

Abstract
To investigate the frequency of heterogeneity among the multiple 16S rRNA genes within a single microorganism, we determined directly the 120-bp nucleotide sequences containing the hypervariable α region of the 16S rRNA gene from 475 Streptomyces strains. Display of the direct sequencing patterns revealed the existence of 136 heterogeneous loci among a total of 33 strains. The heterogeneous loci were detected only in the stem region designated helix 10. All of the substitutions conserved the relevant secondary structure. The 33 strains were divided into two groups: one group, including 22 strains, had less than two heterogeneous bases; the other group, including 11 strains, had five or more heterogeneous bases. The two groups were different in their combinations of heterogeneous bases. The former mainly contained transitional substitutions, and the latter was mainly composed of transversional substitutions, suggesting that at least two mechanisms, possibly misincorporation during DNA replication and horizontal gene transfer, cause rRNA heterogeneity.
Since ribosomes are an indispensable component of the protein synthesis apparatus and the structures are strictly conserved, the DNA component of the small ribosome subunit has been proven extensively to be an important and useful molecular clock for quantitating evolutionary relationships between organisms (2, 12, 29, 30). In virtually all species, the sequences of multicopy rRNA genes are identical or nearly identical and the homogeneity is thought to be governed by concerted evolution (8), which may originate from stringent selective pressure on the primary sequences of rRNA molecules to maintain their precise interactions with components of the complex protein-synthesizing machinery (29). From these characteristics, the DNA sequences of the 16S rRNA genes (rDNA) have become powerful tools in modern prokaryotic taxonomy with the increasing use of PCR technology (21, 25, 26).
The phylogenetic classification of prokaryotes with 16S rDNA sequences is based on the assumption that the differences in sequences reflect the evolution of the organisms that they have been extracted from. Interpretation of these sequence data may be complicated by the presence of several equivalent molecules with some degree of individually different evolutionary patterns within a single organism. Several recent studies using rDNA sequencing have reported the existence of divergent 16S rRNA sequences within a single organism in the eubacterial actinomycete Thermobispora bispora (26), the archaebacterium Halobacterium marismortui (15), and phytoplasma (14). These studies clearly showed the presence of sequence heterogeneity between rRNA operons on single genomes, but it has not been elucidated whether such intra-rRNA heterogeneity is peculiar or general. To clarify this, the extent of such sequence heterogeneity should be systematically studied; this would be important not only for estimation of the consistency of the risk frequency with the phylogenetic relationship reconstructed by using cloned 16S rRNA genes but also for evaluating the mechanism introducing breaks into the rRNA homogeneity fixed by hypothetical concerted evolution.
To address this need, an extensive study was conducted with a partial sequence containing the most variable α region (25) of the 16S rRNA genes from 475 standard type strains belonging to the genus Streptomyces. This is the first study to estimate the extent of heterogeneity in the 16S rRNA genes within a single microorganism. The results presented here may provide important information concerning the application of 16S rDNA sequences for phylogenetic analyses and the evolutionary mechanisms of the redundant multigenes on a single genome.
MATERIALS AND METHODS
Strains, cultivation, and DNA preparation.
The Streptomyces strains used were provided by the Japan Culture Collection of Microorganisms (JCM), and most were standard strains of the International Streptomyces Project (23, 24). The strains were cultivated on specific agar plates, as recommended in the JCM Catalogue of Strains (16). Chromosomal DNA for the PCR template was isolated from a single colony formed on a plate by using the Insta Gene kit (Bio-Rad) according to the supplier’s protocol. To exclude the possibility of template DNA contamination, threefold expansion of single colonies was employed.
PCR amplification and sequencing of part of the 16S rDNA.
