

Abstract
Crosslinking mass spectrometry (XL-MS) is a valuable technique for generating point-to-point distance measurements in protein space. However, cell-based XL-MS experiments require efficient software that can detect crosslinked peptides with sensitivity and controlled error rates. Many algorithms implement a filtering strategy designed to reduce the size of the database prior to mounting a search for crosslinks, but concern has been expressed over the possibility of reduced sensitivity using these strategies. We present a new scoring method that uses a rapid pre-search method and a concept inspired by computer vision algorithms to resolve crosslinks from other conflicting reaction products. Searches of several curated crosslink datasets demonstrate high crosslink detection rates and even the most complex proteome-level searches (using cleavable or noncleavable crosslinkers) can be completed efficiently on a conventional desktop computer. The detection of protein-protein interactions is increased twofold through the inclusion of compositional terms in the scoring equation. The combined functionality is made available as CRIMP 2.0 in the Mass Spec Studio.
Graphical Abstract

Introduction
Affinity-based techniques have dominated the discovery of molecular interactions for many years1,2. However, the data returned from such techniques only indirectly identify binding partners. Direct interactions are in fact very difficult to detect, particularly in a high throughput setting. Crosslinking mass spectrometry (XL-MS) may offer a solution. The basic approach involves chemically installing a bifunctional reagent between two reactive amino acid residues and detecting the linkage points using methods derived from bottom-up proteomics3,4. The major elements of the technique have a long history, but its application to whole cells for structure analysis is relatively recent, enabled by high sensitivity mass spectrometers and new reagents. A number of computational tools support crosslink identification5–7, but we need new approaches as tool comparisons reveal performance gaps5,6,8,9.
The efficient detection of crosslinks installed in whole proteomes is a major unmet need9,10. There are solutions, but they remain difficult to scale and they focus on a narrow selection of crosslinking reagent types. Some of the earliest methods supported searches of limited complexity, rarely involving more than a few 10’s of proteins9,11. The quadratic nature of the search space makes scaling these approaches difficult. Methods that evaluate all possible peptide couplings struggle with whole proteome searches. Even very strict peak matching criteria and improved data structures9 remain inefficient because the search scales as n2 (where n is the number of peptides in a search library). One solution to this problem involves a variant of the crosslinking reaction. Gas-phase cleavable reagents like PIR, DSSO and DSBU generate linear peptides in MS2. These peptides can be selected for fragmentation in MS3 to facilitate a more straightforward proteomics search12,13. Unfortunately, MS3-based detection methods present sensitivity and duty cycle issues14. An MS2-based alternative using stepped HCD fragmentation is an effective work-around14,15 but computational efficiency is lost when defaulting to MS2. The existing whole proteome search tools mostly target cleavable reagents, but there are many noncleavable reagents that can tune crosslink yield and distance measurements, and they provide cross-peptide fragment ions that are very useful in validating IDs. Thus, while we require more tools for XL-MS data analysis in general, the need for noncleavable reagents is particularly great.
Several approaches to the O(n2) time complexity problem have been considered. Strategies include accelerated implementations of the brute-force method described above, but more commonly, software packages use a two-pass search to first restrict the search space, followed by a more refined scoring strategy that acts on the reduced database8,16,17. There are many ways to implement a two-pass method. For gas-phase cleavable crosslinkers, the MS2-liberated peptides provide masses that narrow the search space18. Database reduction can also use an open-modification search inspired by routines used for detecting unknown post-translational modifications19. The exercise is defined as a search for a peptide with a modification of unknown mass, that is, the linked second peptide. Each peptide receives a preliminary score. Candidate peptides with scores above some threshold are selected and ranked, then used to restrict the search for the second peptide8,16. The resulting combinations are rescored in a full search against the MS2 spectrum. It has been suggested that a pre-search strategy may not advance sufficiently high numbers of peptides to be sensitive enough in proteome-scale applications9, where sensitivity is defined as the fraction of the total number of real crosslinks detected in an experiment. This would be unfortunate as brute-force methods may not be practical for proteome-wide searches, especially if post-translational modifications need to be considered.
We updated our CRIMP crosslink detection software20 to determine if whole proteome searches can be sensitive, fast, and independent of the style of crosslinker used. CRIMP 2.0 incorporates an improved library reduction engine and a new scoring algorithm that resolves spectral conflicts across all categories of hits (e.g., free peptides, monolinks and crosslinks). Our revised approach to error estimation accounts for cross-category spectral conflicts, mostly ignored in other search tools, and supports a new method of detecting of protein-protein interactions. We demonstrate that a library reduction strategy can indeed deliver high sensitivity and support whole proteome analysis of noncleavable and cleavable experiment types, with only modest computational resources.
Experimental Section
Synthetic peptide benchmark dataset 1, with variant
Data from a synthetic crosslinked peptide library, which contained a set of crosslinked and monolinked peptides engineered from 95 Cas9 tryptic peptides, was accessed from PRIDE (PXD014337)21. Details on the library and the database searches are contained in Supplementary Information. To simulate interprotein crosslinks, Cas9 was segmented into four subunits in such a manner that subunits A, B, C, and D could form both intra and interprotein crosslinks. Here, interprotein crosslinks are the most abundant: 248 potential interlinks and 178 potential intra-links (which includes homotypic crosslinks). This segmentation, and the parameterization of the CRIMP search, can also be found in Supplementary Information. In the first search strategy, crosslinks were considered correct if crosslinked peptides were assigned to the right peptide group, simulating an intraprotein crosslink search. In the second search strategy, crosslinks were considered correct if they were assigned to the right peptide group and the right category of crosslink.
