

ABSTRACT
Tuberculosis is still the leading cause of death globally from any infectious disease, despite the widespread use of the live attenuated vaccine Bacille Calmette Guerin (BCG). While BCG has some efficacy against disseminated TB disease in children, protection wanes into adulthood resulting in over 1.8 million TB deaths per year. This has led to efforts to develop novel vaccine candidates that either replace or boost BCG, as well as to test novel delivery mechanisms to enhance BCG’s efficacy. Traditional BCG vaccination is performed as an intradermal (ID) injection but delivering BCG by an alternate route may enhance the depth and breadth of protection. Previously, we demonstrated that phenotypically and genotypically disparate Diversity Outbred (DO) mice have heterogenous responses to M. tuberculosis challenge following intradermal BCG vaccination. Here, we utilize DO mice to examine BCG-induced protection when BCG is delivered systemically via intravenous (IV) administration. We find that DO mice vaccinated with IV BCG had a greater distribution of BCG throughout their organs compared to ID-vaccinated animals. However, compared to ID-vaccinated mice, M. tuberculosis burdens in lungs and spleens were not significantly reduced in animals vaccinated with BCG IV, nor was lung inflammation significantly altered. Nonetheless, DO mice that received BCG IV had increased survival over those vaccinated by the traditional ID route. Thus, our results suggest that delivering BCG by the alternate IV route enhances protection as detected in this diverse small animal model.
KEYWORDS: Mycobacterium tuberculosis, vaccine, BCG, route, intravenous, diversity outbred, mycobacterium, tuberculosis
INTRODUCTION
Tuberculosis remains a global public health problem despite the widespread use of the BCG vaccine for over 100 years (1). While BCG can provide protection early in life against disseminated Mycobacterium tuberculosis (M.tb.) infection in children, protection wanes in adulthood. However, M.tb. is still the leading cause of death from any infectious disease despite widespread BCG vaccination, with ~1.8 million deaths annually associated with pulmonary tuberculosis (2). The full range of factors that cause waning protection in the face of BCG vaccination are unknown, as is the absolute efficacy of BCG; efficacy estimates in different areas of the world range from 0 to 80% (2). However, BCG has been administered to over 3 billion people and maintains an excellent safety record. BCG is therefore still the gold standard against which other vaccines against tuberculosis are compared. Given the current widespread use of BCG, finding ways to augment the protection afforded by BCG could have substantial public health implications.
Novel vaccination strategies to enhance immunity primed by BCG include revaccination, vaccination by alternate routes, and utilizing BCG in a prime-boost strategy with a novel vaccine candidate (3). Several of these strategies are currently being employed in various human clinical trials, with some success. For example, a recent phase 2 clinical study in which adolescent subjects who had received BCG vaccination at birth were revaccinated with BCG exhibited about 45% less sustained Quantiferon conversion compared to placebo treatment, interpreted as reduction of tuberculosis infection (4). While such clinical results are promising for the field, room for improvement remains. One approach could be to deliver BCG by an alternate route. BCG is traditionally administered as an intradermal (ID) injection but delivering BCG directly to the lungs may position the vaccine to generate better protective immunity at the site of typical primary infection. Studies examining the protective efficacy of BCG or novel vaccine candidates delivered by the aerosol or intranasal route have had variable success in several animal models, including mice and nonhuman primates (NHP) (5–10).
Another alternative is to deliver BCG or other vaccines by a method that would promote a heightened systemic response, such as intravenous delivery. This method has been studied for the delivery of malaria vaccines, and several human clinical trials have demonstrated the potential to boost efficacy by intravenous (IV) administration (1). A recent study in rhesus macaques demonstrated that the majority of animals that received BCG IV were well protected against M.tb. challenge (11). IV BCG administration has been tested by several investigators over the years in mice (12–16). Most of these studies used inbred C57BL/6J (C57) mice, and enhanced protection against primary M.tb. challenge compared to ID vaccination was not readily observed. Given that IV BCG was successful in providing enhanced protection in NHP, yet not in C57 mice, we hypothesized that the inherent resistance of C57 mice may mask any enhanced protection by BCG given IV.
One potential alternative to traditional inbred mouse strains is Diversity Outbred (DO) mice. DO mice are an outbred population developed by mixed breeding of eight founder mouse lines that included both inbred laboratory and wild-derived strains (17). Iterative outbreeding has produced a population of animals with high genetic diversity between individuals, on the order of that observed in humans. Moreover, a large component of the genetic diversity is contributed by the wild-derived mice (17). Diversity Outbred (DO) mice therefore provide a unique model that is genetically and phenotypically heterogenous, and thus well suited for studying vaccine-induced protection against M.tb.
We and others have demonstrated that DO mice given a low dose aerosol primary M.tb. challenge exhibit a wide array of disease presentations and susceptibilities (18–20). ID BCG vaccination provided protection across the DO population in terms of increased survival and reductions in mean lung and spleen CFU (CFU) (19). However, some animals are not protected by BCG. A wide range of outcomes was found across the population following vaccination and challenge, similar to the wide range of outcomes seen after primary challenge alone (19). This heterogeneity may reflect the broad spectrum of disease observed in humans. Here, we utilized DO mice to test whether vaccination with BCG IV affords better protection than BCG administered ID. The results demonstrate that vaccination of DO mice with BCG IV provides moderately enhanced protection against M.tb. challenge compared to ID BCG vaccination, particularly in terms of improved survival.
