A) An echocardiographic 4-chamber view from a patient with a dilated cardiomyopathy presenting with prominent trabeculation in the LV apex and lateral wall. In this case the criteria for LVNC are not fulfilled. (B) An echocardiographic 4-chamber view from a patient with a typical LVNC presenting with hypertrabeculation in the LV apex and lateral wall. LV = left ventricle; LVNC left ventricular noncompaction.
Figure 4. A) An echocardiographic image from a patient with LVNC.
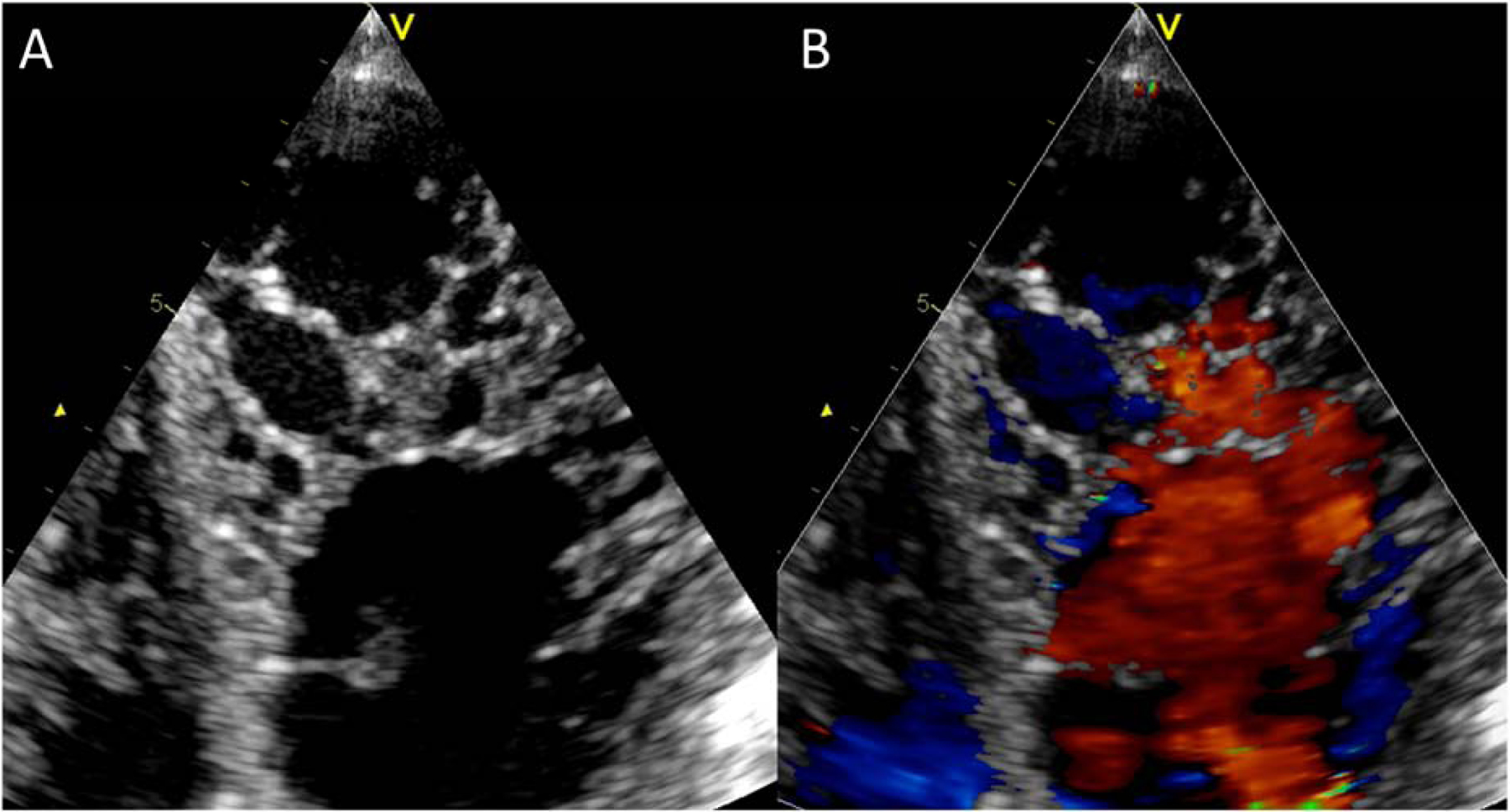
An atypical 4-chamber view was used to better illustrate the non-compaction in the LV apex. B) The same view with color Doppler imaging. This view highlights perfusion of intertrabecular recesses from the left ventricular cavity. Abbreviations as in Figure 3.
In addition to morphological abnormalities, systolic dysfunction is frequently present in LVNC hearts. It was hypothesized that small vessel “dysfunction” with impaired coronary flow reserve and microcirculatory defects, together with a primary myocardial disease, is responsible for the functional abnormalities (52). Thus, in classical LVNC cases, especially in advanced stages, both hypokinetic and akinetic regions can be detected in the diseased segments by wall motion analysis. Recent studies suggest that deformation imaging could better reveal systolic impairment in patients with LVNC, even in those with preserved LV ejection fraction (53,54). In addition, a tissue Doppler-derived strain rate study demonstrated a distinct deformation pattern in LVNC, with significantly higher longitudinal systolic strain rate and strain in the basal segments than in the apex, which could help differentiate LVNC from dilated cardiomyopathy (55). Diastolic dysfunction is another typical echocardiographic feature of LVNC. Thus, most patients (even children) present with abnormal diastolic filling parameters (56). Diastolic dysfunction is attributed in part to abnormal relaxation resulting from extensive trabeculation (57).
