

Abstract
There has been extensive interest in cellular therapies for the treatment of myocardial infarction, but bottlenecks concerning cellular accumulation and retention remain. Here, we report a novel system of in situ crosslinking mesenchymal stem cells (MSCs) for the formation of a living depot at the infarct site. We have engineered bone marrow-derived mesenchymal stem cells that are surface decorated with heterodimerizing leucine zippers, termed ZipperCells. When delivered intravenously in sequential doses, we demonstrated that ZipperCells can migrate to the infarct site, crosslink, and show ~500% enhanced accumulation and ~600% improvement in prolonged retention at 10 days after injection compared to unmodified MSCs. This study introduces an advanced approach to creating non-invasive therapeutics depots using cellular crosslinking and provides the framework for future scaffold-free delivery methods for cardiac repair.
Keywords: Stem Cells, Leucine Zippers, Myocardial Infarction, Accumulation, Retention
Graphical Abstract
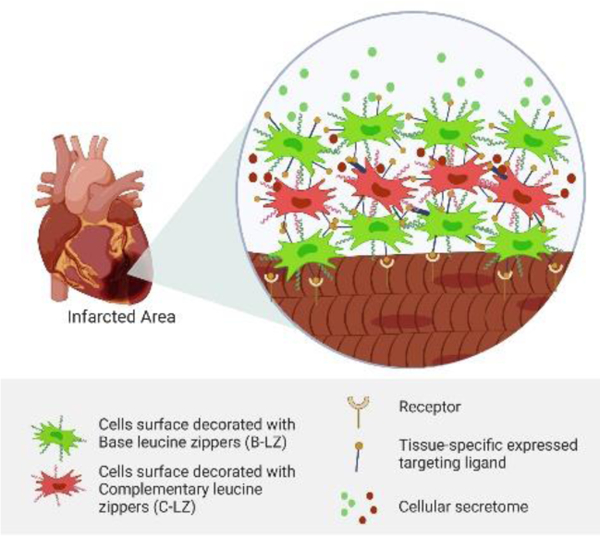
Traditional cell therapies for cardiac repair typically involve invasive intramyocardial injections and continue to suffer from poor cell accumulation and retention. Here, we report a novel, non-invasive system of in situ crosslinking mesenchymal stem cells (MSCs) for the formation of a living depot at the infarct site. We demonstrate a ~500% enhanced accumulation compared to unmodified MSCs.
1. Introduction
Myocardial infarction remains a leading cause of death worldwide [1]. Although the current standard of care for patients focuses on restoring perfusion as quickly as possible with percutaneous coronary interventions [2], patients are often left with dysregulated endogenous infarct repair and chronic severe morbidities, including ventricular arrhythmias and heart failure due to insufficient cardiac contractile function [3–5]. The drug delivery and regenerative medicine fields have focused on cell delivery, particularly mesenchymal stem cell delivery, to combat these downstream effects due to their regenerative abilities [6–8]. Despite these efficacious tools, delivery to the target site after intravenous injection remains highly ineffective, and traditional cell interventions typically require invasive intramyocardial injection or implantation [9,10]. These invasive injections or implantations come with a variety of mechanical and biological risks such as arrhythmia [11], induction of fibrosis [12], and elevated serum myocardial biomarkers, indicative of cardiomyocyte death[13]. Even with local delivery, promising in vitro MSC therapies are often plagued by poor therapeutic accumulation and retention at the infarct site of interest [14–16]. Rapid cellular clearing diminishes effective doses and therefore potential therapeutic efficacy.
Thus, there is an urgent need for non-invasive approaches that enhance the accumulation and retention of cell-based therapeutics at the disease site. To address this gap, here we propose to develop a novel delivery strategy to enhance the accumulation and retention of cells at the site of MI. The goal is to create a modular platform of injectable cells that crosslink to a scaffold at the infarct site to serve as a drug depot. The design criteria for the scaffold are (1) enhanced accumulation at the infarct site, (2) prolonged retention, and (3) in case of drug complications or immunogenic reactions, the possibility to dissolve the scaffold into its individual subunits for elimination.
We hypothesize that cell accumulation and retention can be enhanced at the infarct site by surface-decorating them with heterodimerizing leucine zippers that accumulate at the site of MI and cross-link into scaffold-like materials. Unlike conventional delivery strategies, where cells or carriers can no longer accumulate once the targeted areas at infarct sites are saturated and occupied, each dose of cells in our platform will serve as a capturing surface for the next dose of cells. This will dramatically amplify the accumulation of cells at the desired sites in a layer-by-layer fashion and allow adjustment of the concentration and type of therapy in subsequent doses.
