

Abstract

Acridones present numerous pharmacological activities, including inhibition of microtubule affinity-regulating kinase 4 (MARK4) kinase activity. To investigate structure–activity relationships and develop potent MARK4 inhibitors, derivatives of 2-methylacridone were synthesized and tested for their activity against MARK4 kinase. Selective substitutions at the nitrogen atom were accomplished by treating 2-methylacridone with alkyl halides in the presence of K2CO3. In addition, amidation of acridone acetic acid 11 with piperazine or tryptophan methyl ester followed by derivatization with various amines gave a series of new acridone derivatives. Among the tested compounds, six were identified as possessing high inhibitory activity against MARK4. The molecular modeling studies showed that the derivatives bearing piperazine or tryptophan bind well to the ATP-binding site of MARK4. The antiproliferative activity of six active compounds was evaluated against HeLa and U87MG cancer cells. Tryptophan derivatives 23a, 23b, and 23c showed significant cytotoxicity against both cell lines with EC50 values ranging from 2.13 to 4.22 μM, while derivatives bearing piperazine were found to be not cytotoxic. Additionally, compound 23a decreased the proliferation of human MDA-MB-435 and U251 cancer cells in the low micromolar range; however, it also affects the non-cancerous HGF cells. Due to their high binding affinity against MARK4, the synthesized compounds could be potential agents to target MARK4 against cancer and tauopathies.
Keywords: tryptophan derivatives, acridones, microtubule affinity-regulating kinase 4, piperazine derivatives, MARK4 inhibitors, anticancer compounds, MCF-7 cancer cells, HepG2 cancer cells
Protein kinases comprise a large family of proteins widely targeted for pharmacological intervention of various diseases.1,2 Microtubule affinity-regulating kinases belong to the family of AMP-activated protein kinases (MARKs) and consist of four members in mammals (MARK1-4), with high sequence homology and similar structure.3 MARK4 is a Ser/Thr kinase, first identified by its ability to phosphorylate microtubule-associated proteins tau, MAP2, and MAP4 at Ser residues in the microtubule-binding repeat domain, playing a significant role in microtubule dynamics regulation.4,5 Other cellular functions of MARK4 include the regulation of cell polarity, cell cycle progression, signal transduction, and adipogenesis.6−8
All members of the MARK family are thought to be involved in microtubule stabilization; however, MARK4 is the major isoform identified in brain tissue.5,9 MARK4 may be the homologue responsible for the destabilization of microtubules in neuronal cells and the phosphorylation of tau proteins observed in Alzheimer’s disease.10,11 Overexpression or dysregulation of MARK4 is associated with metabolic disorders, including type-II diabetes, diet-induced obesity, and cardiovascular diseases.12,13 Furthermore, MARK4 is overexpressed in gliomas,14 glioblastomas,15 metastatic breast carcinomas, and hepatocellular carcinomas,16 and it is also found to be a negative regulator of mTORC1,17 and inhibit Hippo signaling,18 indicating MARK4 as a potent target for treating cancer. Therefore, a number of MARK4 inhibitors have been found, with the majority of them possessing low selectivity or bioavailability, such as 5RC,19 pyrrolopyrimidinones,20 bis-indolyl pyrroles,21 acridones,22 and PCC0208017 (Figure 1).23
Figure 1.
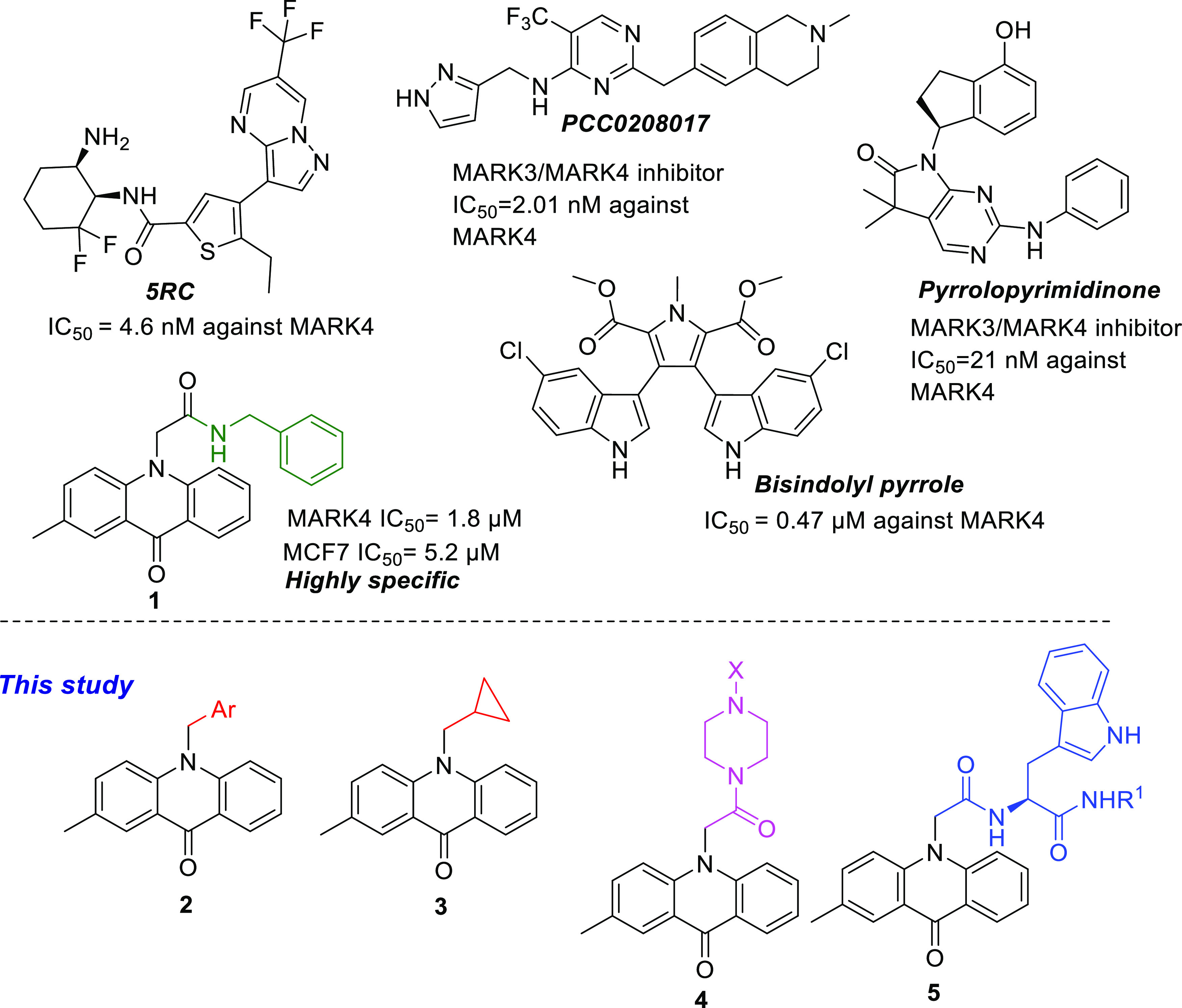
Structures of acridone 1, an overview of its activity, and the general structure of the acridones 2–5 disclosed in the current paper.
Acridones are planar heterocyclic aromatic compounds that have attracted interest due to their various biological activities.24 Acridone-based derivatives have been studied for their antiviral,25 antimicrobial,26 antibacterial,27 anti-inflammatory,28 antimalarial,29 antiparasitic,30 and anticancer activities.31 Acridone alkaloids and synthetic derivatives interact with diverse molecular targets, including topoisomerases II,32 telomerase,33 cathepsins L and V,34 acetylcholinesterase,35 and protein kinases.36,37 Furthermore, N-substituted acridones have been evaluated as anticonvulsant38 and antipsoriatic agents39 and have been found to inhibit p-glycoprotein (p-gp) and modulate p-gp-mediated multidrug resistance (MDR) in cancer cells.40
We have previously reported the synthesis and inhibitory activity of N-substituted acridones and bis-indolyl pyrroles against MARK4.21,22 Several compounds with submicromolar activity against MARK4 could be identified as selective inhibitors (Figure 1). Compound 1 had an IC50 of 1.8 μM against MARK4, exerted anticancer effects, and induced apoptosis against breast cancer cells MCF-7 and human liver cancer cells HepG2. Docking models of 1 in the active site of MARK4 have shown that the benzyl ring extends toward the DFG motif. Therefore, the present study was undertaken to synthesize acridone inhibitors with increased selectivity and potency against MARK4 by introducing extra groups that would engage the P-loop or the DFG motif of the active site of MARK4. Herein, the design, synthesis, and biological evaluation of novel acridone derivatives 2–5 are presented. The synthesized acridones of the general type 4 and 5 were found to bind to MARK4 at its binding site with high affinity. The tryptophan analogues 5 display significant anticancer activity on cervical cancer cells HeLa and glioma cells U87MG, while derivatives bearing piperazine 4 were not cytotoxic. Due to their high binding affinity against MARK4, these compounds could be potential agents to target MARK4 against cancer and tauopathies.
1. Results and Discussion
1.1. Synthesis of Acridone Derivatives
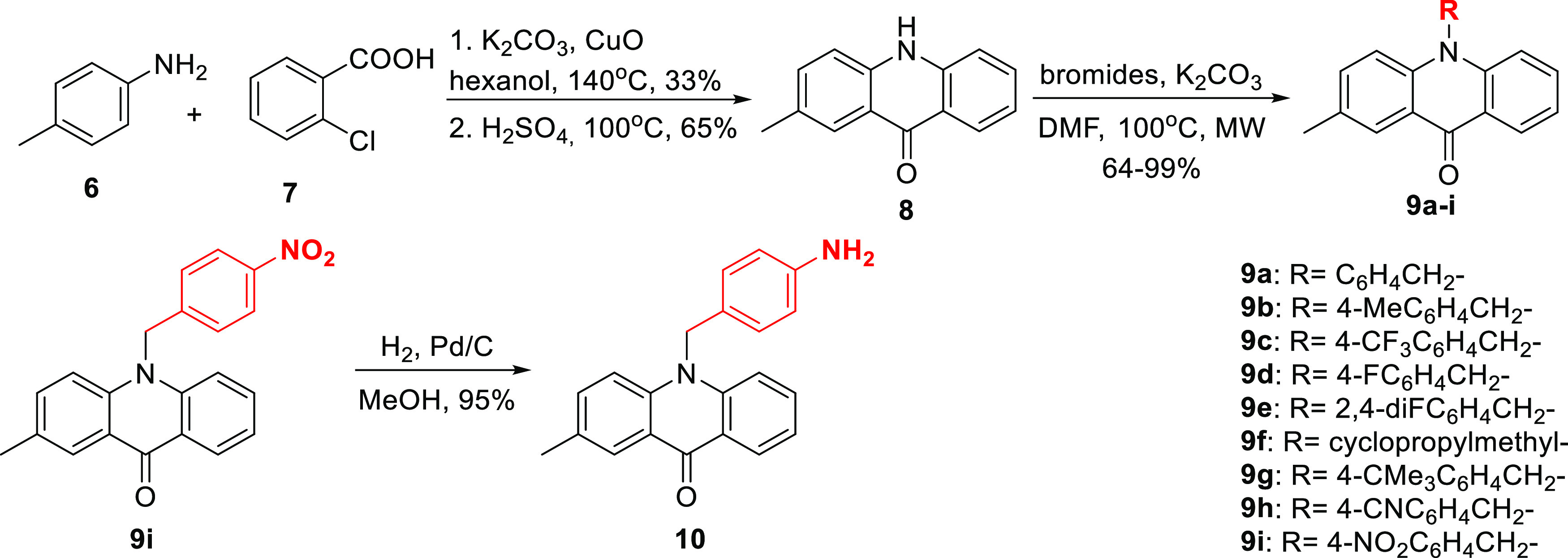
Initially, N-alkylated derivatives of 2-methylacridone were selected for synthesis as inhibitors of MARK4. 2-Methylacridone (8) was prepared according to our previously published methodology (Scheme 1).22 An Ullmann-type condensation of p-toluidine (6) and o-chlorobenzoic acid (7), followed by treatment with H2SO4 gave 2-methylacridin-9(10H)-one (8). Compounds 9a–i were prepared from the N-alkylation of acridone 8 with the appropriate bromides using K2CO3 in dimethylformamide (DMF) at 100 °C in the microwave in good yields ranging from 64 to 99% as demonstrated in Scheme 1. Further catalytic hydrogenation of the nitro compound 9i with Pd/C in methanol afforded amine 10.
Scheme 1. Synthesis of a Series of N-Alkylated Acridones.
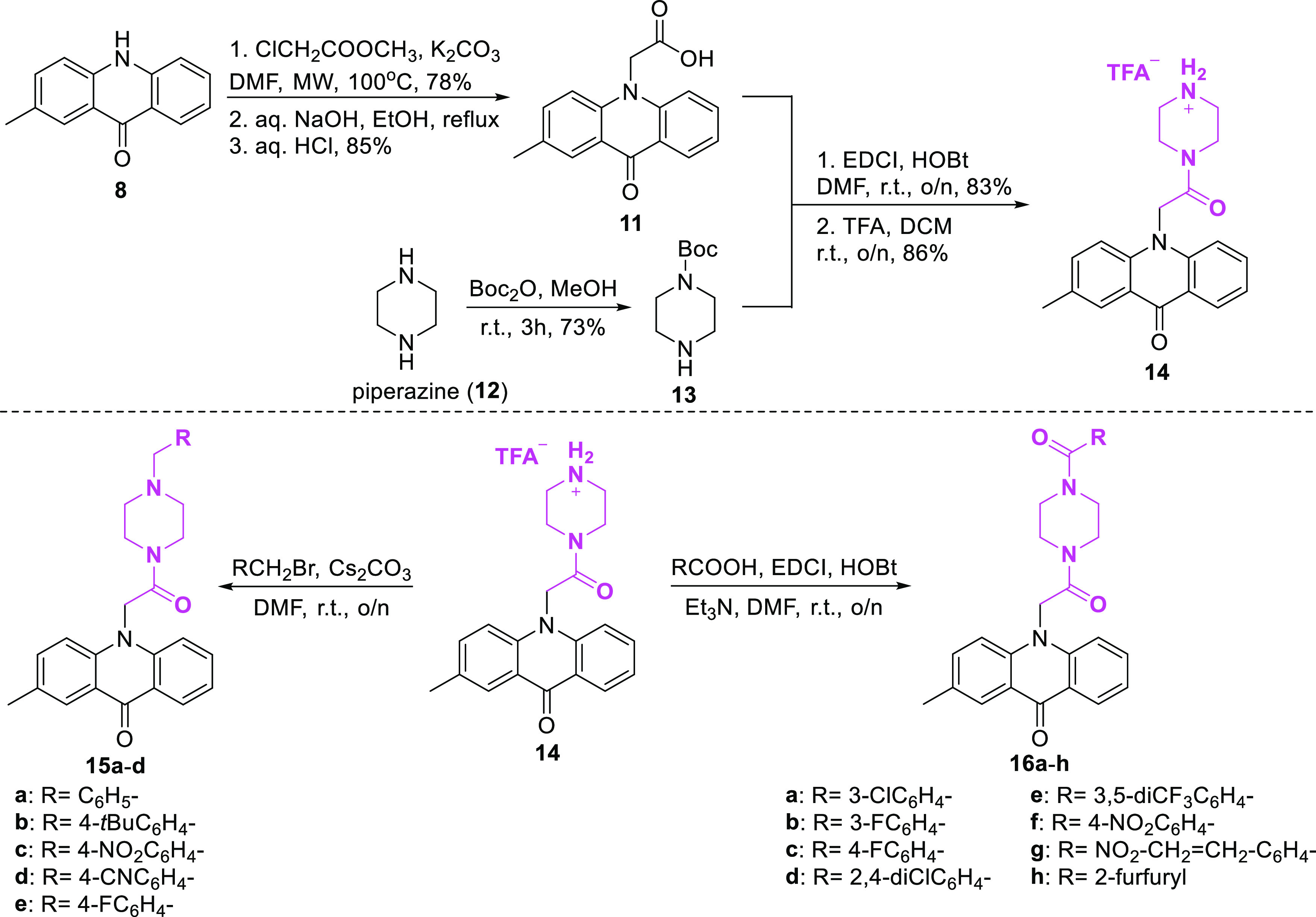
The second category of acridones being selected for synthesis included piperazine on the acridone scaffold. At first, the N-alkylation of 2-methylacridinone with methyl chloroacetate and subsequent ester hydrolysis using NaOH followed by acidification led to the corresponding carboxylic acid 11 (Scheme 2). Mono-boc-protected piperazine 13 was synthesized from the reaction of piperazine with Boc2O in MeOH. Subsequently, the coupling reaction of acridone carboxylic acid 11 and Boc-protected piperazine 13 was accomplished using EDCI and HOBt in DMF, and after deprotection with TFA in DCM, acridone 14 was isolated as a TFA salt. Afterward, alkylation of acridone 14 using benzyl bromides and Cs2CO3 in DMF produced the desired analogues 15a–d in 53–75% yields. While piperazine analogues 16a–h were prepared in moderate to high yields via amidation of 14 with various benzoic acids using EDCI, HOBt, and Et3N in DMF.
Scheme 2. Synthesis of Acridone–Piperazine Derivatives 15a–d and 16a–h.
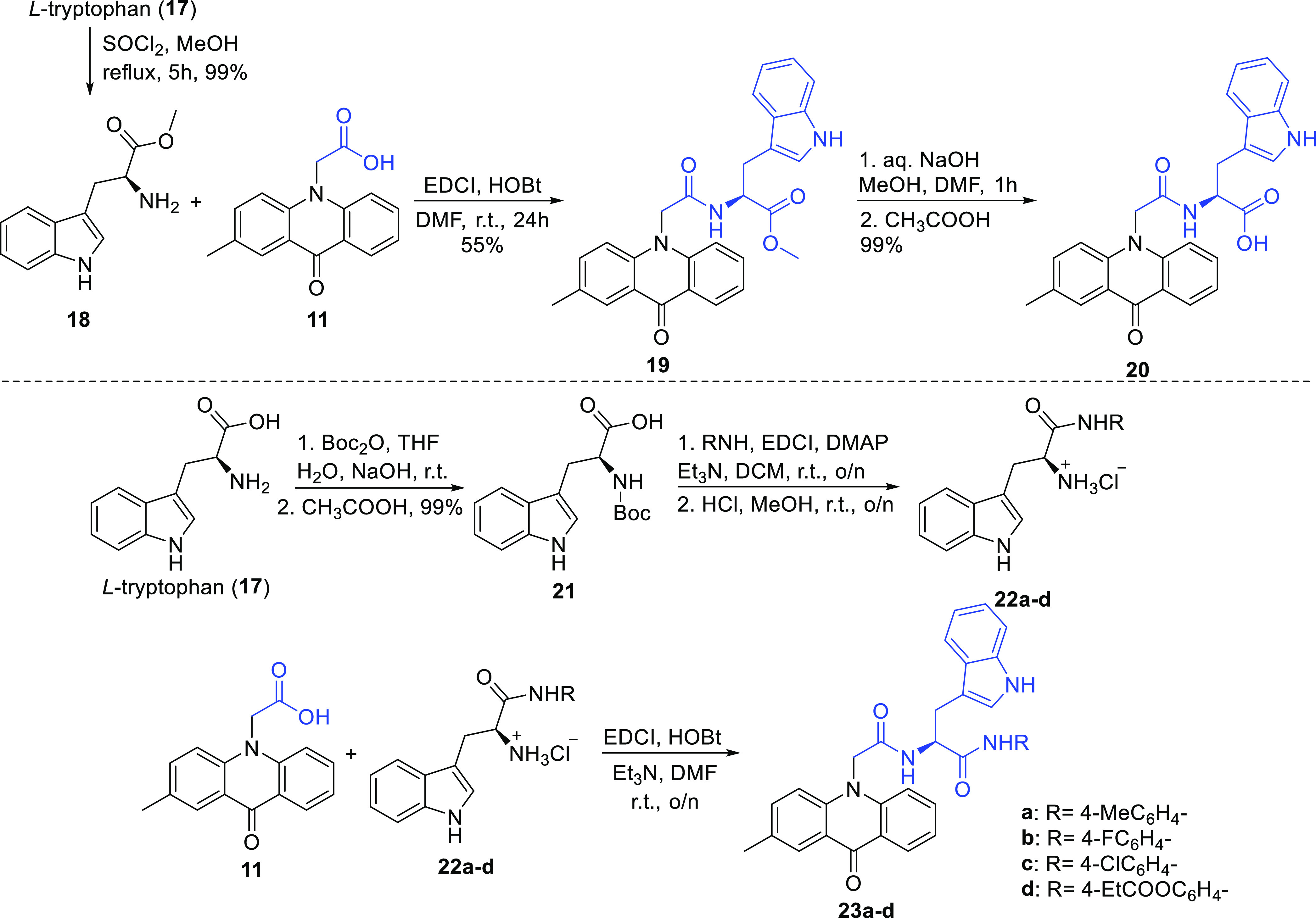
Finally, acridone–tryptophan derivatives were selected to be synthesized as potential MARK4 inhibitors. l-Tryptophan methyl ester 18 was produced via classical esterification from l-tryptophan by reaction with MeOH and thionyl chloride (Scheme 3). The coupling reaction of amine 18 and carboxylic acid 11 was accomplished using EDCI and HOBt in DMF, and compound 19 was isolated in 55% yield. After hydrolysis of the methyl ester 19 and treatment with CH3COOH, the desired compound 20 was furnished. Furthermore, using l-tryptophan as a starting material, compound 21 was synthesized by reaction with Boc2O in the presence of NaOH. Tryptophan hydrochloric salts 22a–d were obtained from Boc-protected-tryptophan 21 by reaction with anilines using EDCI, DMAP, and Et3N and the subsequent deprotection with HCl in MeOH, as illustrated in Scheme 3. Afterward, coupling of acridone carboxylic acid 11 with compounds 22a–d using EDCI, HOBt, and Et3N in DMF afforded the desired derivatives 23a–d in yields ranging from 59 to 69%.
Scheme 3. Synthesis of l-Tryptophan–Acridone Derivatives.
1.2. In Silico Studies
The crystal structure of the catalytic domain of MARK4 with the pyrazolopyrimidine inhibitor 5RC (Figure 1) was retrieved from the protein data bank (PDB ID: 5ES1) (Figure 2A).195RC is located in the ATP-binding site, with the pyrazolopyrimidine group interacting with the hinge region, forming a hydrogen bond with Ala135, while the aminocyclohexane moiety interacts with the catalytic loop (Glu182 and Asn183) and the DFG motif (Asp196-Phe197-Gly198). In our previous work, docking studies of N-substituted acridones were carried out, and the analysis showed that compound 1 occupies the active site with the acridone backbone extending toward the hinge region, interacting with Ala135, while the benzyl group extends toward the DFG motif (Figure 2B).22
Figure 2.