Contrast Echocardiography.
In obese patients or patients with lung disease who may have poor acoustic windows, conventional echocardiography has diagnostic limitations. In these cases, the diagnosis is often missed because of imaging quality limitations, especially in the more apical region of the heart. Echocardiographic contrast imaging with various contrast agents enhances endocardial border definition and could improve detection of this rare cardiomyopathy, which could otherwise be misdiagnosed (58,59). Thus, when conventional echocardiographic images are poor or diagnosis is uncertain, contrast echocardiography can be helpful.
Cardiac magnetic resonance (CMR).
CMR may help to accurately describe and diagnose LVNC and distinguish true LVNC from the prominent hypertrabeculation that can be seen in normal hearts and individuals (Figure 5)(60). The major advantage of CMR is that a 3-dimensional data set with equal image quality can be acquired. Thus, potential trabeculae at any region cannot be missed. The major marker is (as for echocardiography) the presence of several, prominent trabeculations in the LV with topographic involvement of apical and mid segments of the lateral and inferior walls. Prior studies were performed in small clinical series (61,62). A noncompacted/compacted ratio >2.3 on CMR is considered the cutoff for LVNC diagnosis (Figure 6)(61). This criterion yielded >43% of positive subjects in the Multi-Ethnic Study of Atherosclerosis (39). Importantly, to avoid misdiagnosis, compact papillary muscle should be distinguished from prominent trabeculations, which is quite easy to do with the 3-dimensional dataset acquired during CMR.
Figure 5. Cardiac magnetic resonance (CMR) from a patient with ischemic heart disease and ejection fraction = 27%.
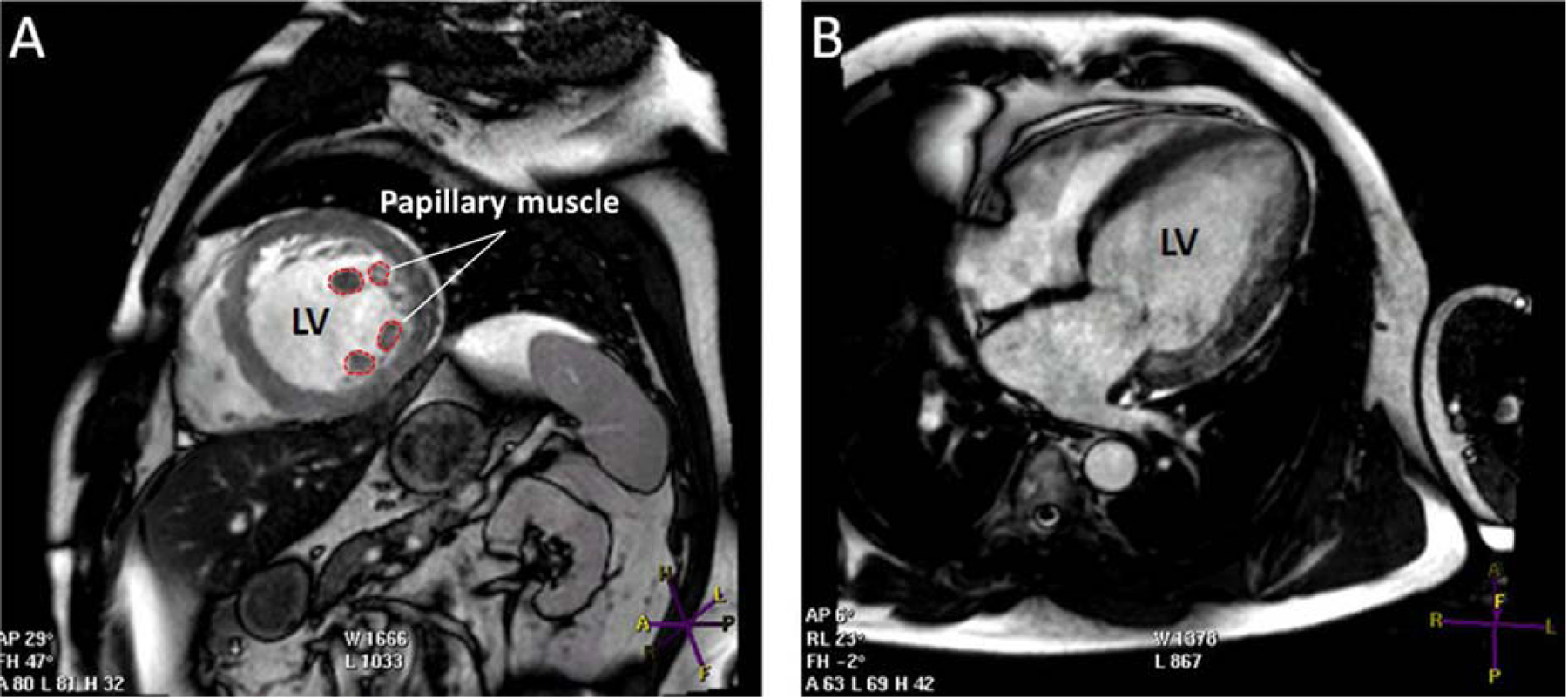
Apart from the ischemic heart disease history, this patient does not meet the CMR criteria for LVNC cardiomyopathy. A) Short axis view showing the papillary muscle with prominent trabeculation in mid left ventricle segments. B) Long axis view showing trabeculation mainly in left ventricular lateral segments. Other abbreviations as in Figure 3.