To detect the heterogeneous 16S rRNA genes within a single strain, we employed direct sequencing of PCR products through a single-stranded template produced by λ exonuclease, as described previously (7, 11). The sequences of the synthesized oligonucleotides used were as follows: sense primer for PCR, 5′-TCACGGAGAGTTTGATCCTG-3′; antisense primer for PCR, 5′-GCGGCTGCTGGCACGTAGTT-3′; sequencing primer, 5′-AGTAACACGTGGGCAATCTG-3′. The sequences for each primer were selected from the conserved region and corresponded to nucleotide positions 1 to 20, 481 to 500, and 105 to 124, respectively, of the S. ambofaciens rDNA sequence (18). A nucleotide sequence covering the variable region of 16S rDNA was amplified by using phosphorylated sense and nonphosphorylated antisense primers. The sense primer was phosphorylated with T4 polynucleotide kinase (Takara) and ATP. PCR was performed in a 50-μl reaction mixture for 30 or 40 cycles of denaturation (for 30 s at 97°C), annealing (for 1 min at 50°C), and extension (for 1 min at 72°C) with AmpliTaq DNA polymerase (Perkin-Elmer). Whole samples were fractionated by agarose gel electrophoresis, the 0.5-kbp PCR products were recovered, and the phosphorylated sense strand was digested for 1 h at 37°C with λ exonuclease (BRL) in the recommended buffer (67 mM glycine-KOH, pH 9.4; 2.5 mM MgCl2). After phenol-chloroform extraction, the remaining antisense strand was used as a sequencing template.
DNA sequences were determined with a 7-deaza-dGTP Sequenase, version 2.0, kit (United States Biochemical Corp.) according to the supplier’s protocol, except that 1 μg of single-stranded DNA binding protein (SSB) (Stratagene) was added to 15 μl of the reaction mixture. After the termination reaction, SSB was digested with 20 μg of proteinase K (Sigma) for 30 min at 37°C in 6 μl of the termination mixture.
Cloning and sequencing of the amplified 16S rDNAs.
The whole 16S rDNA region was amplified by PCR with primers 5′-GGGAAGCTTCACGGAGAGTTTGATCCT-3′ and 5′-CCCTCTAGAAAGGAGGTGATCCAGC-3′, corresponding to nucleotide positions 1 to 18 and 1511 to 1526, respectively, of the S. ambofaciens rDNA sequence. After purification of the products through agarose gel electrophoresis, the PCR products were digested with HindIII and XbaI, recognition sequences of which were included in the primers, followed by cloning into pBluescript SK+ digested with the same enzymes. The transformation of Escherichia coli JM105 and DNA handling were performed according to the standard protocol (22). The DNA sequences of selected clones were determined with the BcaBest DNA sequencing kit (Takara) according to the supplier’s protocol.
Analysis of DNA sequences.
Editing of the determined DNA sequences and prediction of the local secondary structures were performed by the GENETYX program (Software Development) on a PC9801 personal computer (NEC).
RESULTS
Detection of strains carrying heterogeneous 16S rRNA genes.
Direct sequencing patterns from the 475 Streptomyces strains were displayed. Theoretically, if the heterogeneous 16S rRNA genes are present within a single strain, multiple bands at the same electrophoretic distances should appear. As expected, such multiple bands were observed in the sequencing patterns from several strains. The sequencing pattern of S. althioticus JCM4344, representing 11 heterogeneous signals, is shown in Fig. 1 as an example. Among the 475 sequencing patterns displayed, 33 patterns (6.9%) exhibited such heterogeneous sequencing patterns. Table 1 presents strain names with JCM numbers and the numbers of heterogeneous loci. The heterogeneous signals appeared irregularly within the α region and were never observed within the conserved region, suggesting that the multiple bands did not result from mutations during PCR but originated from heterogeneity within the α region of the 16S rRNA genes within a strain. Indeed, the existence of the heterogeneous 16S rRNA genes was confirmed in some strains through cloning and sequencing of the PCR products as mentioned below. These results clearly indicate that the existence of naturally occurring heterogeneous 16S rRNA genes within a single microorganism is not rare.
FIG. 1.
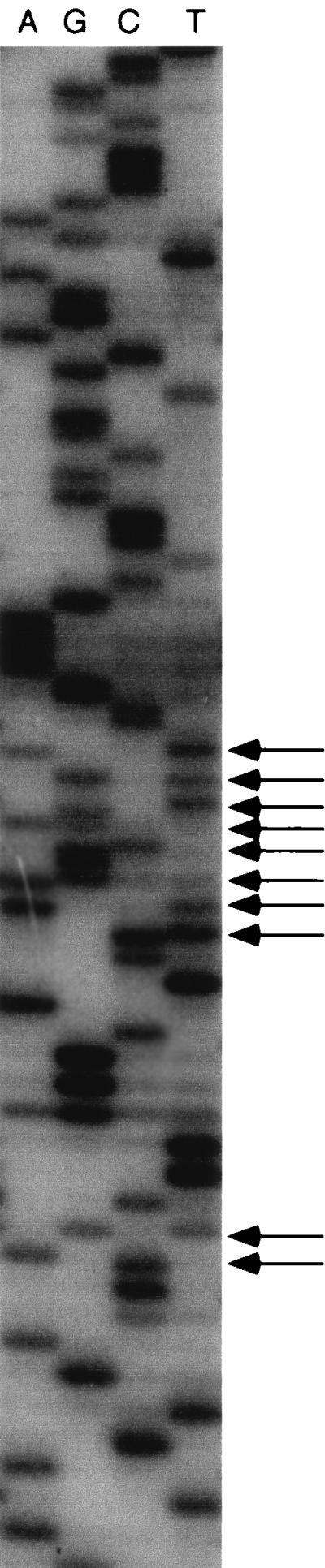
Direct sequencing pattern of the hypervariable α region of S. althioticus JCM4344 16S rDNA. Arrows indicate the loci representing heterogeneity.