(A) Three-dimensional (3D) model of the interaction between MARK4 and the co-crystallized ligand 5RC (PDB ID: 5ES1) and (B) 3D molecular docking model of the interaction between compound 1 in the active site of MARK4.
In an effort to synthesize acridones with better affinity against MARK4, we aimed to increase the interactions with the residues of the DFG motif and/or the P-loop. A number of acridone derivatives were subjected to docking studies to predict the molecular interactions between them and amino acid residues of MARK4 (PDB ID: 5ES1). Molecular docking experiments were carried out using Autodock Vina software; only the lowest energy conformations were considered for analysis. The data from the experiments are demonstrated in Table S1 and Figures 3–9, S1–S21. All tested compounds were found to occupy the ATP-binding site of MARK4 and showed binding affinity scores ranging from −8.6 to −10.8 kcal/mol (Table S1). Analysis of docking results indicates that the presented compounds were stabilized by various noncovalent interactions, including hydrogen bonds with residues occupying the MARK4 catalytic site. In general, the derivatives bearing piperazine (15a–e, and 16a–h) and tryptophan (23a–d) displayed better binding and had higher docking scores than the N-alkylated acridones (9a–i and 10).
Figure 3.

(A) Two-dimensional (2D) molecular docking model of compound 9a in the active site of MARK4 (PDB code: 5ES1). (B) A 3D model of the interaction between compound 9a in the active site of MARK4 (BIOVIA Discovery Studio).
Figure 9.

(A) 2D molecular docking model of compound 23c and (B) a 3D model of the interaction between compound 23c in the active site of MARK4 (PDB ID: 5ES1). Hydrogen bonds are indicated by green dotted lines.
N-Alkylated acridones 9a–i and 10 complexes with MARK4 were stabilized by various interactions such as hydrogen and halogen bonds, pi-sulfur, and van der Waals interactions with the active site residues (Figures S1–S7). The acridone moiety extends toward the hinge region, and in most cases, a hydrogen bond through the carbonyl group of acridone forms one hydrogen bond with the amino group of Ala135. As shown in Figure 3, the phenyl group of compound 9a forms pi-alkyl interactions with Ala195, which is close to the DFG motif. The introduction of various substituents on the phenyl moiety increases interactions with either the P-loop or the DFG motif. For example, fluoro substitution at the p-position on the phenyl ring of compound 9d gives a hydrogen bond with Gly65 and a halogen bond with Ala68 of the hydrophobic region of the P-loop (Figure 4). On the other hand, amine substitution at the p-position on the phenyl of 10 increases interactions with the DFG motif, and the amine group forms hydrogen bonds with Glu182, Asp196, and Asn183 (Figure 5).
Figure 4.

(A) 2D molecular docking model of compound 9d in the active site of MARK4 (PDB code: 5ES1). (B) A 3D model of the interaction between compound 9d in the active site of MARK4 (BIOVIA Discovery Studio).
Figure 5.

(A) 2D molecular docking model of compound 10 in the active site of MARK4 (PDB code: 5ES1). (B) A 3D model of the interaction between compound 10 in the active site of MARK4 (BIOVIA Discovery Studio).
Complexes of compounds 15a–e and 16a–h with MARK4 share similar poses and were stabilized by various interactions (Figures 6, 7, and S8–S18). More specifically, the acridone backbone extends toward the hinge region, forming a hydrogen bond with Ala135 through the carbonyl group, while the piperazine moiety is oriented toward the phosphate pocket and interacts with Glu182 of the catalytic loop. The substituted benzyl groups of compounds 15a–e interact with amino acids of the DFG motif (Asp196 and Gly198) and extend toward Ile87, Phe67, and Glu103 (Figures S8–S12). The substituted phenyl groups of compounds 16a–h extend toward either Asn66, Phe67, and Leu100 or Glu182, Asp178, and Phe199 interacting with the side chain of Lys85 (phosphate pocket) and the carboxylic acid of Asp196 (DFG motif) (Figures 6, 7, and S13–S18).
Figure 6.

(A) 2D molecular docking model of compound 16a and (B) a 3D model of the interaction between compound 16a in the active site of MARK4 (PDB ID: 5ES1). Hydrogen bonds are indicated by green dotted lines.
Figure 7.

(A) 2D molecular docking model of compound 16b and (B) a 3D model of the interaction between compound 16b in the active site of MARK4 (PDB ID: 5ES1). Hydrogen bonds are indicated by green dotted lines.
Detailed analysis of complexes MARK4-20 and MARK4-23a–d showed that tryptophan analogues share quite similar poses with the acridone backbone, forming a hydrogen bond with Ala135 (hinge region), like the rest of synthesized acridones (Figures 8, 9, and S19–S21). Tryptophan analogues 20 and 23a–d were found to form a second hydrogen bond, more specifically, between the NH group of the indole moiety with the carboxylic acid of Asp196 (DFG motif). The substituted benzene rings extend toward Glu139 and Asp142, forming π-ionic interactions with the carboxylic acid of Glu139 (Figures 8 and 9). These observations indicate that the synthesized acridones possess a high binding affinity for MARK4 and may significantly inhibit its kinase activity.
Figure 8.

