


Abstract
Knowledge of protein–ligand binding sites (LBSs) enables research ranging from protein function annotation to structure-based drug design. To this end, we have previously developed a stand-alone tool, P2Rank, and the web server PrankWeb (https://prankweb.cz/) for fast and accurate LBS prediction. Here, we present significant enhancements to PrankWeb. First, a new, more accurate evolutionary conservation estimation pipeline based on the UniRef50 sequence database and the HMMER3 package is introduced. Second, PrankWeb now allows users to enter UniProt ID to carry out LBS predictions in situations where no experimental structure is available by utilizing the AlphaFold model database. Additionally, a range of minor improvements has been implemented. These include the ability to deploy PrankWeb and P2Rank as Docker containers, support for the mmCIF file format, improved public REST API access, or the ability to batch download the LBS predictions for the whole PDB archive and parts of the AlphaFold database.
Graphical Abstract
Graphical Abstract.
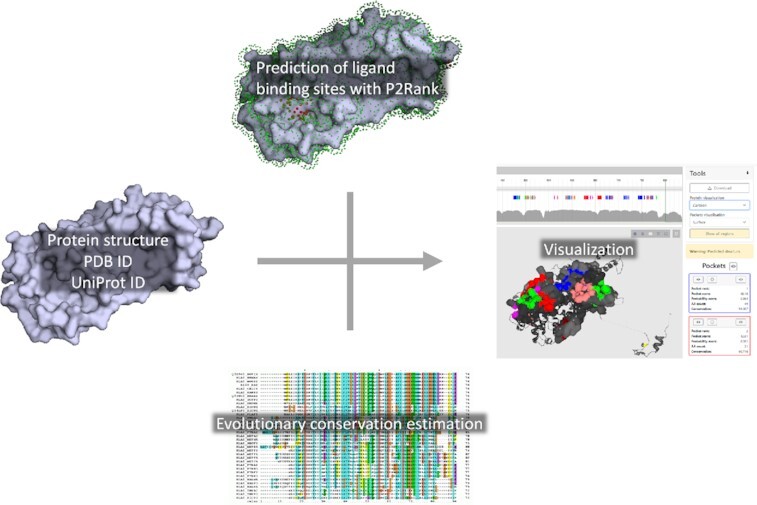
PrankWeb accepts a protein structure on its input, computes evolutionary conservation, and predicts binding sites which are then mapped onto the structure and can be visually inspected.
INTRODUCTION
Interactions of proteins with other molecules drive biological processes at the molecular level. One specific class of such interactions are protein–small molecule (ligand) interactions; identifying the sites and roles of these interactions is crucial for the elucidation of the molecular mechanisms of enzymes, regulation of protein oligomerization, or designing new drugs (e.g., in case drug resistance has occurred) (1,2). In these applications, precise knowledge of the protein’s ligand-binding sites (LBSs) is required. As experimental identification of LBSs is time-consuming and expensive, computational methods have been developed to facilitate LBS identification from the protein three-dimensional (3D) structure. These methods can be broadly categorized as geometric, energetic, evolution-based, and knowledge- or machine learning (ML)-based. Many of the existing methods combine the aforementioned approaches, which is also the case of the P2Rank method (3) developed in our group. P2Rank assigns structural, physico-chemical, and evolutionary features to points on a mesh covering the protein surface and builds an ML model over this representation. The model is used to detect ligandable points, which are then clustered to obtain a list of surface patches corresponding to the predicted LBSs. The approach has achieved state-of-the-art performance and is still on par or outperforming newer deep learning methods (4).
The lack of broadly accessible online resources has historically hindered access to the LBS prediction methods. To this end, we have developed PrankWeb (5), an online tool encapsulating the P2Rank approach. PrankWeb has allowed its users to enter a 3D structure as a Protein Data Bank (6) (PDB) file or using a PDB identifier, carried out evolutionary conservation analysis, predicted the LBSs using P2Rank, and enabled visual examination of the results. This paper introduces PrankWeb 3, an improved version of the resource.