RESULTS
Because BCG infection patterns have not been studied in outbred DO mice, we assessed the dissemination of BCG after ID or IV vaccination. Groups of DO mice were vaccinated either ID or IV with 105 BCG Pasteur, and the distribution of BCG in lungs, livers, spleens, bone marrow, and whole blood was assessed 8 weeks after vaccination. This time point was chosen to evaluate the BCG burden and host environment when mice were challenged with M.tb. at 8 weeks after vaccination in subsequent experiments. BCG distribution in DO mice vaccinated ID followed a pattern similar to that reported previously (21). At this time point after vaccination, only 2/18 ID-vaccinated DO mice had detectable BCG in lungs (Fig. 1A), and no animals had detectable BCG in livers (Fig. 1C), bone marrow (Fig. 1D), or whole blood (unpublished data). However, a majority of spleens of ID-vaccinated DO mice (8/12) contained detectable BCG (Fig. 1B).
FIG 1.

Delivering BCG intravenously increases the deposition of BCG in organs compared to parenteral vaccination. Groups of 10 DO mice were vaccinated intradermally (ID) or intravenously (IV) with 105 BCG Pasteur. Animals were euthanized 8 weeks after vaccination for BCG enumeration in A) lungs, B) spleens, C) livers, and D) bone marrow. Bone marrow comprises eluate from both femurs of each mouse. Data from two independent experiments are combined and are represented as log10 BCG burden from individual mice with bars representing the group mean. The limit of detection for CFU enumeration is denoted with a dotted line (L.O.D.). Results were analyzed Student's t test; **, P < 0.0001, *, P < 0.05.
In contrast to ID vaccination, IV vaccination resulted in a broader dissemination of BCG and in greater quantities of BCG in organs at 8 weeks after vaccination, as has been previously observed in C57 mice (15). These data demonstrated a significant shift in the localization of BCG in IV-vaccinated DO mice, where the majority of animals had BCG in lungs (Fig. 1A), spleens (Fig. 1B), and livers (Fig. 1C), compared to intradermally vaccinated DO mice. As previously demonstrated in C57 mice (16), IV BCG vaccination led to the dissemination of BCG into the bone marrow of 9/18 mice (Fig. 1D). Like ID-vaccinated mice, none of the IV-vaccinated DO mice contained detectable BCG in whole blood. These data demonstrated that, compared to ID vaccination, BCG delivered by the IV route results in larger quantities of BCG in more organs at 8 weeks after vaccination.
We next compared the ability of ID or IV BCG vaccination to protect against an aerogenic M.tb. challenge. In some experiments, ID vaccination with heat-killed BCG (HK-BCG) was also included as a comparator because HK-BCG maintains some of the inflammatory properties of M.tb. but is poorly protective (22). Mice were vaccinated as described above and challenged aerogenically with ~50 CFU of M.tb. 8 weeks after vaccination. M.tb. burdens in the organs were then assessed at 7 weeks after challenge, a time point chosen because supersusceptible DO mice typically succumb to M.tb. by this time (18, 19). Therefore, we were able to evaluate survival through that time period but also to assess bacterial burdens when BCG effects remain maximal. We previously demonstrated that BCG-induced protection in DO mice is heterogenous when assessed at 14 weeks after M.tb (19). Here, ID BCG vaccination also led to heterogeneous outcomes. HK-BCG vaccination ID did not reduce bacterial organ burdens (Fig. 2A). In contrast, across the population we found a modest ~0.4 log10 reduction in M.tb in lungs of mice that received BCG ID compared to naive mice (Fig. 2A). We observed a similarly moderate reduction of ~0.5 log10 M.tb. in lungs of mice given BCG IV compared to naive mice (Fig. 2A), and one DO mouse vaccinated IV had no detectable M.tb. in lungs. ID vaccination of DO mice resulted in a moderate ~0.6 log10 M.tb. reduction in spleens across the DO population compared to naive DO mice (Fig. 2B). IV vaccination of DO mice resulted in a mean reduction of ~1.0 log10 M.tb. in spleens across the population (Fig. 2B), and 6/45 IV-vaccinated DO animals had no detectable M.tb. in spleens. Taken together, these data suggest that IV BCG provided moderately enhanced protection against disseminated M.tb. in spleens but little enhancement in protection against pulmonary M.tb.
FIG 2.

Intravenous BCG provides enhanced M.tb. control compared to intradermal BCG. Groups of 15–20 female DO mice were vaccinated with 105 BCG IV or ID, with heat-killed BCG (HK) or treated with PBS (naive). Mice were aerogenically challenged with ~50 CFU of M.tb. 8 weeks after vaccination and euthanized 7 weeks after challenge for M.tb. CFU enumeration. Data from four experiments of similar design and outcome were combined and presented as individual mice with bars depicting the mean log10 M.tb. burden in A) lungs and B) spleens. The limit of detection for CFU enumeration is denoted with a dotted line (L.O.D.). Results were analyzed by one-way ANOVA with Tukey's posttest; *, P < 0.01.
Several mice in the IV vaccination groups had M.tb. burdens that were below the level of detection by plating, either because the level of M.tb. was in fact low, because M.tb. was cleared from these organs, or because M.tb. never seeded there after challenge. We addressed these possibilities by performing PCR to assess the presence or absence of M.tb. genomes in the organs (23, 24). We chose several control lung and spleen samples with known M.tb. CFU levels for comparison. We performed CEQ PCR on one lung and five spleen samples from the DO mice with < 1.7 log10 M.tb. CFU. Of these, the one lung sample and two of five spleen samples were also below the level of detection by CEQ PCR (Table S1).
In addition to measuring M.tb. burdens in the organs, we also assessed the impact of disease in the lungs by histopathology. We harvested lung tissue at 7 weeks after vaccination/M.tb. challenge and quantitated the proportion of lung with disease involvement. The lungs of the naive DO mice exhibited a wide range of lung pathology and inflammation. DO mice vaccinated with HK-BCG exhibited similar degrees of lung inflammation compared to that found in naive mice (Fig. 3). BCG vaccination reduced the proportion of lung tissue with inflammation across the DO population, whether delivered ID or IV, although the reduction was only significant in the ID-vaccinated DO group (Fig. 3). Of note, some lungs exhibited no signs of inflammation, while some lungs were completely infiltrated with little visible air spaces and substantial tissue damage (Fig. 4).