Synthetic peptide benchmark dataset 2
A second synthetic crosslinked peptide library contained a set of crosslinked and monolinked peptides engineered from the amino acid sequences of 141 peptides derived from 38 proteins of the E.coli ribosomal complex. Data for this library were accessed from PRIDE (PDX029252)15. Data from the DSSO crosslinked libraries were used, with varying degrees of added peptide noise and database entrapment as per the published study. The sets contain a maximum of 1018 crosslinked peptides. The parameterization of the CRIMP search can also be found in Supplementary Information.
Proteome-scale E.coli dataset
Data from a large crosslinking experiment was accessed from the ProteomeXchange Consortium partner repository jPOSTrepo under the accession codes JPST000834 and PXD01912022. In this study, E. coli K12 strain was produced and lysed, followed by separation and crosslinking of the soluble high molecular weight proteome by size exclusion chromatography. Details on crosslinking, sample fractionation, data collection and establishing true and false protein-protein interactions (PPIs) can be found in Supporting Information, along with the parameterization of the CRIMP searches.
Software design and availability
CRIMP2.0 was built within the Mass Spec Studio design framework, a repository of data analysis routines available within the Studio for structural mass spectrometry applications such as HX-MS, covalent labeling, and integrative structure modeling. Software was written in C# and it leverages an extensive repository of reusable content. All the processed results can be regenerated using the datasets listed above, using CRIMP2.0 from www.msstudio.ca (version 2.4.0.3545).
Results and Discussion
Strategies for library reduction
All concepts for crosslink detection use one of two methods to reduce the search space (Figure 1). The brute-force enumeration of all peptide pairs constrains the search through the precursor mass by determining the set of all possible α and β peptide masses that sum to the precursor mass, after accounting for the addition of the linker itself. All relevant combinations are then scored directly against the MS2 spectrum of the crosslink peptide in a 1-pass determination. Precursor mass is a relatively weak constraint as large numbers of peptides need to be exhaustively paired, particularly when the mass tolerance is relaxed. In return, the overall search has the potential to be very sensitive as no combinations will be missed, in theory.
Figure 1.

Schematic outlining the typical approaches to searching MS2 datasets for evidence of peptide crosslinking. The 1-pass approach begins with the precursor mass of the putative crosslinked peptide and constrains a database search through a simple three-term sum involving the α peptide mass, the β peptide mass and the linker mass. Combinations are then searched against the MS2 spectrum. The 2-pass method begins and ends with the MS2 spectrum. First, candidate α and β peptides are found in the MS2 data, and only then is precursor mass used to constrain combinations for a more exhaustive search of the MS2 data.
The other concept invokes a 2-pass approach, where the MS2 spectrum of the crosslinked peptide is mined twice. The first pass attempts to identify options for the dominant α peptide through a linearized fragment set, a subset of the total fragments in the spectrum. That is, a database search looks for fragments arising from the free peptide plus its mass-modified form. Detecting the mass-modified form requires an open modification algorithm that treats the second peptide (plus linker) as a modification whose mass can be calculated by subtracting the candidate α peptide mass from the precursor mass. In this fashion, the α peptide sequence can be read past the crosslink site. This open modification search is also dependent upon the precursor mass, but the selection of α peptide candidates is primarily driven by the quality of the MS2 hits. Most search tools restrict this pre-search to the α peptide alone8,16, relying again on the precursor ion to define the candidate β peptides by subtraction of the α peptides. These tools avoid linearizing the β peptide, arguing that the MS2 spectral data for the β peptide are comparatively sparse because of preferential fragmentation23. Nevertheless, linearizing both peptides has been successfully demonstrated in the Goodlett lab24. The original Kojak search tool also looks for both and then constructs the peptide combinations for rescoring25, but it underperforms tools that restrict reduction to the α peptide, which seemingly validates the concerns expressed over the value of the β peptide sequence information.
We wanted to explore the double peptide pre-search concept further, because search performance can be affected by many elements of the workflow. In whole proteome searching, particularly where the goal is to identify protein-protein interactions from as little as one crosslink match, we argue that both peptides must generate fragment series of sufficient quality to identify two proteins or protein groups. A search tool that finds poor-quality β peptides is of little value in a whole proteome search26, thus extending linearization to both peptides could be the best approach for PPI detection.
Upgrading crosslink analysis in the Mass Spec Studio: CRIMP 2.0
To minimize the computational limitations associated with precursor mass constraints, we added full peptide linearization to the original version of CRIMP26. Improvements were also made to crosslink rescoring and error estimation, supported by improvements in signal processing. Several computational enhancements accelerated search speed and improved the navigation and export of search results. These are described briefly below.