TABLE 1.
Strains with heterogeneity in the sequenced region
| JCM strain no. | Organism | No. of combination of heterogeneous basesa
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R | M | W | S | K | Y | sss | B | H | D | V | Sum | ||
| 4840 | S. thermodiastaticus | 1 | 3 | 1 | 4 | 4 | 1 | 14 | 0 | 0 | 0 | 0 | 14 |
| 3131 | S. ruber | 1 | 1 | 0 | 3 | 5 | 2 | 12 | 0 | 0 | 0 | 0 | 12 |
| 4081 | Streptomyces sp. | 1 | 0 | 0 | 4 | 4 | 2 | 11 | 0 | 0 | 0 | 1 | 12 |
| 4958 | S. ederensis | 1 | 0 | 0 | 6 | 3 | 1 | 11 | 0 | 0 | 0 | 0 | 11 |
| 4771 | S. hydrogenans | 1 | 0 | 0 | 4 | 6 | 0 | 11 | 0 | 0 | 0 | 0 | 11 |
| 4823 | S. roseiscleroticus | 1 | 1 | 0 | 2 | 5 | 2 | 11 | 0 | 0 | 0 | 0 | 11 |
| 3070 | S. armeniacus | 0 | 2 | 1 | 3 | 4 | 0 | 10 | 0 | 0 | 0 | 0 | 10 |
| 4344 | S. althioticus | 2 | 1 | 2 | 1 | 3 | 1 | 10 | 0 | 0 | 0 | 0 | 10 |
| 4862 | S. xanthocidicus | 0 | 0 | 0 | 5 | 3 | 1 | 9 | 0 | 0 | 0 | 0 | 9 |
| 4651 | S. matensis | 1 | 1 | 1 | 0 | 3 | 1 | 7 | 0 | 1 | 0 | 0 | 8 |
| 4845 | S. tropicalensis | 0 | 1 | 0 | 2 | 2 | 0 | 5 | 0 | 0 | 0 | 0 | 5 |
| 4591 | S. luridus | 0 | 0 | 1 | 0 | 0 | 1 | 2 | 0 | 0 | 0 | 0 | 2 |
| 5067 | S. crystallinus | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| 5075 | S. capillispiralis | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| 4508 | S. prunicolor | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 |
| 4523 | S. varsoviensis | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 |
| 4624 | S. aureofaciens | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 |
| 4773 | S. iakyrus | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 |
| 4976 | S. longwoodensis | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 |
| 4276 | S. griseofuscus | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| 4521 | S. umbrinus | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| 4653 | S. misakiensis | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| 4792 | S. moderatus | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| 4856 | S. viridochromogenes | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| 4360 | S. coerulescens | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| 4841 | S. thermonitrificans | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| 5058 | S. anthocyanicus | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| 5065 | S. tricolor | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| 4962 | S. geysiriensis | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 |
| 4350 | S. badius | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| 4832 | S. spectabilis | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 |
| 4620 | S. antibioticus | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 |
| 4785 | S. lusitanus | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 |
| Totals | 12 | 11 | 6 | 36 | 47 | 18 | 130 | 2 | 2 | 1 | 1 | 136 | |
The abbreviations of heterogeneous combinations are according to the nomenculture of the International Union of Pure and Applied Chemistry, as follows: R, A or G; M, A or C; W, A or T; S, G or C; K, G or T; Y, C or T; B, not A; H, not G; D, not C; U, not T. sss and sum indicate the total numbers of duplicate combinations and heterogeneous bases, respectively.