Figure 6. CMR from a patient with LVNC.
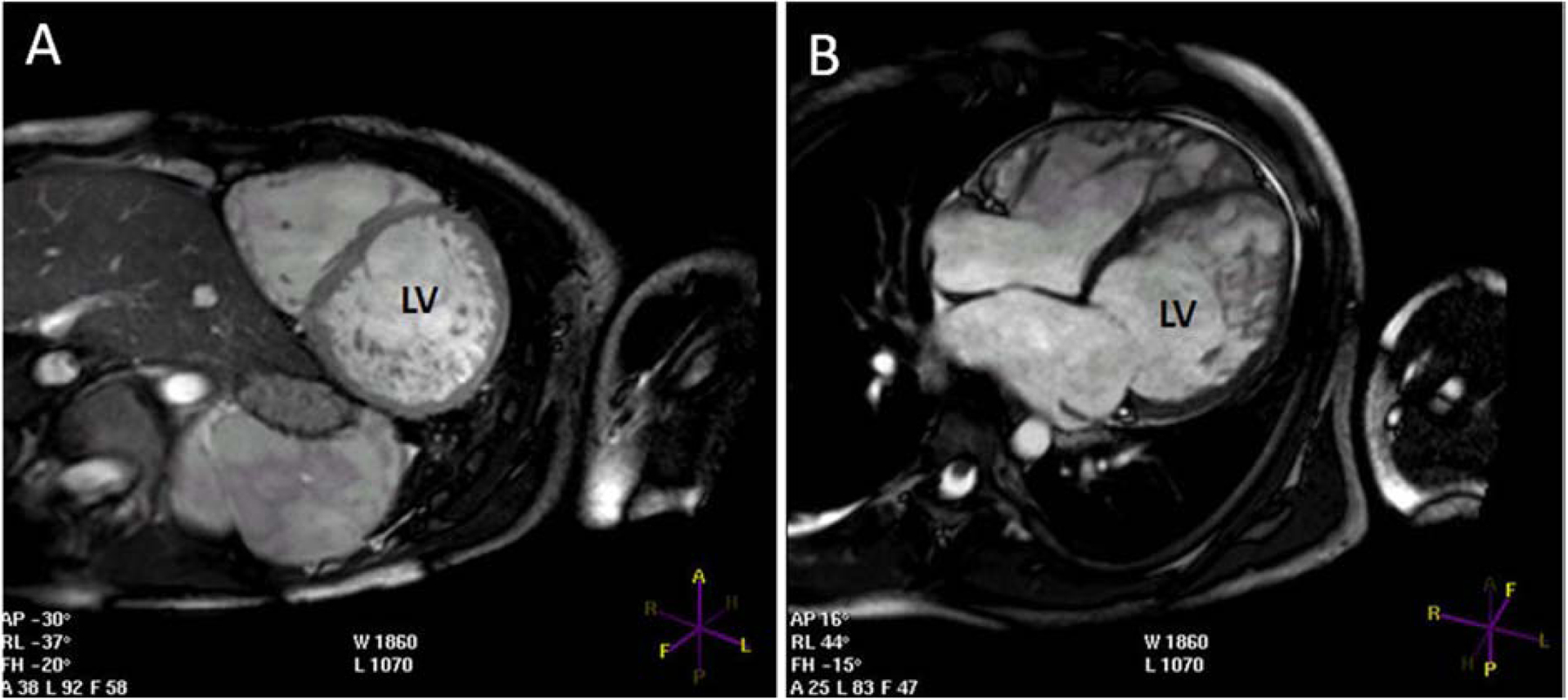
A) Short axis view showing the hypertrabeculation in all mid LV segments apart from the interventricular septum. B) Long axis view showing the hypertrabeculation mainly in the apical and mid LV segments. LV = left ventricular.
Fractal analysis was also used to quantify LV trabeculae (15). In a recent study of 30 patients, the combination of end-diastolic measurements at basal, mid, and apical segments was found to be the best selector of LVNC cases from the normal population (63). When grouping patients according to normal and reduced EF, interpretation of the data was challenged by the unanswered question of whether normal and low EF groups simply represent 2 phases of the same condition diagnosed at different evolutionary stages, or whether they represent the phenotypes of different diseases. The authors concluded that “A gold standard for the diagnosis of LVNC continues to be lacking as no imaging or pathology signature has yet been agreed” (64). While waiting for the ideal definition and diagnostic criteria, a descriptive diagnosis including both LVNC and the LV morphofunctional phenotype (e.g., DCM-like, HCM-like) can be adopted in order to collect data from emerging series.