FIG 3.
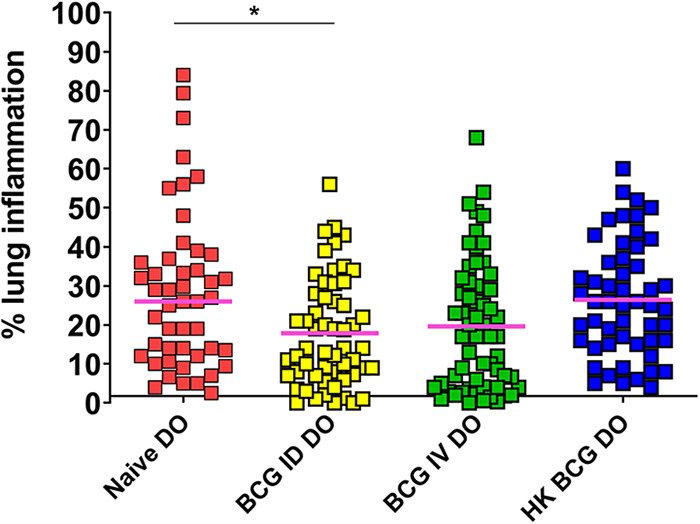
Naïve and BCG-vaccinated DO mice challenged with M.tb. exhibit a range of lung inflammation outcomes. Groups of 15–20 female DO mice were vaccinated with 105 BCG ID or IV, HK-BCG, or treated with PBS (naive). Mice were challenged by aerosol with ~50 CFU M.tb. Animals were euthanized 7 weeks after infection, and lung sections isolated and prepared for slides with H&E staining. Animals that became moribund before 7 weeks were euthanized and lung samples similarly isolated. H&E-stained slides were scanned, and images were analyzed to assess the percentage of lung tissue with disease involvement compared to the total lung area. Dots represent the percentage of each lung that was inflamed and/or had disease involvement in individual animals, and lines represent the median of the group. Data represent the combined results from four independent experiments of similar design. Differences between the groups indicated by the lines were significant by one-way ANOVA with Tukey's posttest; *, P < 0.01.
FIG 4.
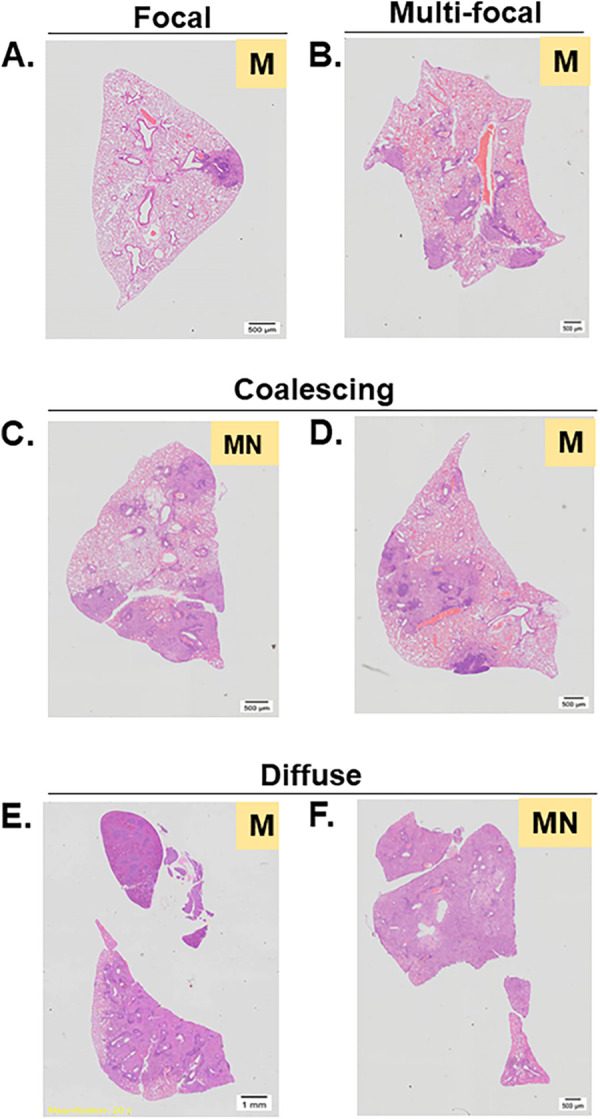
Representative images of lung sections prepared for H&E staining demonstrate qualitatively different disease pathologies. Lung tissues were collected from naive or BCG-vaccinated DO mice 7 weeks after M.tb. challenge. Tissues were formalin-fixed and processed for H&E staining. Slides were scanned at 20× on an Olympus VS200 slide scanner. Sections were scored by a veterinary pathologist who categorized the lesion distribution as focal (A), multifocal (B), coalesced (C, D), or diffuse (E, F). The pathologist also categorized the predominant cell type within lesions as monocytes (M) or monocytes/neutrophils (MN). Images presented are representative of the various types of lung pathology found throughout all groups of mice.