A limiting aspect of the structure-based LBS prediction approaches is the necessity of having the protein 3D structure determined. Although the number of resolved protein structures keeps increasing, it is still far behind the number of known protein sequences (7). However, recent advances in protein structure prediction, namely the introduction of the AlphaFold 2 method (8) and the AlphaFold Protein Structure Database (AlphaFold DB) (9), have opened the door for the application of structure-based approaches also toward proteins for which only the sequence is known. This development has motivated one of the major improvements in PrankWeb 3: the adoption of the AlphaFold DB, allowing PrankWeb users to enter a UniProt accession number as the input. This change significantly increases the number of proteins to which PrankWeb is applicable (section Predicted structures). Another significant improvement is the replacement of the former evolutionary conservation estimation pipeline with a faster, more consistent version (section Evolutionary conservation calculation pipeline). The last major change has been the refactoring of the PrankWeb application resulting in a modular architecture with strictly separated components. Such architecture enables easy utilization of the application or its parts (such as the conservation calculation pipeline) to advanced users via Docker containers (section Other improvements). A detailed description of the changes follows.
EVOLUTIONARY CONSERVATION CALCULATION PIPELINE
Evolutionary conservation (EC) has been identified as a powerful indicator of functionally significant regions of protein structures; for this reason, it has been utilized as an optional feature capable of improving the default P2Rank predictions. Previous versions of PrankWeb utilized a series of sequence databases to construct a multiple sequence alignment (MSA) of sequences similar to the given query, and subsequently quantified the EC of its individual columns using Jensen–Shannon divergence (10). This approach possessed two major drawbacks. First, the use of fallback sequence databases for the construction of an MSA of sufficient size resulted in discontinuities in the conservation scores as the number of sequences in the MSA exceeded the threshold. A single P2Rank model was thus unable to account for the different sequence distributions (and, therefore, conservation scores) intrinsic to the individual sequence databases. Second, and more importantly, the previous EC calculation pipeline could take several hours to complete, severely impacting the user’s experience with PrankWeb.
Starting with PrankWeb 3, the former EC calculation pipeline has been replaced with a simpler, faster, and more consistent one inspired by the recent Amino Acid Interactions web server v2.0 (11). The new pipeline operates as follows. First, polypeptide chain sequences are extracted from the input file using P2Rank. The phmmer tool from the HMMER software package (http://hmmer.org/) is then used to identify and align similar sequences for each respective query; UniRef50 Release 2021_03 (12) is used as the single target sequence database. Up to 1000 sequences are then randomly selected from each MSA to form the respective sample MSAs; weights are assigned to the individual sequences constituting the sample MSAs using the Gerstein/Sonnhammer/Chothia algorithm (13) implemented in the esl-weight miniapp included with the HMMER software. Finally, per-column information content (i.e. conservation score) and gap character frequency values are calculated using the esl-alistat miniapp, taking the individual sequence weights into account; positions containing the gap character in >50 % of sequences are masked to appear as possessing no conservation at all. The pipeline utilizes a fixed seed value for any random selection, making the output deterministic for a given query.
Table 1 shows the runtimes of the new EC calculation pipeline measured on the datasets used for the training, validation, and testing of P2Rank models. It can be seen that for 50% of queries, the EC calculation pipeline (which constitutes most of the time required for PrankWeb predictions) finishes in about 2 min, while nearly all queries finish within 5 min. In comparison, for the previous EC conservation pipeline on the CHEN11 dataset, the median of runtimes was 275 s (4.6 min) while 95th percentile was 854 s (14.2 min).
Table 1.
The runtimes of the new EC calculation pipeline (in seconds) measured on the datasets used for the training (CHEN11), validation (JOINED), and testing (COACH420 and HOLO4K) of P2Rank models. The computations were performed on a desktop computer running Ubuntu 20.04, HMMER v3.3.2, and using the i7-3770K processor. The numbers in parentheses indicate the number of polypeptide chains in the respective datasets. See the original P2Rank publication (3) for a detailed description of the datasets
| CHEN11 (251) | JOINED (643) | COACH420 (420) | HOLO4K (8588) | |
|---|---|---|---|---|
| Runtime (50th percentile; s) | 107 | 109 | 108 | 127 |
| Runtime (95th percentile; s) | 139 | 244 | 193 | 324 |
The adoption of the new EC calculation pipeline necessitated the preparation of a new EC-aware P2Rank model. Table 2 presents the evaluation of all the new P2Rank models prepared for PrankWeb 3, as well as their comparison with the former models; it can be seen that the new Default models exceed the performance of the corresponding old models when evaluated on the representative HOLO4K dataset.
Table 2.