The typical 2-layered structure of the LV wall can be better measured in CMR, where the thinner, compacted layer can be precisely measured in affected ventricular segments. As in echocardiography, functional data (hypokinesis of the noncompacted segments vs. normal kinesis of unaffected segments) may further strengthen the diagnostic hypothesis. Advanced CMR modalities can provide additional information. For example, high-intensity endocardial T2 signals, subendocardial perfusion defects, and delayed enhancement of the subendocardial layer can add information about function and fibrosis of the affected segments and the possibility of assessing whether abnormalities coincide with noncompacted versus compacted segments (65,66). Advances in imaging are contributing to the ability to distinguish pathologic LVNC from nonpathologic hypertrabeculation. The correct diagnosis may prevent unneeded restrictions for athletes (61). A current gap is the inability to establish the thickness and functionality of the thin, compacted LVNC heart layers. This knowledge may lead to improved clinical management.
Management of LVNC
There are no specific guidelines for management of LVNC. Management includes confirmation of the echocardiographic or CMR diagnosis. Differential diagnoses include prominent hypertrabeculation with normal compacted LV layer, apical hypertrophic cardiomyopathy, dilated cardiomyopathy, endocardial fibroelastosis, and LV apical thrombus.
Clinical management of LVNC depends on the presence or absence of cardiac dysfunction or arrhythmias. Patients with normal LV size and function undergo clinical monitoring, while symptomatic patients with LV dilation and dysfunction or hypertrophy may be clinically managed according to phenotype. Guidelines suggest that familial LVNC should be diagnosed by echocardiographic screening of family members (45). Echocardiographic screening is recommended for family members, given that the symptoms are variable and the risks include heart failure and sudden cardiac death. Genetic testing for LVNC does not change clinical management of the disease; however, it may be helpful for confirming diagnoses in family members and/or determining potential development in family members to aid in the timing of screening (see Central Illustration)(67).
Clinical monitoring may include clinical history, physical examination, echocardiography, Holter monitoring, and measurement of high-sensitivity troponin (Central Illustration).
Currently, there are no specific treatments for LVNC. Depending on the phenotype, patients are managed according to their clinical needs and corresponding guidelines (e.g., for congestive heart failure, arrhythmias). Oral anticoagulation is a debated issue in subjects with normal LV function and absence of LV hypertrophy: patients are either treated on the basis of the phenotype (oral anticoagulation given independently on arrhythmias or LV dysfunction for primary prevention of embolic episodes) or in the presence of LV dysfunction, arrhythmias, prior embolic events, or proven atrial or ventricular thrombi.
Complications with LVNC include heart failure, arrhythmias including sudden cardiac death, and systemic embolic events (16). Atrial tachycardia and fibrillation are common. Ventricular tachyarrhythmias have been reported in up to 47% of symptomatic patients referred to a tertiary referral center, and SCD has been reported in 13% to 18% of (mostly adult) patients with LVNC. Whether the risk of ventricular arrhythmias is higher than that seen in patients with corresponding functional phenotypes (DCM, HCM, and so forth) is not clear. As anticipated (68), LVNC has been considered a reason to restrict athletic participation (33,69,70). However, a 48.6 ± 14.6-month follow-up in athletes fulfilling LVNC criteria did not reveal adverse events (6), thus advising caution before introducing restrictions based on isolated LVNC.
It is unknown whether or not the small compacted layer and the deep recesses of the heart in patients with LVNC increases the risk of complications, such as ventricular perforation in interventional occasions or implantation of devices. This issue is not governed by guidelines, and decisions may be eventually supported by tailored evaluations of families, including evidence of sudden death in affected relatives. In 30 patients with LVNC who underwent implantable cardioverter defibrillator (ICD) implantation for secondary or primary prevention, 11 patients (37%) had appropriate ICD therapies in a mean follow-up period of 40 ± 34 months: 3 with antitachycardia pacing, 4 with ICD shocks, and 4 with both antitachycardia pacing and ICD shocks (69). Although clinical predictors for appropriate ICD therapy are not available, this single study suggests that ICD therapy may be effective in patients with LVNC.
Cardiac resynchronization therapy improves functional NYHA class in patients with LVNC and may hence be considered in patients with a LV ejection fraction ≤35% and signs of ventricular dyssynchrony (71,72). More studies need to be completed to determine the safety and efficacy of the use of ICDs in patients with LVNC.
