

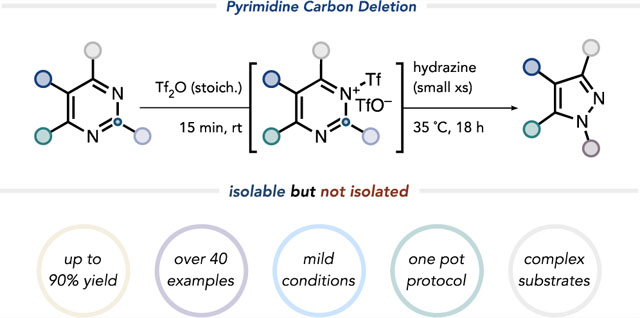
Abstract
A method for the conversion of pyrimidines into pyrazoles by a formal carbon deletion has been achieved guided by computational analysis. The pyrimidine heterocycle is the most common diazine in FDA-approved drugs, and pyrazoles are the most common diazole. An efficient method to convert pyrimidines into pyrazoles would therefore be valuable by leveraging the chemistries unique to pyrimidines to access diversified pyrazoles. One method for the conversion of pyrimidines into pyrazoles is known, though it proceeds in low yields and requires harsh conditions. The transformation reported here proceeds under milder conditions, tolerates a wide range of functional groups, and enables the simultaneous regioselective introduction of N-substitution on the resulting pyrazole. Key to the success of this formal one-carbon deletion method is a room-temperature triflylation of the pyrimidine core, followed by hydrazine-mediated skeletal remodeling.
Graphical Abstract
INTRODUCTION
Of the top 200 selling U.S. FDA-approved drug compounds as of 2014, 59% contain at least one N-heterocycle.1 While monoazacycles are the most prevalent,1 di-azacycles also feature prominently. Of the latter, pyrimidines are the most common diazine (and the 6th most abundant N-heterocycle overall), and pyrazoles are the most common diazole.1–5 These heterocycles are privileged by virtue of the vectors they present for interactions (e.g., H-bonding) with biological targets4 and the numerous synthetic methods for their peripheral functionalization.6,7 Moreover, their late-stage peripheral diversification can accelerate the pace of drug and agrochemical discovery. This type of molecular editing8 can circumvent the need to effect a change on the periphery of a target heterocycle at the outset of a drug derivative synthesis or even a complete redesign of the route.
In comparison, direct, late-stage modifications of the core framework of heterocycles are challenging to accomplish. To expand the chemical space accessible from a given compound beyond peripheral modifications, interest in the skeletal editing of N-heterocycles (illustrated in Figure 2A) has increased in recent years.9–13 In combination with peripheral editing, the continued development of skeletal editing would facilitate the late stage, omnidirectional exploration of chemical space around a central molecule.
Figure 2.
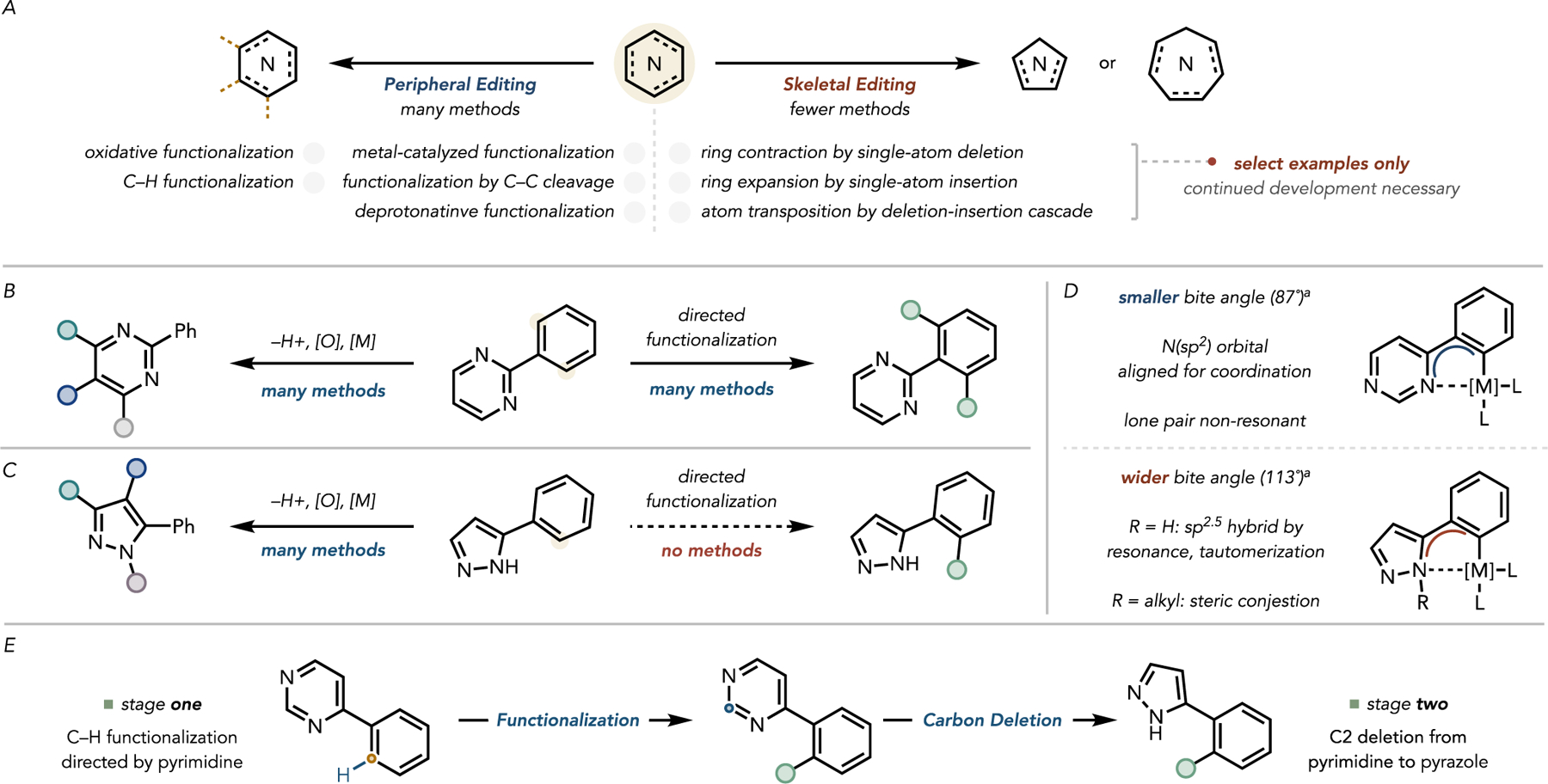
(A) Conceptual outline of peripheral and skeletal editing strategies. (B) Peripheral edits available about the pyrimidine heterocycle, including its use as a directing group for C–H functionalization. (C) Peripheral edits available for pyrazoles, noting its key inability to direct C–H functionalizations. (D) Rationale behind the poor directing group strength of pyrazoles relative to pyrimidines. a ωB97x-D, 6–31G(d,p). (E) Conceptual outline to leverage both peripheral and skeletal editing to access C–H functionalized pyrazoles.