Spectral annotation and fragment assignment
We developed an adaptable method of precursor ion detection that is sensitive to the S/N of the given feature. The goodness-of-fit of an isotopic distribution for a candidate peptide can be relaxed when the S/N is poorer, allowing the natural advantages of tandem mass spectrometry to override poor MS ion statistics. Improvements in the identification of monoisotopic peaks support both centroid and profile-mode datasets and a new approach to fragment ion assignment was adopted. Fragment types are numerous and complex in crosslinked peptide spectra, and certain fragment types will draw from a much larger distribution of possible values than others. For example, the number of possible internal fragment ions for crosslinked peptides (i.e., generated by two cleavage events) is much larger than simple single cleavage events and it is inappropriate to allow for equal weighting of these internal fragments during assignment, as they are less abundant than other sequence ions. We developed a greedy assignment algorithm based on a hierarchy of fragment types and the resulting identifications and weights are used in place of the number of detected peaks for crosslink scoring.
Scoring and conflict resolution
CRIMP uses a probabilistic scoring of α and β peptide candidates to assemble a list of candidates, based on OMSSA+ E values20. A scoring and ranking strategy was developed to ensure that high-probability candidate peptides are enriched within the top N groups of hits, where groups are defined as candidate peptides with similar scores. Top N is adjustable, but experimentation has shown that a value of 10 is sufficient in most every scenario, and the dependency on the α and β peptide scores is well-behaved (Figure S1). Peptides are then paired and exhaustively scored against the MS2 data.
Crosslinking reactions produce crosslinks in relatively low abundance, and the reaction products contain comparatively high amounts of monolinked, looplinked and free peptides. Many annotated fragment peaks can be shared between these products, particularly when they share sequence. Resolving conflicts is not always straightforward as different products tend to generate unique scoring ranges and noise distributions. We developed a strategy that manages conflicts and normalizes scores across products to determine the best overall match to a spectrum. Briefly, a multi-term scoring vector inspired by computer vision algorithms was created that contains several scores calculated from overlapping subsets of fragments identified in the MS2 spectrum. These subsets are selected based on their ability to discriminate between different digestion products. Scoring vectors are determined for all possible matches and a given identification is penalized using the shared fragment assignments from possible conflicts. This penalized scoring vector is transformed using q-values and then a final CLAM (Competitive Label Assignment Method) score is determined from the inner product of the transformed scoring vector (see Supplementary Information).
False discovery rate (FDR) estimation and aggregation
We use FDR calculations to assess the total error in search results, involving decoy database searches to estimate the error. Database searches and FDR calculations are usually performed in a category-based manner and are not able to address the impact of cross-category misidentifications on FDR (where category refers to the various products of a crosslinking reaction). Additionally, there can be sparse numbers of decoy hits for any given category, which makes FDR estimation less precise. All categories should be integrated in a joint FDR calculation, but it requires a method for normalizing score distributions. There is considerable variation in database size for the different categories and thus in the underlying search noise. For example, the search space for interprotein crosslinks is considerably greater than the search space for intraprotein crosslinks, necessitating a much different decoy database and creating a different scoring distribution. To integrate all categories of data, we standardize CLAM scores according to the specific error distributions of each, and then all categories of scores are combined. A scaling step transforms these scores to a uniform range of 1–100 to permit direct score comparisons between categories (e.g., monolinks and crosslinks). Only then is the final FDR estimate determined, and it is calculated as a geometric average of the global and the local FDR estimates as a concession to the validity of both approaches (see Supplementary Information).
Search results are then aggregated within runs, and between runs in the case of replicates. The first step of the aggregation workflow is to reduce crosslink spectrum matches (CSMs) to unique peptides and peptide pairs, by collapsing redundancies arising from charge states, retention times and variable modifications. Next, higher level aggregations to unique residue pairs (URPs), proteins, and PPIs are made, with each level supported by a separate error calculation. However, PPIs are not sensitively determined with these crosslink-centric methods of detection and error estimation. We reasoned that PPIs should possess evidence for their component proteins in the form of free peptides, monolinks, looplinks and intra-protein links. To this end, we rescore PPIs before error estimation by calculating a transformed value that includes evidence of the presence of component proteins and the success of the labeling chemistry (see Supplementary Information).
Computational enhancements
We significantly improved the way searches are supported computationally, particularly in database reduction. We adapted an indexing approach used by MSFragger for whole proteome searching, and by pLink2 for α peptide searches8,27. Here we apply the fragment index concept to the whole protein level, so that every unique fragment from every protein is only scored once per spectrum during the library reduction step. That is, we drop redundant fragment across the database. The increase in computational efficiency is considerable, and the approach even limits database expansion when incorporating variable modifications and missed cleavages. To generate stable lists of candidate α and β peptides, the reduced library is propagated between replicate searches in a manner that is reminiscent of “match between runs” in proteomics28. It means that we share the reduced library between replicates for any given precursor mass. Replicate DDA runs can generate some variation in crosslinker sampling, leading to unequal sets of candidate peptides during reduction. Propagation therefore combines the candidate lists of peptides across all replicates, followed by a precursor assignment pass using the complete set of peptides.