Summary and Conclusions
In summary, evidence that LVNC is a cardiomyopathy includes the following: 1) specific mutations in genes in the Notch1 pathway in mice and humans leading to dysregulated signaling and hypertrabeculation and noncompaction; and 2) specific mutations in G4.5 in mice and humans disrupting the TAZ protein leading to dysregulated remodeling of cardiolipin and Barth syndrome, characterized by hypertrabeculation and noncompaction in utero and failure to thrive. In contrast, evidence that LVNC is a trait shared by multiple cardiac diseases has not been ruled out. The data presented on mechanical load from pregnancy and athletes is compelling. However, Notch1 signaling is involved in mechanosensation (73–75), suggesting that individuals who develop LVNC may have an underlying mutation in a gene that disrupts Notch signaling or in other endocardially-expressed mechanosensing genes. In these patients, an additional modifier, such as stress or increased load, may be needed for the phenotype to present. Although echocardiography and CMR are useful for LVNC diagnosis, these approaches are indirect and present limitations of interobserver and intraobserver variability. Guidelines for clinical management of LVNC suggest that familial LVNC should be diagnosed by echocardiographic screening of family members. Genetic testing does not change clinical management of the disease, but may be helpful for confirming diagnosis in family members and/or determining potential development in family members to aid in the timing of screening. Anticoagulation is the only medication that can be administered in addition to therapies commonly used in phenotype-based management of cardiomyopathies. We suggest that the AHA, WHO, and the ESC form a working group in the near future, and agree on guidelines to specifically define:
LVNC as primary pathology, which can be isolated or associated with cardiomyopathy. It may be clinically useful to indicate the cardiomyopathy phenotype and the LVNC (HCM-LVNC, RCM-LVNC, DCM-LVNC, ARVC-LVNC) to distinguish I-LVNC with normal LV size and function.
the role of LVNC as a marker for addressing clinical and genetic diagnostic hypotheses.
reproducible and unified imaging-based diagnostic criteria for LVNC.
the risk of thrombosis in patients with I-LVNC, especially when LV size and function are normal.
In parallel, to establish real-world data and outcomes in LVNC patients, we recommend increased collection of LVNC electronic health record data, with imaging data and genetic information, if possible (76).
Acknowledgements:
Dr. Arbustini was supported by the European Union INHERITANCE project #241924 and Italian Ministry of Health “Diagnosis and Treatment of Hypertrophic Cardiomyopathies” (#RF-OSM-2008-1145809) IRCCS Policlinico San Matteo, Pavia. Dr. Hall was supported by the John and Nancy Lindahl Foundation and NHLBI 1R21DK078029-01.
Abbreviations
- ARVC
arrhythmogenic right ventricular cardiomyopathy
- CMR
cardiac magnetic resonance
- DCM
dilated cardiomyopathy
- FAK
focal adhesion kinase
- HCM
hypertrophic cardiomyopathy
- ICD
internal cardioverter defibrillator
- LV
left ventricle/ventricular
- LVNC
left ventricular noncompaction
- MIB1
mindbomb, Drosophila homolog of 1
- TAZ
tafazzin
Footnotes
Disclosures. JLH consults for USF Health. The other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Jenni R, Oechslin EN, van der Loo B. Isolated ventricular non-compaction of the myocardium in adults. Heart 2007;93:11–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maron BJ, Towbin JA, Thiene G, et al. ; American Heart Association; Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; Council on Epidemiology and Prevention. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006;113:1807–16. [DOI] [PubMed] [Google Scholar]
- 3.Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2008;29:270–6. [DOI] [PubMed] [Google Scholar]
- 4.The Fuster V. 3 pathways of translational medicine: an evolution to a call-and-response method. J Am Coll Cardiol 2014;64:223–5. [DOI] [PubMed] [Google Scholar]
- 5.Zaragoza MV, Arbustini E, Narula J. Noncompaction of the left ventricle: primary cardiomyopathy with an elusive genetic etiology. Curr Opinion Pediatrics 2007;19:619–27. [DOI] [PubMed] [Google Scholar]
- 6.Gati S, Chandra N, Bennett RL, et al. Increased left ventricular trabeculation in highly trained athletes: do we need more stringent criteria for the diagnosis of left ventricular non-compaction in athletes? Heart 2013;99:401–8. [DOI] [PubMed] [Google Scholar]
- 7.Gati S, Papadakis M, Van Niekerk N, et al. Increased left ventricular trabeculation in individuals with sickle cell anaemia: physiology or pathology? Int J Cardiol 2013;168:1658–60. [DOI] [PubMed] [Google Scholar]
- 8.Gati S, Papadakis M, Papamichael ND, et al. Reversible de novo left ventricular trabeculations in pregnant women: implications for the diagnosis of left ventricular noncompaction in low-risk populations. Circulation 2014;130:475–83. [DOI] [PubMed] [Google Scholar]