Leucine zippers are a well-established class of protein dimerization domains with a wide range of binding affinities from low picomolar to micromolar ranges [17–19] and are therefore ideal for use in the presented platform. These α-helical proteins are characterized by a series of leucines spaced 7 residues apart. Through a series of hydrophobic, hydrophilic, and ionic interactions, dimerization between zippers is mediated. While present endogenously, specifically designed synthetic leucine zippers are stable at a wide range of pH values and salt concentrations and can survive the acidic environments of infarcts, and many other physiological conditions. Depending on their structure, specific zippers do not homodimerize but instead form heterodimers with specific counterparts [17] which minimizes off-target crosslinking.
Here, we have developed a novel platform to improve cell accumulation and retention at the site of myocardial infarction. We selected heterodimerizing zippers to achieve selective crosslinking while preventing self-aggregation via homodimerization. We then characterized and tested the ability of leucine zipper decorated cells, termed ZipperCells, to form stable, but reversible physical crosslinking under in vitro conditions. Next, we assessed whether this crosslinking allows for enhancement in accumulation and prolonged retention under systemic administration in a preclinical model of MI. Finally, we tested the capacity of ZipperCells to be exchanged or outcompeted, which would make them ideal for use in minimally invasive cell therapy.
2. Results
Design and Characterization of the ZipperCell Depot System
The ZipperCell depot system centers around the use of a small library of heterodimerizing, synthetic leucine zippers for physical crosslinking mediated by competition-based dimerization. In vivo, sequential administration of leucine zipper decorated cells facilitates the physical crosslinking and therefore retention and accumulation of cells at the infarct site (Fig. 1). Leucine zippers were generated from previously established SynZip sequences [20] which have been further customized to include a Gly-Ser linker chain, a single cysteine for site-specific thiol maleimide conjugation, and a polyhistidine tag for purification (Fig. 2A & Fig. S1 A). These customized constructs can be expressed and purified with high yield in an E. Coli system (Fig. 2B). The leucine zipper set includes 3 pairs of leucine zippers with varying affinities (10, 80, and 200 nM) and scramble control (Fig. 2C & Fig. S1 B).
Figure 1.
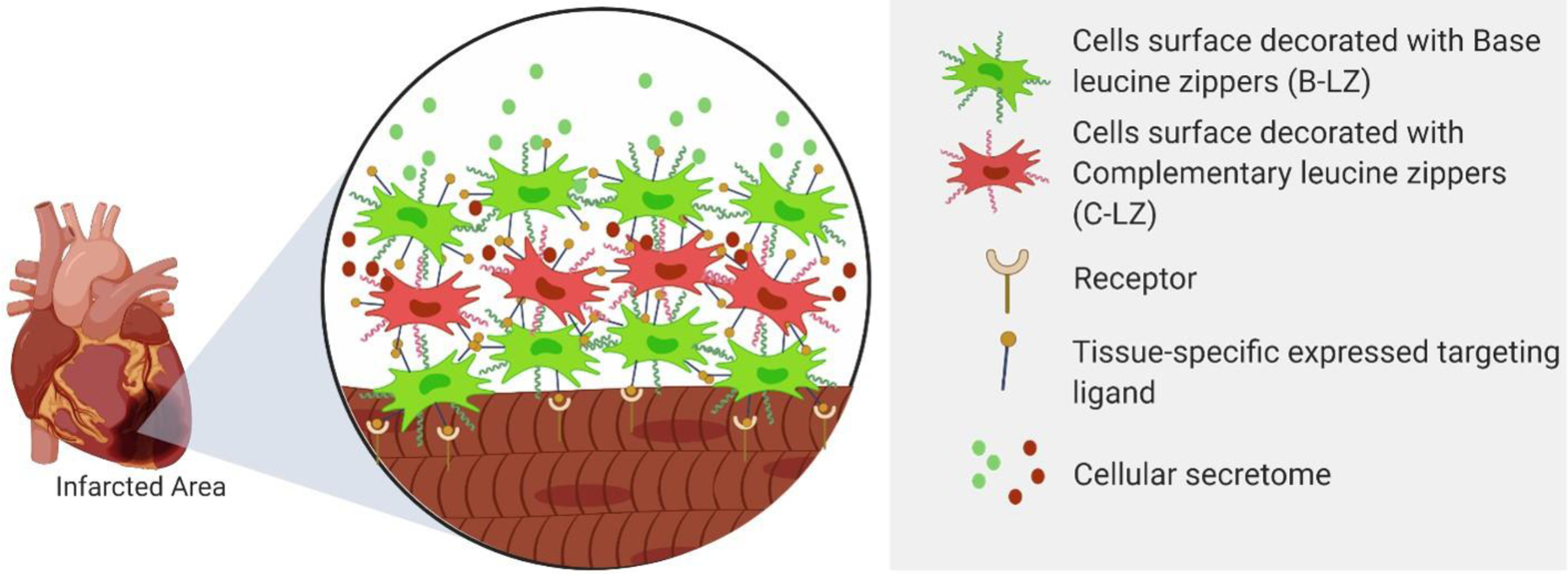
Schematic representation of layer-by-layer in situ cellular depot formation. Cells are initially decorated with either a Base Leucine Zipper (B-LZ) or Complementary Leucine Zipper (C-LZ) on the cell surface. Alternating doses allow for cellular crosslinking for enhanced accumulation and retention at the site of infarction.