A set of changes was made to manage resources on more limited platforms like desktop and laptop computers. The user can adjust the pre-searching stringency for α and β peptides (as well as top N), creating the opportunity to conduct a search that approximates a brute-force strategy for samples of limited complexity, or one that applies strong reduction for whole proteomes. This allows the user to balance concerns over sensitivity with computational performance, however we will demonstrate below that the tradeoff is not a problematic one, and optimal searches converge on a very narrow set of parameters. Finally, numerous improvements were introduced to support the full range of crosslinking experiments and it anticipates new crosslinker designs. The new version preserves the validation-based routines and spectral navigation tools of version 1, which are particularly useful in structure modeling applications of XL-MS (Figure S2). We include visualization and export utilities for many downstream applications. Cleavable crosslinkers are accommodated through an MS2-based workflow, as opposed to the less efficient MS3-based routine, and tool tips are included throughout, aided by an extensive glossary of terms.
Evaluation of search sensitivity
We then reanalyzed the triplicate datasets from the synthetically generated crosslinked peptides provided by Beveridge et al.21 For the DSS noncleavable linker set, we detected 225±7 unique crosslinks at a real FDR of 3.1% (for an expected FDR of 5%). The real FDR is calculated based on the known composition of the peptide set. Sensitivity exceeds several other tools that have operated on this dataset (Figure S3). We used an Eα peptide setting of 10% to reflect the higher quality of the dominant peptide and allowed the Eβ setting to be 99%, relaxing the requirement for a high-quality β peptide. These are the default values for all searches. To compare with a brute-force approach, CRIMP 2.0 identified 1341 CSMs with a real FDR of 2.0%, compared to 885 CSMs and a real FDR of 3.1% for OpenPepXL. The lower sensitivity of the brute force approach is likely related to the higher level of noise that accompanies a search of all possible pairwise combinations.
We then searched the replicate noncleavable crosslinker data with various levels of database entrapment (Figure 2A,B). The sensitivity decreases, but approximately 35% of the detectable unique crosslinks are still found with almost 80,000 proteins as entrapment. To evaluate our approach for interprotein detection, we altered the database from its original design by segmenting the Cas9 protein into four smaller “proteins” (see Experimental Section). This segmentation turns many intraprotein crosslinks into interprotein crosslinks. The results show a diminishing sensitivity for interprotein crosslinks at higher levels of entrapment, but the rate of reduction is only slightly greater than intraprotein crosslinks (Figure 2C,D). Error estimation begins to destabilize at higher entrapment because of the very low number of hits returned in the searches. These searches are computationally efficient even with entrapment, highlighting the utility of a strong database reduction concept (Figure S4).
Figure 2.

Crosslink sensitivity determination using the replicate dataset from Beveridge et al.21 for the DSS crosslinker. The analysis for the Cas9 database at (A) 5% and (B) 1% calculated FDR. The results for the segmented Cas9 database at (C) 5% and (D) 1% FDR, presented as both intraprotein and an interprotein search results. Effect of entrapment is shown using multiple added databases with the noted protein complexity. Real % FDR posted as callouts.