A subset of lung tissues from 12 to 15 DO mice from each vaccine group were further analyzed by a veterinary pathologist to determine the types of lesions and inflammation and the predominant cell types within each lung section in the region of inflammation. In general, lesion distribution was varied in all groups, consistent with heterogenous disease presentation (Table S2). A subset of lungs contained classical focal lesions predominantly associated with mononuclear cells (Fig. 4A and B) or more advanced disease with coalescing lesions (Fig. 4C and D), while other animals had diffuse pneumonia (Fig. 4E and F) (18, 19). Only seven animals contained lesions with monocytes as the predominant cell type, with minimal or mild neutrophil aggregates (MN) (Table S2); interestingly, all of these animals were either naive or HK-BCG-vaccinated mice. However, because DO mice are inherently heterogeneous, it is difficult to associate pathological patterns with vaccination status. In general, we did not find any distinct pattern of histopathological changes associated with either the ID or IV vaccine groups compared with the naive or HK-BCG-vaccinated animals. A limitation to this analysis is that pathology was only captured in a single lobe, and therefore potential heterogeneity in inflammation presentation across different lung lobes was not evaluated. However, our analyses suggest that while vaccination had a modest impact on the overall proportion of inflammation, it had minimal effect on the nature of inflammation.
Weight loss is a hallmark of advanced tuberculosis in people and wasting and cachexia are associated with tuberculosis disease in animals (25). Ultimately, a combination of disease factors such as lung damage and weight loss contribute to survival. Because our results supported enhanced systemic protection from IV BCG vaccination, we compared IV BCG administration to ID vaccination in terms of protecting animals from wasting and ultimately extending survival of DO mice after challenge. Groups of DO mice were vaccinated and challenged as described above and followed for weight loss and survival outcomes. We found no significant differences in the percentage of peak body weights at the time of euthanasia between the naive animals and the vaccine groups (Fig. 5A). The naive M.tb. challenged animals only gained on average ~15% body weight over the course of the infection (Fig. 5B), which may reflect a failure to thrive due to infection. When we compared the peak body weight to initial weight, we saw a significant increase in the weight gain of the IV-vaccinated DO mice but not the ID-vaccinated mice compared to the naive group (Fig. 5B). When weights were plotted over time, the difference in body weight was also readily observable between IV-vaccinated mice compared to naive mice (Fig. S1). There were no significant differences in weight gain between any other groups. Thus, while eventually all animals lost a similar proportion of weight by the time of euthanasia, the IV-vaccinated animals thrived with substantial weight gains during the infection.
FIG 5.

Weight gain, but not overall weight loss, is impacted by BCG vaccination. Groups of 15–20 DO mice were vaccinated with 105 BCG ID or IV, HK-BCG, or treated with PBS (naive). Mice were aerogenically challenged with ~50 CFU of M.tb. 8 weeks after vaccination and followed for survival. Animals were weighed weekly throughout the experiment, and daily if they demonstrated signs of illness, such as ruffled fur or cachexia. Animals were euthanized when they met established humane endpoint criteria. Animal weights were used to determine several weight related traits. A) The weight at euthanasia was divided by peak body weight to generate the “percentage of peak body weight” at euthanasia. B) The peak body weight was divided by the initial body weight to determine “percentage weight gain at peak.”
IV-vaccinated DO mice survived significantly longer than naive, ID-vaccinated, or HK-BCG-vaccinated DO mice after M.tb. challenge (Fig. 6; P < 0.05, Log-rank test). Average survival times were 193 days for naive DO mice, 229 days for ID-vaccinated DO mice and 230 days for HK-BCG vaccinated DO mice, and 351 days for IV-vaccinated DO mice. Taken together, these results demonstrate that IV BCG provides moderately enhanced protection compared to ID BCG vaccination in DO mice.
FIG 6.
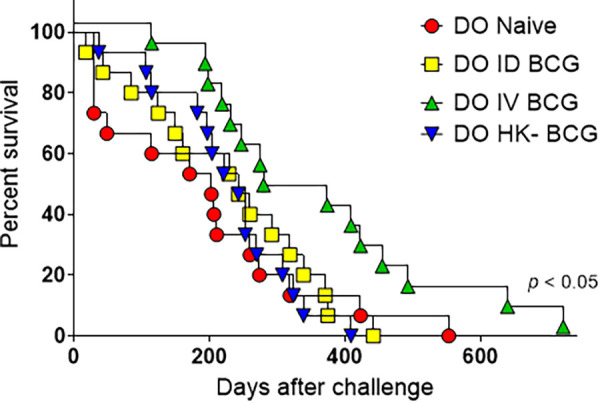
Intravenous BCG extends survival of DO mice following M.tb. challenge. Groups of 15–20 DO mice were vaccinated with 105 BCG ID or IV, HK-BCG, or treated with PBS (naive). Mice were aerogenically challenged with ~50 CFU of M.tb. 8 weeks after vaccination and followed for survival. Animals were euthanized when they met established humane endpoint criteria. DO IV-BCG mice survived longer than any of the other three vaccine groups; P < 0.05 by the Log-rank Mantel-Cox test.
DISCUSSION
We recently demonstrated that DO mice are a useful small animal model for vaccine development for tuberculosis. This new resource provides a heterogenous genetic and phenotypic population with a wide variety of outcomes following M.tb. challenge, similar to the heterogenous outcomes observed in humans. Here, we utilized that heterogenous model to test the capacity of BCG delivered intravenously to protect against M.tb. We found that IV BCG affords modestly improved protection in terms of reduction of M.tb. CFU and improved survival compared to BCG vaccination ID.
The moderately enhanced survival observed in the DO mice may stem from the changes in BCG deposition within the host. IV BCG administration resulted in greater quantities BCG persisting in more organs at the time of M.tb. challenge, including BCG migrating into the bone marrow compartment. BCG in the bone marrow following IV vaccination has been observed in mice and NHP. This localization may lead to conditioning of myeloid cells, which in some studies has been linked to the generation of trained immunity (16), as reviewed in reference (26). This epigenetic reprogramming allows myeloid cells to better control M.tb. intracellular replication (16). However, in the case of enhanced protection of NHPs by IV BCG, evidence suggests that trained immunity may not be a major player in driving protection (11). Other data suggest that the location and endurance of BCG within systemic organs may contribute to protection. However, we also observed wider dissemination of BCG in C57 mice but did not observe enhanced protection, suggesting this is not a mechanism that drives enhanced protection. The core mechanism of the reduction in M.tb. CFU and pathologies in NHP, and enhanced survival in the DO mice following IV BCG administration, remains to be revealed.