Two classes of strains possessing heterogeneous 16S rRNA genes.
If the heterogeneity of the rRNA genes is caused by a simple mechanism, the frequency distribution of strains with different numbers of heterogeneous bases in the 16S rRNA genes must be simple. As summarized in Table 2, the frequency of appearance reveals that the distribution of the strains is discontinuous and biphasic. Twenty-two strains, 21 of 33 strains with only one heterogeneous base and 1 strain with two heterogeneous bases, were placed in one group (the small group); the other strains, having more than five heterogeneous bases, were classified into another group (the large group), indicating the possibility that rRNA heterogeneity in a single genome is not governed by simple mechanisms. Interestingly, the large group exhibited typical gamma distribution. The maximum number of heterogeneous loci is 14, in S. thermodiasticus JCM4840, and strains with 11 heterogeneous loci are the most frequent in the large group.
TABLE 2.
Frequency of sequence heterogeneity detected in the 16S rDNA variable region (120 bp) of Streptomyces strains
| No. of heterogeneous basesa | No. (%) of strains |
|---|---|
| 14 | 1 (0.2) |
| 13 | 0 (0) |
| 12 | 2 (0.4) |
| 11 | 3 (0.6) |
| 10 | 2 (0.4) |
| 9 | 1 (0.2) |
| 8 | 1 (0.2) |
| 7 | 0 (0) |
| 6 | 0 (0) |
| 5 | 1 (0.2) |
| 4 | 0 (0) |
| 3 | 0 (0) |
| 2 | 1 (0.2) |
| 1 | 21 (4.4) |
| 0 | 442 (93.1) |
| Total | 475 (100) |
Number of heterogeneous loci detected.
To characterize the heterogeneity in detail, the combination patterns of bases in the heterogeneous loci were studied. Two transversional combinations, K (G and T) and S (G and C), appeared frequently (61% of all heterogeneous loci), and the transitional combination frequencies, Y (C and T) and R (A and G), were 22% (Table 3). As mentioned above, the two groups may have distinct origins. Thus, there is the possibility that their heterogeneous base combinations are different. In the small group, triplet heterogeneous combinations were often found at a frequency of 17%. The transitional combinations were present at frequencies of 52.5% of total combinations without triplet combinations, and the frequency of Y was almost twofold higher than R. In contrast to the small group, the extent of transversional combinations was 80%, that of transitional combinations was only 18%, and the frequency of Y was almost identical to that of R in the large group. It is noteworthy that the triplet combinations are rare (1.8%) in the large group. These results strongly suggest that rRNA heterogeneity is governed by at least two distinct mechanisms.
TABLE 3.
Base composition at heterogeneous loci
| Base combination | No. (%) of strains
|
||
|---|---|---|---|
| Small groupa | Large groupb | Total | |
| K (GT) | 5 (21.7) | 42 (37.2) | 47 (34.6) |
| S (GC) | 2 (8.7) | 34 (30.1) | 36 (26.5) |
| Y (CT) | 7 (30.4) | 11 (9.7) | 18 (13.2) |
| R (AG) | 3 (13.0) | 9 (8.0) | 12 (8.8) |
| M (AC) | 1 (4.3) | 10 (8.8) | 11 (8.1) |
| W (TA) | 1 (4.3) | 5 (4.4) | 6 (4.4) |
| H (ACT) | 1 (4.3) | 1 (0.9) | 2 (1.5) |
| B (GCT) | 2 (8.7) | 0 (0) | 2 (1.5) |
| V (AGC) | 0 (0) | 1 (0.9) | 1 (0.7) |
| D (AGT) | 1 (4.3) | 0 (0) | 1 (0.7) |
| Totals | 23 (100) | 113 (100) | 136 (100) |
Sequences having less than two heterogeneous bases.
Sequences having more than five heterogeneous bases.
Structural conservation of the heterogeneous 16S rRNAs.