While pyrimidines are competent directing groups for peripheral functionalization by C–H functionalization (typically of substituents at C2 of the pyrimidine, vide infra),14–19 few methods for their skeletal editing are known (Figure 2B); however, reports describing the conversion of other hetero-cycles into pyrimidines have begun to appear. Recently, Morandi and coworkers reported a nitrogen insertion into indole C–C bonds to generate quinazolines.20 Similarly, Levin and coworkers recently disclosed an analogous carbene insertion into pyrazole N–N bonds to generate 2-aryl pyrimidines.21 Here, we report a reverse of the latter process—a method for the conversion of pyrimidines into pyrazoles by a formal carbon deletion (Figure 1B).
Figure 1.
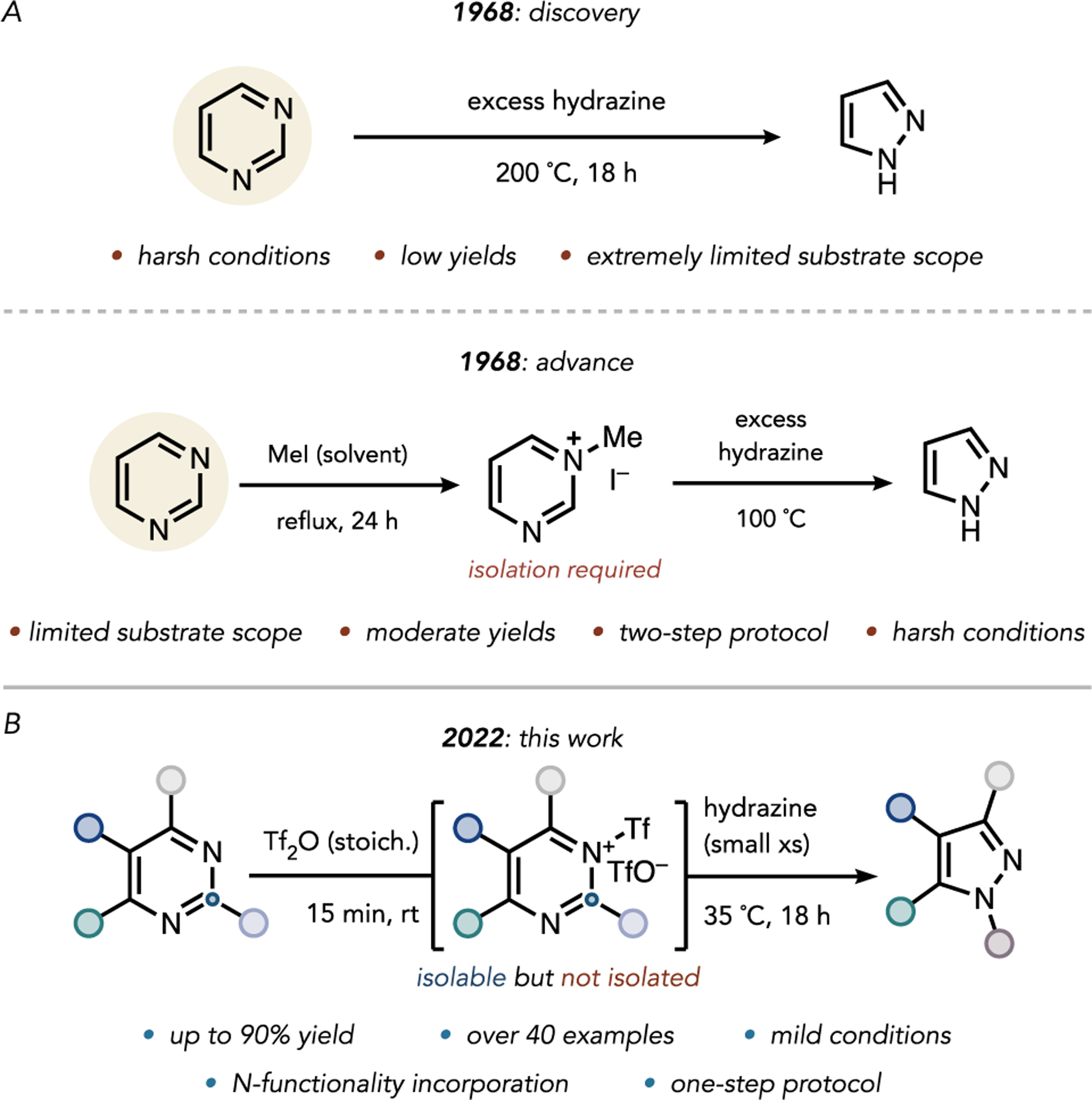
(A) Original disclosure of the hydrazine-mediated contraction of pyrimidine to pyrazole. (B) Summary of present disclosure.
Unlike pyrimidines, pyrazoles are relatively poor directing groups for the C–H functionalization of their C-bound substituents, especially when the nitrogen adjacent to the substituent is itself substituted (Figure 2C,D).22 Because both NH - and N-functionalized pyrazoles are found in many biologically relevant molecules, we reasoned that a general, functional group tolerant method for the transformation of pyrimidines into pyrazoles would enable access to C–H functionalized pyrazole-containing compounds by leveraging the directing ability of the parent pyrimidine (Figure 2E). This example of skeletal editing would thus constitute a rapid diversification of the heterocyclic moiety and would complement typical peripheral diversification tactics.