Previous attempts to examine protection following intravenous BCG vaccination in mice have not demonstrated superior protection over intradermal vaccination (13, 15), which contrasts results observed in NHPs (11, 27–31). However, previous mouse studies utilized inbred mouse strains, such as C57BL/6, which are relatively resistant to M.tb. infection itself. Previous studies did not examine survival, so additive benefits from BCG delivered IV may have been missed. Our demonstration of only moderately enhanced protection in DO mice is discordant with the impressive protection demonstrated in the rhesus macaques by Darrah et al. (11) and others. However, mouse and NHP studies differ in several aspects of design, including dosages of BCG, BCG strain, and challenge route and doses. In the future, the contributions of these variables on the outcomes of IV BCG-induced protection in DO mice can be investigated.
Other difficulties in comparing the mouse to NHP are differing metrics used to evaluate disease progression in each species. In an NHP, M.tb. disease progression can be quantified in lungs by PET/CT; animals can be tracked for symptomatic disease; and M.tb. bacterial burdens can be assessed, often before an animal becomes moribund. In mice it is difficult to track disease progression in the lungs of live animals with similar imaging techniques, and protection in mice is predominantly evaluated by samples collected at necropsy. Thus, the differences in measures of “protection” could also contribute to disparate results observed between the DO mice and NHP. Ultimately the question of which animal model systems, if any, should be used for tuberculosis vaccine development is still a matter of debate and awaits validation by comparison with human clinical studies. The present work supports the value of DO mice as a small animal population with high genetic diversity that may be a useful complement to resource-intensive studies in NHPs.
Despite subtle bacteriological and pathological differences, we observed improved survival in DO mice that received IV BCG compared to all other vaccine groups (Fig. 6). Given that neither lung nor spleen CFU were significantly different between mice vaccinated with BCG IV and ID at 7 weeks after challenge (Fig. 2), the improved survival likely cannot simply be attributed to initial reductions in CFU. Notably, however, several animals in the group vaccinated with BCG IV had undetectable CFU in organs by either colony enumeration on agar (Fig. 2) or by CEQ PCR (Table S1). This suggests that IV BCG may have either prevented dissemination of M.tb., promoted bacterial clearance in a small subset of animals, or both. The degree of lung inflammation was also equivalent between the IV and ID BCG groups (Fig. 3), and similar histological disease presentations were observed in both ID BCG and IV BCG vaccine groups (Fig. 4 and Table S2). Therefore, enhanced survival cannot be ascribed directly to reduced lung pathology, nor to differences in weight loss (Fig. 5A and 5B). Survival of an animal following M.tb. infection is obviously multifactorial, and a better understanding of factors that ultimately lead to death is needed.
Taken together, therefore, these data demonstrate that delivering BCG intravenously to DO mice provides moderately enhanced protection compared to traditional parenteral vaccination. Although it is not yet possible to know how well these outcomes model vaccination in people, the results support further exploration of this genetically diverse small animal model as a means to evaluate tuberculosis vaccines and to investigate immune correlates of protection.
MATERIALS AND METHODS
Mice.
Female Diversity Outbred (DO) mice were purchased from Jackson Laboratories (Bar Harbor, ME) and were used when 6 to 10 weeks of age, from breeding generations 34 to 38. Within each experiment, all animals were age matched. Only female DO mice were used in these studies because of excessive aggression in male mice, with wounds confounding infection studies. All animal studies were performed under protocols approved by the Animal Care and Use Committee (ACUC) of CBER/FDA. Mice were housed in microisolator cages and were given autoclaved food and water ad libitum. Animal protocols stressed practices and procedures designed to strictly minimize any suffering.
Bacteria and growth conditions.
As described previously (32), Mycobacterium bovis BCG Pasteur (BCG) and M. tuberculosis Erdman (M.tb. Erdman) were derived from the mycobacterial culture collection of the Trudeau Institute and propagated once to prepare working seed stocks that were then used to prepare all infection stocks, without further passage. M.tb. Erdman and BCG Pasteur were grown in 7H9 media supplemented with OADC, glycerol, and 0.05% Tween 80 (Difco Laboratories, Detroit, MI) to midlogarithmic phase as previously described. Vials for single infection use were then frozen in 0.5 mL aliquots at −80°C until use. A sample from each batch of bacterial stock was subjected to quality control experiments to determine the number of CFU (CFU), to confirm typical colony morphologies, and to confirm vaccination efficacy or infection in mice. Heat-killed BCG was prepared by thawing a stock vial of BCG and heating it to 100°C for 30 min as described previously (22).
BCG vaccination and M.tb. challenge.
For each experiment, a single frozen vial of BCG was thawed and diluted to the desired concentration in sterile PBS. HK-BCG was diluted to the same concentration as live BCG. Groups of 10 to 20 DO mice were vaccinated intradermally or intravenously via the tail vein with 105 CFU of BCG or HK-BCG or were sham-vaccinated with phosphate-buffered saline (PBS, low endotoxin, Lonza, Walkersville, MD). Animals were challenged by aerosol with M.tb. Erdman 8 weeks after vaccination. For each experiment, a frozen vial of M.tb. was thawed and diluted in PBS to the desired concentration, and the suspension added to the nebulizer of a Middlebrook chamber (GlasCol, Terre Haute, IN). Mice were challenged over a 30-minute exposure, targeting administration of a dose of ~50 CFU. To determine actual delivered dose, five C57 mice/challenge run were euthanized 4 to 5 h after challenge, and the entire lung for each animal homogenized and plated to determine uptake CFU. Mice were monitored and euthanized when clearly unable to reach food and water, following preestablished humane endpoint criteria.