It is known that the α region possibly forms a stem and loop structure designated helix 10 (4) and that one of the criteria for whether the rRNA gene-like sequence is a functional gene is structural conservation. To confirm this, the secondary structures of the sequences were predicted. All of the heterogeneous loci in the small group occurred in the stem region of helix 10, and the predicted secondary structure was not affected by one- or two-base heterogeneity. Since predicting the secondary structure is not possible when the sequencing pattern exhibits multiheterogeneous bases, it is necessary to determine the sequence of each 16S rRNA gene of such strains. Thus, we cloned and sequenced the PCR products from the strains of the large group. The PCR product from the DNA of S. thermodiastaticus JCM4840, in which direct sequencing revealed 14 heterogeneous bases, was cloned into pBluescript. Ten clones were selected randomly, and the DNA sequences of the 120-bp region were determined. As shown in Fig. 2A, in two distinct sequences that were predicted from the heterogeneous sequencing pattern displayed by the direct method (data not shown), the heterogeneity was limited to within nucleotides 179 to 197, corresponding to the stem region of helix 10. The predicted secondary structure of each sequence formed the stem and loop structure as expected (Fig. 2B). The same result was obtained from the sequencing analysis of the clones from S. armeniacus JCM3070, the direct sequencing of which revealed 11 heterogeneous bases (data not shown). Thus, all heterogeneous loci were located in the stem part of the α region without alteration of the secondary structure, as demonstrated by comparison of different strains (11), indicating that the heterogeneous rDNA sequences in these strains encode functional 16S rRNAs.
FIG. 2.

(A) Secondary structures of S. thermodiastaticus JCM4840 16S rRNA regions used this study. Helix numbering is according to the nomenclature (4), and the base position numbering is according to that of S. ambofaciens (18). The arrow indicates the region used as a sequencing primer. Heterogeneous loci are indicated by the abbreviations given in Table 1, footnote a. (B) Secondary structures of helix 10 of two distinct sequences in S. thermodiastaticus 16S rRNAs. Bars and dots indicate Watson-Crick pairing and non-Watson-Crick pairing, respectively.
DISCUSSION
Heterogeneity among rRNA genes within a single organism has been investigated with denaturing gel electrophoresis (17) and DNA sequencing of the cloned gene (14, 15, 28). Our extensive direct sequencing approach, using the most variable region of Streptomyces 16S rDNA, revealed the general existence of this heterogeneity. How is the heterogeneity generated? It is generally considered that mutations are introduced by misincorporation and misrepair by DNA polymerase during DNA replication. If the mutations occurred equally on each substitutions, the expected value of each heterogeneous base combination would be 17%, and thus the frequency of the transition set must be 33%. Estimation of the substitution pattern using pseudogenes which might not be exposed to selective pressure (6) showed that transition is more frequent (53%) than transversion (47%). This is considered to reflect the tendency for base substitutions during DNA replication during the evolutionary process of the genome. In the small group, the frequency of transition, except for triplet bases, was 52.5%, which is almost identical to that of the pseudogenes, suggesting that the heterogeneity observed in the small group might be unbalanced in the steady state of molecular evolution of the redundant 16S rRNA genes that, during DNA replication, might be exposed to the selective pressure of concerted evolution.
In the large group, the frequency of transitional combination was only 18%, which is much lower than the expected value (33%), suggesting that the driving force for the origin of the heterogeneity in the group is not mutations during DNA replication, as in the small group. It is known that conjugative plasmids are generally found in Streptomyces, and the plasmids can mobilize the host chromosome at high frequency (9). One possible explanation for the mechanism that generates the heterogeneity in the large group is horizontal gene transfer mediated by conjugative plasmids. This consideration is not limited to the genus Streptomyces. In other microorganisms, gene flux mediated by conjugative plasmids is thought to have an important role in increasing genetic diversity (1, 27), suggesting that it is a mechanism common to many microorganisms.
All of the heterogeneous loci were located in the stem region of helix 10. The same result was obtained from the comparison of different strains in previous studies (11). Evolutional selective pressure probably operates on the secondary structure of the region, and the base composition of helix 10 may not affect rRNA function. The substitution rate at the stem region is estimated to be twofold higher than that at the loop region (20). The helix 10 stem of Streptomyces 16S rRNA is considered to be a typical tolerance region against mutations which do not alter the secondary structure.