Figure 2.
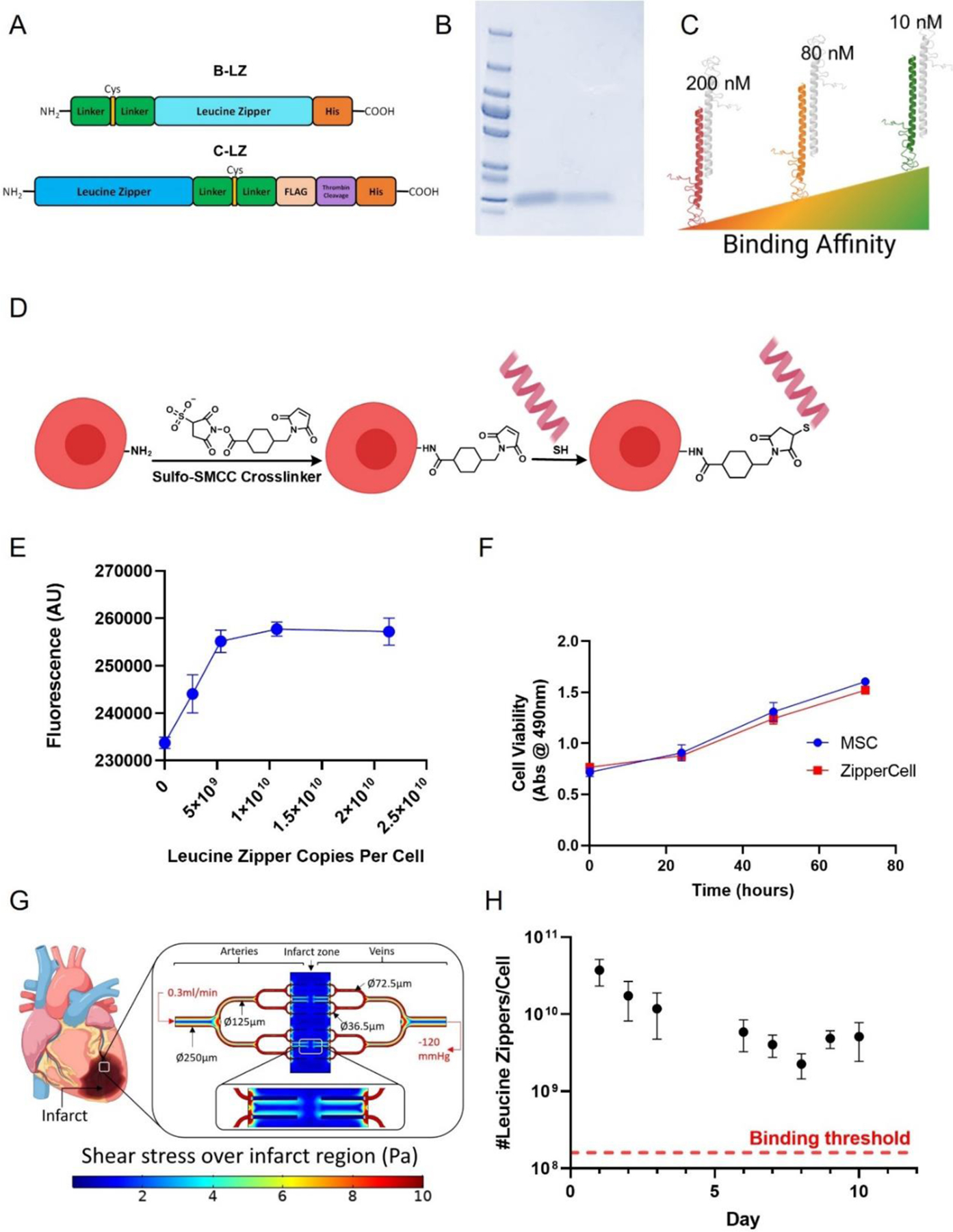
Design of leucine zippers and ZipperCell conjugation. (A) Structures of customized synthetic leucine zipper constructs. (B) Coomassie Brilliant Blue stained SDS polyacrylamide gel with ladder, complementary leucine zipper (C-LZ) 10nM, and base leucine zipper (B-LZ), respectively. (C) Three orthogonal pairs of leucine zippers with varying binding affinities of 10, 80, and 200 nM. (D) Schematic representation of cellular leucine zipper decoration via heterobifunctional crosslinker (Sulfo SMCC), followed by maleimide-thiol conjugation. (E) Leucine zipper density on cells is controlled by varying the leucine zipper concentration. (F) Viability of ZipperCells. (G) Computational model in COMSOL Multiphysics to calculate the required leucine zipper densities for a stable network under cardiac flow conditions. (H) Detection of leucine zippers on cells as a function of time. Data are presented as mean ± SD with *p <0.05, **p<0.01, ****p<0.0001 by one-way ANOVA followed by Tukey’s post-test, n=3.
Once expressed, leucine zippers were conjugated to the surface of mesenchymal stem cells. First, a heterobifunctional crosslinker, sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (Sulfo SMCC) was covalently bonded to amines of the cell surface. Next, thiol-containing leucine zippers were conjugated to the crosslinker (Fig. 2D). By varying concentrations of fluorescently labeled leucine zippers, we were able to determine the saturating dose of leucine zipper and control leucine zipper density on the cell surface (Fig. 2E). We then determined, via MTS assay, that ZipperCell surface decoration did not significantly affect cell viability (Fig. 2F). Next, we used computational modeling to predict the number of leucine zippers required per cell to allow for stable network formation under in vivo blood flow conditions (Fig. 2G). By accounting for factors such as flow rate, blood