Determination of bacterial burden in organs and tissue pathology.
Bacterial burdens in organs were determined at the indicated time points after infection, either BCG enumeration at 8 weeks after vaccine, or M.tb. enumeration at 7 weeks after challenge. Mice were euthanized, and organs were removed aseptically and transferred to sterile homogenizer bags containing 5 mL of sterile PBS per organ. Organs were disrupted using a Stomacher (Seward, England), and the homogenates were serially diluted and plated for CFU enumeration on 7H11 plates containing 10% OADC enrichment (Becton, Dickinson, Sparks, MD) medium, 10 μg/mL ampicillin, 50 μg/mL cycloheximide, and for M.tb. enumeration 2 μg/mL 2-thiophenecarboxylic acid hydrazide (TCH) (Sigma). The addition of TCH to the agar plates inhibits BCG growth but has no effect on M. tuberculosis growth (33). Organ homogenates were also frozen and stored at −80°C to be used for later analyses. A portion of each lung was removed and preserved in 10% formalin for histological analyses. Formalin fixed samples were then sent to American Histolabs, Inc. (Gaithersburg, MD), where the tissues were embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin (H&E). Slides were scanned and digitized and subjected to densitometry analyses to assess the level of inflammation in the tissue using the Image-Pro Plus software (Media Cybernetics, Rockville, MD). Lung inflammation was quantitated by assigning areas with dark pink and purple color staining as inflamed (34). The percentage of dark pink and purple colored areas (compared to light pink and open areas) from a lung section of each mouse was determined by the software and reported as percentage lung inflammation per sample. Gross histological examination was performed by a veterinary pathologist (IDEXX Bioanalytics, Colombia, MO). Tissues were scored based on lesion distribution (focal, multifocal, multifocal coalescing, or diffuse), and on the predominant inflammatory cell type present in lesions (mononuclear cells, neutrophils, or mixed mononuclear cell prominence with neutrophil aggregates). For experiments in which BCG was enumerated from bone marrow, both femurs from each mouse were excised, the femur heads cut to expose the bone marrow. Each femur was flushed with 0.5 mL PBS 0.05% Tween 80. The eluate from both femurs was combined for each mouse, and the entire eluate was plated onto 7H11 agar for CFU enumeration.
Chromosomal equivalents (CEQ) PCR.
Amplification of M. tuberculosis genomic DNA to quantify CEQ was performed as described in Muñoz-Elias et al. (24) and Lin et al. (23). Briefly, 0.2 mL of lung homogenate generated above was transferred into a tube with 0.1 mm zirconia-silica beads (MP Biomedicals) beads and tris-EDTA, and 25:24:1 phenol: chloroform: isoamyl alcohol was added. The samples were further homogenized using a FastPrep-24 tissue homogenizer (MP Biomedical) and were subjected to subsequent rounds of DNA purification with isopropanol and 3 M sodium acetate. Precipitated DNA was washed with 70% ethanol and resuspended in 100 μL sterile water. DNA was quantified using a Nanodrop spectrophotometer. Quantitative real-time PCR was performed using 1 μL of DNA from each sample, representing approximately 0.04% of the total genomic DNA extracted from a single mouse organ. DNA was amplified using a sigF primer/probe combination (Integrated DNA Technologies) on a Viia7 thermocycler (Life Technologies) using reaction conditions described previously (23). Samples were run in duplicate reactions. Chromosomal equivalents were quantified based on a standard curve which was generated using serial dilutions of genomic DNA derived from known quantities of broth grown M. tuberculosis.
Statistical analyses.
The statistical significance of differences within parameters was assessed using Student's t test, one-way ANOVA with Tukey's posttest, the Log-rank Mantel-Cox test, or others as described in the text (GraphPad Prism, San Diego, CA).
Footnotes
Supplemental material is available online only.
Contributor Information
Sherry L. Kurtz, Email: sherry.kurtz@fda.hhs.gov.
Karen L. Elkins, Email: karen.elkins@fda.hhs.gov.
Sabine Ehrt, Weill Cornell Medicine.