Identification success rates (in %) measured using the DCA criterion utilizing a 4.0 Å threshold for the distance between the center of the predicted LBS and any ligand atom; only the n or (n + 2), respectively, top-ranking predicted LBSs are considered in the evaluation, where n is the number of ligands in the respective 3D structure. Values for Default (old) and Default + conservation (old) are taken from the original PrankWeb publication (5) and are shown only for comparison, as these models are no longer used. B-factor-free are used with AlphaFold predictions which utilize the B-factor field for confidence scores. Please note that old models were generated by the older version of P2Rank, which used older versions of BioJava and CDK. Using newer versions changed how certain PDB files are parsed, and an upgrade of the CDK library fixed a bug in the algorithm that generates SAS points. This, together with bug fixes in P2Rank itself, causes the scores for the Default (old) and Default models to differ
| COACH420 | HOLO4K | |||
|---|---|---|---|---|
| Top-n | Top-(n + 2) | Top-n | Top-(n + 2) | |
| Default (old) | 72.0 | 78.3 | 68.6 | 74.0 |
| Default + conservation (old) | 73.2 | 77.9 | 72.1 | 76.7 |
| Default | 71.6 | 76.8 | 72.7 | 78.0 |
| Default + conservation | 74.3 | 77.2 | 74.5 | 78.4 |
| B-factor-free | 71.2 | 77.5 | 72.1 | 77.2 |
| B-factor-free + conservation | 74.9 | 78.5 | 73.9 | 77.7 |
PREDICTED STRUCTURES
The AlphaFold DB (9) is a freely and openly accessible resource housing 3D structure models for a selection of biomedically significant proteins predicted using AlphaFold 2 (8). In PrankWeb 3, we have precomputed the P2Rank LBS predictions for two components of the AlphaFold DB—the ‘model organism proteomes’ and ‘Swiss-Prot’—totalling over 800 000 proteins. As the AlphaFold 3D structure models utilize the B-factor fields of the structure files to store the per-residue confidence scores, computing these LBS predictions necessitated the preparation of two additional, B-factor field-agnostic P2Rank models (Table 2); it can be seen that the performance of these on the representative HOLO4K dataset (consisting of experimentally resolved 3D structures) is only marginally worse compared to the models utilizing B-factor as a feature.
To show how PrankWeb can be used to predict and visualize binding sites for predicted structures, we chose a protein from the G protein-coupled receptors (GPCR) family. The GPCR family is not only the largest protein family (with over 800 members), but also a family with >160 validated drug targets. GPCRs are membrane proteins and as such have represented a major challenge for structural biology. Advances in cryoEM methodology have brought a revolution in our understanding of intricate differences among GPCR proteins with more than 450 structures of over 80 proteins (14) solved so far, but many proteins indicated in human disease are still without an experimentally solved structure. The availability of high-quality 3D structure models in the AlphaFold DB, however, massively expands the number of proteins that can be investigated with PrankWeb. We used PrankWeb to show predicted binding sites on the AlphaFold model of succinate receptor 1 (uniprot code Q9BXA5), a protein suspected as a major player in the development of kidney hypertension and possibly also metabolic syndrome and thus potential drug target (15) without known experimentally solved 3D structure. The structure submission interface of PrankWeb has been extended to enable fetching predicted structures from the AlphaFold DB via the UniProt accession. After the accession is entered, the structure is downloaded from the AlphaFold DB (if not cached) and binding sites are predicted with P2Rank. Once the results are available, they are visualized in the PrankWeb interface. For AlphaFold predictions, the structure is color-coded by the confidence score. Moreover, PrankWeb enables visualization of only high-confidence regions (pLDDT > 70).
The results for the succinate receptor 1 are shown in Figure 1. Figure 1 A displays the best predicted pocket in blue on top. As the experimental structure with, or even without a ligand, is not known, the predicted structure was aligned using PyMOL with the structure of a closely related P2Y12 receptor (PDB ID 4NTJ (16)). The structural alignment (Figure 1B) shows that the best predicted succinate receptor binding pocket is different from ligand binding pocket of P2Y12 receptor as expected due to different properties and size of these ligands, although we can not be completely sure that the predicted binding site is correct as there is no experimentally solved structure of this receptor. This shows that using AlphaFold models for prediction of binding sites provides information that can not be extracted from experimentally solved structures of closely related proteins.