We then expanded the tool comparison using a new set of replicate data published by Matzinger et al.,15 involving an expanded set of synthetic crosslinked peptides. We focused on cleavable crosslinkers and used our default search conditions. Again, CRIMP 2.0 outperformed all tools at both the expected and real FDRs (Figure 3), detecting 673 crosslinked peptides on average (66% of all possible crosslinks), and 760 crosslinks (75% of all possible) when all replicates were aggregated after the search. Under search conditions where more free peptides are present, CRIMP 2.0 performed equally well, particularly in high complexity searches with extensive entrapment (Figure S5). FDR estimation is reasonably accurate in these peptide-contaminated datasets (1–3.4%), even with entrapment. Much larger datasets are likely required to generate a more precise estimate.
Figure 3.
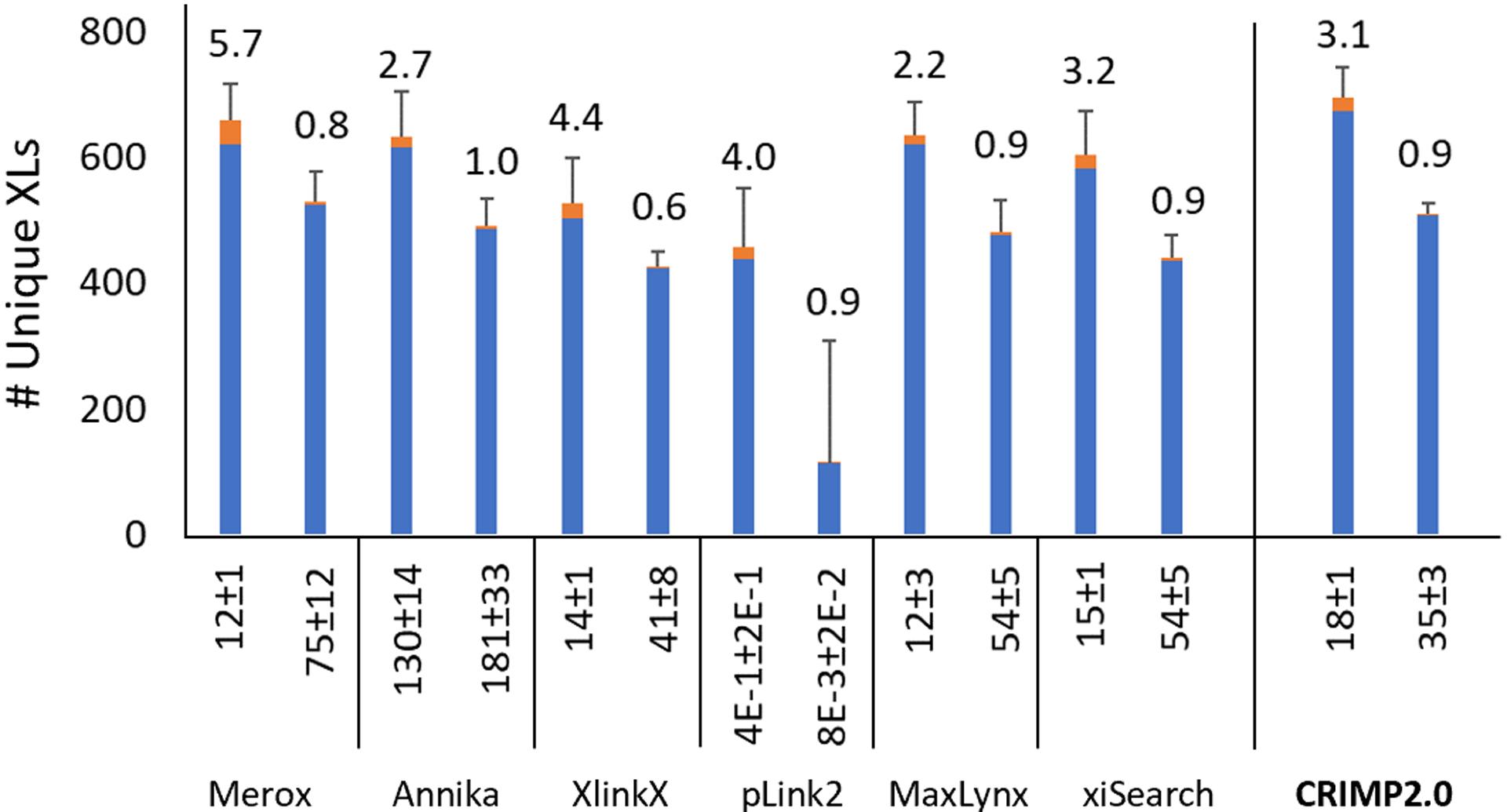
Average crosslinked peptide numbers using DSSO as the crosslinking reagent and a stepped HCD MS2 method for data acquisition. All results for the noted algorithms are derived from Matzinger et al.15 with the addition of CRIMP 2.0, at an expected FDR of 1% (left bar in pair), and corrected results are shown using a post-score cutoff to reach an experimentally validated FDR of 1% (right bar in pair). True positives in blue, false positives in orange. Real % FDR posted as callouts. Error bars indicate standard deviation of the total hits, n=3.
CRIMP 2.0 uses a new strategy for detecting PPIs. It has been the practice in the community to restrict a database search to only those proteins that are detected in the sample being crosslinked22. This detection is achieved with a separate proteomics analysis. The approach helps constrain the search space (and thus accelerate the search) and it also limits the generation of false positive identifications. Other methods use some element of compositional analysis to restrict the peptide search space29,30. CRIMP’s PPI scoring method incorporates all internal evidence for the existence of a protein in the score but does not demand it (Supplementary Information). That is, the detection of other crosslinks and all other possible reaction products for a given PPI are incorporated into the score. We and others have observed that bona fide PPIs often present multiple interprotein crosslinks, and thus should be granted a more confident identification, but single interprotein crosslinks do occur and should not be penalized. Our scoring method is applied during the aggregation of peptide identifications and is designed to boost PPI scores but not override these high-quality individual interprotein crosslinks. To demonstrate, we allocated all the possible crosslinked peptides from the synthetic peptide library of Matzinger et al.15 to their ribosomal proteins of origin. PPIs generated from searching this library will not be physically meaningful, but they will still be valid hits. This exercise generates 331 PPIs after removing redundancies, 300 of which are heterotypic and 31 that are homotypic. At 1% FDR, we detected 313 (95%) of the possible PPIs in a database search restricted to 171 proteins, which diminishes to 151 (46%) with heavy entrapment (20,334 human proteins) (Figure 4). These results indicate a robust scoring method. We see strong FDR control when searching crosslinks “contaminated” with linear peptide data, which shows that compositional data (in the form of free peptides at least) do not overwhelm the requirement for quality crosslink identification. Activating the compositional boost for this dataset has a negligible effect on FDR estimation (Figure S6).
Figure 4.

Number of detected PPIs in synthetic peptide benchmark dataset 2, from Matzinger et al.15 Blue bars represent a search conducted at an estimated 5% FDR and orange bars a search conducted at an estimated 1% FDR. All three sets of data from the benchmark were searched, with the indicated numbers of proteins in the search database to explore the effect of entrapment. Real FDR values are indicated as callouts.