REFERENCES
- 1.World Health Organization. 2022. Global tuberculosis report 2022. [Google Scholar]
- 2.Ahmed A, Rakshit S, Adiga V, Dias M, Dwarkanath P, D’Souza G, Vyakarnam A. 2021. A century of BCG: impact on tuberculosis control and beyond. Immunol Rev 301:98–121. doi: 10.1111/imr.12968. [DOI] [PubMed] [Google Scholar]
- 3.Scriba TJ, Netea MG, Ginsberg AM. 2020. Key recent advances in TB vaccine development and understanding of protective immune responses against Mycobacterium tuberculosis. Semin Immunol 50:101431. doi: 10.1016/j.smim.2020.101431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nemes E, Geldenhuys H, Rozot V, Rutkowski KT, Ratangee F, Bilek N, Mabwe S, Makhethe L, Erasmus M, Toefy A, Mulenga H, Hanekom WA, Self SG, Bekker LG, Ryall R, Gurunathan S, DiazGranados CA, Andersen P, Kromann I, Evans T, Ellis RD, Landry B, Hokey DA, Hopkins R, Ginsberg AM, Scriba TJ, Hatherill M, Team C–S . 2018. Prevention of M. tuberculosis infection with H4:IC31 vaccine or BCG revaccination. N Engl J Med 379:138–149. doi: 10.1056/NEJMoa1714021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Derrick SC, Kolibab K, Yang A, Morris SL. 2014. Intranasal administration of Mycobacterium bovis BCG induces superior protection against aerosol infection with Mycobacterium tuberculosis in mice. Clin Vaccine Immunol 21:1443–1451. doi: 10.1128/CVI.00394-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.White AD, Sibley L, Dennis MJ, Gooch K, Betts G, Edwards N, Reyes-Sandoval A, Carroll MW, Williams A, Marsh PD, McShane H, Sharpe SA. 2013. Evaluation of the safety and immunogenicity of a candidate tuberculosis vaccine, MVA85A, delivered by aerosol to the lungs of macaques. Clin Vaccine Immunol 20:663–672. doi: 10.1128/CVI.00690-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Darrah PA, Bolton DL, Lackner AA, Kaushal D, Aye PP, Mehra S, Blanchard JL, Didier PJ, Roy CJ, Rao SS, Hokey DA, Scanga CA, Sizemore DR, Sadoff JC, Roederer M, Seder RA. 2014. Aerosol vaccination with AERAS-402 elicits robust cellular immune responses in the lungs of rrhesus macaques but fails to protect against high-dose Mycobacterium tuberculosis challenge. J Immunol 193:1799–1811. doi: 10.4049/jimmunol.1400676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hokey DA, Wachholder R, Darrah PA, Bolton DL, Barouch DH, Hill K, Dheenadhayalan V, Schwander S, Godin CS, Douoguih M, Pau MG, Seder RA, Roederer M, Sadoff JC, Sizemore D. 2014. A nonhuman primate toxicology and immunogenicity study evaluating aerosol delivery of AERAS-402/Ad35 vaccine Evidence for transient t cell responses in peripheral blood and robust sustained responses in the lungs. Human Vaccines & Immunotherapeutics 10:2199–2210. doi: 10.4161/hv.29108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aguilo N, Toledo AM, Lopez-Roman EM, Perez-Herran E, Gormley E, Rullas-Trincado J, Angulo-Barturen I, Martin C. 2014. Pulmonary Mycobacterium bovis BCG vaccination confers dose-dependent superior protection compared to that of subcutaneous vaccination. Clin Vaccine Immunol 21:594–597. doi: 10.1128/CVI.00700-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verreck FAW, Tchilian EZ, Vervenne RAW, Sombroek CC, Kondova I, Eissen OA, Sommandas V, van der Werff NM, Verschoor E, Braskamp G, Bakker J, Langermans JAM, Heidt PJ, Ottenhoff THM, van Kralingen KW, Thomas AW, Beverley PCL, Kocken CHM. 2017. Variable BCG efficacy in rhesus populations: pulmonary BCG provides protection where standard intra-dermal vaccination fails. Tuberculosis (Edinb) 104:46–57. doi: 10.1016/j.tube.2017.02.003. [DOI] [PubMed] [Google Scholar]
- 11.Darrah PA, Zeppa JJ, Maiello P, Hackney JA, Wadsworth MH, Hughes TK, Pokkali S, Swanson PA, Grant NL, Rodgers MA, Kamath M, Causgrove CM, Laddy DJ, Bonavia A, Casimiro D, Lin PL, Klein E, White AG, Scanga CA, Shalek AK, Roederer M, Flynn JL, Seder RA. 2020. Prevention of tuberculosis in macaques after intravenous BCG immunization. Nature 577:95–102. doi: 10.1038/s41586-019-1817-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lefford MJ. 1977. Induction and expression of immunity after BCG immunization. Infect Immun 18:646–653. doi: 10.1128/iai.18.3.646-653.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nuermberger EL, Yoshimatsu T, Tyagi S, Bishai WR, Grosset JH. 2004. Paucibacillary tuberculosis in mice after prior aerosol immunization with Mycobacterium bovis BCG. Infect Immun 72:1065–1071. doi: 10.1128/IAI.72.2.1065-1071.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bickett TE, McLean J, Creissen E, Izzo L, Hagan C, Izzo AJ, Silva Angulo F, Izzo AA. 2020. Characterizing the BCG induced macrophage and neutrophil mechanisms for defense against Mycobacterium tuberculosis. Front Immunol 11:1202. doi: 10.3389/fimmu.2020.01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mittrücker HW, Steinhoff U, Köhler A, Krause M, Lazar D, Mex P, Miekley D, Kaufmann SH. 2007. Poor correlation between BCG vaccination-induced T cell responses and protection against tuberculosis. Proc Natl Acad Sci USA 104:12434–12439. doi: 10.1073/pnas.0703510104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonca LE, Pacis A, Tzelepis F, Pernet E, Dumaine A, Grenier JC, Mailhot-Leonard F, Ahmed E, Belle J, Besla R, Mazer B, King IL, Nijnik A, Robbins CS, Barreiro LB, Divangahi M. 2018. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell 172:176–190. doi: 10.1016/j.cell.2017.12.031. [DOI] [PubMed] [Google Scholar]