The heterogeneous base combinations of the large and small groups exhibited significant differences (Table 2). In the 120-bp region, the conservation of the helix 10 stem structure is obviously due to selective pressure. Therefore, mutations occurring in the stem region are individually deleterious if they destabilize the important structure; fitness can be restored, however, when a compensation occurs that reestablishes the pairing potential. Among all non-Watson-Crick nucleotide pairs, the U-G pair appears to be the least deleterious (10) and in some cases even offers a selective advantage (19), suggesting that the base combinations in the small group result in the compensation of substitutions due to U-G pairing. It is thought that exchange between heterogeneous 16S rRNA genes, or the consolidation of heterogeneous 16S rRNA genes, occurred in the large group. If the heterogeneity of the group is established through these mechanisms, there is no need for compensation mutations by non-Watson-Crick pairing, and the restriction on heterogeneous base combination is not severe. This is probably the reason for the differences in the heterogeneous base combinations between the two groups.
Some of the results of analysis of 16S rRNA genes are critical for phylogenetic analysis or prokaryotic taxonomy. Recent comparative analysis of sequences deposited in GenBank revealed a level of intraspecific and intrastrain sequence variation that cannot be explained exclusively by errors in laboratory procedures (3), and almost identical 16S rRNA sequences have been reported in phenotypically divergent bacteria (5). Despite these results, sequence analysis of the 16S rRNA gene is of great importance for modern bacterial taxonomy, and the 16S rRNA sequence database is the most complete (13). From the viewpoint of phylogenetic analysis, it is difficult to consider that one- or two-base heterogeneity within the α region affects the topology of the phylogenetic relationship reconstructed with 16S rRNA sequences. Indeed, in our previous study, one- or two-base heterogeneity within the α region did not influence the phylogenetic relationship reconstructed with the 120-bp region used in this study (11). In contrast, the number of heterogeneous bases in the large group was enough to alter the topology of the phylogenetic tree constructed with 16S rDNA sequences. Thus, the frequency of the risk of using single 16S rRNA genes for phylogenetic comparison would be estimated as more than 2% per strain, based on the frequency of appearance of the strains belonging to the large group.
REFERENCES
- 1.Amabile-Cuevas C F, Chicurel M E. Bacterial plasmids and gene flux. Cell. 1992;70:189–199. doi: 10.1016/0092-8674(92)90095-t. [DOI] [PubMed] [Google Scholar]
- 2.Amann R I, Ludwig W, Schleifer K-H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev. 1995;59:143–169. doi: 10.1128/mr.59.1.143-169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clayton R A, Sutton G, Hinkle P S, Jr, Bult C, Fields C. Intraspecific variation in small-subunit rRNA sequences in GenBank: why single sequences may not adequately represent prokaryotic taxa. Int J Syst Bacteriol. 1995;45:595–599. doi: 10.1099/00207713-45-3-595. [DOI] [PubMed] [Google Scholar]
- 4.De Rijk P, Neefs J M, Van de Peer Y, De Wachter R. Compilation of small ribosomal subunit RNA sequences. Nucleic Acids Res. 1992;20:2075–2089. doi: 10.1093/nar/20.suppl.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fox G E, Wisotzkey J D, Jurtshuk P., Jr How close is close: 16S rRNA sequence identity may not be sufficient to guarantee species identity. Int J Syst Bacteriol. 1992;42:166–170. doi: 10.1099/00207713-42-1-166. [DOI] [PubMed] [Google Scholar]
- 6.Gojobori T, Li W-H, Graur D. Patterns of nucleotide substitution in pseudogenes and functional genes. J Mol Evol. 1982;18:360–369. doi: 10.1007/BF01733904. [DOI] [PubMed] [Google Scholar]
- 7.Higuchi R G, Ochman H. Production of single-stranded DNA templates by exonuclease digestion following the polymerase chain reaction. Nucleic Acids Res. 1989;17:5865. doi: 10.1093/nar/17.14.5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hillis D M, Moritz C, Porter C A, Baker R J. Evidence for biased gene conversion in concerted evolution of ribosomal DNA. Science. 1991;251:308–310. doi: 10.1126/science.1987647. [DOI] [PubMed] [Google Scholar]
- 9.Hopwood D A, Kieser T. Conjugative plasmid in Streptomyces. In: Clewell D B, editor. Bacterial conjugation. New York, N.Y: Plenum Press; 1993. pp. 293–311. [Google Scholar]