- 9.Pignatelli RH, McMahon CJ, Dreyer WJ, et al. Clinical characterization of left ventricular noncompaction in children: a relatively common form of cardiomyopathy. Circulation 2003;108:2672–8. [DOI] [PubMed] [Google Scholar]
- 10.Oechslin E, Jenni R. Left ventricular non-compaction revisited: a distinct phenotype with genetic heterogeneity? Eur Heart J 2011;32:1446–56. [DOI] [PubMed] [Google Scholar]
- 11.Pantazis AA, Elliott PM. Left ventricular noncompaction. Current Opin Cardiol 2009;24:209–13. [DOI] [PubMed] [Google Scholar]
- 12.Paterick TE, Umland MM, Jan MF, et al. Left ventricular noncompaction: a 25-year odyssey. J Am Soc Echocardiogr 2012;25:363–75. [DOI] [PubMed] [Google Scholar]
- 13.Roberts WC, Karia SJ, Ko JM, et al. Examination of isolated ventricular noncompaction (hypertrabeculation) as a distinct entity in adults. Am J Cardiol 2011;108:747–52. [DOI] [PubMed] [Google Scholar]
- 14.Saleeb SF, Margossian R, Spencer CT, et al. Reproducibility of echocardiographic diagnosis of left ventricular noncompaction. J Am Soc Echocardiogr 2012;25:194–202. [DOI] [PubMed] [Google Scholar]
- 15.Jacquier A, Thuny F, Jop B, et al. Measurement of trabeculated left ventricular mass using cardiac magnetic resonance imaging in the diagnosis of left ventricular non-compaction. Eur Heart J 2010;31:1098–104. [DOI] [PubMed] [Google Scholar]
- 16.Udeoji DU, Philip KJ, Morrissey RP, et al. Left ventricular noncompaction cardiomyopathy: updated review. Ther Adv Cardiovasc Dis 2013;7:260–73. [DOI] [PubMed] [Google Scholar]
- 17.Arunamata A, Punn R, Cuneo B, et al. Echocardiographic diagnosis and prognosis of fetal left ventricular noncompaction. J Am Soc Echocardiogr 2012;25:112–20. [DOI] [PubMed] [Google Scholar]
- 18.Vinograd CA, Srivastava S, Panesar LE. Fetal diagnosis of left-ventricular noncompaction cardiomyopathy in identical twins with discordant congenital heart disease. Pediatr Cardiol 2013;34:1503–7. [DOI] [PubMed] [Google Scholar]
- 19.Liu J, Bressan M, Hassel D, et al. A dual role for ErbB2 signaling in cardiac trabeculation. Development 2010;137:3867–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gupta V, Poss KD. Clonally dominant cardiomyocytes direct heart morphogenesis. Nature 2012;484:479–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Risebro CA, Riley PR. Formation of the ventricles. ScientificWorldJournal 2006;6:1862–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sedmera D, Thompson RP. Myocyte proliferation in the developing heart. Dev Dynam 2011;240:1322–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gassmann M, Casagranda F, Orioli D, et al. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature 1995;378:390–4. [DOI] [PubMed] [Google Scholar]
- 24.Jones FE, Golding JP, Gassmann M. ErbB4 signaling during breast and neural development: novel genetic models reveal unique ErbB4 activities. Cell Cycle 2003;2:555–9. [PubMed] [Google Scholar]
- 25.Kramer R, Bucay N, Kane DJ, et al. Neuregulins with an Ig-like domain are essential for mouse myocardial and neuronal development. Proc Natl Acad Sci U S A 1996;93:4833–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee KF, Simon H, Chen H, et al. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature 1995;378:394–8. [DOI] [PubMed] [Google Scholar]
- 27.Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature 1995;378:386–90. [DOI] [PubMed] [Google Scholar]
- 28.Furuta Y, Ilic D, Kanazawa S, et al. Mesodermal defect in late phase of gastrulation by a targeted mutation of focal adhesion kinase, FAK. Oncogene 1995;11:1989–95. [PubMed] [Google Scholar]
- 29.Pentassuglia L, Sawyer DB. ErbB/integrin signaling interactions in regulation of myocardial cell-cell and cell-matrix interactions. Biochim Biophys Acta 2013;1833:909–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luxan G, Casanova JC, Martinez-Poveda B, et al. Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat Med 2013;19:193–201. [DOI] [PubMed] [Google Scholar]
- 31.Chen H, Zhang W, Sun X, et al. Fkbp1a controls ventricular myocardium trabeculation and compaction by regulating endocardial Notch1 activity. Development 2013;140:1946–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ulusoy RE, Kucukarslan N, Kirilmaz A, et al. Noncompaction of ventricular myocardium involving both ventricles. Eur J Echocardiogr 2006;7:457–60. [DOI] [PubMed] [Google Scholar]
- 33.Oechslin EN, Attenhofer Jost CH, Rojas JR, et al. Long-term follow-up of 34 adults with isolated left ventricular noncompaction: a distinct cardiomyopathy with poor prognosis. J Am Coll Cardiol 2000;36:493–500. [DOI] [PubMed] [Google Scholar]
- 34.Angelini A, Melacini P, Barbero F, et al. Evolutionary persistence of spongy myocardium in humans. Circulation 1999;99:2475. [DOI] [PubMed] [Google Scholar]
- 35.Burke A, Mont E, Kutys R, et al. Left ventricular noncompaction: a pathological study of 14 cases. Hum Pathol 2005;36:403–11. [DOI] [PubMed] [Google Scholar]
- 36.Stahli BE, Gebhard C, Biaggi P, et al. Left ventricular non-compaction: prevalence in congenital heart disease. Int J Cardiol 2013;167:2477–81. [DOI] [PubMed] [Google Scholar]