In fact, such a transformation was reported in 1968, when van der Plas and Jongejan found that heating pyrimidine to 200°C in the presence of solvent quantities of aqueous hydrazine affected its conversion to pyrazole, as outlined in Figure 1A. However, this reaction failed on substituted pyrimidines.23 N-Methylation of the pyrimidine nitrogen by iodomethane lowered the LUMO of the pyrimidine ring such that the nucleophilic attack of hydrazine to effect the contraction could occur at 100°C, though the scope remained limited, and this two-step protocol required solvent quantities of iodomethane.
To broaden the scope of this transformation and generalize the conditions, we sought to achieve even more pronounced LUMO lowering of the pyrimidine unit in a one-pot process. Computations indicated that pyrimidine N-triflylation would sufficiently lower the pyrimidine LUMO(−3.88vs −3.13eV for N–Me pyridinium, corresponding to a difference of 13.98 kcal/mol; see the Supporting Information for details) to facilitate the nucleophilic attack of hydrazine at 23°C. We experimentally confirmed triflic anhydride to be a competent equimolar activator. This discovery ultimately enabled the contraction of a broad range of substituted pyrimidines to the corresponding pyrazoles under mild conditions (Figure 1B). This formal carbon deletion converts the most prevalent diazine into the most common diazole found in FDA-approved pharmaceuticals. Importantly, the pyrimidine-to-pyrazole conversion sets the stage for using well-established pyrimidinedirected peripheral C–H functionalization for the diversification of the eventual pyrazole-containing products.
RESULTS AND DISCUSSION
We first sought to identify conditions for the catalytic activation of the pyrimidine core; however, no reactivity was observed using sub-stoichiometric amounts of Brønsted or Lewis acids, likely due to competing protonation or binding to the hydrazine nucleophile. We instead reasoned, with support from computations (see the Supporting Information for details), that successful activation might be achieved by N-acylation or N-sulfonylation.24–27 The activators evaluated on the basis of this hypothesis are summarized in Table 1. This investigation led to our discovery that triflylation of the pyrimidine nitrogen prior to addition of hydrazine effected the desired contraction in 37% yield at 23 °C (Table 1, entry 8). This preactivation stage is necessary to prevent hydrazine triflylation, which was found to be irreversible at temperatures up to 100 °C. A significant increase in yield to 72% was observed when the reaction was conducted at 35 °C (Table 1, entry 9); at this temperature, nucleophilic attack on the ring by hydrazine could outcompete detriflylation (Figure 3A). A solvent screen led to the identification of anhydrous 1,4-dioxane as the optimal solvent, giving 5-phenyl-1H-pyrazole in 90% yield (Table 1, entry 10; see the Supporting Information for details). While water quenched the activated N-triflylpyrimidinium triflate species, exposure to air did not impede the reaction. Furthermore, N-triflylation could be quantitatively achieved at temperatures as low as −78° C, reflected in the calculated low barrier of this step (Figure 3). As anticipated, hydrazine addition at −78° C led to quantitative detriflylation of the N-triflylpyrimidinium species.
Table 1.
Effect of Pyrimidine Activator
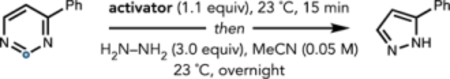
| ||
|---|---|---|
| entry | activator | yield (%)a |
| 1 | BzCl | 0 |
| 2 | AcCl | 0 |
| 3 | EtOCOCl | 0 |
| 4 | PhOCOCl | 17 |
| 5 | 1:1 PhOCOCl, NaI | 62 |
| 6 | MsCl | 0 |
| 7 | TsCl | 0 |
| 8 | Tf2O | 37 |
| 9b | Tf2O | 72 |
| 10 c | Tf 2 O | 90 |
Determined by 1H NMR.
35°C.
Dioxane used as solvent.
Figure 3.
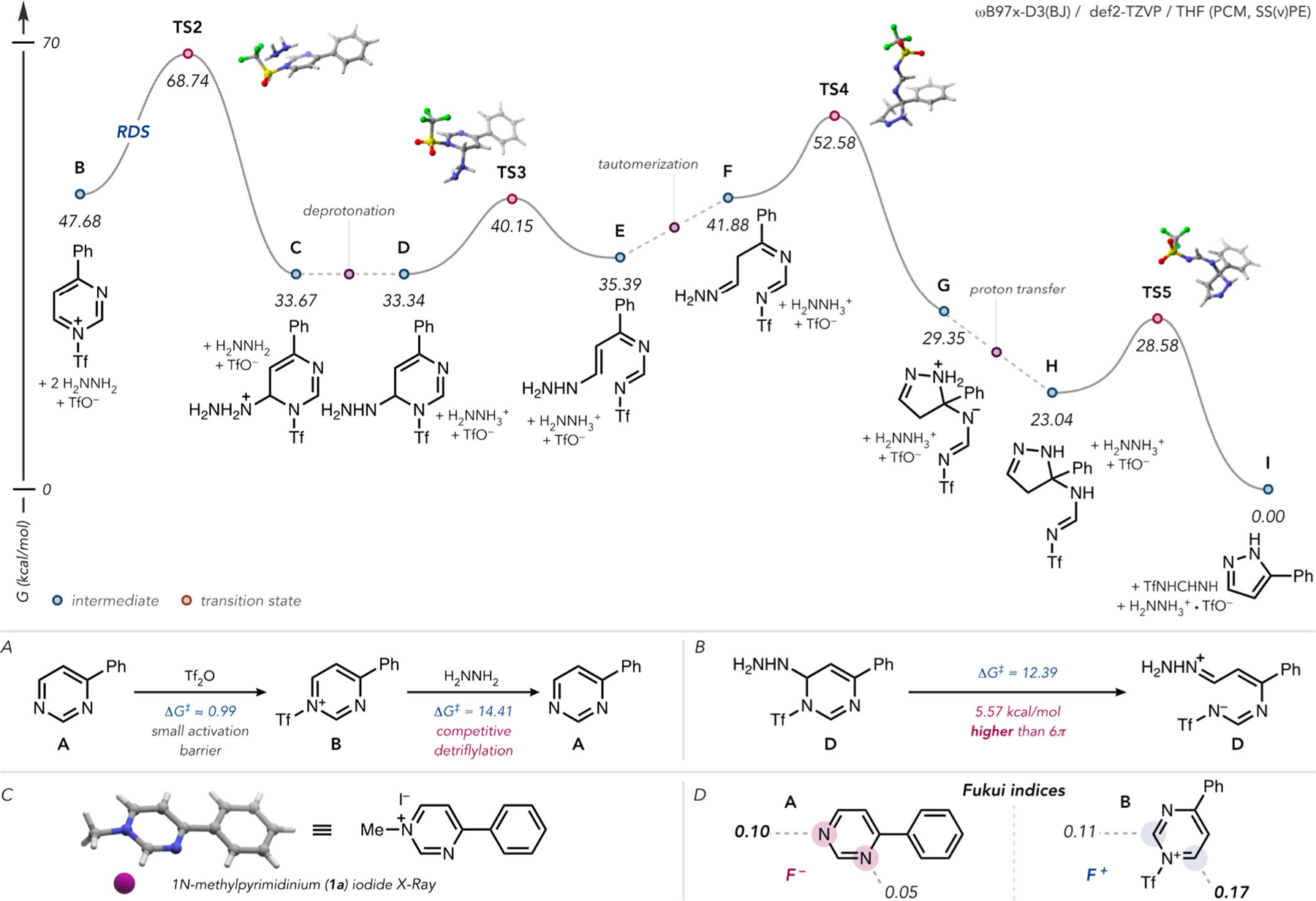
Mechanism of the triflylation-promoted, hydrazine-mediated ring contraction of pyrimidines. (A) Competitive detriflylation of B. (B) Calculations for aminal collapse pathway. (C) X-ray crystal structure of N-methyl-4-phenylpyrimidinium iodide (MeCN molecule omitted for clarity). (D) Fukui nucleophilicity (left) and electrophilicity (right) indices for A and B, respectively.