- 10.James B D, Olsen G J, Liu J, Pace N R. The secondary structure of ribonuclease P RNA, the catalytic element of ribonucleoprotein enzymes. Cell. 1988;52:19–26. doi: 10.1016/0092-8674(88)90527-2. [DOI] [PubMed] [Google Scholar]
- 11.Kataoka M, Ueda K, Kudo T, Seki T, Yoshida T. Application of the variable region in 16S rDNA to create an index for rapid species identification in the genus Streptomyces. FEMS Microbiol Lett. 1997;151:249–255. doi: 10.1111/j.1574-6968.1997.tb12578.x. [DOI] [PubMed] [Google Scholar]
- 12.Lane D, Pace B, Olson G J, Stahl D A, Sogin M L, Pace N R. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci USA. 1985;82:6955–6959. doi: 10.1073/pnas.82.20.6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Larsen N, Olsen G J, Maidak B L, McCaughey M J, Overbeek R, Macke T J, Marsh T L, Woese C R. The ribosomal database project. Nucleic Acids Res. 1993;21:3021–3023. doi: 10.1093/nar/21.13.3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liefting L W, Andersen M T, Beever R E, Gardner R C, Forster R L S. Sequence heterogeneity in the two 16S rRNA genes of Phormium yellow leaf phytoplasma. Appl Environ Microbiol. 1996;62:3133–3139. doi: 10.1128/aem.62.9.3133-3139.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mylvaganam S, Dennis P P. Sequence heterogeneity between the two genes encoding 16S rRNA from the halophilic archaebacterium Haloarcula marismortui. Genetics. 1992;130:399–410. doi: 10.1093/genetics/130.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakase T. JCM catalogue of strains. 5th ed. Saitama, Japan: RIKEN; 1992. [Google Scholar]
- 17.Nübel U, Engelen B, Felske A, Snaidr J, Wieshuber A, Amann R I, Ludwig W, Backhaus H. Sequence heterogeneities of genes encoding 16S rRNAs in Paenibacillus polymyxa detected by temperature gradient gel electrophoresis. J Bacteriol. 1996;178:5636–5643. doi: 10.1128/jb.178.19.5636-5643.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pernodet J-L, Boccard F, Alegre M T, Gagnat J, Guérineau M. Organization and nucleotide sequence analysis of a ribosomal RNA gene cluster from Streptomyces ambofaciens. Gene. 1989;79:33–46. doi: 10.1016/0378-1119(89)90090-5. [DOI] [PubMed] [Google Scholar]
- 19.Rousset F, Pélandakis M, Solignac M. Evolution of compensatory substitutions through G · U intermediate state in Drosophila rRNA. Proc Natl Acad Sci USA. 1991;88:10032–10036. doi: 10.1073/pnas.88.22.10032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rzhetsky A. Estimating substitution rates in ribosomal RNA genes. Genetics. 1995;141:771–783. doi: 10.1093/genetics/141.2.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saiki R K, Gelfand D H, Stoffel S, Scharf S J, Higuchi R, Horn G T, Mullis K B, Erlich H A. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. 1988;239:487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- 22.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 23.Shirling E B, Gottlieb D. Cooperative description of type cultures of Streptomyces. III. Additional species descriptions from first and second studies. Int J Syst Bacteriol. 1968;18:279–391. [Google Scholar]
- 24.Shirling E B, Gottlieb D. Cooperative description of type cultures of Streptomyces. IV. Species descriptions from second, third, and fourth studies. Int J Syst Bacteriol. 1969;19:392–512. [Google Scholar]
- 25.Stackebrandt E, Witt D, Kemmerling C, Kroppenstedt R, Liesack W. Designation of streptomycete 16S and 23S rRNA-based target regions for oligonucleotide probes. Appl Environ Microbiol. 1991;57:1468–1477. doi: 10.1128/aem.57.5.1468-1477.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stackebrandt E, Liesack W, Witt D. Ribosomal RNA and rDNA sequence analyses. Gene. 1992;115:255–260. doi: 10.1016/0378-1119(92)90567-9. [DOI] [PubMed] [Google Scholar]
- 27.Syvanen M. Horizontal gene transfer: evidence and possible consequences. Annu Rev Genet. 1994;28:237–261. doi: 10.1146/annurev.ge.28.120194.001321. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Zhang Z, Ramanan N. The actinomycete Thermobispora bispora contains two distinct types of transcriptionally active 16S rRNA genes. J Bacteriol. 1997;179:3270–3276. doi: 10.1128/jb.179.10.3270-3276.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woese C R. Bacterial evolution. Microbiol Rev. 1987;51:221–271. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woese C R, Kandler O, Wheelis M L. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria and Eucarya. Proc Natl Acad Sci USA. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]