(A) 2D molecular docking model of compound 23a and (B) a 3D model of the interaction between compound 23a in the active site of MARK4 (PDB ID: 5ES1). Hydrogen bonds are indicated by green dotted lines.
Furthermore, the physiochemical properties of synthesized acridones were predicted through the SwissADME platform (http://www.swissadme.ch). Molecular weight, MlogP, hydrogen bond donors and acceptors, rotatable bonds, and topological polar surface area were calculated to define Lipinski’s rule of five and Veber’s rules violations, to evaluate their drug-likeness. All tested compounds are well qualified in Lipinski’s rules (0 or 1 violation), possessing molecular weights below 500 Dalton, MlogP values below five, hydrogen bond donors below five, and hydrogen bond acceptors below 10 (Table S2). From the synthesized compounds, only 23d has more than 10 rotational bonds and does not obey Veber’s rules (rotatable bonds ≤ 10 and topological polar surface area ≤ 140 Å2).
1.3. Enzyme Inhibition Assay
All of the synthesized compounds were tested for their ability to inhibit the protein kinase, MARK4. Increasing concentrations of the ligands were used to estimate the IC50 value of each compound. From the synthesized compounds, six showed excellent inhibitory potential against MARK4. MARK4, free of any ligand, was used as a reference representing the completely active enzyme with 100% enzyme activity. Most N-alkylated derivatives of 2-methylacridone were inactive; only 9c and 10 revealed a small inhibitory activity of 13.50 and 12.50 μM, respectively. On the other hand, most of the amide derivatives of piperazine and tryptophan were found to be more potent. The compounds that showed appreciable inhibition were 23a, 23b, 23c, 16c, 16a, and 16b with IC50 values of 0.82, 1.01, 1.49, 2, 0.93, and 0.91 μM, respectively, presented in Tables 12–3. Figure 10 depicts that the loss in protein activity incurred is directly proportional to the concentration of the ligand. The results show the efficacy of the ligands as potent inhibitors of MARK4 protein. Using 15a as a reference point, it can be observed that the addition of a fluorine substituent at a para position on the phenyl ring slightly enhanced the activity giving derivative 15e with an IC50 value of 3.20 μM. Comparing the activity of 15e with the halogenated derivatives 16a, 16c, and 16b, it was observed that the introduction of a second amide group is beneficial for activity. The addition of one fluorine or chloride at the meta position of the phenyl ring affords two compounds 16a and 16b possessing similar activities with IC50 ∼0.91 μM. In contrast, the addition of a second chloride, a nitro, or two CF3 substituents results in reduced activities. Amidation of compound 11 with tryptophan or tryptophan methyl ester yields two inactive derivatives 19 and 20. Interestingly, the formation of a second amide group from 19 with various anilines affords potent MARK4 inhibitors. The most active compound, 23a, has an IC50 value of 0.82 μM and carries a para methyl group on aniline. Compounds 23b and 23c, bearing fluorine and chlorine substituents, respectively, were slightly less potent (IC50 = 1.01 and 1.49 μM) compared to compound 23a.
Table 1. Binding and Enzyme Inhibition Parameters of MARK4 with the N-Alkylated Derivatives of 2-Methylacridones.
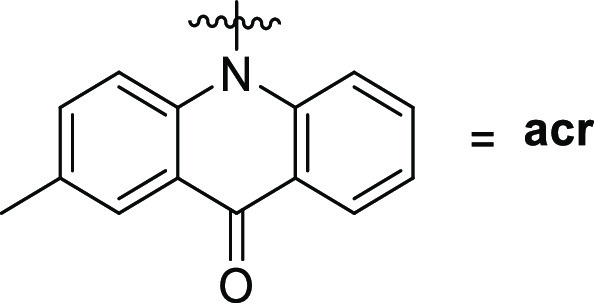
Table 2. Binding and Enzyme Inhibition Parameters of MARK4 with Piperazine Bearing Acridones.
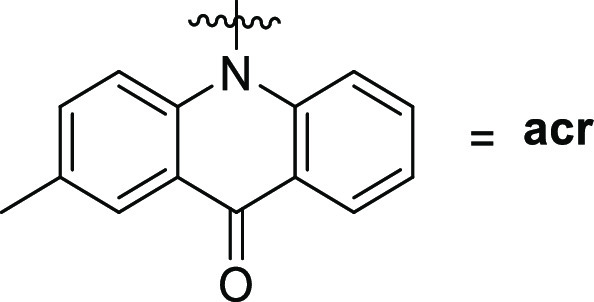
Table 3. Binding and Enzyme Inhibition Parameters of MARK4 with the Tryptophan Compounds.
Figure 10.
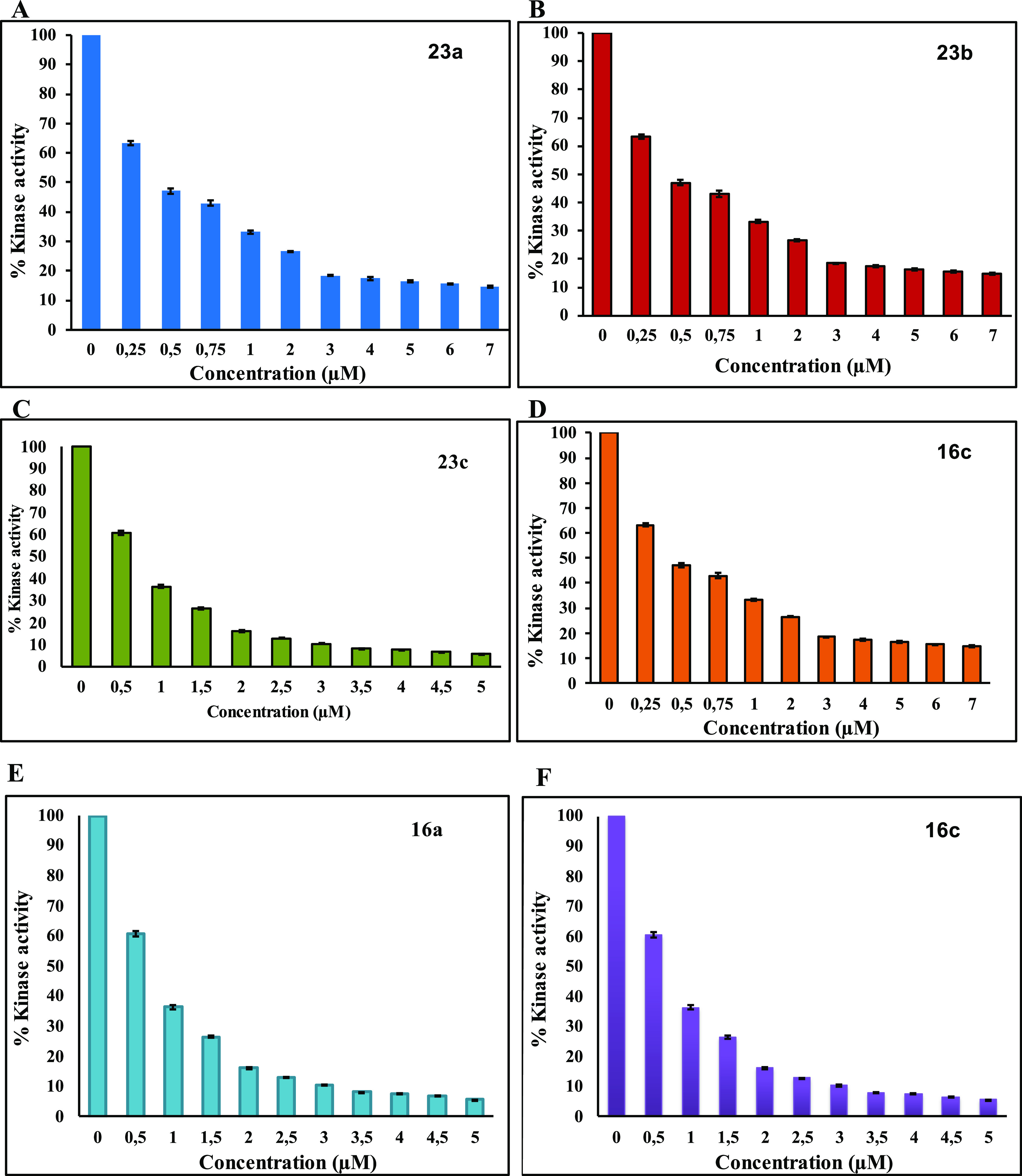
Enzyme inhibition assay of MARK4 with different ligands: (A) 23a, (B) 23b, (C) 23c, (D) 16c, (E) 16a, (F) 16b.
1.4. Fluorescence-Based Binding Assay
The intrinsic fluorescence (IF) of proteins aromatic amino acids (phenylalanine, tyrosine, and tryptophan) has been used extensively to monitor folding and unfolding and changes related to pH, temperature, denaturants, and pressure. Moreover, the binding of protein with an externally provided ligand can also be measured to estimate various binding and thermodynamic parameters.41,42 Tryptophan residue is exquisitely sensitive to any changes in the protein structure.43 IF of the protein is an important tool to investigate the protein–ligand binding affinities.44,45 The quenching observed in fluorescence measurements can result from various reasons such as shuffling of the molecules, interactions amongst them, charge transfer, and formation of complexes.
To validate our enzyme inhibition results, MARK4 was studied for the fluorescence-based binding assay. Increased concentration of ligands was measured with the native protein, and decreased intensity was observed, as shown in Figure 11. The compounds exhibiting appreciable binding interactions with MARK4 showed a remarkable decrease in fluorescence intensity. The quenching in fluorescence intensity was used to find the value of the binding constant (K). The binding constants of the protein–ligand association are presented in Table 4 for each ligand.
Figure 11.
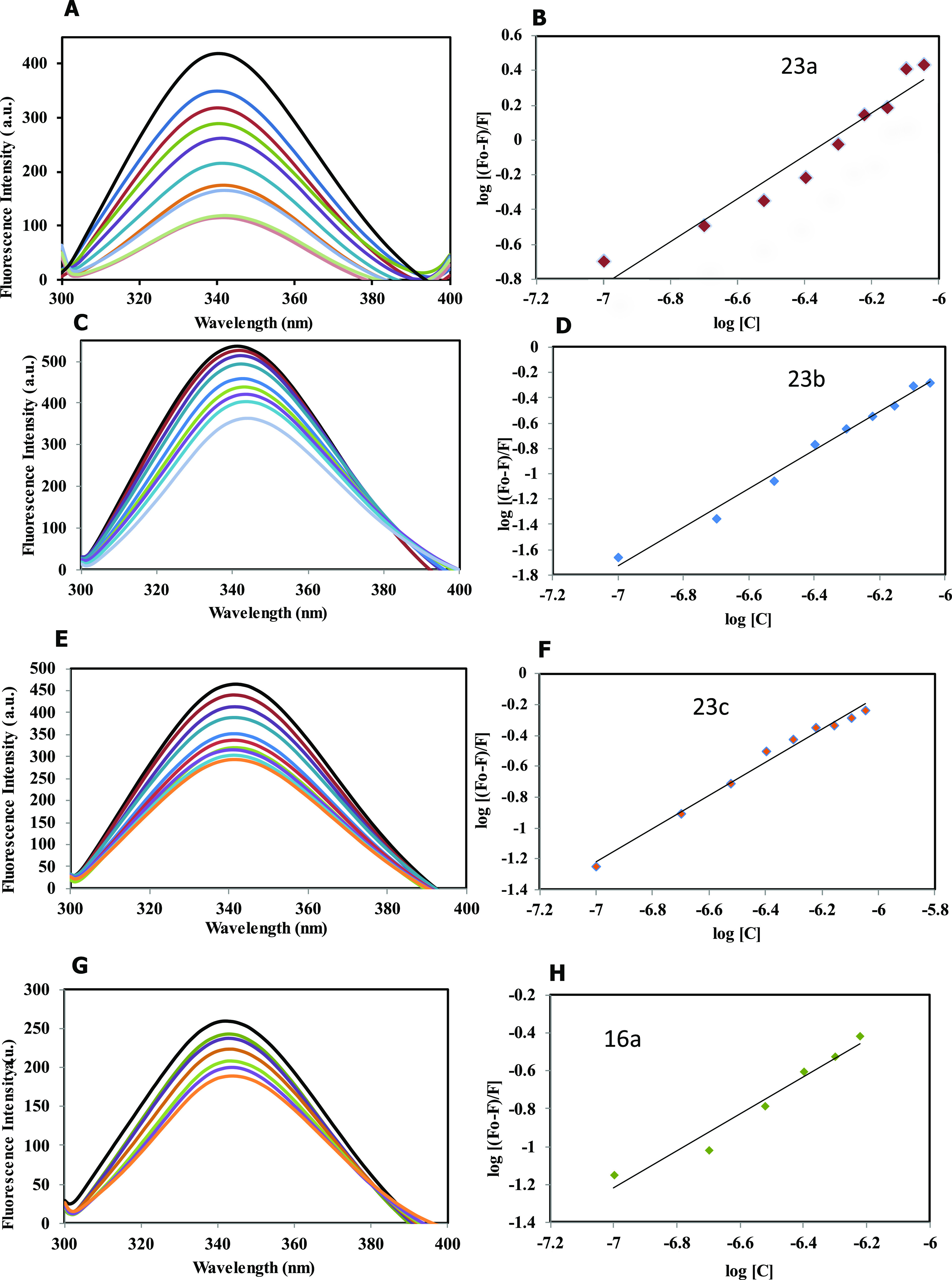
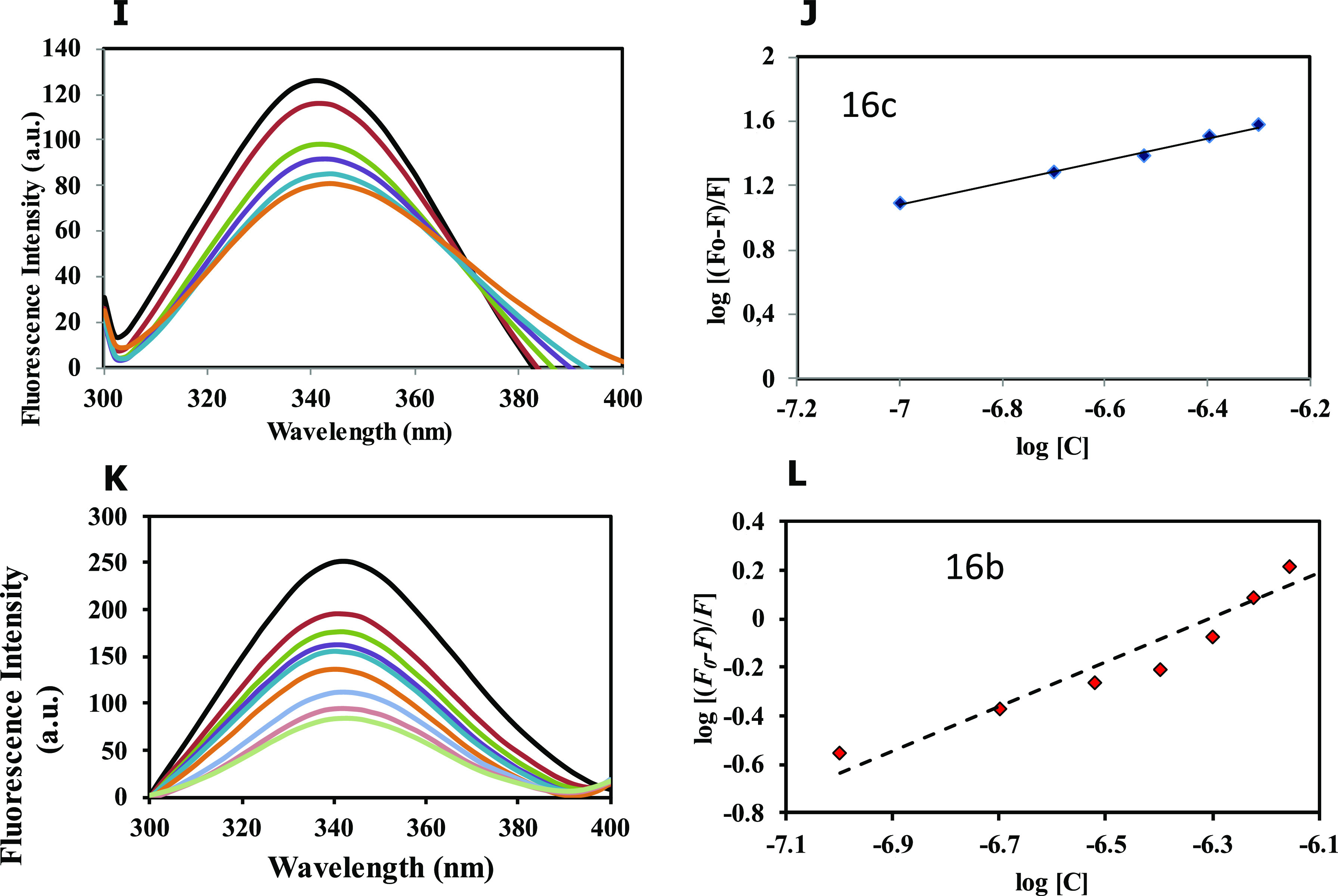
Fluorescence binding studies of MARK4 with the compounds. (A) Fluorescence spectra of MARK4 with increasing concentrations of compound 23a (0.1–0.9 μM). (B) The modified Stern–Volmer (MSV) plot displaying fluorescence quenching of MARK4 by 23a. (C) Fluorescence spectra of MARK4 with increasing concentrations of compound 23b (0.1–0.9 μM). (D) The modified Stern–Volmer (MSV) plot displaying fluorescence quenching of MARK4 by 23b. (E) Fluorescence spectra of MARK4 with increasing concentrations of compound 23c (0.1–0.9 μM). (F) The modified Stern–Volmer (MSV) plot displaying fluorescence quenching of MARK4 by 23c. (G) Fluorescence spectra of MARK4 with increasing concentrations of compound 16a (0.1–0.9 μM). (H) The modified Stern–Volmer (MSV) plot displaying fluorescence quenching of MARK4 by 16a. (I) Fluorescence spectra of MARK4 with increasing concentrations of compound 16c (0.1–0.9 μM). (J) The modified Stern–Volmer (MSV) plot displaying fluorescence quenching of MARK4 by 16c. (K) Fluorescence spectra of MARK4 with increasing concentrations of compound 16b (0.1–0.9 μM). (L) The modified Stern–Volmer (SV) plot displaying fluorescence quenching of MARK4 by 16b.
Table 4. Binding and Enzyme Inhibition Parameters of MARK4 with the Compoundsa.
| compound | €binding constant K, M–1 | *IC50 (μM) |
|---|---|---|
| 23a | 6.3 × 107 | 0.82 |
| 23b | 7.3 ×108 | 1.01 |
| 23c | 1.9 × 106 | 1.49 |
| 16a | 4.6 × 105 | 0.93 |
| 16b | 6.7 × 105 | 0.912 |
| 16c | 7.9 × 105 | 2 |
Values obtained from € Fluorescence and *Enzyme Inhibition Assay.
1.5. Cell Culture and Viability Studies
As overexpression of MARK4 has been associated with the growth and proliferation of cancer cells, the effect of the synthesized acridones on the cell viability of two carcinoma cell lines was determined. Compounds 23a, 23b, 23c, 16a, 16b, and 16c were the more potent inhibitors of MARK4 (IC50 < 2 μM). The cervical cancer cells HeLa, the glioblastoma cells U87MG and U251, the melanoma MDA-MB-435, and the human gingival fibroblasts HGFs were selected to assess the cytotoxic activity of the synthesized compounds by using the MTT assay at a concentration range of 1–20 μM for 24 h (Figures 12–16). U87MG and U251 were chosen based on previous studies, which demonstrated that MARK4 is overexpressed in gliomas.46,47 The tested cell lines express different levels of MARK4. According to The Human Protein Atlas.org, MARK4 kinase has low cancer expression specificity. Specifically, the RNA expression score for cell lines tested was 7.4 (HeLa), 15.3 (U87), 11.7 (MDA-MB-435), 12.1 (U251), and 15 (HGFs).48
Figure 12.

Growth inhibition (sensitivity curves) of HeLa cells generated after treatment with 23a, 23b, 23c, 16c, 16a, and 16b.
Figure 16.

Growth inhibition (sensitivity curves) of HGF cells generated after treatment with 23a and 16a.
Figure 13.

Growth inhibition (sensitivity curves) of U87MG cells generated after treatment with 23a, 23b, 23c, 16c, 16a, and 16b.
Figure 14.

Growth inhibition (sensitivity curves) of MDA-MB-435 cells generated after treatment with 23a and 16a.
Figure 15.