- 37.Engberding R, Bender F. Identification of a rare congenital anomaly of the myocardium by two-dimensional echocardiography: persistence of isolated myocardial sinusoids. Am J Cardiol 1984;53:1733–4. [DOI] [PubMed] [Google Scholar]
- 38.Bellet S, Gouley B. Congenital heart disease with multiple cardiac anomolies: report of a case showing aortic atresia, fibrous scar in myocardium and embryonal sinusoidal remains. Am J Med Sci 1932;183:458–65. [Google Scholar]
- 39.Kawel N, Nacif M, Arai AE, et al. Trabeculated (noncompacted) and compact myocardium in adults: the multi-ethnic study of atherosclerosis. Circ Cardiovasc Imaging 2012;5:357–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peters F, Dos Santos C, Essop R. Isolated left ventricular non-compaction with normal ejection fraction. Cardiovasc J Afr 2011;22:90–3. [DOI] [PubMed] [Google Scholar]
- 41.Clarke SL, Bowron A, Gonzalez IL, et al. Barth syndrome. Orphanet J Rare Dis 2013;8:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marziliano N, Mannarino S, Nespoli L, et al. Barth syndrome associated with compound hemizygosity and heterozygosity of the TAZ and LDB3 genes. Am J Med Genet A 2007;143A:907–15. [DOI] [PubMed] [Google Scholar]
- 43.Phoon CK, Acehan D, Schlame M, et al. Tafazzin knockdown in mice leads to a developmental cardiomyopathy with early diastolic dysfunction preceding myocardial noncompaction. J Am Heart Assoc 2012;1:jah3-e000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Towbin JA. Left ventricular noncompaction: a new form of heart failure. Heart Fail Clin 2010;6:453–69, viii. [DOI] [PubMed] [Google Scholar]
- 45.Hershberger RE, Lindenfeld J, Mestroni L, et al. ; Heart Failure Society of America. Genetic evaluation of cardiomyopathy--a Heart Failure Society of America practice guideline. J Card Fail 2009;15:83–97. [DOI] [PubMed] [Google Scholar]
- 46.Lu JT, Campeau PM, Lee BH. Genotype-phenotype correlation--promiscuity in the era of next-generation sequencing. New Engl J Med 2014;371:593–6. [DOI] [PubMed] [Google Scholar]
- 47.Captur G, Wilson R, Bennett M, et al. B Embryogenesis of ventricular myocardial trabeculae - novel insights from episcopic 3D imaging and fractal analysis of wild-type and Notch MIB1 noncompaction mouse models. Heart 2014;100(Suppl 3):A125–A128. [Google Scholar]
- 48.Chin TK, Perloff JK, Williams RG, et al. Isolated noncompaction of left ventricular myocardium. A study of eight cases. Circulation 1990;82:507–13. [DOI] [PubMed] [Google Scholar]
- 49.Jenni R, Oechslin E, Schneider J, et al. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart 2001;86:666–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ritter M, Oechslin E, Sutsch G, et al. Isolated noncompaction of the myocardium in adults. Mayo Clin Proc 1997;72:26–31. [DOI] [PubMed] [Google Scholar]
- 51.Stollberger C, Finsterer J, Blazek G. Left ventricular hypertrabeculation/noncompaction and association with additional cardiac abnormalities and neuromuscular disorders. Am J Cardiol 2002;90:899–902. [DOI] [PubMed] [Google Scholar]
- 52.Jenni R, Wyss CA, Oechslin EN, et al. Isolated ventricular noncompaction is associated with coronary microcirculatory dysfunction. J Am Coll Cardiol 2002;39:450–4. [DOI] [PubMed] [Google Scholar]
- 53.Bellavia D, Michelena HI, Martinez M, et al. Speckle myocardial imaging modalities for early detection of myocardial impairment in isolated left ventricular non-compaction. Heart 2010;96:440–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Dalen BM, Caliskan K, Soliman OI, et al. Left ventricular solid body rotation in non-compaction cardiomyopathy: a potential new objective and quantitative functional diagnostic criterion? Eur J Heart Fail 2008;10:1088–93. [DOI] [PubMed] [Google Scholar]
- 55.Niemann M, Liu D, Hu K, et al. Echocardiographic quantification of regional deformation helps to distinguish isolated left ventricular non-compaction from dilated cardiomyopathy. Eur J Heart Fail 2012;14:155–61. [DOI] [PubMed] [Google Scholar]
- 56.McMahon CJ, Pignatelli RH, Nagueh SF, et al. Left ventricular non-compaction cardiomyopathy in children: characterisation of clinical status using tissue Doppler-derived indices of left ventricular diastolic relaxation. Heart 2007;93:676–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Agmon Y, Connolly HM, Olson LJ, Khandheria BK, Seward JB. Noncompaction of the ventricular myocardium. J Am Soc Echocardiogr 1999;12:859–63. [DOI] [PubMed] [Google Scholar]
- 58.Andresen H, Kaag N, Potratz J. Non-compaction of ventricular myocardium and contrast-enhanced echocardiography. Z Kardiol 2005;94:483–5. [DOI] [PubMed] [Google Scholar]
- 59.Chow CM, Lim KD, Wu L, et al. Images in cardiovascular medicine. Isolated left ventricular noncompaction enhanced by echocontrast agent. Circulation 2007;116:e90–1. [DOI] [PubMed] [Google Scholar]
- 60.Peritz DC, Vaughn A, Ciocca M, et al. Hypertrabeculation vs Left Ventricular Noncompaction on Echocardiogram: A Reason to Restrict Athletic Participation? JAMA Intern Med 2014;174:1379–82. [DOI] [PubMed] [Google Scholar]