A test on a dataset of high quality and limited complexity is not a perfect mimic of a full in situ crosslinking experiment. Therefore, we next analyzed the E. coli BS3 and DSSO crosslinking experiments from the Rappsilber group, involving 314 samples total, each processed with a 90 min LC-MS/MS runs on a QExactive22. Because no enrichment was used for this analysis beyond restricting the charge states selected during the DDA experiments, the dataset presents the full range of reaction products, both in terms of the type of reaction product and their yields. Using a single high-abundance fraction from the dataset, we conducted a grid search to determine the optimal database search settings. Interestingly, we did not see a need to expand N beyond 10 in the library reduction phase. True peptides were almost always found at the highest ranks of the list, indicating that our scoring method is effective for pre-searching linearized crosslinks in high-complexity states. Similarly, we found that an Eα of 10% and an Eβ of 99% provides near-optimal sensitivity, the same values used in the smaller datasets. Using these settings, all runs were searched as a single set for each crosslinker on a desktop, against the restricted search space used in the original study (approximately half of the proteome)22. The search generated identifications for all product types, where, not surprisingly, the interprotein links were detected at the lowest abundance (Table 1).
Table 1.
Identification of reaction products from the combined search of LC-MS/MS runs from an extensively fractionated in situ crosslinking experiment applied to E. coli lysate, for both BS3 and DSSO, at a 1% FDR.
| Interlinks | Homotypic links# | Intra-links | looplinks | Monolinks | free peptides | |
|---|---|---|---|---|---|---|
| BS3 | 12,415 | 3048 | 61,032 | 13,436 | 53,723 | 416,465 |
| DSSO | 20,104 | 5149 | 78,153 | 10,270 | 61,412 | 331,577 |
indicates a crosslink to two identical or overlapping sequences
The CRIMP runs completed in 43 hrs for BS3 (168 raw datafiles) and 39 hours for DSSO (146 raw datafiles) using modest hardware (10-core Intel i9–10850K 3.60GHz CPU with 32 GB of RAM), which is a reasonable time commitment given that each dataset required over 30 days to collect. To gauge the success of the crosslinking reaction, we have found it useful to conduct an ultrafast search with detuned parameters (e.g., increasing the minimum number of fragments in reduction, setting Eα and Eβ to 0.01% and 25%, and/or restricting the peptide size range). These searches will find high quality crosslinks. When preliminary results look promising, the samples can then be processed with greater sensitivity using the default settings.
Finally, the searches reveal a higher number of PPIs than first reported22. At 5% FDR, we calculate a total of 1820 PPIs for a nominal 5% FDR (compared with 756 in the original study) and 1254 PPIs at a nominal 1% FDR (compared with 590 in the original study). The calculated FDR seems to provide a reasonable estimate of the real error rate, given the approximate nature of the true PPI space (Figure 5). We note that the sensitivity arises from the compositional boost, as it increases the number of hits over two-fold compared to a composition-naïve scoring method. It supports the notion that the PPI search space is likely being overestimated in constructing an “all with all” interaction database. That is, adding an additional layer of information raise the scores of true interactors above a noise distribution that is likely wider than it should be. Interestingly, the overlap between the two reagents is 33% at 5% FDR, increasing slightly to 40% at 1% FDR, highlighting how differences in reagent design can affect sampling of the interactome.
Figure 5.

PPI search results for the in-situ crosslinking of the E. coli proteome using two crosslinking reagents. (A) searches conducted at a targeted 5% FDR and (B) searches conducted with a targeted 1% FDR. Results are based on the approximate PPI database established in Lenz et al.22, using the composition-informed PPI scoring method. Percentages at the bottom of the figure show calculated FDR values based on the composition of the library.
Conclusions
Our results demonstrate that a 2-pass database reduction method can return a sensitive measure of crosslinking composition in complex samples. Controlling the degree of database restriction allows the user to tune the search speed to meet the needs of the experiment, without causing great concern over lost sensitivity as the dependency on search parameters is modest and predictable. With minor adjustments to key search terms such as N, Eα and Eβ, even a human proteome and a dense multi-hour LC-MS/MS run can be processed in a day or less on a single desktop computer like the one used in this study. It is possible that brute force methods may prove to be less sensitive than 2-pass methods for highly complex systems. An unnecessary expansion of the database may generate a noisy search, much like proteomics searches do when they are parameterized with excessive numbers of variable modifications. CRIMP allows for a robust search of both cleavable and noncleavable crosslinkers alike and noncleavable reagents should get more attention for in situ applications. These reagents are easier to synthesize and are clearly complementary at this scale. Additionally, these reagents generate cross-peptide fragment ions that may be essential in validating hits, particularly when exploring highly complex states where interactions are defined by post-translational modifications. CRIMP 2.0 offers the sensitivity and search speed required for such activities.
Supplementary Material
Figure S1. 3D sensitivity plots for N, Eα and Eβ.
Figure S2. A screenshot of the post-search validation viewer in CRIMP 2.0.
Figure S3. A comparison of several crosslink search tools used on the synthetic peptide dataset benchmark 1, from Beveridge et al.21
Figure S4. Effect of entrapment on database search times.
Figure S5. Benchmarking the performance of XL search tools on complex samples, with database entrapment.
Figure S6. Analysis of the DSSO crosslinking data from the synthetic peptide benchmark dataset 2, for detection of PPIs.
ACKNOWLEDGMENTS
The authors would like to thank the many members of the community for extensive feedback provided on the use of CRIMP 2.0 for crosslinking applications. This work was funded by the Natural Sciences and Engineering Research Council of Canada Discovery Grant (RGPIN 2017-04879 and CANARIE grant RS3-084.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
- Additional experimental details
- Database segmentation for synthetic peptide benchmark dataset 1.