Mechanistically, as initially proposed by van Veldhuizen,28 this transformation may proceed, as shown in Figure 3. N-Triflylation of the least sterically hindered (and most π-excessive) pyrimidine nitrogen (ΔG‡ ≈ 0.99 kcal/mol; Figure 3A) serves to activate the pyrimidine ring toward nucleophilic attack (see the Supporting Information for details). The site-selectivity for N-triflylation is supported by the formation of 1-methyl–4-phenylpyrimidinium iodide (1a, Figure 3C) upon methylation of A and corroborated by the higher Fukui nucleophilicity index that was calculated for this atom (Figure 3D). The activated pyrimidinium is then attacked at C6 by hydrazine to give C. Computations found this step to be rate limiting, as shown in Figure 3. Detriflylation at this stage was found to be competitive at a lower temperature (Figure 3A), reflected in the yield increase when the reaction temperature was increased to 35 °C. Deprotonation of highly acidic hydrazinium species C gives D, followed by 6π electrocyclic ring opening to give E. The barrier to electrocyclic ring opening from charged species C was calculated to be 16.18 kcal/mol higher than from neutral species D. Additionally, the barrier for aminal collapse from D was calculated to be 5.57 kcal/mol higher than 6π electrocyclic ring opening from the same intermediate (Figure 3B). Thereafter, the terminal hydrazone nitrogen engages the ring-opened species (after tautomerization) at C4 to give charge-separated species G. Subsequent proton transfer to give H and elimination of N-triflylformamidine through a 1,5-sigmatropic H-shift results in the pyrazole product. In total, the reaction of 4-phenylpyrimdine (A) with hydrazine to give 5-phenyl-1H-pyrazole (I) and formamidine was calculated to be exergonic by 11.64 kcal/mol.
The scope of the formal carbon deletion is summarized in Figure 4. Because some substrates were observed to react more slowly than others, and product decomposition did not occur over prolonged reaction times, reactions were run over 18 h for consistency. The contraction protocol was found to be tolerant of electronically diverse arenes, as shown in Figure 4A (compare yield for 2a and 3b). Ortho- and meta-phenyl derivatives displayed a similar trend (see 3a and b). Only a slight difference in yield was observed for ortho- and meta-methyl derivatives (see 4a and 4b). Halogenated substrates were tolerated as evidenced by the formation of 5a and 5b as well as 6a and 6b.
Figure 4.
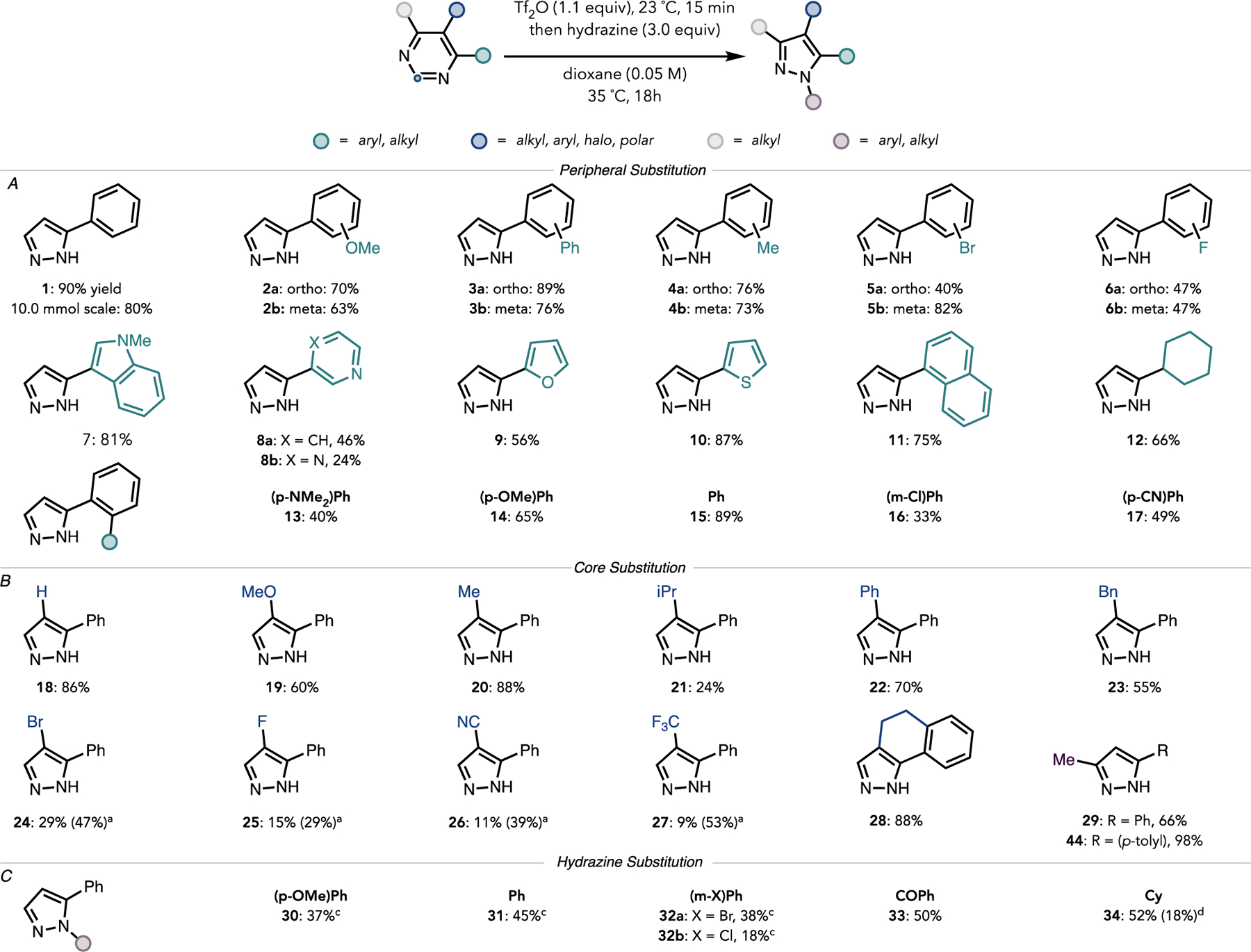
Ring contraction scope. Reactions performed on 0.10 mmol of substrate. Percentages reported are isolated yields. (A) Scope of substrates substituted with arenes and heteroarenes at C4. (B) Scope of substrates substituted at C5. aTriflylation stage run at 60 °C for 2 h before the reaction mixture was cooled to 35 °C for the addition of hydrazine. (C) Scope of hydrazines. CHydrochloride salt of hydrazine derivative was used together with 4.0 equiv of Na2 CO3. dThe products were isolated as a 3:1 N2/N1 substitution mixture.
Heteroarene substitution on the pyrimidine ring was also well-tolerated. For example, 7 was formed in 81% yield. 4-(3-Pyridyl)pyrimidine and 4-pyrazinylpyrimidine, where competing triflylation of the heterocycle substituents was anticipated, contracted to 8a and 8b in poorer yields of 47% and 24%, respectively. Mass recovery in these cases was modest, and distinct competing reactivity was observed at the pyrazine unit. 2-Furanyl and 2-thiophenyl derivatives also participated in the ring contraction, giving 9 and 10 in 56 and 87% yield, respectively.