Growth inhibition (sensitivity curves) of U251 cells generated after treatment with 23a and 16a.
The results were expressed as EC50 (the concentration that causes 50% loss of cell viability) and are presented in Table 5. Treatment of Hela and U87MG cells with various concentrations of piperazine analogues 16a, 16b, and 16c for 24 h had a rather limited effect on cell viability (EC50 more than 50 μM). On the contrary, tryptophan derivatives 23a, 23b, and 23c showed significant cytotoxicity against both cell lines. These compounds potently inhibited the growth of Hela and U87MG cells with EC50 values ranging from 2.13 to 4.22 μM. Additionally, 23a was tested against MDA-MB-435, U251, and physiological HGF cells. The results demonstrate that 23a decreased the proliferation of human MDA-MB-435 and U251 cancer cells and exhibited significant antiproliferative properties. The compound acts in a nonselective manner since it as well affects the growth of non-cancerous HGF cells. Of all treated cells, MDA-MB-435 cells were the most sensitive to 23a.
Table 5. Effective Concentrations of MARK4 Inhibitors Causing 50% Loss of Cell Viability (EC50 Values) in HeLa and U87MG Cancer Cell Linesa.
| EC50 (μM) |
|||||
|---|---|---|---|---|---|
| compound | HeLa | U87MG | MDA-MB-435 | U251 | HGFs |
| 23a | 2.13 ± 0.08 | 4.13 ± 0.06 | 1.76 ± 0.06 | 7.98 ± 0.21 | 3.55 ± 0.08 |
| 23b | 2.52 ± 0.07 | 4.22 ± 0.12 | ND | ND | ND |
| 23c | 2.16 ± 0.03 | 3.51 ± 0.09 | ND | ND | ND |
| 16a | >50 | >50 | >50 | >50 | >50 |
| 16b | >50 | >50 | ND | ND | ND |
| 16c | >50 | >50 | ND | ND | ND |
Results are presented as mean ± SE of three independent experiments. ND = Not determined.
2. Conclusions
We have previously reported the synthesis and inhibitory activity of N-substituted acridones against MARK4 kinase. Continuing our optimization program beyond compound 1, we discovered new MARK4 inhibitors with higher potency. Several acridone derivatives were synthesized to bear three types of variations on the acridone core: N-alkylated derivatives of 2-methylacridone and amides bearing a piperazine ring or tryptophan. All of the newly synthesized compounds were screened for their inhibitory activities against MARK4. In accordance with the in silico studies, the N-alkylated derivatives of 2-methylacridone were less active than the other tested compounds. Six derivatives exhibited activities below 2 μM in an enzyme inhibition assay. To validate our enzyme inhibition results, MARK4 was studied using a fluorescence-based binding assay. Derivatives 23a and 23b had binding constants 6.3 × 107 and 7.3 × 108 M–1, respectively. In general, the synthesized molecules are in accordance with the Lipinski and Veber rules. Compound 23a demonstrated high antiproliferative effects against 4 different cancer cell lines in the low micromolar range; however, it also affects the non-cancerous HGF cells. Surprisingly, the tryptophan analogues display significant anticancer activity on HeLa and U87MG cell lines, while derivatives bearing piperazine were not cytotoxic. The role of substituents in the activity of the acridones is obviously crucial. A plausible explanation for this observation is that the tryptophan or piperazine moiety may modulate the permeability of the inhibitors through the cell membranes or exert additional cytotoxicity mechanisms by targeting other proteins that control cancer cell growth. Due to their high binding affinity against MARK4, these compounds could be potential agents to target MARK4 against cancer and tauopathies.
3. Materials and Methods
3.1. Synthesis of Acridone Derivatives
3.1.1. General Experimental Details
Commercial reagents and solvents of high purity were purchased and used without further purification. All reactions were carried out under an atmosphere of argon in dried flasks or vials unless otherwise specified. Reactions were monitored by thin-layer chromatography on silica gel plates and visualized by a UV lamp. Aqueous ceric sulfate/phosphomolybdic acid, ethanolic p-anisaldehyde solution, ninhydrin, and heat were used as developing agents. NMR spectra were recorded on an Agilent 500 spectrometer with 1H at 500 MHz and 13C at 125 MHz, using the tetramethylsilane internal standard at 25 °C, unless otherwise noted. Chemical shifts are reported in δ values (ppm) from internal reference peaks (CDCl31H 7.26, 13C 77.00; DMSO-d61H 2.50, 13C 39.51; CD3COCD31H 2.05, 13C 29.85). Coupling constants (J) are reported in Hz. The LC-MS spectra were recorded on an LC20AD Shimadzu connected to Shimadzu LCMS-2010EV equipped with a C18 analytical column [SUPELCO Discovery C18 (25 cm × 4.6 mm, 5 μm) or Poroshell 120 EC-C18 (4.6 mm × 100 mm, 2.7 μm)]. HRMS experiments were carried out on a Q Exactive Plus Mass Spectrometer; flow rate 0.5Ml/min; 90% ACN + 0.1% HCOOH; spray voltage = 3 kV capillary; temperature = 300 °C. Melting points (mp) are uncorrected. Optical rotation was measured on a Kruss P-3000 polarimeter with a sodium lamp at the solvent, temperature, and concentration indicated for each compound.
Compounds 8 and 11 were prepared according to our previously published procedure.22
3.1.2. General Procedure for the Synthesis of Acridone Derivatives 9a–i, 10
To a suspension of 2-methylacridin-9(10H)-one (150 mg, 0.717 mmol) in dry DMF (6 mL), potassium carbonate (995 mg, 7.20 mmol) and 3.74 mmol of bromides were added, and the mixture was stirred under microwave irradiation at 100 °C for 1.5–4 h. The reaction mixture was extracted with dichloromethane, and the combined organic extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography.
3.1.2.1. 10-Benzyl-2-methylacridin-9(10H)-one, 9a
Reaction time: 1.5 h; 99% yield; mp = 162–164 °C; 1H NMR (500 MHz, CDCl3) δ 8.60 (d, J = 7.9 Hz, 1H), 8.39 (s, 1H), 7.61 (dd, J = 7.5 Hz, 1H), 7.45 (d, J = 8.7 Hz, 1H), 7.37–7.27 (m, 6H), 7.19 (d, J = 7.2 Hz, 2H), 5.59 (s, 2H), 2.46 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 178.2, 142.4, 140.6, 135.6, 135.5, 133.9, 131.3, 129.2, 127.8, 127.7, 127.1, 125.6, 122.5, 122.4, 121.3, 115.1, 115.0, 50.7, 20.6; 354.15; ESI-HRMS m/z for C21H18NO [M + H]+ calcd 300.1388 found 300.1379.
3.1.2.2. 2-Methyl-10-(4-methylbenzyl)acridin-9(10H)-one, 9b
Reaction time: 2.5 h, 80% yield; mp = 209–211 °C; 1H NMR (500 MHz, CDCl3) δ 8.58 (dd, J = 8.0, 1.2 Hz, 1H), 8.37 (s, 1H), 7.62–7.54 (m, 1H), 7.42 (dd, J = 8.7, 1.7 Hz, 1H), 7.32 (d, J = 8.7 Hz, 1H), 7.27–7.23 (m, 2H), 7.14 (d, J = 7.9 Hz, 2H), 7.07 (d, J = 7.9 Hz, 2H), 5.52 (s, 2H), 2.44 (s, 3H), 2.33 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 178.1, 142.3, 140.6, 137.4, 135.4, 133.8, 132.5, 131.1, 129.8, 127.7, 126.9, 125.5, 122.4, 122.3, 121.2, 115.2, 115.0, 50.4, 21.0, 20.6; ESI-HRMS m/z for C22H20NO [M + H]+ calcd 314.1545 found 314.1540.
3.1.2.3. 2-Methyl-10-(4-(trifluoromethyl)benzyl)acridin-9(10H)-one, 9c
Reaction time: 4 h; 64% yield; mp = 217–219 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.38 (dd, J = 8.0, 1.5 Hz, 1H), 8.18 (s, 1H), 7.74 (ddd, J = 8.7, 7.0, 1.6 Hz, 1H), 7.70 (d, J = 8.2 Hz, 2H), 7.62–7.57 (m, 2H), 7.54 (d, J = 8.9 Hz, 1H), 7.37–7.31 (m, 3H), 5.91 (s, 2H), 2.42 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ; 176.5, 141.8, 141.5, 140.1, 135.8, 134.3, 131.0, 127.9, 126.8, 126.0, 125.8, 124.2, 121.6, 121.5 (2C), 116.1, 115.9, 48.6, 20.2 (one carbon is missing due to overlapping); ESI-HRMS m/z for C22H17F3NO [M + H]+ calcd 368.1262; found 368.1254.
3.1.2.4. 10-(4-Fluorobenzyl)-2-methylacridin-9(10H)-one, 9d
Reaction time: 2.5 h; 84% yield; mp = 193–195 °C; 1H NMR (500 MHz, CDCl3) δ 8.55 (d, J = 7.8 Hz, 1H), 8.34 (s, 1H), 7.58 (dd, J = 7.5, 7.6 Hz, 1H), 7.41 (d, J = 8.4 Hz, 1H), 7.29–7.22 (m, 2H), 7.20 (d, J = 8.7 Hz, 1H), 7.16–7.11 (m, 2H), 7.01 (dd, J = 8.7, 8.6 Hz, 2H), 5.51 (s, 2H), 2.42 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 178.0, 162.2, 142.1, 140.4, 135.3, 133.8, 131.3, 131.2, 127.7, 127.3, 127.0, 122.4, 122.3, 121.2, 116.0, 114.9, 114.7, 49.9, 20.6; ESI-HRMS m/z for C21H17FNO [M + H]+ calcd 318.1294; found 318.1289.
3.1.2.5. 10-(2,4-Difluorobenzyl)-2-methylacridin-9(10H)-one, 9e
Reaction time: 2.5 h; 82% yield; mp = 228–230 °C; 1H NMR (500 MHz, CDCl3) δ 8.61 (d, J = 7.9 Hz, 1H), 8.41 (s, 1H), 7.63 (dd, J = 8.2, 7.3 Hz, 1H), 7.49 (d, J = 8.6 Hz, 1H), 7.31 (d, J = 7.9 Hz, 1H), 7.28 (d, J = 8.7 Hz, 1H), 7.21 (d, J = 8.7 Hz, 1H), 7.01–6.96 (dd, J= 10.2, 8.6 Hz, 1H), 6.80–6.74 (dd, J =15.1, 8.1 Hz, 1H), 6.74–6.69 (dd, J =8.4, 8.3 Hz, 1H), 5.58 (s, 2H), 2.48 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 178.0, 162.3, 160.3, 142.1, 140.3, 135.6, 134.0, 131.6, 128.0, 127.9, 127.3, 122.5, 121.6, 118.5, 118.4, 114.6, 114.5, 111.9, 104.4, 44.4, 20.6; ESI-HRMS m/z for C21H16F2NO [M + H]+ calcd 336.1200; found 336.1192.
3.1.2.6. 10-(Cyclopropylmethyl)-2-methylacridin-9(10H)-one, 9f
Reaction time: 3 h; 72% yield; mp = 224–226 °C; 1H NMR (500 MHz, CDCl3) δ 8.56 (dd, J = 8.0, 1.2 Hz, 1H), 8.35 (s, 1H), 7.70–7.65 (m, 1H), 7.55 (d, J = 8.8 Hz, 1H), 7.51 (dd, J = 8.8, 1.8 Hz, 1H), 7.48 (d, J = 8.8 Hz, 1H), 7.24 (d, J = 6.9 Hz, 1H), 4.35 (d, J = 5.7 Hz, 2H), 2.45 (s, 3H), 1.33–1.26 (m, 1H), 0.66–0.61 (m, 2H), 0.05–0.46 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 177.9, 142.1, 140.3, 135.2, 133.5, 130.9, 128.0, 127.2, 122.4, 120.9, 115.2, 115.1, 48.6, 20.6, 9.8, 4.3 (1 carbon is missing due to overlapping); ESI-HRMS m/z for C18H18NO [M + H]+ calcd 264.1388; found 264.1381.
3.1.2.7. 10-(4-(tert-Butyl)benzyl)-2-methylacridin-9(10H)-one, 9g
Reaction time: 2.5 h; 71% yield; mp = 146–148 °C, 1H NMR (500 MHz, CDCl3) δ 8.59 (d, J = 7.7 Hz, 1H), 8.37 (s, 1H), 7.59 (dd, J = 7.5, 7.4 Hz, 1H), 7.52–7.40 (m, 2H), 7.34 (d, J = 8.1 Hz, 3H), 7.29–7.24 (m, 1H), 7.10 (d, J = 7.7 Hz, 2H), 5.53 (s, 2H), 2.44 (s, 3H), 1.29 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 178.1, 150.7, 142.4, 140.6, 135.4, 133.8, 132.4, 131.2, 127.7, 127.0, 126.0, 125.3, 122.4, 122.3, 121.2, 115.3, 115.1, 50.3, 34.5, 31.3, 20.6; ESI-HRMS m/z for C25H26NO [M + H]+ calcd 356.2014; found 356.2003.
3.1.2.8. 4-((2-Methyl-9-oxoacridin-10(9H)-yl)methyl)benzonitrile, 9h
Reaction time: 2.5 h; 75% yield; mp = 298–300 °C; 1H NMR (500 MHz, CDCl3) δ 8.60 (d, J = 7.7 Hz, 1H), 8.39 (s, 1H), 7.65 (m, 2H), 7.62 (d, J = 7.8 Hz, 1H), 7.46 (d, J = 8.6 Hz, 1H), 7.32 (d, J = 7.8 Hz, 2H), 7.30 (d, J = 7.7 Hz, 1H), 7.21 (d, J = 8.6 Hz, 1H), 7.14 (d, J = 8.7 Hz, 1H), 5.63 (s, 2H), 2.47 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 178.0, 142.1, 141.3, 140.2, 135.6, 134.1, 133.1, 131.8, 128.1, 127.4, 126.6, 122.6, 122.5, 121.7, 118.3, 114.5, 114.4, 112.0, 50.3, 20.6; ESI-HRMS m/z for C22H17N2O [M + H]+ calcd 325.1341; found 325.1336.
3.1.2.9. 2-Methyl-10-(4-nitrobenzyl)acridin-9(10H)-one, 9i
Reaction time: 2.5 h; 96% yield; mp = 303–305 °C; 1H NMR (500 MHz, CDCl3) δ 8.61 (d, J = 7.6 Hz, 1H), 8.40 (s, 1H), 8.22 (d, J = 8.3 Hz, 2H), 7.64 (dd, J = 8.1, 7.4 Hz, 1H), 7.47 (d, J = 8.6 Hz, 1H), 7.39 (d, J = 8.3 Hz, 2H), 7.32 (dd, J = 7.6, 7.4 Hz, 1H), 7.22 (d, J = 8.6 Hz, 1H), 7.15 (d, J = 8.8 Hz, 1H), 5.67 (s, 2H), 2.47 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 178.0, 147.7, 143.3, 142.0, 140.2, 138.6, 135.6, 134.1, 131.9, 128.1, 127.5, 126.7, 124.5, 122.6, 121.8, 114.4, 114.3, 50.2, 20.6; ESI-HRMS m/z for C21H17N2O3 [M + H]+ calcd 345.1239; found 345.1245.
3.1.2.10. 10-(4-Aminobenzyl)-2-methylacridin-9(10H)-one, 10
To the solution of 2-methyl-10-(4-nitrobenzyl)acridin-9(10H)-one (19.4 mg, 0.056 mmol) in MeOH (1.35 mL), a catalytic amount of Pd/C (≈10%) was added. A hydrogen-filled balloon was adjusted, the system was degassed, and the reaction was stirred for 1.5 h at room temperature. The solution was filtered through Celite and concentrated under reduced pressure to afford compound 10 in 95% yield (17.8 mg). 10: mp = 289–291 °C; 1H NMR (500 MHz, CDCl3) δ 8.59 (dd, J = 8.0, 1.4 Hz, 1H), 8.38 (s, 1H), 7.61 (ddd, J = 8.6, 7.0, 1.6 Hz, 1H), 7.45 (dd, J = 8.7, 2.0 Hz, 1H), 7.38 (d, J = 8.7 Hz, 1H), 7.31 (d, J = 8.7 Hz, 1H), 7.29–7.25 (m, 1H), 6.97 (d, J = 8.3 Hz, 2H), 6.65 (d, J = 8.3 Hz, 2H), 5.48 (s, 2H), 2.46 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 178.2, 146.0, 142.5, 140.7, 135.4, 133.8, 131.2, 127.7, 127.0, 126.7, 125.1, 122.4, 122.4, 121.2, 115.6, 115.3, 115.2, 50.3, 20.6; ESI-HRMS m/z for C21H19N2O [M + H]+ calcd 315.1497; found 315.1493.
3.1.2.11. tert-Butyl piperazine-1-carboxylate, 13
To the solution of piperazine (500 mg, 5.81 mmol) in abs MeOH (14 mL), a solution of di-tert-butyl dicarbonate (0.61 mL, 2.66 mmol) in abs MeOH (7 mL) was added dropwise during 0.5 h with stirring at 0 °C. The reaction mixture was stirred at room temperature for 3 h, and then H2O was added, followed by extractions with ethyl acetate. The organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography (eluent; DCM/MeOH = 5/1) to afford compound 13 as a white solid in 73% yield (790 mg). The spectral data were in accordance with the literature.4913: 1H NMR (500 MHz, CDCl3) δ 3.55–3.21 (m, 4H), 2.94–2.69 (m, 4H), 1.46 (s, 9H).
3.1.2.12. tert-Butyl 4-(2-(2-Methyl-9-oxoacridin-10(9H)-yl)acetyl)piperazine-1-carboxylate, S1