- CRIMP search settings for synthetic peptide benchmark dataset 1.
- CRIMP search settings for synthetic peptide benchmark dataset 2.
- CRIMP search settings for proteome-scale E. coli dataset
- Scoring XLs in CRIMP 2.0
- Core OMSSA++ score.
- Multiple Perspectives Scoring Strategy.
- Calculating the Competitive Label Assignment Method (CLAM) Composite Score
- Error estimation in CRIMP 2.0
- Aggregation
- Composition-informed PPI scoring
References
- (1).Budayeva HG; Cristea IM A Mass Spectrometry View of Stable and Transient Protein Interactions. Adv. Exp. Med. Biol 2014, 806, 263–282. 10.1007/978-3-319-06068-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Gingras AC; Abe KT; Raught B Getting to Know the Neighborhood: Using Proximity-Dependent Biotinylation to Characterize Protein Complexes and Map Organelles. Curr. Opin. Chem. Biol 2019, 48, 44–54. 10.1016/J.CBPA.2018.10.017. [DOI] [PubMed] [Google Scholar]
- (3).Belsom A; Rappsilber J Anatomy of a Crosslinker. Curr. Opin. Chem. Biol 2020, 60, 39–46. 10.1016/j.cbpa.2020.07.008. [DOI] [PubMed] [Google Scholar]
- (4).Sinz A Investigation of Protein-Protein Interactions in Living Cells by Chemical Crosslinking and Mass Spectrometry. Analytical and Bioanalytical Chemistry. 2010. 10.1007/s00216-009-3405-5. [DOI] [PubMed] [Google Scholar]
- (5).Pirklbauer GJ; Stieger CE; Matzinger M; Winkler S; Mechtler K; Dorfer V MS Annika: A New Cross-Linking Search Engine. J. Proteome Res 2021, 20 (5), 2560–2569. 10.1021/ACS.JPROTEOME.0C01000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Yllmaz Ş; Busch F; Nagaraj N; Cox J Accurate and Automated High-Coverage Identification of Chemically Cross-Linked Peptides with MaxLynx. Anal. Chem 2022, 94 (3), 1608–1617. https://doi.org/ 10.1021/ACS.ANALCHEM.1C03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Yu C; Huang L Cross-Linking Mass Spectrometry (XL-MS): An Emerging Technology for Interactomics and Structural Biology. Anal. Chem 2018, 90 (1), 144. 10.1021/ACS.ANALCHEM.7B04431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Chen ZL; Meng JM; Cao Y; Yin JL; Fang RQ; Fan SB; Liu C; Zeng WF; Ding YH; Tan D; Wu L; Zhou WJ; Chi H; Sun RX; Dong MQ; He SM A High-Speed Search Engine PLink 2 with Systematic Evaluation for Proteome-Scale Identification of Cross-Linked Peptides. Nat. Commun 2019, 10 (1). 10.1038/S41467-019-11337-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Netz E; Dijkstra TMH; Sachsenberg T; Zimmermann L; Walzer M; Monecke T; Ficner R; Dybkov O; Urlaub H; Kohlbacher O OpenPepXL: An Open-Source Tool for Sensitive Identification of Cross-Linked Peptides in XL-MS. Mol. Cell. Proteomics 2020, mcp.TIR120.002186. 10.1074/mcp.tir120.002186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Dai J; Jiang W; Yu F; Yu W Xolik: Finding Cross-Linked Peptides with Maximum Paired Scores in Linear Time. Bioinformatics 2019, 35 (2), 251–257. 10.1093/BIOINFORMATICS/BTY526. [DOI] [PubMed] [Google Scholar]
- (11).Götze M; Pettelkau J; Schaks S; Bosse K; Ihling CH; Krauth F; Fritzsche R; Kühn U; Sinz A StavroX-A Software for Analyzing Crosslinked Products in Protein Interaction Studies. J. Am. Soc. Mass Spectrom 2012. 10.1007/s13361-011-0261-2. [DOI] [PubMed] [Google Scholar]
- (12).Tang X; Bruce JE A New Cross-Linking Strategy: Protein Interaction Reporter (PIR) Technology for Protein-Protein Interaction Studies. Mol. Biosyst 2010, 6 (6), 939–947. 10.1039/B920876C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Iacobucci C; Götze M; Ihling CH; Piotrowski C; Arlt C; Schäfer M; Hage C; Schmidt R; Sinz A A Cross-Linking/Mass Spectrometry Workflow Based on MS-Cleavable Cross-Linkers and the MeroX Software for Studying Protein Structures and Protein-Protein Interactions. Nat. Protoc 2018, 13 (12), 2864–2889. 10.1038/S41596-018-0068-8. [DOI] [PubMed] [Google Scholar]
- (14).Kolbowski L; Lenz S; Fischer L; Sinn LR; O’Reilly FJ; Rappsilber J Improved Peptide Backbone Fragmentation Is the Primary Advantage of MS-Cleavable Crosslinkers. Anal. Chem 2022, 94 (22), 7779–7786. 10.1021/ACS.ANALCHEM.1C05266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Matzinger M; Vasiu A; Madalinski M; Müller F; Stanek F; Mechtler K Mimicked Synthetic Ribosomal Protein Complex for Benchmarking Crosslinking Mass Spectrometry Workflows. Nat. Commun. 2022 131 2022, 13 (1), 1–13. 10.1038/s41467-022-31701-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Mendes ML; Fischer L; Chen ZA; Barbon M; O’Reilly FJ; Giese SH; Bohlke‐Schneider M; Belsom A; Dau T; Combe CW; Graham M; Eisele MR; Baumeister W; Speck C; Rappsilber J An Integrated Workflow for Crosslinking Mass Spectrometry. Mol. Syst. Biol 2019, 15 (9). 