Because directed C–H functionalization is often employed to introduce aromatic groups15,29,30 we also evaluated the effect of biaryl substituents on the pyrimidine contraction. These results are summarized in Figure 4A. Contraction of a para-dimethylaminophenyl derivative led to 13 in a modest yield of 40%, possibly due to competing N-triflylation of the more nucleophilic dimethylaniline nitrogen; 59% of the starting material was recovered in this case. A para-methoxyphenyl derivative gave 14 in an appreciable 65% yield, though some demethylation was observed. meta-Chlorophenyl and para-cyanophenyl derivatives gave 16 and 17 in 33 and 49% yield, respectively, along with a significant amount of recovered starting material.
We then sought to investigate the effect of functional groups appended directly to the pyrimidine ring. These results are summarized in Figure 4B. Hydrocarbon groups at C5 of the pyrimidine were well tolerated, giving yields of 50–88% (see 20, 22, or 23). A fused pyrimidine participated readily, giving 28 in 88% yield. Steric encumbrance at C5 adversely affected the contraction; isopropyl appended 21 was formed in only 24% yield. Though electron rich polar groups were tolerated (e.g., methoxypyrazole 19 is formed in 60% yield), electrondeficient pyrimidines performed poorly (see 24–27), likely due to incomplete triflylation or competing detriflylation of these substrates upon the addition of the hydrazine nucleophile. Heating these substrates to 60 °C for 2 h during the activation stage (to increase triflylation) then cooling to 35 °C prior to hydrazine introduction increased the yield significantly (e.g., from 9 to 53% for trifluoromethyl derivative 27).
Because N1-functionalized pyrazoles, such as 31, are incapable of directing C–H functionalizations (vide supra), we also evaluated the formation of N1-substituted pyrazoles (Figure 4C). Phenylhydrazine provided N1-phenylpyrazole (31) in 50% yield, which was a lower yield than when unsubstituted hydrazine was used likely due to the reduced nucleophilicity of the phenyl-bearing nitrogen. The constitution of 31 is consistent with a faster initial attack of the terminal phenylhydrazine nitrogen. Phenylhydrazine hydrochloride derivatives also affected the contraction smoothly to give 30 and 32a-b when excess Na2 CO3 was added (see the Supporting Information for details). Notably, these hydrazines performed poorly in their free base forms. Electron-poor benzohydrazide contracted to 33 in 50% yield. A reverse in regioselectivity was observed when cyclohexylhydrazine was used as the contraction coupling partner, generating a 2.5:1 mixture of isomeric pyrazoles 34a-b in 70% combined yield favoring substitution at N2 (confirmed by NOESY). This switch in selectivity likely arose from the increased nucleophilicity of the alkyl-bearing nitrogen in this case, favoring its initial addition to the activated pyrimidinium. C2-Aryl derivatives gave only trace amounts of the desired contraction products (see the Supporting Information for details). In these cases, because the C2-aryl group would be excised upon contraction, these substrates are not particularly well-aligned with the goal of diversification of substituted pyrimidines to afford the corresponding substituted pyrazoles.