To the solution of 2-(2-methyl-9-oxoacridin-10(9H)-yl)acetic acid (100 mg, 0.374 mmol) in dry DMF (2.5 mL), tert-butyl piperazine-1-carboxylate (76.6 mg, 0.411 mmol), EDCI (107.5 mg, 0.561 mmol), and HOBt hydrate (86 mg, 0.561 mmol) were successively added. The reaction mixture was stirred at room temperature overnight, and then it was concentrated under reduced pressure. The residue was purified by flash column chromatography (eluent; DCM/MeOH = 18/1) to afford compound S1 as a yellow solid in 83% yield (135 mg). S1: mp = 201–203 °C; 1H NMR (500 MHz, CDCl3) δ 8.46 (d, J = 7.8 Hz, 1H), 8.23 (s, 1H), 7.56 (dd, J = 7.8, 7.5 Hz, 1H), 7.37 (d, J = 8.4 Hz, 1H), 7.20 (dd, J = 7.5, 7.4 Hz, 1H), 7.00 (d, J = 8.6 Hz, 1H), 6.92 (d, J = 8.6 Hz, 1H), 4.91 (s, 2H), 3.68–3.63 (m, 2H), 3.59 (bs, 4H), 3.52–3.47 (m, 2H), 2.38 (s, 3H), 1.51 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 178.1, 164.9, 154.4, 142.2, 140.3, 135.2, 133.7, 131.3, 127.7, 127.0, 122.4, 122.3, 121.3, 114.1, 114.0, 80.7, 48.1, 44.8, 42.1, 28.4, 20.5; ESI-MS m/z for C25H29N3O4Na [M + Na]+ calcd 458.20; found 457.90.
3.1.2.13. 4-(2-(2-Methyl-9-oxoacridin-10(9H)-yl)acetyl)piperazin-1-ium 2,2,2-trifluoroacetate, 14
To the solution of tert-butyl 4-(2-(2-methyl-9-oxoacridin-10(9H)-yl)acetyl)piperazine-1-carboxylate (50 mg, 0.115 mmol) in dry DCM (0.6 mL), TFA (0.6 mL) was added, and the mixture was stirred at room temperature for 3 h. The reaction mixture was concentrated under reduced pressure, and the residue was washed with CHCl3. The product was collected by filtration and washed with petroleum ether. Compound 14 was afforded as a yellow solid in 86% yield (33 mg). 14: mp > 230 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.08 (s, 2H), 8.34 (dd, J = 8.0, 1.5 Hz, 1H), 8.14 (s, 1H), 7.79–7.70 (m, 1H), 7.62–7.57 (m, 2H), 7.53 (d, J = 8.9 Hz, 1H), 7.31 (dd, J = 7.6, 7.3 Hz, 1H), 5.53 (s, 2H), 3.96–3.91 (m, 2H), 3.72–3.67 (m, 2H), 3.40–3.35 (m, 2H), 3.21–3.15 (m, 2H), 2.44 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.6, 165.1, 142.3, 140.6, 135.2, 133.7, 130.6, 126.5, 125.7, 121.4 (2C), 121.1, 116.3, 116.1, 47.3, 42.9, 42.7, 41.6, 38.5, 20.2 ESI-MS m/z for C20H22N3O2 [M + H]+ calcd 336.17; found 335.90.
3.1.3. General Procedure for the Synthesis of Acridone Derivatives 15a–e
The solution of 4-(2-(2-methyl-9-oxoacridin-10(9H)-yl)acetyl)piperazin-1-ium 2,2,2-trifluoroacetate 14 (30 mg, 0.067 mmol) in dry DMF (0.45 mL) was cooled to 0 °C and Cs2CO3 (33 mg, 0.101 mmol) and 0.067 mmol of the corresponding bromide were successively added. The reaction mixture was stirred at room temperature overnight, and then H2O was added, followed by extractions with dichloromethane. The organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography (eluent; gradient PS/EA = 3/1 - EA).
3.1.3.1. 10-(2-(4-Benzylpiperazin-1-yl)-2-oxoethyl)-2-methylacridin-9(10H)-one, 15a
Yellowish solid; 67% yield; mp = 184–186 °C; 1H NMR (500 MHz, DMSO-d6, 40 °C) δ 8.34 (dd, J = 8.0, 1.5 Hz, 1H), 8.14 (s, 1H), 7.77–7.72 (m, 1H), 7.59 (dd, J = 8.8, 2.2 Hz, 1H), 7.49 (d, J = 8.7 Hz, 1H), 7.43 (d, J = 8.7 Hz, 1H), 7.40–7.35 (m, 4H), 7.34–7.26 (m, 2H), 5.45 (s, 2H), 3.75 (brs, 2H), 3.60 (s, 2H), 3.51 (brs, 2H), 2.60 (brs, 2H), 2.43 (s, 5H); 13C NMR (125 MHz, DMSO-d6, 40 °C) δ 176.4, 164.6, 142.2, 140.5, 135.0, 133.5, 130.3, 128.8, 128.1, 126.9, 126.3, 125.5, 121.4, 121.4, 120.8, 115.9, 115.7, 61.7, 52.7, 52.1, 47.1, 44.3, 41.6, 20.1 (one carbon is missing due to overlapping); ESI-HRMS m/z calcd for C27H28N3O2 [M + H]+ = 426.2182; found 426.2171.
3.1.3.2. 10-(2-(4-(4-(tert-Butyl)benzyl)piperazin-1-yl)-2-oxoethyl)-2-methylacridin-9(10H)-one, 15b
Yellowish solid; 75% yield; mp = 198–200 °C; 1H NMR (500 MHz, DMSO-d6, 40 °C) δ 8.34 (dd, J = 7.9, 1.4 Hz, 1H), 8.14 (s, 1H), 7.86–7.69 (m, 1H), 7.59 (dd, J = 8.8, 2.0 Hz, 1H), 7.49 (d, J = 8.8 Hz, 1H), 7.43 (d, J = 8.8 Hz, 1H), 7.38 (d, J = 8.2 Hz, 2H), 7.32–7.27 (m, 3H), 5.45 (s, 2H), 3.74 (brs, 2H), 3.55 (s, 2H), 3.51 (brs, 2H), 2.59 (brs, 2H), 2.43 (s, 3H), 2.41 (brs, 2H), 1.30 (s, 9H); 13C NMR (125 MHz, DMSO-d6, 40 °C) δ 176.4, 164.6, 149.3, 142.2, 140.5, 135.0, 134.6, 133.5, 130.3, 128.6, 126.3, 125.5, 124.8, 121.4, 121.3, 120.8, 115.9, 115.7, 61.4, 52.8, 52.1, 47.1, 44.3, 41.6, 34.1, 31.1, 20.0; ESI-HRMS m/z calcd for C31H36N3O2 [M + H]+ = 482.2808; found 482.2803.
3.1.3.3. 2-Methyl-10-(2-(4-(4-nitrobenzyl)piperazin-1-yl)-2-oxoethyl)acridin-9(10H)-one, 15c
Yellow solid; 53% yield; mp = 208–210 °C; 1H NMR (500 MHz, DMSO-d6, 40 °C) δ 8.34 (dd, J = 8.0, 1.6 Hz, 1H), 8.24 (d, J = 8.7 Hz, 2H), 8.14 (s, 1H), 7.75 (ddd, J = 8.7, 7.0, 1.6 Hz, 1H), 7.67 (d, J = 8.7 Hz, 2H), 7.60 (dd, J = 8.8, 2.1 Hz, 1H), 7.50 (d, J = 8.7 Hz, 1H), 7.44 (d, J = 8.7 Hz, 1H), 7.30 (dd, J = 7.6, 7.3 Hz, 1H), 5.47 (s, 2H), 3.77 (brs, 2H), 3.74 (s, 2H), 3.53 (brs, 2H), 2.63 (brs, 2H), 2.45 (brs, 2H), 2.44 (s, 3H); 13C NMR (125 MHz, DMSO-d6, 40 °C) δ 176.4, 164.7, 146.6, 146.2, 142.3, 140.6, 135.1, 133.6, 130.3, 129.7, 126.3, 125.6, 123.3, 121.4 (2C), 120.9, 115.9, 115.7, 60.7, 52.7, 52.2, 47.2, 44.3, 41.6, 20.1; ESI-HRMS m/z calcd for C27H27N4O4 [M + H]+ = 471.2032; found 471.2025.
3.1.3.4. 4-((4-(2-(2-Methyl-9-oxoacridin-10(9H)-yl)acetyl)piperazin-1-yl)methyl)benzonitrile, 15d
Yellowish solid; 58% yield; mp = 177–179 °C; 1H NMR (500 MHz, DMSO-d6, 40 °C) δ 8.34 (d, J = 7.9 Hz, 1H), 8.14 (s, 1H), 7.83 (d, J = 7.9 Hz, 2H), 7.78–7.70 (m, 1H), 7.61–7.57 (m, 3H), 7.50 (d, J = 8.7 Hz, 1H), 7.43 (d, J = 8.8 Hz, 1H), 7.30 (dd, J = 7.6, 7.2 Hz, 1H), 5.46 (s, 2H), 3.75 (brs, 2H), 3.68 (s, 2H), 3.52 (brs, 2H), 2.61 (brs, 2H), 2.43 (brs, 5H); 13C NMR (125 MHz, DMSO-d6, 40 °C) δ 176.4, 164.7, 144.0, 142.3, 140.5, 135.0, 133.6, 132.1, 130.3, 129.5, 126.3, 125.6, 121.4 (2C), 120.9, 118.7, 115.9, 115.7, 109.8, 61.0, 52.7, 52.1, 47.1, 44.3, 41.6, 20.1; ESI-HRMS m/z calcd for C28H27N4O2 [M + H]+ = 451.2134; found 451.2128.
3.1.3.5. 10-(2-(4-(4-Fluorobenzyl)piperazin-1-yl)-2-oxoethyl)-2-methylacridin-9(10H)-one, 15e
Yellowish solid; 68% yield; mp = 200–202 °C; 1H NMR (500 MHz, DMSO-d6, 40 °C) δ. 34 (dd, J = 8.0, 1.4 Hz, 1H), 8.14 (s, 1H), 7.83–7.68 (m, 1H), 7.59 (dd, J = 8.8, 1.8 Hz, 1H), 7.49 (d, J = 8.7 Hz, 1H), 7.43 (d, J = 8.9 Hz, 1H), 7.40 (dd, J = 8.3, 5.8 Hz, 2H), 7.29 (dd, J = 7.4, 7.5 Hz, 1H), 7.21–7.14 (m, 2H), 5.44 (s, 2H), 3.73 (brs, 2H), 3.57 (s, 2H), 3.51 (brs, 2H), 2.58 (brs, 2H), 2.43 (s, 3H), 2.40 (brs, 2H); 13C NMR (125 MHz, DMSO-d6, 40 °C) δ 176.4, 164.6, 161.2, 142.2, 140.5, 135.0, 133.9, 133.5, 130.6, 130.3, 126.3, 125.5, 121.4, 121.3, 120.8, 115.9, 115.7, 114.8, 60.8, 52.6, 52.0, 47.1, 44.3, 41.6, 20.0; ESI-HRMS m/z calcd for C27H27FN3O2 [M + H]+ = 444.2087; found 443.2087.
3.1.4. General Procedure for the Synthesis of Acridone Derivatives 16a–h
The solution of 4-(2-(2-methyl-9-oxoacridin-10(9H)-yl)acetyl)piperazin-1-ium 2,2,2-trifluoroacetate (30 mg, 0.067 mmol) in dry DMF (0.45 mL) was cooled to 0 °C and Et3N (10 μL, 0.074 mmol), EDCI (15.4 mg, 0.080 mmol), HOBt hydrate (12.3 mg, 0.080 mmol) and 0.067 mmol of the corresponding carboxylic acid were successively added. The reaction mixture was stirred at room temperature overnight, and then H2O was added, followed by extractions with dichloromethane. The organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash column chromatography (eluent; gradient PS/EA = 3/1 - EA).
3.1.4.1. 10-(2-(4-(3-Chlorobenzoyl)piperazin-1-yl)-2-oxoethyl)-2-methylacridin-9(10H)-one, 16a
Yellowish solid; 67% yield; mp > 230 °C; 1H NMR (500 MHz, DMSO-d6, 40 °C) δ 8.32 (d, J = 7.9 Hz, 1H), 8.12 (s, 1H), 7.74 (m, 1H), 7.59 (dd, J = 8.8, 1.5 Hz, 1H), 7.56–7.48 (m, 4H), 7.46 (d, J = 8.8 Hz, 1H), 7.43 (d, J = 7.2 Hz, 1H), 7.29 (dd, J = 7.4 Hz, 1H), 5.49 (s, 2H), 3.92–3.45 (m, 8H), 2.42 (s, 3H); 13C NMR (125 MHz, DMSO-d6, 40 °C) δ 176.4, 167.6, 165.1, 142.2, 140.5, 137.7, 135.1, 133.6, 133.2, 130.4, 130.4, 129.5, 126.7, 126.3, 125.6, 125.5, 121.4 (2C), 120.9, 116.0, 115.8, 47.2, 44.1, 41.6, 20.1; ESI-HRMS m/z calcd for C27H25ClN3O3 [M + H]+ = 474.1584; found 474.1577.
3.1.4.2. 10-(2-(4-(3-Fluorobenzoyl)piperazin-1-yl)-2-oxoethyl)-2-methylacridin-9(10H)-one, 16b
Yellowish solid; 71% yield; mp > 230 °C; 1H NMR (500 MHz, DMSO-d6, 40 °C) δ 8.34 (dd, J = 8.0, 1.3 Hz, 1H), 8.14 (s, 1H), 7.79–7.72 (m, 1H), 7.60 (dd, J = 8.8, 1.9 Hz, 1H), 7.57–7.52 (m, 2H), 7.48 (d, J = 8.8 Hz, 1H), 7.37–7.28 (m, 4H), 5.50 (s, 2H), 3.92–3.40 (m, 8H), 2.44 (s, 3H); 13C NMR (125 MHz, DMSO-d6, 40 °C) δ 176.4, 167.7, 165.1, 161.7, 142.2, 140.5, 137.9, 135.0, 133.6, 130.7, 130.4, 126.3, 125.6, 122.9, 121.4 (2C), 120.9, 116.4, 115.9, 115.8, 113.9, 47.2, 44.1, 41.5, 20.1; ESI-HRMS m/z calcd for C27H25FN3O3 [M + H]+ = 458.1880; found 458.1868.
3.1.4.3. 10-(2-(4-(4-Fluorobenzoyl)piperazin-1-yl)-2-oxoethyl)-2-methylacridin-9(10H)-one, 16c
Yellowish solid; 78% yield; mp > 230 °C; 1H NMR (500 MHz, DMSO-d6, 40 °C) δ 8.34 (dd, J = 8.0, 1.6 Hz, 1H), 8.14 (s, 1H), 7.75 (ddd, J = 8.6, 7.0, 1.7 Hz, 1H), 7.62–7.51 (m, 4H), 7.47 (d, J = 8.8 Hz, 1H), 7.34–7.28 (m, 3H), 5.49 (s, 2H), 3.85–3.55 (m, 8H), 2.43 (s, 3H); 13C NMR (125 MHz, DMSO-d6, 40 °C) δ 176.4, 168.4, 165.1, 162.5, 142.2, 140.5, 135.0, 133.6, 132.0, 130.3, 129.5, 126.3, 125.6, 121.4, 121.4, 120.9, 115.9, 115.8, 115.3, 47.2, 44.1, 41.5, 20.1; ESI-HRMS m/z calcd for C27H25FN3O3 [M + H]+ = 458.1880; found 458.1873.
3.1.4.4. 10-(2-(4-(2,4-Dichlorobenzoyl)piperazin-1-yl)-2-oxoethyl)-2-methylacridin-9(10H)-one, 16d
Yellowish solid; 64% yield; mp > 230 °C; 1H NMR (500 MHz, DMSO-d6, 40 °C) δ 8.34 (dd, J = 8.0, 1.4 Hz, 1H), 8.14 (s, 1H), 7.79–7.73 (m, 2H), 7.61–7.76 (m, 5H), 7.30 (dd, J = 7.5, 7.4 Hz, 1H), 5.54 (s, 1H), 5.45 (s, 1H), 3.92 (s, 1H), 3.87 (s, 1H), 3.73 (s, 2H), 3.63 (s, 1H), 3.51 (s, 1H), 3.41 (s, 1H), 3.24 (s, 1H), 2.44 (s, 3H); 13C NMR (125 MHz, DMSO-d6, 40 °C) δ 176.4, 165.0, 164.8, 142.2, 140.5, 135.0, 134.4, 134.3, 133.6, 130.4, 130.3, 129.3, 129.0, 127.9, 126.3, 125.5, 121.4 (2C), 120.9, 116.0, 115.8, 47.2, 46.10, 45.82, 44.31, 43.85, 41.72, 41.32, 41.10, 40.99, 20.1; ESI-HRMS m/z calcd for C27H24Cl2N3O3 [M + H]+ = 508.1195 found 508.1188.
3.1.4.5. 10-(2-(4-(3,5-Bis(trifluoromethyl)benzoyl)piperazin-1-yl)-2-oxoethyl)-2-methylacridin-9(10H)-one, 16e
Yellowish solid; 55% yield; mp > 230 °C; 1H NMR (500 MHz, DMSO-d6, 40 °C) δ 8.35 (d, J = 7.9 Hz, 1H), 8.28–8.18 (m, 3H), 8.15 (s, 1H), 7.76 (m, 1H), 7.60 (dd, J = 8.8, 1.6 Hz, 1H), 7.54 (d, J = 8.7 Hz, 1H), 7.48 (d, J = 8.8 Hz, 1H), 7.31 (dd, J = 7.5, 7.4 Hz, 1H), 5.52 (s, 2H), 3.94–3.54 (m, 7H), 3.42 (s, 1H), 2.44 (s, 3H); 13C NMR (125 MHz, DMSO-d6, 40 °C) δ 176.4, 166.2, 165.0, 142.3, 140.6, 138.4, 135.0, 133.5, 130.5, 130.4, 127.84, 126.4, 125.6, 123.1, 122.8, 121.4 (2C), 120.9, 115.9, 115.8, 47.2, 43.9, 41.4, 20.1; ESI-HRMS m/z calcd for C29H24F6N3O3 [M + H]+ = 576.1722; found 576.1717.
3.1.4.6. 2-Methyl-10-(2-(4-(4-nitrobenzoyl)piperazin-1-yl)-2-oxoethyl)acridin-9(10H)-one, 16f
Yellow solid; 64% yield; mp = 189–191 °C; 1H NMR (500 MHz, DMSO-d6, 40 °C) δ 8.36–8.31 (m, 2H), 8.14 (s, 1H), 8.79–8.73 (m, 3H), 7.60 (dd, J = 8.8, 1.9 Hz, 1H), 7.54 (d, J = 8.5 Hz, 1H), 7.48 (d, J = 8.7 Hz, 1H), 7.30 (dd, J = 7.4 Hz, 1H), 5.51 (d, J = 18.1 Hz, 2H), 3.95–3.50 (m, 7H), 3.38 (s, 1H), 2.43 (s, 3H); 13C NMR (125 MHz, DMSO-d6, 40 °C) δ 176.4, 167.3, 165.1, 147.9, 142.2, 141.9, 140.5, 135.0, 133.6, 130.4, 128.3, 126.4, 125.6, 123.7, 121.4 (2C), 120.9, 115.9, 115.8, 47.2, 44.1, 41.6, 20.1; ESI-HRMS m/z calcd for C27H25N4O5 [M + H]+ = 485.1825; found 485.1820.
3.1.4.7. (E)-2-Methyl-10-(2-(4-(4-(2-nitrovinyl)benzoyl)piperazin-1-yl)-2-oxoethyl)acridin-9(10H)-one, 16g
Yellow solid; 50% yield; mp > 230 °C; 1H NMR (500 MHz, DMSO-d6, 40 °C) δ 8.34 (dd, J = 8.0, 1.1 Hz, 1H), 8.27 (d, J = 13.6 Hz, 1H), 8.20–8.11 (m, 2H), 7.96 (d, J = 7.7 Hz, 2H), 7.80–7.71 (m, 1H), 7.62–7.56 (m, 3H), 7.54 (d, J = 8.7 Hz, 1H), 7.48 (d, J = 8.8 Hz, 1H), 7.31 (dd, J = 7.5, 7.4 Hz, 1H), 5.50 (s, 2H), 3.98–3.36 (m, 8H), 2.44 (s, 3H); 13C NMR (125 MHz, DMSO-d6, 40 °C) δ 176.4, 168.3, 165.1, 142.2, 140.5, 138.8, 138.1, 135.1, 133.6, 131.4, 130.4, 129.8, 127.6, 126.4, 125.6, 121.4 (2C), 120.9, 115.9, 115.8, 47.3, 44.1, 41.4, 20.1 (one carbon is missing due to overlapping); ESI-HRMS m/z calcd for C29H27N4O5 [M + H]+ = 511.1981; found 511.1980.
3.1.4.8. 10-(2-(4-(Furan-2-carbonyl)piperazin-1-yl)-2-oxoethyl)-2-methylacridin-9(10H)-one, 16h
Yellowish solid; 70% yield; mp = 222–224 °C; 1H NMR (500 MHz, DMSO-d6, 40 °C) δ 8.34 (d, J = 8.0 Hz, 1H), 8.14 (s, 1H), 7.88 (s, 1H), 7.80–7.68 (m, 1H), 7.60 (dd, J = 8.8, 1.7 Hz, 1H), 7.55 (d, J = 8.7 Hz, 1H), 7.50 (d, J = 8.8 Hz, 1H), 7.31 (dd, J = 7.4 Hz, 1H), 7.08 (d, J = 3.4 Hz, 1H), 6.66 (dd, J = 3.0, 1.6 Hz, 1H), 5.50 (s, 2H), 3.96–3.91 (m, 2H), 3.89–3.85 (m, 2H), 3.80–3.75 (m, 2H), 3.64–3.59 (m, 2H), 2.44 (s, 3H); 13C NMR (125 MHz, DMSO-d6, 40 °C) δ 176.5, 165.1, 158.5, 146.8, 144.8, 142.3, 140.6, 135.1, 133.6, 130.4, 126.3, 125.6, 121.4, 121.4, 120.9, 116.0, 115.9, 115.8, 111.3, 47.3, 44.1, 41.5, 20.1; ESI-HRMS m/z calcd for C25H24N3O4 [M + H]+ = 430.1767; found 430.1760.
3.1.4.9. Methyl l-Tryptophanate, 18