10.15252/MSB.20198994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Rinner O; Seebacher J; Walzthoeni T; Mueller L; Beck M; Schmidt A; Mueller M; Aebersold R Identification of Cross-Linked Peptides from Large Sequence Databases. Nat. Methods 2008, 5 (4), 315–318. 10.1038/NMETH.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sinz A Divide and Conquer: Cleavable Cross-Linkers to Study Protein Conformation and Protein–Protein Interactions. Anal. Bioanal. Chem 2017, 409 (1), 33–44. https://doi.org/ 10.1007/s00216-016-9941-x. [DOI] [PubMed] [Google Scholar]
- (19).Chick JM; Kolippakkam D; Nusinow DP; Zhai B; Rad R; Huttlin EL; Gygi SP A Mass-Tolerant Database Search Identifies a Large Proportion of Unassigned Spectra in Shotgun Proteomics as Modified Peptides. Nat. Biotechnol 2015, 33 (7), 743–749. 10.1038/NBT.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sarpe V; Rafiei A; Hepburn M; Ostan N; Schryvers AB; Schriemer DC High Sensitivity Crosslink Detection Coupled With Integrative Structure Modeling in the Mass Spec Studio. Mol. Cell. Proteomics 2016, 15 (9), 3071. 10.1074/MCP.O116.058685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Beveridge R; Stadlmann J; Penninger JM; Mechtler K A Synthetic Peptide Library for Benchmarking Crosslinking-Mass Spectrometry Search Engines for Proteins and Protein Complexes. Nat. Commun 2020, 11 (1). 10.1038/S41467-020-14608-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lenz S; Sinn LR; O’Reilly FJ; Fischer L; Wegner F; Rappsilber J Reliable Identification of Protein-Protein Interactions by Crosslinking Mass Spectrometry. Nat. Commun 2021, 12 (1). 10.1038/S41467-021-23666-Z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Trnka MJ; Baker PR; Robinson PJJ; Burlingame AL; Chalkley RJ Matching Cross-Linked Peptide Spectra: Only as Good as the Worse Identification. Mol. Cell. Proteomics 2014, 13 (2), 420–434. 10.1074/MCP.M113.034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Singh P; Shaffer SA; Scherl A; Holman C; Pfuetzner RA; Larson Freeman TJ; Miller SI; Hernandez P; Appel RD; Goodlett DR Characterization of Protein Cross-Links via Mass Spectrometry and an Open-Modification Search Strategy. Anal. Chem 2008, 80 (22), 8799–8806. https://doi.org/ 10.1021/ac801646f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Hoopmann MR; Zelter A; Johnson RS; Riffle M; Maccoss MJ; Davis TN; Moritz RL Kojak: Efficient Analysis of Chemically Cross-Linked Protein Complexes. J. Proteome Res 2015, 14 (5), 2190–2198. 10.1021/PR501321H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Giese SH; Fischer L; Rappsilber J A Study into the Collision-Induced Dissociation (CID) Behavior of Cross-Linked Peptides. Mol. Cell. Proteomics 2016, 15 (3), 1094. 10.1074/MCP.M115.049296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kong AT; Leprevost FV; Avtonomov DM; Mellacheruvu D; Nesvizhskii AI MSFragger: Ultrafast and Comprehensive Peptide Identification in Mass Spectrometry–Based Proteomics. Nat. Methods 2017 145 2017, 14 (5), 513–520. 10.1038/NMETH.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Cox J; Hein MY; Luber CA; Paron I; Nagaraj N; Mann M Accurate Proteome-Wide Label-Free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteomics 2014, 13 (9), 2513–2526. 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zhou C; Dai S; Lin Y; Lian S; Fan X; Li N; Yu W Exhaustive Cross-Linking Search with Protein Feedback. J. Proteome Res 2022, 22 (1), 101–113. 10.1021/ACS.JPROTEOME.2C00500. [DOI] [PubMed] [Google Scholar]
- (30).Lima DB; de Lima TB; Balbuena TS; Neves-Ferreira AGC; Barbosa VC; Gozzo FC; Carvalho PC SIM-XL: A Powerful and User-Friendly Tool for Peptide Cross-Linking Analysis. J. Proteomics 2015, 129, 51–55. 10.1016/J.JPROT.2015.01.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. 3D sensitivity plots for N, Eα and Eβ.
Figure S2. A screenshot of the post-search validation viewer in CRIMP 2.0.
Figure S3. A comparison of several crosslink search tools used on the synthetic peptide dataset benchmark 1, from Beveridge et al.21
Figure S4. Effect of entrapment on database search times.
Figure S5. Benchmarking the performance of XL search tools on complex samples, with database entrapment.
Figure S6. Analysis of the DSSO crosslinking data from the synthetic peptide benchmark dataset 2, for detection of PPIs.