Figure 1.

P2Rank prediction on an AlphaFold model of human succinate receptor (Q9BXA5). (A) Visualization of the pockets from PrankWeb (available at https://prankweb.cz/analyze?database=v3-alphafold&code=Q9BXA5). The main pocket is in blue on the top of the structure. The structure is colored-coded by AlphaFold confidence (darker being more confident). (B) The predicted succinate receptor structure (in cyan) is aligned with closely related P2Y receptor (in grey, PDB ID 4NTJ) and its ligand (in magenta). The best binding pocket predicted for succinate receptor is shown in blue and is clearly outside of the binding pocket of P2Y receptor (visualized with PyMOL, http://www.pymol.org/pymol).
OTHER IMPROVEMENTS
Additional updates focus on improving the user experience and usability. The updates range from small quality of life improvements to complete redesign of the PrankWeb architecture.
The most noticeable change is in the results visualization page (Figure 2). First, the user can now select a visualization mode for the inspected protein and the predicted binding sites. The modes available are surface, cartoon, and balls and sticks. Second, when a pocket prediction is carried out on a predicted structure, the user can hide low-confident regions, i.e. regions with pLDDT score <70. Finally, the protein surface is colored by conservation score for the experimental structures, and by residue-level confidence scores for the predicted structures.
Figure 2.

PrankWeb results visualization page. The view shows predicted LBSs on the AlphaFold model of the human striatin-interacting protein (Q5VSL9), available at https://prankweb.cz/analyze?database=v3-alphafold&code=Q5VSL9. Pockets are displayed using surface visualization while the rest of hte structure is shown as cartoon. Different putative pockets are distinguished by color. The parts of the structure which are not part of any pocket are color-coded by tha AlphaFold confidence score, with darker regions being more confident. Finally, the visualization shows only high-confident parts of the structure (pLDDT score > 70) which are connected by dotted lines. Switching between full structure and confident regions only can be controlled by the user.
Another addition to the results visualization page is the pocket’s probability score. By default, the pockets are sorted using the P2Rank’s raw pocket score. However, as this value is not bound, it is hard to interpret by a user. To tackle this we added the pocket’s probability score that has a clearly defined maximum value and thus should provide easier interpretation to a user. The pocket probability score is calculated as a monotonous transformation of a raw pocket score to the interval [0,1]. The transformation is calibrated for each model on the HOLO4K dataset in such a way that the probability score represents a ratio of true binding sites among all predicted sites with a comparable raw score.
We have also updated the HTTP-based API to v2, indicating breaking changes. The core idea was to shift the API closer to the REST ideas. The change allows users to easily create new prediction tasks for custom structures using POST. GET requests can be used to retrieve prediction status, log, structure or prediction archive. The prediction archive can be also downloaded from the user interface and contains visualizations of the protein in PyMOL, parameters used to run P2rank, prediction log file and information about the predicted pockets in the CSV format. In addition, the archive can contain conservation scores if the user has chosen to use conservation in the prediction.
We also added links to the pre-computed predictions described in the section Evolutionary conservation calculation pipeline. Users can thus download all predictions computed for PDB and AlphaFold. For each database, we provide predictions computed with and without the use of conservation. The archive has similar content to the archive for a single prediction, the main difference is in the structure as the archives house multiple predictions.
Another modification in PrankWeb 3 is added support for the mmCIF format as the structure definition format. This was necessary as the PDB format has been deprecated due to its limitations.
Finally, under the hood, PrankWeb’s architecture has been completely redesigned. The new modular architecture strictly separates web-based user interface, data storage, and an execution component. The execution component is responsible for running the predictions from start to end. Starting with a protein file or UniProt ID, it will compute conservation and produce pocket predictions. Each component corresponds to a Docker image. Combined with docker-compose, it is easy to deploy and update PrankWeb instances. Thanks to the modular architecture, users can deploy only the execution component, using Docker, on their hardware. As a result, it is possible to run predictions on private data without exposing them to third-party servers. Another advantage is that such deployment allows users to run as many predictions as their computation resources allow. On the other hand, we are aware that not every user has the capacity to run the predictions on a large scale database such as PDB and parts of the AlphaFold.
DATA AVAILABILITY
The PrankWeb web server is publicly available at https://prankweb.cz/. The source codes are available at https://github.com/cusbg/p2rank-framework.