We also modified the protocol so that the ring contraction could be carried out in the presence of free OH and NH groups (Figure 5A). To this end, transient OH triflylation was achieved by using an excess of triflic anhydride along with Na2 CO3 as an acid scavenger, enabling the contraction of an OH-containing pyrimidine to give 35 in 65% yield after basic workup (to unveil the in situ-triflylated phenol-OH). When the basic detriflylation step was omitted, O-triflylated product 36 was isolated in 61% yield along with 35 in 39% yield, presumably arising through hydrazine-mediated detriflylation of the phenol. This approach was applied to NH-containing 4-(para-aminophenyl)pyrimidine to give 37 in 47% yield along with a 33% recovery of starting material. A substituted quinazoline (a structural motif common in pharmaceuticals) contracted to give 38 in 41% yield (Figure 5B).
Figure 5.
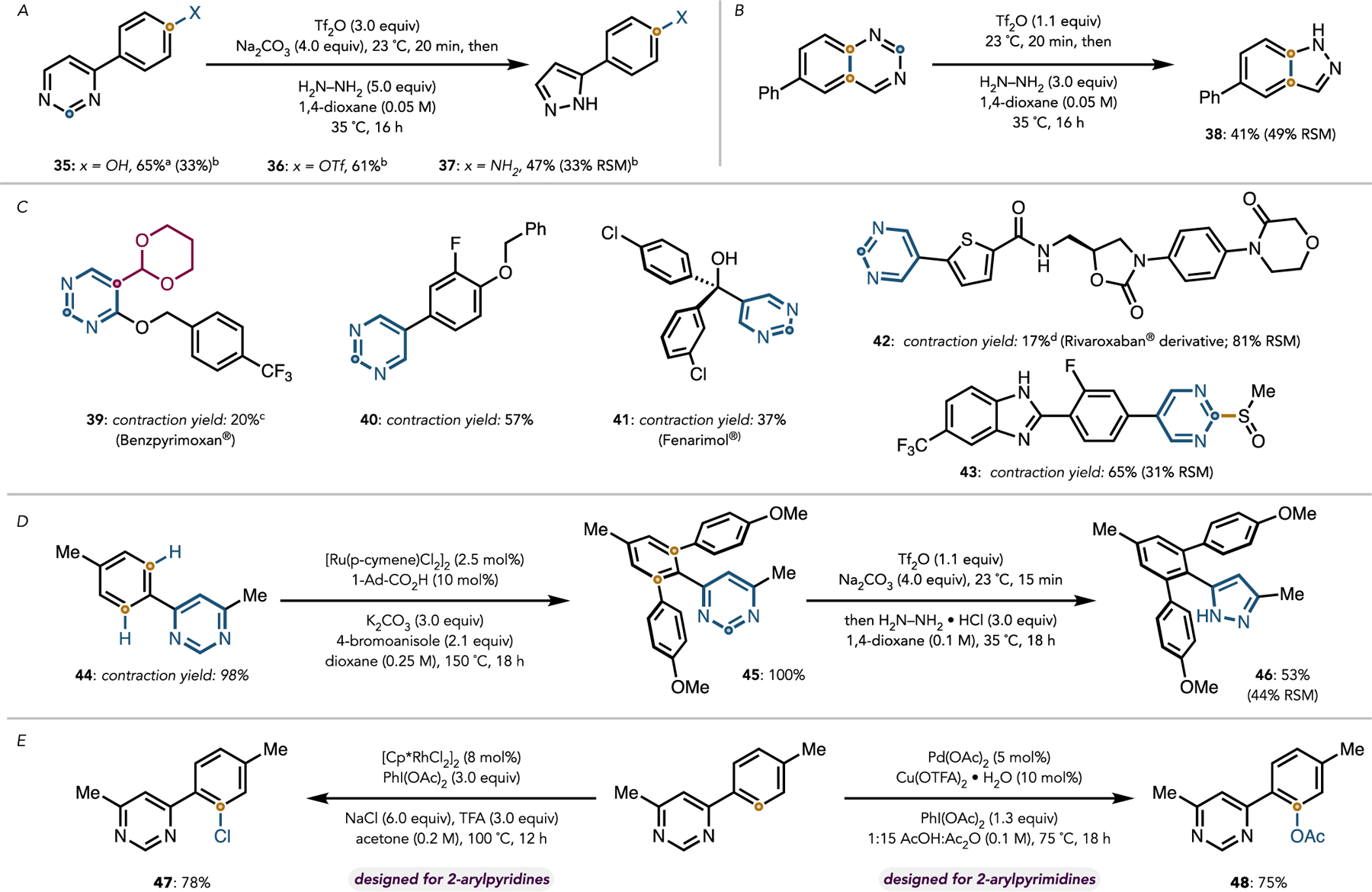
(A,B) Scope of specialized substrates. a3.0 equiv Tf2O was employed together with 4.0 equiv Na2 CO3; the reaction mixture was stirred with 10.0 equiv aqueous NaOH in 2:1 1,4-dioxane:methanol (0.33M) at 23 °C after completion to affect detriflylation. bThe basic workup step was omitted. (C) Scope of druglike substrates. cThe reaction was run at 60 °C. dHFIP was used as the reaction solvent, and triflylation was carried out at 0°C. (D) Synthesis of a C–H functionalized late-stage intermediate of Celecoxib. (E) Demonstrative pyrimidine-directed C–H functionalization reactions.
We also investigated the contraction of the pyrimidine group in complex molecules of pharmaceutical relevance, such as those shown in Figure 5C. Sterically hindered pyrimidine 39 (benzpyrimoxan)31 failed to undergo contraction under the typical conditions. However, the established protocol could be adapted to overcome the steric hindrance in 39 by increasing the reaction temperature to 60 °C, which led to a 20% yield of the desired contraction product. In comparison, the pyrimidine moiety of 40 readily underwent contraction in 57% yield with 41% starting material recovery.32 The successful contractions of 40–42 demonstrate the applicability of the method to C5-substituted pyrimidines. The agrochemical Fenarimol (41) underwent contraction in 37% yield along with 51% of the starting material recovered. Pyrimidinylated Rivaroxaban (Xarelto) derivative 42 underwent ring contraction in HFIP in 17% yield with 79% starting material recovery when triflylation carried was out at 0 °C. Notably, amidinecontaining methylsulfinyl pyrimidine 43 underwent contraction in 65% yield with 31% starting material recovery. The successful contraction of this compound demonstrates that substrates bearing non-aryl C2-substitution can participate successfully in the transformation.