The solution of l-tryptophan (50 mg, 0.245 mmol) in dry MeOH (0.7 mL) was cooled to 0 °C. Then, SOCl2 (0.04 mL) was added dropwise at the same temperature, and the reaction mixture was heated at 66 °C for 5 h. The reaction was quenched with Na2CO3 to pH = 8–9, followed by extractions with ethyl acetate. The organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography (eluent; EA) to afford compound 18 as a white solid in quantitative yield (54 mg). The spectral data were in accordance with the literature.5018: 1H NMR (500 MHz, CDCl3) δ 8.07 (brs, 1H), 7.62 (d, J = 7.9 Hz, 1H), 7.37 (d, J = 8.1 Hz, 1H), 7.20 (dd, J = 7.9, 7.2 Hz, 1H), 7.13 (dd, J = 8.1, 7.2 Hz, 1H), 7.08 (brs, 1H), 3.84 (dd, J = 7.3, 5.1 Hz, 1H), 3.71 (s, 3H), 3.29 (dd, J = 14.4, 4.8 Hz, 1H), 3.06 (dd, J = 14.4, 7.7 Hz, 1H), 1.60 (brs, 2H).
3.1.4.10. Methyl (2-(2-Methyl-9-oxoacridin-10(9H)-yl)acetyl)-l-tryptophanate, 19
To the solution of methyl l-tryptophanate (57.6 mg, 0.264 mmol) in dry DMF (1 mL) 2-(2-methyl-9-oxoacridin-10(9H)-yl)acetic acid (47 mg, 0.176 mmol), EDCI (50.6 mg, 0.264 mmol) and HOBt hydrate (40.4 mg, 0.264 mmol) were successively added. The reaction mixture was stirred at room temperature overnight and then concentrated under reduced pressure. The residue was purified by flash column chromatography (eluent; 5% acetone in DCM) to afford compound 19 as a yellow solid in 55% yield (45 mg). 19: mp = 182–184 °C; 1H NMR (500 MHz, DMSO-d6) δ 10.97 (brs, 1H), 8.96 (d, J = 7.3 Hz, 1H), 8.31 (d, J = 7.8 Hz, 1H), 8.10 (s, 1H), 7.70 (m, 1H), 7.54 (d, J = 6.8 Hz, 2H), 7.40 (d, J = 7.7 Hz, 2H), 7.32–7.26 (m, 2H), 7.23 (s, 1H), 7.10 (dd, J = 7.4, 6.9 Hz, 1H), 7.01 (dd, J = 7.2, 6.9 Hz, 1H), 5.16 (d, J = 18.1 Hz, 1H), 5.08 (d, J = 18.1 Hz, 1H), 4.63 (dd, J = 13.0, 8.4 Hz, 1H), 3.63 (s, 3H), 3.24 (dd, J = 14.4, 4.8 Hz, 1H), 3.14 (dd, J = 14.4, 7.7 Hz, 1H), 2.43 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.5, 172.0, 167.3, 142.3, 140.5, 136.2, 135.2, 133.7, 130.6, 127.1, 126.4, 125.6, 123.9, 121.4, 121.4, 121.1, 121.0, 118.5, 118.0, 115.8, 115.7, 111.5, 109.3, 53.2, 52.0, 48.5, 27.1, 20.2; ESI-MS m/z calcd for C28H25N3O4Na [M + Na]+ = 490.17; found 490.05.
3.1.4.11. (2-(2-Methyl-9-oxoacridin-10(9H)-yl)acetyl)-l-tryptophan, 20
To the solution of methyl (2-(2-methyl-9-oxoacridin-10(9H)-yl)acetyl)-l-tryptophanate (20 mg, 0.043 mmol) in a mixed solvent of MeOH (0.5 mL) and DMF (0.5 mL), an aqueous solution of 1 N NaOH (0.5 mL) was added. The reaction mixture was stirred at room temperature for 1 h and then concentrated under reduced pressure. The resulting residue was dissolved in ethyl acetate and acidified with conc. HCl to pH = 1. The mixture was extracted with ethyl acetate, dried over Na2SO4, and concentrated under reduced pressure to obtain compound 20 as a yellow solid in 99% yield (19 mg). 20: mp = 202–204 °C; 1H NMR (500 MHz, DMSO-d6) δ 12.91 (brs, 1H), 10.94 (s, 1H), 8.81 (d, J = 7.9 Hz, 1H), 8.30 (d, J = 7.9 Hz, 1H), 8.09 (s, 1H), 7.71–7.62 (m, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.50 (d, J = 8.4 Hz, 1H), 7.39 (d, J = 7.9 Hz, 1H), 7.31–7.24 (m, 2H), 7.21 (s, 1H), 7.10 (dd, J = 7.6, 7.3 Hz, 1H), 7.00 (dd, J = 7.5, 7.3 Hz, 1H), 5.15 (d, J = 18.0 Hz, 1H), 5.04 (d, J = 18.0 Hz, 1H), 4.57 (dd, J = 13.0, 8.4 Hz, 1H), 3.26 (dd, J = 14.7, 4.4 Hz, 1H), 3.11 (dd, J = 14.3, 8.8 Hz, 1H), 2.42 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.5, 173.1, 167.0, 142.3, 140.5, 136.1, 135.2, 133.7, 130.5, 127.3, 126.4, 125.6, 123.8, 121.4, 121.4, 121.1, 120.9, 118.4, 118.2, 115.9, 115.8, 111.4, 109.8, 53.3, 48.6, 27.3, 20.2; ESI-HRMS m/z calcd for C27H24N3O4 [M + H]+ = 454.1767; found 454.1761.
3.1.4.12. (tert-Butoxycarbonyl)-l-tryptophan, 21
To the solution of l-tryptophan (1 g, 4.90 mmol) in a mixed solvent of THF (45 mL) and H2O (49 mL), NaOH (430 mg, 10.8 mmol) and di-tert-butyl dicarbonate (2.5 mL, 10.8 mmol) were added and the reaction mixture was stirred at room temperature for 36 h. THF was removed under reduced pressure, and the aqueous layer was acidified with CH3COOH to pH = 4. The mixture was extracted with ethyl acetate, dried over Na2SO4, and concentrated under reduced pressure to afford compound 21 as a white solid in quantitative yield (1.49 g). The spectral data were in accordance with the literature.5121: 1H NMR (500 MHz, CDCl3) δ 8.11 (brs, 1H), 7.60 (d, J = 7.9 Hz, 1H), 7.34 (d, J = 7.7 Hz, 1H), 7.20 (dd, J = 7.9, 7.0 Hz, 1H), 7.12 (dd, J = 7.7, 7.0 Hz, 1H), 6.99 (s, 1H), 5.07 (d, J = 7.3 Hz, 1H), 4.67 (dd, J = 13.0, 8.4 Hz, 1H), 3.33 (m, 2H), 1.43 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 176.6, 155.6, 136.1, 127.7, 123.0, 122.2, 119.7, 118.8, 111.2, 109.9, 80.3, 54.2, 28.3, 27.5.
3.1.5. General Procedure for the Synthesis of Tryptophan Derivatives S2–S5
To a solution of (tert-butoxycarbonyl)-l-tryptophan 21 (200 mg, 0.658 mmol) in dry DCM (16 mL), Et3N (0.18 mL, 0.657 mmol), EDCI (252 mg, 1.31 mmol), and DMAP (160.6 mg, 1.31 mmol) were successively added. After the mixture was stirred at 0 °C for 0.5 h, 0.987 mmol of the corresponding amine was added, and the reaction mixture was stirred at room temperature overnight. The reaction was quenched with saturated NH4Cl, and the layers were separated. The organic phase was washed with saturated brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography (eluent; gradient PS/EA = 6/1 - EA).
3.1.5.1. tert-Butyl (S)-(3-(1H-Indol-3-yl)-1-oxo-1-(p-tolylamino)propan-2-yl)carbamate, S2
White solid; 72% yield; 1H NMR (500 MHz, CDCl3) δ 8.24 (s, 1H), 7.73 (brs, 1H), 7.69 (d, J = 7.8 Hz, 1H), 7.35 (d, J = 8.1 Hz, 1H), 7.20 (dd, J = 7.8, 7.3 Hz, 1H), 7.16 (d, J = 8.0 Hz, 2H), 7.12 (dd, J = 7.6, 7.3 Hz, 1H), 7.04 (d, J = 8.0 Hz, 2H), 7.00 (s, 1H), 5.34 (d, J = 4.5 Hz, 1H), 4.62 (d, J = 4.5 Hz, 1H), 3.38–3.32 (m, 1H), 3.28–3.22 (m, 1H), 2.28 (s, 3H), 1.43 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 170.0, 155.8, 136.2, 134.7, 134.1, 129.3, 127.3, 123.3, 122.3, 120.2, 119.8, 118.8, 111.3, 110.5, 80.4, 55.8, 28.4, 28.3, 20.8.
3.1.5.2. tert-Butyl (S)-(1-((4-Fluorophenyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)carbamate, S3
White solid; 75% yield; 1H NMR (500 MHz, CDCl3) δ 8.23 (s, 1H), 8.05 (brs, 1H), 7.65 (d, J = 7.9 Hz, 1H), 7.35 (d, J = 8.1 Hz, 1H), 7.20 (m, 3H), 7.09 (dd, J = 7.9, 7.4 Hz, 1H), 7.00 (s, 1H), 6.89 (dd, J = 8.1, 7.4 Hz, 2H), 5.36 (brs, 1H), 4.63 (brs, 1H), 3.36–3.29 (m, 1H), 3.28–3.22 (m, 1H), 1.41 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 170.2, 159.3, 155.9, 136.2, 133.3, 127.2, 123.3, 122.4, 121.8, 119.9, 118.8, 115.4, 111.4, 110.4, 80.5, 55.7, 28.3, 28.2.
3.1.5.3. tert-Butyl (S)-(1-((4-Chlorophenyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-yl)carbamate, S4
White solid; 69% yield; 1H NMR (500 MHz, CD3COCD3) δ 10.06 (brs, 1H), 9.34 (s, 1H), 7.63 (m, 3H), 7.37 (d, J = 8.1 Hz, 2H), 7.29 (d, J = 8.8 Hz, 1H), 7.20 (s, 1H), 7.09 (dd, J = 7.6, 7.3 Hz, 1H), 7.00 (dd, J = 8.8, 7.3 Hz, 1H), 6.10 (d, J = 5.7 Hz, 1H), 4.58–4.51 (m, 1H), 3.34 (dd, J = 14.6, 6.2 Hz, 1H), 3.22 (dd, J = 14.2, 6.9 Hz, 1H), 1.37 (s, 9H); 13C NMR (125 MHz, CD3COCD3) δ 171.7, 156.3, 138.7, 137.6, 129.4, 128.7, 124.5, 122.2, 122.0, 121.9, 119.6, 119.4, 112.2, 111.1, 79.5, 57.0, 28.9, 28.5.
3.1.5.4. Ethyl (S)-4-(2-((tert-Butoxycarbonyl)amino)-3-(1H-indol-3-yl)propanamido)benzoate, S5
White solid; 71% yield; 1H NMR (500 MHz, CDCl3) δ 8.19 (s, 1H), 8.14 (s, 1H), 7.91 (d, J = 8.5 Hz, 2H), 7.66 (d, J = 7.8 Hz, 1H), 7.35 (d, J = 8.5 Hz, 3H), 7.20 (dd, J = 7.8, 7.4 Hz, 1H), 7.10 (dd, J = 7.5, 7.4 Hz, 1H), 7.04 (s, 1H), 5.30 (d, J = 5.8 Hz, 1H), 4.63 (s, 1H), 4.34 (q, J = 7.1 Hz, 2H), 3.39–3.23 (m, 2H), 1.42 (s, 9H), 1.37 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 170.4, 166.1, 155.9, 141.4, 136.2, 130.6, 127.2, 126.0, 123.3, 122.5, 119.9, 118.9, 118.7, 111.3, 110.3, 80.7, 60.8, 55.9, 28.3, 28.1, 14.3.
3.1.6. General Procedure for the Synthesis of Tryptophan Derivatives 22a–d
To a solution of Boc-protected-tryptophans S2–S5 (0.40 mmol) in MeOH (3 mL), conc. HCl (0.25 mL) was added, and the mixture was stirred at room temperature overnight. The reaction mixture was concentrated under reduced pressure, and the residue was washed with CHCl3. The product was collected by filtration and washed with petroleum ether.
3.1.6.1. (S)-3-(1H-Indol-3-yl)-1-oxo-1-(p-tolylamino)propan-2-aminium Chloride, 22a
White solid; 99% yield; mp = 186–188 °C; [α]D28 = +130.4 (c 0.5, MeOH); 1H NMR (500 MHz, DMSO-d6) δ 11.07 (s, 1H), 10.89 (s, 1H), 8.42 (brs, 3H), 7.72 (d, J = 7.9 Hz, 1H), 7.49 (d, J = 8.3 Hz, 2H), 7.35 (d, J = 8.1 Hz, 1H), 7.27 (d, J = 2.3 Hz, 1H), 7.12 (d, J = 8.3 Hz, 2H), 7.08–7.04 (m, 1H), 6.97–6.92 (m, 1H), 4.26 (brs, 1H), 3.38–3.33 (m, 1H), 3.30 (dd, J = 14.6, 6.9 Hz, 1H), 2.26 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 167.0, 136.2, 135.7, 132.9, 129.1, 127.1, 124.8, 121.1, 119.6, 118.7, 118.4, 111.4, 106.9, 53.4, 27.2, 20.5; ESI-MS m/z calcd for C18H20N3O [M + H]+ = 294.16; found 293.95.
3.1.6.2. (S)-1-((4-Fluorophenyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-aminium Chloride, 22b
White solid; 99% yield; mp = 187–189 °C; [α]D28 = +120 (c 0.5, MeOH); 1H NMR (500 MHz, DMSO-d6) δ 11.14 (s, 1H), 11.07 (s, 1H), 8.44 (brs, 3H), 7.72 (d, J = 7.9 Hz, 1H), 7.67–7.61 (m, 2H), 7.35 (d, J = 8.1 Hz, 1H), 7.28 (d, J = 1.9 Hz, 1H), 7.17 (dd, J = 8.9, 8.8 Hz, 2H), 7.06 (dd, J = 7.9, 7.4 Hz, 1H), 6.94 (dd, J = 8.1, 7.4 Hz, 1H), 4.25–4.31 (m, 1H), 3.37 (dd, J = 14.6, 6.5 Hz, 1H), 3.31 (dd, J = 14.6, 6.9 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 167.2, 158.3, 136.2, 134.7, 127.1, 124.9, 121.43, 121.1, 118.7, 118.4, 115.4, 111.4, 106.8, 53.4, 27.2; ESI-MS m/z calcd for C17H17FN3O [M + H]+ = 298.14; found 297.95.
3.1.6.3. (S)-1-((4-Chlorophenyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-aminium Chloride, 22c
White solid; 99% yield; mp = 197–199 °C; [α]D28 = +116 (c 0.5, MeOH); 1H NMR (500 MHz, DMSO-d6) δ 11.22 (s, 1H), 11.06 (s, 1H), 8.43 (brs, 3H), 7.71 (d, J = 7.9 Hz, 1H), 7.66 (d, J = 8.8 Hz, 2H), 7.39 (d, J = 8.8 Hz, 2H), 7.34 (d, J = 8.1 Hz, 1H), 7.27 (d, J = 1.6 Hz, 1H), 7.06 (dd, J = 7.9, 7.2 Hz, 1H), 6.94 (dd, J = 8.1, 7.2 Hz, 1H), 4.29 (brs, 1H), 3.40–3.34 (m, 1H), 3.30 (dd, J = 14.6, 7.0 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 167.4, 137.2, 136.2, 128.7, 127.5, 127.1, 124.9, 121.1, 121.1, 118.6, 118.4, 111.4, 106.7, 53.5, 27.1; ESI-MS m/z calcd for C17H17ClN3O [M + H]+ = 314.11; found 313.90.
3.1.6.4. (S)-1-((4-(Ethoxycarbonyl)phenyl)amino)-3-(1H-indol-3-yl)-1-oxopropan-2-aminium Chloride, 22d
White solid; 99% yield; mp = 162–164 °C; [α]D28 = +88 (c 0.5, MeOH); 1H NMR (500 MHz, DMSO-d6) δ 11.40 (s, 1H), 11.06 (d, J = 1.2 Hz, 1H), 8.45 (s, 3H), 7.93 (d, J = 8.7 Hz, 2H), 7.77 (d, J = 8.7 Hz, 2H), 7.71 (d, J = 7.9 Hz, 1H), 7.34 (d, J = 8.1 Hz, 1H), 7.28 (d, J = 2.1 Hz, 1H), 7.06 (dd, J = 8.7, 7.4 Hz, 1H), 6.93 (dd, J = 7.9, 7.4 Hz, 1H), 4.38–4.32 (m, 1H), 4.29 (q, J = 7.1 Hz, 2H), 3.43–3.38 (dd in H2O, 1H), 3.31 (dd, J = 14.6, 7.0 Hz, 1H), 1.31 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 167.9, 165.2, 142.5, 136.2, 130.2, 127.1, 124.9, 121.1, 119.0, 118.6, 118.4, 111.4, 106.7, 60.5, 53.6, 27.1, 14.2 (one carbon is missing due to overlapping); ESI-MS m/z calcd for C20H22N3O3 [M + H]+ = 352.17; found 351.95.
3.1.7. General Procedure for the Synthesis of Acridone Derivatives 23a–d
The solution of 2-(2-methyl-9-oxoacridin-10(9H)-yl)acetic acid (30 mg, 0.112 mmol) in dry DMF (0.75 mL) was cooled to 0 °C, and Et3N (18 μL, 0.132 mmol), EDCI (25.7 mg, 0.134 mmol), HOBt hydrate (20.5 mg, 0.134 mmol), and 0.123 mmol of hydrochloric salts 22a–d were successively added. The reaction mixture was stirred at room temperature overnight, and then H2O was added, followed by extractions with dichloromethane. The organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. MeOH (2 mL) was added to the residue, and the product was collected by filtration and washed with petroleum ether.
3.1.7.1. (S)-3-(1H-Indol-3-yl)-2-(2-(2-methyl-9-oxoacridin-10(9H)-yl)acetamido)-N-(p-tolyl)propenamide, 23a
Yellow solid; 69% yield; mp = 223–225 °C; [α]D28 = −17,6 (c 0.5, DMF); 1H NMR (500 MHz, DMSO-d6) δ 10.92 (s, 1H), 10.12 (s, 1H), 8.89 (d, J = 8.3 Hz, 1H), 8.30 (dd, J = 8.0, 1.3 Hz, 1H), 8.09 (s, 1H), 7.70 (d, J = 7.9 Hz, 1H), 7.66 (t, J = 7.5 Hz, 1H), 7.52–7.46 (m, 3H), 7.38 (d, J = 8.1 Hz, 1H), 7.32 (brs, 1H), 7.27 (dd, J = 7.5, 7.4 Hz, 1H), 7.24 (d, J = 1.7 Hz, 1H), 7.13 (d, J = 8.2 Hz, 2H), 7.09 (dd, J = 7.5, 7.4 Hz, 1H), 6.99 (dd, J = 7.5, 7.2 Hz, 1H), 5.23 (d, J = 18.1 Hz, 1H), 5.04 (d, J = 18.1 Hz, 1H), 4.83 (m, 1H), 3.26 (dd, J = 14.4, 5.0 Hz, 1H), 3.12 (dd, J = 14.4, 9.1 Hz, 1H), 2.41 (s, 3H), 2.26 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.5, 170.0, 166.9, 142.3, 140.6, 136.3, 136.1, 135.2, 133.7, 132.4, 130.5, 129.1, 127.3, 126.4, 125.6, 123.9, 121.4, 121.3, 121.0, 120.9, 119.6, 118.6, 118.3, 115.9, 115.8, 111.3, 109.7, 54.3, 48.5, 28.5, 20.5, 20.2; ESI-HRMS m/z calcd for C34H31N4O3 [M + H]+ = 543.2396; found 543.2389.
3.1.7.2. (S)-N-(4-Fluorophenyl)-3-(1H-indol-3-yl)-2-(2-(2-methyl-9-oxoacridin-10(9H)-yl)acetamido)propenamide, 23b