ACKNOWLEDGEMENTS
Computational resources were supplied by the project ‘e-Infrastruktura CZ’ (e-INFRA CZ LM2018140) supported by the Ministry of Education, Youth and Sports of the Czech Republic. This work was also carried out with the support of the Charles University grant SVV-260588 and the ELIXIR CZ Research Infrastructure (ID LM2018131, MEYS CR), including access to the computational resources.
Contributor Information
David Jakubec, Department of Software Engineering, Faculty of Mathematics and Physics, Charles University, Czech Republic.
Petr Skoda, Department of Software Engineering, Faculty of Mathematics and Physics, Charles University, Czech Republic.
Radoslav Krivak, Department of Software Engineering, Faculty of Mathematics and Physics, Charles University, Czech Republic.
Marian Novotny, Department of Cell Biology, Faculty of Science, Charles University, Czech Republic.
David Hoksza, Department of Software Engineering, Faculty of Mathematics and Physics, Charles University, Czech Republic.
FUNDING
Funding for open access charge: ELIXIR CZ Research Infrastructure (ID LM2018131, MEYS CR).
Conflict of interest statement. None declared.
REFERENCES
- 1. Konc J., Lešnik S., Janežič D. Modeling enzyme-ligand binding in drug discovery. J. Cheminform. 2015; 7:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Imamura A., Okada T., Mase H., Otani T., Kobayashi T., Tamura M., Kubata B.K., Inoue K., Rambo R.P., Uchiyama S. et al. Allosteric regulation accompanied by oligomeric state changes of Trypanosoma brucei GMP reductase through cystathionine-β-synthase domain. Nat. Commun. 2020; 11:1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Krivák R., Hoksza D. P2Rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure. J. Cheminf. 2018; 10:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mylonas S.K., Axenopoulos A., Daras P. DeepSurf: a surface-based deep learning approach for the prediction of ligand binding sites on proteins. Bioinformatics. 2021; 37:1681–1690. [DOI] [PubMed] [Google Scholar]
- 5. Jendele L., Krivak R., Skoda P., Novotny M., Hoksza D. PrankWeb: a web server for ligand binding site prediction and visualization. Nucleic Acids Res. 2019; 47:W345–W349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein data bank. Nucleic Acids Res. 2000; 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mitchell A.L., Almeida A., Beracochea M., Boland M., Burgin J., Cochrane G., Crusoe M.R., Kale V., Potter S.C., Richardson L.J. et al. MGnify: the microbiome analysis resource in 2020. Nucleic Acids Res. 2020; 48:D570–D578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A. et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Varadi M., Anyango S., Deshpande M., Nair S., Natassia C., Yordanova G., Yuan D., Stroe O., Wood G., Laydon A. et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022; 50:D439–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Capra J.A., Singh M. Predicting functionally important residues from sequence conservation. Bioinformatics. 2007; 23:1875–1882. [DOI] [PubMed] [Google Scholar]
- 11. Vymětal J., Jakubec D., Galgonek J., Vondrášek J. Amino Acid Interactions (INTAA) web server v2.0: a single service for computation of energetics and conservation in biomolecular 3D structures. Nucleic Acids Res. 2021; 49:W15–W20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Suzek B.E., Wang Y., Huang H., McGarvey P.B., Wu C.H. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics. 2015; 31:926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gerstein M., Sonnhammer E.L., Chothia C. Volume changes in protein evolution. J. Mol. Biol. 1994; 236:1067–1078. [DOI] [PubMed] [Google Scholar]
- 14. Yang D., Zhou Q., Labroska V., Qin S., Darbalaei S., Wu Y., Yuliantie E., Xie L., Tao H., Cheng J. et al. G protein-coupled receptors: structure- and function-based drug discovery. Signal Transduct. Target Ther. 2021; 6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ariza A.C., Deen P.M., Robben J.H. The succinate receptor as a novel therapeutic target for oxidative and metabolic stress-related conditions. Front. Endocrinol. (Lausanne). 2012; 3:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang K., Zhang J., Gao Z.G., Zhang D., Zhu L., Han G.W., Moss S.M., Paoletta S., Kiselev E., Lu W. et al. Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature. 2014; 509:115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The PrankWeb web server is publicly available at https://prankweb.cz/. The source codes are available at https://github.com/cusbg/p2rank-framework.