Finally, pyrimidine-directed C–H functionalization of 44 followed by contraction to the corresponding pyrazole gave a C–H functionalized derivative of Celecoxib (Celebrex) precursor 46.33 Specifically, pyrimidine-directed rutheniumcatalyzed C–H bis-arylation14 of 44 gave 45 in quantitative yield. Alternatively, directed C–H mono-arylation of 44 could be achieved in 75% yield (see the Supporting Information for details). Notably, no C–H functionalized products were formed under the same conditions with the analogous pyrazole substrate. Arylpyrimidine 45 underwent contraction to the corresponding pyrazole in 53% yield with 44% starting material recovery. The sequence shown in Figure 5D highlights the power of this transformation for heterocycle swaps, enabling the use of a pyrimidine as a surrogate for a pyrazole. Additionally, directed C–H functionalization reactions of 2-arylpyrimidines, such as ortho-acetoxylation17 and ortho-chlorination,34 were successfully applied to 4-arylpyrimidine 44 to give 47 and 48 in good yields (Figure 5E; see the Supporting Information for details). Finally, in a large-scale demonstration, the contraction of 4-phenylpyrimidine was found to proceed in 80% yield on a 10.0 mmol(1.56 g) scale.
CONCLUSIONS
The approach reported here accomplishes the conversion of pyrimidines into pyrazoles under milder conditions and with a much broader substrate scope than was previously possible. While a pyrimidine to pyrazole conversion has been reported previously, it required high temperatures or methylation of the starting substrates with solvent quantities of the methylating reagents, thus limiting the utility of that previously reported method. Furthermore, we have established a new platform for the preparation of diversified pyrazole compounds by leveraging the pyrimidine core as a strong directing group for C–H functionalization, which is then followed by scaffold hopping9,12,13 to the corresponding pyrazole. Our approach also provides access to a diverse range of N2-substituted pyrazoles, which are not easily accessed at a late stage due to the poor directing ability of these substituted diazoles in C–H functionalizations and the difficulty of their preparation through regioselective functionalization of substituted NH-pyrazoles. This report serves as a demonstration of the concept of “single atom” skeletal editing, examples of which continue to emerge.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Hasan Celik and UC Berkeley’s NMR facility in the College of Chemistry (CoC-NMR) for spectroscopic assistance. Instruments in CoC-NMR are supported in part by NIH S10OD024998. We thank Dr. Nicholas Settineri (UC Berkeley) for X-ray crystallographic studies of 1a. We are grateful to Dr. Kathy Durkin and Dr. Dave Small for computational guidance. We are grateful to Prof. Dean Tantillo (UC Davis) for detailed and insightful discussions regarding the calculations described in Figure 3. The CoC-MGCF was supported in part by NIH S10OD023532. We thank Dr. Eric Simmons [BristolMyersSquibb Company, New Brunswick, NJ, USA (BMS)] for a gift of Rivaroxaban (the precursor to 42). We thank Dr. Charles Yeung [Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA (MSD)] for a gift of 43.
Funding
R.S. is grateful to the National Instituted of General Medical Sciences (NIGMS R35 GM130345) for financial support. G. L. B. thanks the National Science Foundation for a graduate fellowship (NSF GRFP 2021294420). F. C. thanks Bio4-Dreams for funding a visiting student stay at UC Berkeley.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c10746.
Experimental procedures, characterization data, spectra for all new compounds, crystallographic data and Cartesian coordinates of DFT-optimized structures, and computational details (PDF)
Accession Codes
CCDC 2210673 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223336033.
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.2c10746
The authors declare no competing financial interest.
Contributor Information
G. Logan Bartholomew, Department of Chemistry, University of California, Berkeley, Berkeley, California 94720, United States.
Filippo Carpaneto, Department of Chemistry, University of California, Berkeley, Berkeley, California 94720, United States; Present Address: Dipartimento di Chimica, Università degli Studi di Torino, Torino, 10124, Italia.
Richmond Sarpong, Department of Chemistry, University of California, Berkeley, Berkeley, California 94720, United States.
REFERENCES
- (1).Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals: Miniperspective. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- (2).Faria JV; Vegi PF; Miguita AGC; dos Santos MS; Boechat N; Bernardino AMR Recently Reported Biological Activities of Pyrazole Compounds. Bioorg. Med. Chem 2017, 25, 5891–5903. [DOI] [PubMed] [Google Scholar]
- (3).Ansari A; Ali A; Asif M; Shamsuzzaman S Review: Biologically Active Pyrazole Derivatives. New J. Chem 2017, 41, 16–41. [Google Scholar]
- (4).Kumar S; Narasimhan B Therapeutic Potential of Heterocyclic Pyrimidine Scaffolds. Chem. Cent. J 2018, 12, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Shearer J; Castro JL; Lawson ADG; MacCoss M; Taylor RD Rings in Clinical Trials and Drugs: Present and Future. J. Med. Chem 2022, 65, 8699–8712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Verbitskiy E; Rusinov G; Chupakhin O; Charushin V Recent Advances in Direct C–H Functionalization of Pyrimidines. Synthesis 2018, 50, 193–210. [Google Scholar]
- (7).Kang E; Kim HT; Joo JM Transition-Metal-Catalyzed C-H Functionalization of Pyrazoles. Org. Biomol. Chem 2020, 18, 6192–6210. [DOI] [PubMed] [Google Scholar]