Yellow solid; 67% yield; mp = 220–222 °C; [α]D28 = −10,8 (c 0.5, DMF); 1H NMR (500 MHz, DMSO-d6) δ 10.93 (d, J = 1.2 Hz, 1H), 10.28 (s, 1H), 8.93 (d, J = 8.3 Hz, 1H), 8.29 (dd, J = 8.0, 1.6 Hz, 1H), 8.09 (s, 1H), 7.69 (d, J = 7.9 Hz, 1H), 7.67–7.60 (m, 3H), 7.49 (d, J = 8.3 Hz, 1H), 7.38 (d, J = 8.1 Hz, 1H), 7.31 (brs, 1H), 7.28 (dd, J = 7.7, 7.3 Hz, 1H), 7.25 (d, J = 2.1 Hz, 1H), 7.20–7.13 (m, 2H), 7.09 (dd, J = 7.9. 7.2 Hz, 1H), 6.98 (dd, J = 7.8, 7.2 Hz, 1H), 5.24 (d, J = 18.1 Hz, 1H), 5.06 (d, J = 18.1 Hz, 1H), 4.82 (td, J = 8.7, 5.3 Hz, 1H), 3.26 (dd, J = 14.5, 5.1 Hz, 1H), 3.13 (dd, J = 14.5, 9.1 Hz, 1H), 2.41 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.5, 170.1, 167.0, 158.1, 142.3, 140.6, 136.1, 135.2, 135.2, 133.7, 130.5, 127.3, 126.4, 125.6, 123.9, 121.4, 121.3, 121.3, 121.0, 120.9, 118.5, 118.3, 115.9, 115.8, 115.3, 111.3, 109.6, 54.4, 48.5, 28.4, 20.2; ESI-HRMS m/z calcd for C33H28FN4O3 [M + H]+ = 547.2145; found 547.2146.
3.1.7.3. (S)-N-(4-Chlorophenyl)-3-(1H-indol-3-yl)-2-(2-(2-methyl-9-oxoacridin-10(9H)-yl)acetamido)propenamide, 23c
Yellow solid; 59% yield; mp = 234–236 °C; [α]D28 = −11.2 (c 0.5, DMF); 1H NMR (500 MHz, DMSO-d6) δ 10.93 (s, 1H), 10.36 (s, 1H), 8.94 (d, J = 8.2 Hz, 1H), 8.30 (dd, J = 7.8, 1.0 Hz, 1H), 8.09 (s, 1H), 7.70–7.63 (m, 4H), 7.49 (d, J = 8.1 Hz, 1H), 7.38 (d, J = 8.7 Hz, 3H), 7.32 (brs, 1H), 7.28 (dd, J = 7.6, 7.4 Hz, 1H), 7.26 (d, J = 1.6 Hz, 1H), 7.09 (dd, J = 7.8, 7.4 Hz, 1H), 6.98 (dd, J = 7.4 Hz, 1H), 5.23 (d, J = 18.1 Hz, 1H), 5.06 (d, J = 18.1 Hz, 1H), 4.83 (td, J = 8.5, 5.7 Hz, 1H), 3.26 (dd, J = 14.5, 5.0 Hz, 1H), 3.13 (dd, J = 14.5, 9.1 Hz, 1H), 2.41 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.5, 170.4, 167.0, 142.3, 140.6, 137.7, 136.1, 135.2, 133.7, 130.5, 128.6, 127.3, 127.1, 126.4, 125.6, 123.9, 121.4, 121.3, 121.0, 121.0, 118.5, 118.3, 115.9, 115.8, 111.3, 109.6, 54.4, 48.5, 28.3, 20.2 (one carbon is missing due to overlapping); ESI-HRMS m/z calcd for C33H28ClN4O3 [M + H]+ = 563.1850; found 563.1849.
3.1.7.4. Ethyl (S)-4-(3-(1H-Indol-3-yl)-2-(2-(2-methyl-9-oxoacridin-10(9H)-yl)acetamido)propanamido)benzoate, 23d
Yellow solid; 65% yield; mp = 194–196 °C; [α]D28 = −16.8 (c 0.5, DMF); 1H NMR (500 MHz, DMSO-d6) δ 10.94 (d, J = 1.3 Hz, 1H), 10.57 (s, 1H), 8.96 (d, J = 8.2 Hz, 1H), 8.30 (dd, J = 8.0, 1.6 Hz, 1H), 8.09 (s, 1H), 7.94 (d, J = 8.8 Hz, 2H), 7.76 (d, J = 8.8 Hz, 2H), 7.70 (d, J = 7.9 Hz, 1H), 7.68–7.62 (m, 1H), 7.49 (d, J = 8.0 Hz, 1H), 7.43–7.37 (m, 2H), 7.32 (s, 1H), 7.30–7.25 (m, 2H), 7.09 (dd, J = 7.7, 7.4 Hz, 1H), 6.99 (dd, J = 7.4 Hz, 1H), 5.23 (d, J = 18.1 Hz, 1H), 5.07 (d, J = 18.1 Hz, 1H), 4.87 (td, J = 8.6, 5.3 Hz, 1H), 4.29 (q, J = 7.1 Hz, 2H), 3.28 (dd, J = 14.5, 5.0 Hz, 1H), 3.14 (dd, J = 14.5, 9.2 Hz, 1H), 2.41 (s, 3H), 1.31 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, DMSO-d6) δ 176.5, 170.9, 167.1, 165.3, 143.1, 142.3, 140.6, 136.1, 135.2, 133.7, 130.5, 130.2, 127.3, 126.4, 125.6, 124.5, 123.9, 121.4, 121.3, 121.1, 121.0, 118.9, 118.5, 118.3, 115.9, 115.8, 111.3, 109.5, 60.5, 54.5, 48.5, 28.2, 20.2, 14.2; ESI-HRMS m/z calcd for C36H33N4O5 [M + H]+ = 601.2451; found 601.2444.
3.2. In Silico Studies
3.2.1. Molecular Docking
Autodock Vina package52 was used for molecular docking, and the crystal structure of MARK4 (PDB ID: 5ES1)19 was retrieved from the RCSB Protein Data Bank (www.rcsb.org). Through AutoDockTools (1.5.6 package), water molecules and the co-crystalized ligand were removed; the appropriate Gasteiger charges and polar hydrogen atoms were added, while the nonpolar hydrogens were merged. Acridone derivatives were generated and optimized through energy minimization (MM2 force field) by ChemBio3D Ultra 12.0. The coordinate files (pdbqt format) of MARK4 and acridone ligands were prepared using AutoDockTools. Following the standard docking procedure, the three-dimensional grid box was centered on the active site with size 20 × 20 × 20 Å3 and 1 Å spacing. The exhaustiveness value was set to 100 instead of the default 8. The docking poses with the lowest binding affinity were selected for presentation, and figures of the docked complexes were generated by PyMOL 2.1.1 (Schrödinger, LLC) and Discovery Studio (BIOVIA).
3.2.2. Predicted Pharmacokinetic Properties
Compounds were subjected to molecular property prediction and drug-likeness by the SwissADME platform (http://www.swissadme.ch). The chemical structures and SMILES notations of synthesized acridones were entered in the online SwissADME platform to calculate the molecular weight (MW), lipophilicity (MlogP), hydrogen bond donors (HBD), hydrogen bond acceptors (HBA), rotatable bonds (r.b.), and topological polar surface area (TPSA).
3.3. Expression and Purification of MARK4
Human MARK4 was cloned, expressed, and purified as per previously published laboratory protocols.53
3.4. Enzyme Inhibition Assay
Protein kinases are enzymes capable of catalyzing the transfer of the phosphate group from ATP to specific target protein. The kinases specifically phosphorylate Ser/Thr/Tyr residues of the proteins. The phosphorylating capability of the kinases is generally termed kinase activity or enzyme activity of the protein kinase. The kinase activity of MARK4 was measured using a malachite green reagent (BIOMOL Green reagent). MARK4 (5 μM) was purified and incubated with freshly prepared ATP (200 μM), and the sample was mixed and incubated for 1 h at 25 °C. The inhibitory potential of the synthesized compounds was also estimated by incubating the protein with different concentrations of the ligands for 30 min at 37 °C. After completion of incubation, the sample was further incubated with ATP (200 μM) for 1 h. Finally, proteins free of ligand and protein samples with different ligand concentrations were mixed with malachite green to terminate the reaction. The samples were plated in 96-well plates and read on an ELISA reader at 620 nm.
3.5. Fluorescence Binding Assay
A fluorescence protein–ligand binding assay uses fluorescent molecules to detect and measure the interaction between a protein and a ligand by exploiting the intrinsic fluorescence (I.F) of the protein. The protein’s IF refers to the fluorescence emitted by the aromatic amino acids, Trp, Tyr, and Phe, when excited at a specific wavelength. The intensity (I) and wavelength (λ) of the fluorescence emission can assist in interpreting the structure and environment of the protein. The IF of a protein has been used extensively in assessing the protein structure, dynamics, folding, and interactions with other proteins and ligands. Fluctuations in I and λmax of the protein can suggest confirmation alterations in the structure of the protein, as in the case of aggregation and folding. Likewise, the binding of a protein–ligand can alter the fluorescence properties of the protein, providing information about the binding affinity and specificity.
A fluorescence-based binding assay was carried out to estimate the binding of ligands to MARK4. The protein kinase (5–6 μM) was used, and it was titrated against different ligand concentrations.54 Jasco spectrofluorometer (FP-6200, Japan) was used to carry out the quenching experiment. Experiments were carried out in triplicates, blank spectra were subtracted, and the inner filter effect was considered in the experiments55 Stern–Volmer equation (eq 1) (SV) and modified Stern–Volmer equation (eq 2) (MSV) was used to estimate binding parameters.
| 1 |
| 2 |
F0: Fluorescence intensity (protein); F: Fluorescence intensity of protein + ligands; K: Binding constant; n: binding sites; C: concentration (ligand).
3.6. Cell Toxicity
3.6.1. Cell Culture
HeLa (cervical cancer), U87MG (glioblastoma), U251 (glioblastoma), and MDA-MB-435 (melanoma) cancer cell lines were provided by ATCC (American Type Culture Collection) and were maintained in DMEM medium supplemented with 10% (v/v) fetal bovine serum (FBS) and antibiotics/antimycotics. Normal cell line human gingival fibroblasts (HGFs), which have been cultured from gingival tissue and are embryo-like cells with the capacity of self-renewal, were maintained in DMEM low glucose medium supplemented with 10% (v/v) fetal bovine serum (FBS) and antibiotics/antimycotics. All cells were grown as monolayers at 37 °C in a 5% CO2 incubator.
3.6.2. MTT Assays
HeLa and U87MG cells were seeded in 96-well plates (4 × 103 cells per well) and grown in a medium supplemented with 10% FBS. The day after, cells were exposed in the fresh medium with increasing concentrations of the inhibitors (1–20 μM) for 24 h. The viability of the cells was measured by colorimetric viability assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide), as described previously.56 Values shown represent the means ± SE of three independent experiments.
Acknowledgments
The research work was supported by the Hellenic Foundation for Research and Innovation (HFRI) under the HFRI PhD Fellowship grant (Fellowship Number: 409). Human gingival fibroblasts (HGFs) were kindly provided by Assistant Professor A. Bakopoulou from the School of Dentistry, Aristotle University of Thessaloniki.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.3c00084.
Docking scores and interactions between MARK4 and synthesized acridones; 1H NMR and 13C NMR spectra of the synthesized acridones; LC/ESI-MS analysis for the synthesized compounds (PDF)
Author Contributions
Conceptualization, V.S, M.V., and M.I.H.; methodology, V.S. and M.I.H.; software and formal analysis, M.V., V.S., and M.I.H.; investigation, S.A., I.S., E.P., S.F.,M.V., M.I.H., and V.S; resources, V.S. and M.I.H.; data curation, M.V., S.A., M.I.H., and V.S.; writing—original draft preparation, M.V., S.A., M.I.H., and V.S.; writing—review and editing, M.V., S.A., M.I.H., and V.S.; supervision, M.I.H. and V.S.; funding acquisition, M.I.H. and V.S. All authors have read and agreed to the published version of the manuscript
The authors declare no competing financial interest.
Supplementary Material
References
- Cohen P.; Cross D.; Jänne P. A. Kinase drug discovery 20 years after imatinib: progress and future directions. Nat. Rev. Drug Discovery 2021, 20, 551–569. 10.1038/s41573-021-00195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P. Protein kinases — the major drug targets of the twenty-first century?. Nat. Rev. Drug Discovery 2002, 1, 309–315. 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- Naz F.; Anjum F.; Islam A.; Ahmad F.; Hassan M. I. Microtubule affinity-regulating kinase 4: Structure, function, and regulation. Cell Biochem. Biophys. 2013, 67, 485–499. 10.1007/s12013-013-9550-7. [DOI] [PubMed] [Google Scholar]
- Drewes G.; Ebneth A.; Preuss U.; Mandelkow E. M.; Mandelkow E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell 1997, 89, 297–308. 10.1016/S0092-8674(00)80208-1. [DOI] [PubMed] [Google Scholar]
- Trinczek B.; Brajenovic M.; Ebneth A.; Drewes G. MARK4 is a novel microtubule-associated proteins/microtubule affinity-regulating kinase that binds to the cellular microtubule network and to centrosomes. J. Biol. Chem. 2004, 279, 5915–5923. 10.1074/jbc.M304528200. [DOI] [PubMed] [Google Scholar]
- Saadat I.; Higashi H.; Obuse C.; Umeda M.; Murata-Kamiya N.; Saito Y.; Lu H.; Ohnishi N.; Azuma T.; Suzuki A.; Ohno S.; Hatakeyama M. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature 2007, 447, 330–333. 10.1038/nature05765. [DOI] [PubMed] [Google Scholar]
- Rovina D.; Fontana L.; Monti L.; Novielli C.; Panini N.; Sirchia S. M.; Erba E.; Magnani I.; Larizza L. Microtubule-associated protein/microtubule affinity-regulating kinase 4 (MARK4) plays a role in cell cycle progression and cytoskeletal dynamics. Eur. J. Cell Biol. 2014, 93, 355–365. 10.1016/j.ejcb.2014.07.004. [DOI] [PubMed] [Google Scholar]
- Feng M.; Tian L.; Gan L.; Liu Z.; Sun C. Mark4 promotes adipogenesis and triggers apoptosis in 3T3-L1 adipocytes by activating JNK1 and inhibiting p38MAPK pathways. Biol. Cell. 2014, 106, 294–307. 10.1111/boc.201400004. [DOI] [PubMed] [Google Scholar]
- Yu W.; Polepalli J.; Wagh D.; Rajadas J.; Malenka R.; Lu B. A Critical Role for the PAR-1/MARK-Tau Axis in Mediating the Toxic Effects of Aβ on Synapses and Dendritic Spines. Hum. Mol. Genet. 2012, 21, 1384–1390. 10.1093/hmg/ddr576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu G. J.; Lund H.; Wu D.; Blokzijl A.; Classon C.; von Euler G.; Landegren U.; Sunnemark D.; Kamali-Moghaddam M. Role of individual MARK isoforms in phosphorylation of tau at Ser262 in Alzheimer’s Disease. NeuroMol. Med. 2013, 15, 458–469. 10.1007/s12017-013-8232-3. [DOI] [PubMed] [Google Scholar]