- 61.Petersen SE, Selvanayagam JB, Wiesmann F, et al. Left ventricular non-compaction: insights from cardiovascular magnetic resonance imaging. J Am Coll Cardiol 2005;46:101–5. [DOI] [PubMed] [Google Scholar]
- 62.Shieh JT, Jefferies JL, Chin AJ. Disorders of left ventricular trabeculation/compaction or right ventricular wall formation. Am J Med Genet C Semin Med Genet 2013;163C:141–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Captur G, Muthurangu V, Cook C, et al. Quantification of left ventricular trabeculae using fractal analysis. J Cardiovasc Magn Reson 2013;15:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dawson DK, McLernon DJ, Raj VJ, et al. Cardiovascular magnetic resonance determinants of left ventricular noncompaction. Am J Cardiol 2014;114:456–62. [DOI] [PubMed] [Google Scholar]
- 65.Fazio G, Novo G, D’Angelo L, et al. Magnetic resonance in isolated noncompaction of the ventricular myocardium. Int J Cardiol 2010;140:367–9. [DOI] [PubMed] [Google Scholar]
- 66.Nucifora G, Aquaro GD, Pingitore A, et al. Myocardial fibrosis in isolated left ventricular non-compaction and its relation to disease severity. Eur J Heart Fail 2011;13:170–6. [DOI] [PubMed] [Google Scholar]
- 67.Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 2011;8:1308–39. [DOI] [PubMed] [Google Scholar]
- 68.Ilyas S, Ganote C, Lajoie D, et al. Sudden death and isolated right ventricular noncompaction cardiomyopathy: report of 2 autopsied adult cases. Am J Forensic Med Pathol 2013;34:225–7. [DOI] [PubMed] [Google Scholar]
- 69.Kobza R, Steffel J, Erne P, et al. Implantable cardioverter-defibrillator and cardiac resynchronization therapy in patients with left ventricular noncompaction. Heart Rhythm 2010;7:1545–9. [DOI] [PubMed] [Google Scholar]
- 70.Thavendiranathan P, Dahiya A, Phelan D, et al. Isolated left ventricular non-compaction controversies in diagnostic criteria, adverse outcomes and management. Heart 2013;99:681–9. [DOI] [PubMed] [Google Scholar]
- 71.Barth PG, Valianpour F, Bowen VM, et al. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): an update. Am J Med Genet A 2004;126A:349–54. [DOI] [PubMed] [Google Scholar]
- 72.Bertini M, Ziacchi M, Biffi M, et al. Effects of cardiac resynchronisation therapy on dilated cardiomyopathy with isolated ventricular non-compaction. Heart 2011;97:295–300. [DOI] [PubMed] [Google Scholar]
- 73.Jiang WR, Cady G, Hossain MM, et al. Mechanoregulation of h2-calponin gene expression and the role of Notch signaling. J Biol Chem 2014;289:1617–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boopathy AV, Che PL, Somasuntharam I, et al. The modulation of cardiac progenitor cell function by hydrogel-dependent Notch1 activation. Biomaterials 2014;35:8103–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Meloty-Kapella L, Shergill B, Kuon J, et al. Notch ligand endocytosis generates mechanical pulling force dependent on dynamin, epsins, and actin. Dev Cell 2012;22:1299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rasmussen LV. The Electronic Health Record for Translational Research. J Cardiovasc Transl Res 2014;7:607–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ichida F, Tsubata S, Bowles KR, et al. Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation 2001;103:1256–63. [DOI] [PubMed] [Google Scholar]
- 78.Arndt AK, Schafer S, Drenckhahn JD, et al. Fine mapping of the 1p36 deletion syndrome identifies mutation of PRDM16 as a cause of cardiomyopathy. Am J Hum Genet 2013;93:67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Luedde M, Ehlermann P, Weichenhan D, et al. Severe familial left ventricular non-compaction cardiomyopathy due to a novel troponin T (TNNT2) mutation. Cardiovasc Res 2010;86:452–60. [DOI] [PubMed] [Google Scholar]
- 80.Vatta M, Mohapatra B, Jimenez S, et al. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol 2003;42:2014–27. [DOI] [PubMed] [Google Scholar]
- 81.Sasse-Klaassen S, Gerull B, Oechslin E, et al. Isolated noncompaction of the left ventricular myocardium in the adult is an autosomal dominant disorder in the majority of patients. Am J Med Genet A 2003;119A:162–7. [DOI] [PubMed] [Google Scholar]
- 82.Probst S, Oechslin E, Schuler P, et al. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circulation Cardiovasc Genet 2011;4:367–74. [DOI] [PubMed] [Google Scholar]
- 83.Klaassen S, Probst S, Oechslin E, et al. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation 2008;117:2893–901. [DOI] [PubMed] [Google Scholar]
- 84.Monserrat L, Hermida-Prieto M, Fernandez X, et al. Mutation in the alpha-cardiac actin gene associated with apical hypertrophic cardiomyopathy, left ventricular non-compaction, and septal defects. Eur Heart J 2007;28:1953–61. [DOI] [PubMed] [Google Scholar]
- 85.Online Mendelian Inheritance in Man, OMIM. 2014. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University; (Baltimore, MD: ). Available at: http://omim.org/. Accessed August 31, 2014. [Google Scholar]