- (8).Campos KR; Coleman PJ; Alvarez JC; Dreher SD; Garbaccio RM; Terrett NK; Tillyer RD; Truppo MD; Parmee ER The Importance of Synthetic Chemistry in the Pharmaceutical Industry. Science 2019, 363, No. eaat0805. [DOI] [PubMed] [Google Scholar]
- (9).Roque JB; Kuroda Y; Göttemann LT; Sarpong R Deconstructive Diversification of Cyclic Amines. Nature 2018, 564, 244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kennedy SH; Dherange BD; Berger KJ; Levin MD Skeletal Editing through Direct Nitrogen Deletion of Secondary Amines. Nature 2021, 593, 223–227. [DOI] [PubMed] [Google Scholar]
- (11).Jurczyk J; Lux MC; Adpressa D; Kim SF; Lam Y; Yeung CS; Sarpong R Photomediated Ring Contraction of Saturated Heterocycles. Science 2021, 373, 1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Woo J; Christian AH; Burgess SA; Jiang Y; Mansoor UF; Levin MD Scaffold Hopping by Net Photochemical Carbon Deletion of Azaarenes. Science 2022, 376, 527–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Jurczyk J; Woo J; Kim SF; Dherange BD; Sarpong R; Levin MD Single-Atom Logic for Heterocycle Editing. Nat. Synth 2022, 1, 352–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Stefane B; Fabris J; Požgan F C–H Bond Functionalization of Arylpyrimidines Catalyzed by an in Situ Generated Ruthenium(II) Carboxylate System and the Construction of Tris(Heteroaryl)Substituted Benzenes. Eur. J. Org. Chem 2011, 2011, 3474–3481. [Google Scholar]
- (15).Norinder J; Matsumoto A; Yoshikai N; Nakamura E IronCatalyzed Direct Arylation through Directed C–H Bond Activation. J. Am. Chem. Soc 2008, 130, 5858–5859. [DOI] [PubMed] [Google Scholar]
- (16).Song B; Zheng X; Mo J; Xu B Palladium-Catalyzed Monoselective Halogenation of C·H Bonds: Efficient Access to Halogenated Arylpyrimidines Using Calcium Halides. Adv. Synth. Catal 2010, 352, 329–335. [Google Scholar]
- (17).Zheng X; Song B; Xu B Palladium-Catalyzed Regioselective C–H Bond Ortho-Acetoxylation of Arylpyrimidines. Eur. J. Org. Chem 2010, 4376–4380. [Google Scholar]
- (18).Wang X; Truesdale L; Yu J-Q Pd(II)-Catalyzed Ortho-Trifluoromethylation of Arenes Using TFA as a Promoter. J. Am. Chem. Soc 2010, 132, 3648–3649. [DOI] [PubMed] [Google Scholar]
- (19).Chatani N; Ie Y; Kakiuchi F; Murai S Ru3(CO)12-Catalyzed Reaction of Pyridylbenzenes with Carbon Monoxide and Olefins. Carbonylation at a C–H Bond in the Benzene Ring. J. Org. Chem 1997, 62, 2604–2610. [DOI] [PubMed] [Google Scholar]
- (20).Reisenbauer JC; Green O; Franchino A; Finkelstein P; Morandi B Late-Stage Diversification of Indole Skeletons through Nitrogen Atom Insertion. Science 2022, 377, 1104–1109. [DOI] [PubMed] [Google Scholar]
- (21).Hyland EE; Kelly PQ; McKillop AM; Dherange BD; Levin MD Unified Access to Pyrimidines and Quinazolines Enabled by N-N Cleaving Carbon Atom Insertion. J. Am. Chem. Soc 2022, 144,19258–19264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Xiao X; Jia G; Liu F; Ou G; Xie Y RuHCl(CO)(PPh3)3-Catalyzed Direct Amidation of Arene C–H Bond with Azides. J. Org. Chem 2018, 83, 13811–13820. [DOI] [PubMed] [Google Scholar]
- (23).van der Plas HC; Jongejan H Ring Transformations in Reactions of Heterocyclic Compounds with Nucleophiles (III): Conversion of Pyrimidine and Some of Its Methyl Derivatives by Hydrazine and by Methylhydrazine Sulfate into Pyrazoles and Methylpyrazoles. Recl. Trav. Chim. Pays-Bas 1968, 87, 1065–1072. [Google Scholar]
- (24).Kuethe JT; Comins DL Addition of Indolyl and Pyrrolyl Grignard Reagents to 1-Acylpyridinium Salts. J. Org. Chem 2004, 69, 2863–2866. [DOI] [PubMed] [Google Scholar]
- (25).Kuethe JT; Comins DL Addition of Metallo Enolates to Chiral 1-Acylpyridinium Salts: Total Synthesis of (+)-Cannabisativine. Org. Lett 2000, 2, 855–857. [DOI] [PubMed] [Google Scholar]
- (26).Comins DL; Abdullah AH Regioselective Addition of Grignard Reagents to 1-Acylpyridinium Salts. A Convenient Method for the Synthesis of 4-Alkyl(Aryl)Pyridines. J. Org. Chem 1982, 47, 4315–4319. [Google Scholar]
- (27).Comins DL; Hong H The Addition of Metallo Enolates to Chiral 1-Acylpyridinium Salts. An Asymmetric Synthesis of (−)-Sedamine. J. Org. Chem 1993, 58, 5035–5036. [Google Scholar]
- (28).Brouwer MS; van der Plas HC; van Veldhuizen A A NMR Study on the Ring Contraction of N-Methylpyrimidinium Salts into Pyrazoles. Recl. Trav. Chim. Pays-Bas 2010, 97, 110–112. [Google Scholar]
- (29).Štefane B; Fabris J; Požgan F C–H Bond Functionalization of Arylpyrimidines Catalyzed by an in Situ Generated Ruthenium(II) Carboxylate System and the Construction of Tris(Heteroaryl)Substituted Benzenes. Eur. J. Org. Chem 2011, 2011, 3474–3481. [Google Scholar]
- (30).Zhou J; Mao Z; Pan H; Zhang X Pd-Catalyzed Highly Selective and Direct Ortho C–H Arylation of Pyrrolo[2,3-d]Pyrimidine Derivatives. Org. Chem. Front 2020, 7, 324–328. [Google Scholar]
- (31).Satoh E; Kasahara R; Fukatsu K; Aoki T; Harayama H; Murata T Benzpyrimoxan: Design, Synthesis, and Biological Activity of a Novel Insecticide. J. Pestic. Sci 2021, 46, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Zhang X; Nottingham KG; Patel C; Alegre-Requena JV; Levy JN; Paton RS; McNally A Phosphorus-Mediated Sp2-Sp3 Couplings for C–H Fluoroalkylation of Azines. Nature 2021, 594, 217–222. [DOI] [PubMed] [Google Scholar]
- (33).Gong X; Wang R; Zheng J; Feng Y; Kang X Synthesis Method of Celecoxib. CN 111484453 A, 2020. [Google Scholar]
- (34).Zhang P; Hong L; Li G; Wang R Sodium Halides as Halogenating Reagents: Rhodium(III)-Catalyzed Versatile and Practical Halogenation of Aryl Compounds. Adv. Synth. Catal 2015, 357, 345–349. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.