- 17.Churchill GA, Gatti DM, Munger SC, Svenson KL. 2012. The diversity outbred mouse population. Mamm Genome 23:713–718. doi: 10.1007/s00335-012-9414-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niazi MK, Dhulekar N, Schmidt D, Major S, Cooper R, Abeijon C, Gatti DM, Kramnik I, Yener B, Gurcan M, Beamer G. 2015. Lung necrosis and neutrophils reflect common pathways of susceptibility to Mycobacterium tuberculosis in genetically diverse, immune-competent mice. Dis Model Mech 8:1141–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurtz SL, Gardina PJ, Myers TG, Ryden P, Elkins KL. 2020. Whole genome profiling refines a panel of correlates to predict vaccine efficacy against Mycobacterium tuberculosis. Tuberculosis (Edinb) 120:101895. doi: 10.1016/j.tube.2019.101895. [DOI] [PubMed] [Google Scholar]
- 20.Gopal R, Monin L, Torres D, Slight S, Mehra S, McKenna KC, Fallert Junecko BA, Reinhart TA, Kolls J, Baez-Saldana R, Cruz-Lagunas A, Rodriguez-Reyna TS, Kumar NP, Tessier P, Roth J, Selman M, Becerril-Villanueva E, Baquera-Heredia J, Cumming B, Kasprowicz VO, Steyn AJ, Babu S, Kaushal D, Zuniga J, Vogl T, Rangel-Moreno J, Khader SA. 2013. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am J Respir Crit Care Med 188:1137–1146. doi: 10.1164/rccm.201304-0803OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Irwin SM, Goodyear A, Keyser A, Christensen R, Troudt JM, Taylor JL, Bohsali A, Briken V, Izzo AA. 2008. Immune response induced by three Mycobacterium bovis BCG substrains with diverse regions of deletion in a C57BL/6 mouse model. Clin Vaccine Immunol 15:750–756. doi: 10.1128/CVI.00018-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurtz SL, Elkins KL. 2015. Correlates of vaccine-induced protection against Mycobacterium tuberculosis revealed in comparative analyses of lymphocyte populations. Clin Vaccine Immunol 22:1096–1108. doi: 10.1128/CVI.00301-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin PL, Ford CB, Coleman MT, Myers AJ, Gawande R, Ioerger T, Sacchettini J, Fortune SM, Flynn JL. 2014. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nature Medicine 20:75–79. doi: 10.1038/nm.3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munoz-Elias EJ, Timm J, Botha T, Chan WT, Gomez JE, McKinney JD. 2005. Replication dynamics of Mycobacterium tuberculosis in chronically infected mice? Infect Immun 73:546–551. doi: 10.1128/IAI.73.1.546-551.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cegielski JP, McMurray DN. 2004. The relationship between malnutrition and tuberculosis: evidence from studies in humans and experimental animals. Int J Tuberculosis and Lung Dis 8:286–298. [PubMed] [Google Scholar]
- 26.Koeken V, Verrall AJ, Netea MG, Hill PC, van Crevel R. 2019. Trained innate immunity and resistance to Mycobacterium tuberculosis infection. Clin Microbiol Infect 25:1468–1472. doi: 10.1016/j.cmi.2019.02.015. [DOI] [PubMed] [Google Scholar]
- 27.Barclay WR, Anacker RL, Brehmer W, Leif W, Ribi E. 1970. Aerosol-induced tuberculosis in subhuman primates and the course of the disease After intravenous BCG vaccination. Infect Immun 2:574–582. doi: 10.1128/iai.2.5.574-582.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barclay WR, Busey WM, Dalgard DW, Good RC, Janicki BW, Kasik JE, Ribi E, Ulrich CE, Wolinsky E. 1973. Protection of monkeys against airborne tuberculosis by aerosol vaccination with bacillus Calmette-Guerin. Am Rev Respir Dis 107:351–358. [DOI] [PubMed] [Google Scholar]
- 29.Sharpe S, White A, Sarfas C, Sibley L, Gleeson F, McIntyre A, Basaraba R, Clark S, Hall G, Rayner E, Williams A, Marsh PD, Dennis M. 2016. Alternative BCG delivery strategies improve protection against Mycobacterium tuberculosis in non-human primates: protection associated with mycobacterial antigen-specific CD4 effector memory T-cell populations. Tuberculosis (Edinb) 101:174–190. doi: 10.1016/j.tube.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anacker RL, Brehmer W, Barclay WR, Leif WR, Ribi E, Simmons JH, Smith AW. 1972. Superiority of intravenously administered BCG and BCG cell walls in protecting rhesus monkeys (Macaca mulatta) against airborne tuberculosis. Z Immunitatsforsch Exp Klin Immunol 143:363–376. [PubMed] [Google Scholar]
- 31.Ribi E, Anacker RL, Barclay WR, Brehmer W, Harris SC, Leif WR, Simmons J. 1971. Efficacy of mycobacterial cell walls as a vaccine against airborne tuberculosis in the Rheusus monkey. J Infect Dis 123:527–538. doi: 10.1093/infdis/123.5.527. [DOI] [PubMed] [Google Scholar]
- 32.Larsen MH, Biermann K, Jacobs WR. Jr, 2007. Laboratory maintenance of Mycobacterium tuberculosis. Curr Protoc Microbiol Chapter 10:Unit 10A 1. [DOI] [PubMed] [Google Scholar]
- 33.Collins T, Levett PN. 1989. Radiometric studies on the use of selective inhibitors in the identification of Mycobacterium spp. J Med Microbiol 30:175–181. doi: 10.1099/00222615-30-3-175. [DOI] [PubMed] [Google Scholar]
- 34.Derrick SC, Perera LP, Dheenadhayalan V, Yang A, Kolibab K, Morris SL. 2008. The safety of post-exposure vaccination of mice infected with Mycobacterium tuberculosis. Vaccine 26:6092–6098. doi: 10.1016/j.vaccine.2008.09.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Download iai.00168-23-s0001.tif, TIF file, 0.1 MB (74.9KB, tif)
Table S1. Download iai.00168-23-s0002.xlsx, XLSX file, 0.01 MB (9.8KB, xlsx)
Table S2. Download iai.00168-23-s0003.xlsx, XLSX file, 0.01 MB (13.9KB, xlsx)
Legends of Fig. S1 and Tables S1 and S2. Download iai.00168-23-s0004.docx, DOCX file, 0.01 MB (13.6KB, docx)