- Lund H.; Gustafsson E.; Svensson A.; Nilsson M.; Berg M.; Sunnemark D.; von Euler G. MARK4 and MARK3 associate with early tau phosphorylation in Alzheimer’s Disease granulovacuolar degeneration bodies. Acta Neuropathol. Commun. 2014, 2, 22 10.1186/2051-5960-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C.; Tian L.; Nie J.; Zhang H.; Han X.; Shi Y. Inactivation of MARK4, an AMP-activated protein kinase (AMPK)-related kinase, leads to insulin hypersensitivity and resistance to diet-induced obesity. J. Biol. Chem. 2012, 287, 38305–38315. 10.1074/jbc.M112.388934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Gan L.; Chen Y.; Luo D.; Zhang Z.; Cao W.; Zhou Z.; Lin X.; Sun C. Mark4 promotes oxidative stress and inflammation via binding to PPARγ and activating NF-κB pathway in mice adipocytes. Sci. Rep. 2016, 6, 21382 10.1038/srep21382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnani I.; Novielli C.; Fontana L.; Tabano S.; Rovina D.; Moroni R. F.; Bauer D.; Mazzoleni S.; Colombo E. A.; Tedeschi G.; Monti L.; Porta G.; Bosari S.; Frassoni C.; Galli R.; Bello L.; Larizza L. Differential signature of the centrosomal MARK4 isoforms in glioma. Anal. Cell Pathol. 2011, 34, 319–338. 10.1155/2011/206756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beghini A.; Magnani I.; Roversi G.; Piepoli T.; Di Terlizzi S.; Moroni R. F.; Pollo B.; Fuhrman Conti A. M.; Cowell J. K.; Finocchiaro G.; Larizza L. The neural progenitor-restricted isoform of the MARK4 gene in 19q13.2 is upregulated in human gliomas and overexpressed in a subset of glioblastoma cell lines. Oncogene 2003, 22, 2581–2591. 10.1038/sj.onc.1206336. [DOI] [PubMed] [Google Scholar]
- Kato T.; Satoh S.; Okabe H.; Kitahara O.; Ono K.; Kihara C.; Tanaka T.; Tsunoda T.; Yamaoka Y.; Nakamura Y.; Furukawa Y. Isolation of a novel human gene, MARKL1, homologous to MARK3 and its involvement in hepatocellular carcinogenesis. Neoplasia 2001, 3, 4–9. 10.1038/sj.neo.7900132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Guan K. L. Microtubule-associated protein/microtubule affinity-regulating kinase 4 (MARK4) is a negative regulator of the mammalian target of rapamycin complex 1 (mTORC1). J. Biol. Chem. 2013, 288, 703–708. 10.1074/jbc.C112.396903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidary Arash E.; Shiban A.; Song S.; Attisano L. MARK4 inhibits Hippo signaling to promote proliferation and migration of breast cancer cells. EMBO Rep. 2017, 18, 420–436. 10.15252/embr.201642455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack J. S.; Gao M.; Kiefer S. E.; Myers J. E. Jr; Newitt J. A.; Wu S.; Yan C. Crystal structure of microtubule affinity-regulating kinase 4 catalytic domain in complex with a pyrazolopyrimidine inhibitor. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2016, 72, 129–134. 10.1107/S2053230X15024747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naqvi A. A. T.; Jairajpuri D. S.; Noman O.M.A.; Hussain A.; Islam A.; Ahmad F.; Alajmi M. F.; Hassan M. I. Evaluation of pyrazolopyrimidine derivatives as microtubule affinity regulating kinase 4 inhibitors: Towards therapeutic management of Alzheimer’s disease. J. Biomol. Struct. Dyn. 2020, 38, 3892–3907. 10.1080/07391102.2019.1666745. [DOI] [PubMed] [Google Scholar]
- Voura M.; Anwar S.; Thysiadis S.; Khan P.; Dalezis P.; Trafalis D. T.; Hassan M. I.; Sarli V. Synthesis and biological activity of bisindole derivatives as novel MARK4 inhibitors. Eur. J. Med. Chem. Rep. 2022, 6, 100076 10.1016/j.ejmcr.2022.100076. [DOI] [Google Scholar]
- Voura M.; Khan P.; Thysiadis S.; Katsamakas S.; Queen A.; Hasan G. M.; Ali S.; Sarli V.; Hassan M. I. Probing the Inhibition of Microtubule Affinity Regulating Kinase 4 by N-Substituted Acridones. Sci. Rep. 2019, 9, 1676 10.1038/s41598-018-38217-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Liu Z.; Sun H.; Li C.; Wang W.; Ye L.; Yan C.; Tian J.; Wang H. PCC0208017, a novel small-molecule inhibitor of MARK3/MARK4, suppresses glioma progression in vitro and in vivo. Acta Pharm. Sin. B 2020, 10, 289–300. 10.1016/j.apsb.2019.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasher P.; Sharma M. Medicinal chemistry of acridine and its analogues. MedChemComm 2018, 9, 1589–1618. 10.1039/C8MD00384J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepúlveda C. S.; Fascio M. L.; García C. C.; D’Accorso N. B.; Damonte E. B. Acridones as antiviral agents: synthesis, chemical and biological properties. Curr. Med. Chem. 2013, 20, 2402–2414. 10.2174/0929867311320190002. [DOI] [PubMed] [Google Scholar]
- Kudryavtseva T. N.; Sysoev P. I.; Popkov S. V.; Nazarov G. V.; Klimova L. G. Synthesis and antimicrobial activity of some acridone derivatives bearing 1,3,4-thiadiazole and 1,2,4-triazole moieties. Russ. Chem. Bull. 2015, 64, 445–450. 10.1007/s11172-015-0884-8. [DOI] [Google Scholar]
- Aarjane M.; Slassi S.; Tazi B.; Maouloua M.; Amine A. Synthesis, antibacterial evaluation and molecular docking studies of novel series of acridone- 1,2,3-triazole derivatives. Struct. Chem. 2020, 31, 1523–1531. 10.1007/s11224-020-01512-0. [DOI] [Google Scholar]
- Naidoo D.; Coombes P. H.; Mulholland D. A.; Crouch N. R.; van den Bergh A. J. N-Substituted acridone alkaloids from Toddaliopsis bremekampii (Rutaceae: Toddalioideae) of south-central Africa. Phytochemistry 2005, 66, 1724–1728. 10.1016/j.phytochem.2005.04.036. [DOI] [PubMed] [Google Scholar]
- Biagini G. A.; Fisher N.; Berry N.; Stocks P. A.; Meunier B.; Williams D. P.; Bonar-Law R.; Bray P. G.; Owen A.; O’Neill P. M.; Ward S. A. Acridinediones: selective and potent inhibitors of the malaria parasite mitochondrial bc1 complex. Mol. Pharmacol. 2008, 73, 1347–1355. 10.1124/mol.108.045120. [DOI] [PubMed] [Google Scholar]
- Lacroix D.; Prado S.; Kamoga D.; Kasenene J.; Bodo B. Structure and in vitro antiparasitic activity of constituents of Citropsis articulata root bark. J. Nat. Prod. 2011, 74, 2286–2289. 10.1021/np2004825. [DOI] [PubMed] [Google Scholar]
- Yadav T. T.; Murahari M.; Peters G. J.; Yc M. A comprehensive review on acridone based derivatives as future anti-cancer agents and their structure activity relationships. Eur. J. Med. Chem. 2022, 239, 114527 10.1016/j.ejmech.2022.114527. [DOI] [PubMed] [Google Scholar]
- Raza A.; Jacobson B. A.; Benoit A.; Patel M. R.; Jay-Dixon J.; Hiasa H.; Ferguson D. M.; Kratzke R. A. Novel acridine-based agents with topoisomerase II inhibitor activity suppress mesothelioma cell proliferation and induce apoptosis. Invest. New Drugs 2012, 30, 1443–1448. 10.1007/s10637-011-9720-7. [DOI] [PubMed] [Google Scholar]
- Harrison R. J.; Reszka A. P.; Haider S. M.; Romagnoli B.; Morrell J.; Read M. A.; Gowan S. M.; Incles C. M.; Kelland L. R.; Neidle S. Evaluation of by disubstituted acridone derivatives as telomerase inhibitors: the importance of G-quadruplex binding. Bioorg. Med. Chem. Lett. 2004, 14, 5845–5849. 10.1016/j.bmcl.2004.09.037. [DOI] [PubMed] [Google Scholar]
- Marques E. F.; Bueno M. A.; Duarte P. D.; Silva L. R.; Martinelli A. M.; dos Santos C. Y.; Severino R. P.; Brömme D.; Vieira P. C.; Corrêa A. G. Evaluation of synthetic acridones and 4-quinolinones as potent inhibitors of cathepsins L and V. Eur. J. Med. Chem. 2012, 54, 10–21. 10.1016/j.ejmech.2012.04.002. [DOI] [PubMed] [Google Scholar]
- Parveen M.; Aslam A.; Nami S. A.; Malla A. M.; Alam M.; Lee D. U.; Rehman S.; Silva P. S.; Silva M. R. Potent acetylcholinesterase inhibitors: Synthesis, biological assay and docking study of nitro acridone derivatives. J. Photochem. Photobiol. B. 2016, 161, 304–311. 10.1016/j.jphotobiol.2016.05.028. [DOI] [PubMed] [Google Scholar]
- Beniddir M. A.; Le Borgne E.; Iorga B. I.; Loaëc N.; Lozach O.; Meijer L.; Awang K.; Litaudon M. Acridone alkaloids from Glycosmis chlorosperma as DYRK1A inhibitors. J. Nat. Prod. 2014, 77, 1117–1122. 10.1021/np400856h. [DOI] [PubMed] [Google Scholar]
- Murahari M.; Prakash K. V.; Peters G. J.; Mayur Y. C. Acridone-pyrimidine hybrids- design, synthesis, cytotoxicity studies in resistant and sensitive cancer cells and molecular docking studies. Eur. J. Med. Chem. 2017, 139, 961–981. 10.1016/j.ejmech.2017.08.023. [DOI] [PubMed] [Google Scholar]
- Mohammadi-Khanaposhtani M.; Shabani M.; Faizi M.; Aghaei I.; Jahani R.; Sharafi Z.; Shamsaei Zafarghandi N.; Mahdavi M.; Akbarzadeh T.; Emami S.; Shafiee A.; Foroumadi A. Design, synthesis, pharmacological evaluation, and docking study of new acridone-based 1,2,4-oxadiazoles as potential anticonvulsant agents. Eur. J. Med. Chem. 2016, 112, 91–98. 10.1016/j.ejmech.2016.01.054. [DOI] [PubMed] [Google Scholar]
- Putic A.; Stecher L.; Prinz H.; Müller K. Structure-activity relationship studies of acridones as potential antipsoriatic agents. 2. Synthesis and antiproliferative activity of 10-substituted hydroxy-10H-acridin-9-ones against human keratinocyte growth. Eur. J. Med. Chem. 2010, 45, 5345–5352. 10.1016/j.ejmech.2010.08.059. [DOI] [PubMed] [Google Scholar]
- Mayur Y. C.; Ahmad O.; Prasad V. V.; Purohit M. N.; Srinivasulu N.; Kumar S. M. Synthesis of 2-methyl N10-substituted acridones as selective inhibitors of multidrug resistance (MDR) associated protein in cancer cells. Med. Chem. 2008, 4, 457–465. 10.2174/157340608785700180. [DOI] [PubMed] [Google Scholar]
- Anwar S.; Kar R. K.; Haque M. A.; Dahiya R.; Gupta P.; Islam A.; Ahmad F.; Hassan M. I. Effect of pH on the structure and function of pyruvate dehydrogenase kinase 3: Combined spectroscopic and MD simulation studies. Int. J. Biol. Macromol. 2020, 147, 768–777. 10.1016/j.ijbiomac.2020.01.218. [DOI] [PubMed] [Google Scholar]
- Waseem R.; Yadav N. S.; Khan T.; Ahmad F.; Kazim S. N.; Hassan M. I.; Prakash A.; Islam A. Molecular basis of structural stability of Irisin: A combined molecular dynamics simulation and in vitro studies for Urea-induced denaturation. J. Mol. Liq. 2023, 372, 121120 10.1016/j.molliq.2022.121120. [DOI] [Google Scholar]
- Royer C. A.Fluorescence Spectroscopy. In Protein Stability and Folding, Methods in Molecular Biology; Springer, 1995; Vol. 40, pp 65–89. [DOI] [PubMed] [Google Scholar]
- Soares S.; Mateus N.; Freitas Vd. Interaction of different polyphenols with bovine serum albumin (BSA) and human salivary alpha-amylase (HSA) by fluorescence quenching. J. Agric. Food Chem. 2007, 55, 6726–6735. 10.1021/jf070905x. [DOI] [PubMed] [Google Scholar]
- Klajnert B.; Stanisławska L.; Bryszewska M.; Pałecz B. Interactions between PAMAM dendrimers and bovine serum albumin. Biochim. Biophys. Acta, Proteins Proteomics 2003, 1648, 115–126. 10.1016/S1570-9639(03)00117-1. [DOI] [PubMed] [Google Scholar]
- Beghini A.; Magnani I.; Roversi G.; Piepoli T.; Terlizzi S. D.; Moroni R. F.; et al. The neural progenitor-restricted isoform of the MARK4 gene in 19q13.2 is upregulated in human gliomas and overexpressed in a subset of glioblastoma cell lines. Oncogene 2003, 22, 2581–2591. 10.1038/sj.onc.1206336. [DOI] [PubMed] [Google Scholar]
- Magnani I.; Novielli C.; Fontana L.; Tabano S.; Rovina D.; Moroni R. F.; Bauer D.; Mazzoleni S.; Colombo E. A.; Tedeschi G.; Monti L.; Porta G.; Bosari S.; Frassoni C.; Galli R.; Bello L.; Larizza L. Differential signature of the centrosomal MARK4 isoforms in glioma. Anal. Cell Pathol. 2011, 34, 319–338. 10.1155/2011/206756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M.; Zhang C.; Lee S.; Sjöstedt E.; Fagerberg L.; Bidkhori G.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507 10.1126/science.aan2507. [DOI] [PubMed] [Google Scholar]
- Hotra A.; Ragunathan P.; Ng P. S.; Seankongsuk P.; Harikishore A.; Sarathy J. P.; Saw W. G.; Lakshmanan U.; Sae-Lao P.; Kalia N. P.; Shin J.; Kalyanasundaram R.; Anbarasu S.; Parthasarathy K.; Pradeep C. N.; Makhija H.; Dröge P.; Poulsen A.; Tan J. H. L.; Pethe K.; Dick T.; Bates R. W.; Grüber G. Discovery of a novel mycobacterial F-ATP synthase inhibitor and its potency in combination with diarylquinolines. Angew. Chem., Int. Ed. 2020, 59, 13295–13304. 10.1002/anie.202002546. [DOI] [PubMed] [Google Scholar]
- Ruchti J.; Carreira E. M. Ir-catalyzed reverse prenylation of 3-substituted indoles: total synthesis of (+)-aszonalenin and (−)-brevicompanine B. J. Am. Chem. Soc. 2014, 136, 16756–16759. 10.1021/ja509893s. [DOI] [PubMed] [Google Scholar]
- Appleton D. R.; Babcockb R. C.; Coppa B. R. Novel tryptophan-derived dipeptides and bioactive metabolites from the sea hare Aplysia dactylomela. Tetrahedron 2001, 57, 10181–10189. 10.1016/S0040-4020(01)01044-4. [DOI] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naz F.; Asad M.; Malhotra P.; Islam A.; Ahmad F.; Hassan M. I. Cloning, expression, purification and refolding of microtubule affinity-regulating kinase 4 expressed in Escherichia coli. Appl. Biochem. Biotechnol. 2014, 172, 2838–2848. 10.1007/s12010-014-0733-5. [DOI] [PubMed] [Google Scholar]
- Dahiya R.; Mohammad T.; Gupta P.; Haque A.; Alajmi M. F.; Hussain A.; Hassan M. I. Molecular interaction studies on ellagic acid for its anticancer potential targeting pyruvate dehydrogenase kinase 3. RSC Adv. 2019, 9, 23302–23315. 10.1039/C9RA02864A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi Z.; Liu R. Phenotypic characterization of the binding of tetracycline to human serum albumin. Biomacromolecules 2011, 12, 203–209. 10.1021/bm1011568. [DOI] [PubMed] [Google Scholar]
- Sigala I.; Tsamis K. I.; Gousia A.; Alexiou G.; Voulgaris S.; Giannakouros T.; Kyritsis A. P.; Nikolakaki E. Expression of SRPK1 in gliomas and its role in glioma cell lines viability. Tumour Biol. 2016, 37, 8699–8707. 10.1007/s13277-015-4738-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.