pressure, leucine zipper length, and affinity, we determined the minimum number of leucine zippers required per cell. For zipper dimerization at 200, 80, and 10 nM binding affinity (Kd), the number of zippers required was 2.8×108, 2.4×108, and 2.2×108/cell, respectively (Fig. 2G-H). To verify that the extent of our leucine zipper decoration surpassed this calculated threshold and could be maintained as the cells expanded, resulting in ligand dilution, we tracked leucine zipper expression on cells over time in vitro. We found that even after 10 days, leucine zippers were still present at ~10 fold the required density (Fig. 2H).
To verify that our custom-modified synthetic leucine zippers could heterodimerize, FRET assays were utilized (Fig.3A-C). When dimerized in the expected parallel orientation, C- terminally labeled AF555 B-LZ leucine zippers transfer energy to cysteine labeled AF647 C-LZ leucine zippers. This resulted in a greater FRET efficiency than mixtures of B-LZ with scrambled control sequences, pointing to the specificity of heterodimerization in our selected pairs. Finally, we assessed if the presence of heterodimerizing leucine zippers on the surface of cells could facilitate the physical crosslinking of specific cells to form cell sheets. We observed that when base leucine (B-LZ) ZipperCells were seeded in a monolayer, only complementary (C-LZ) ZipperCells bound, resulting in multilayered cell layer sheets with increased thickness (Fig. 3F). However, cultures with scrambled control or only a single leucine zipper were unable to dimerize and were unable to form multi-layered cell sheets (Fig. 3D-E). Quantification of layer thickness confirmed that the successful layer formation resulted in a three-fold greater thickness compared with only a cell monolayer from the scrambled (Scr) protein (Scr Cells/B-LZ Cells/Scr Cells) or only the C-LZ 10nM protein (C-LZ Cells/C-LZ Cells/ C-LZ Cells) (Fig. 3G).
Figure 3.
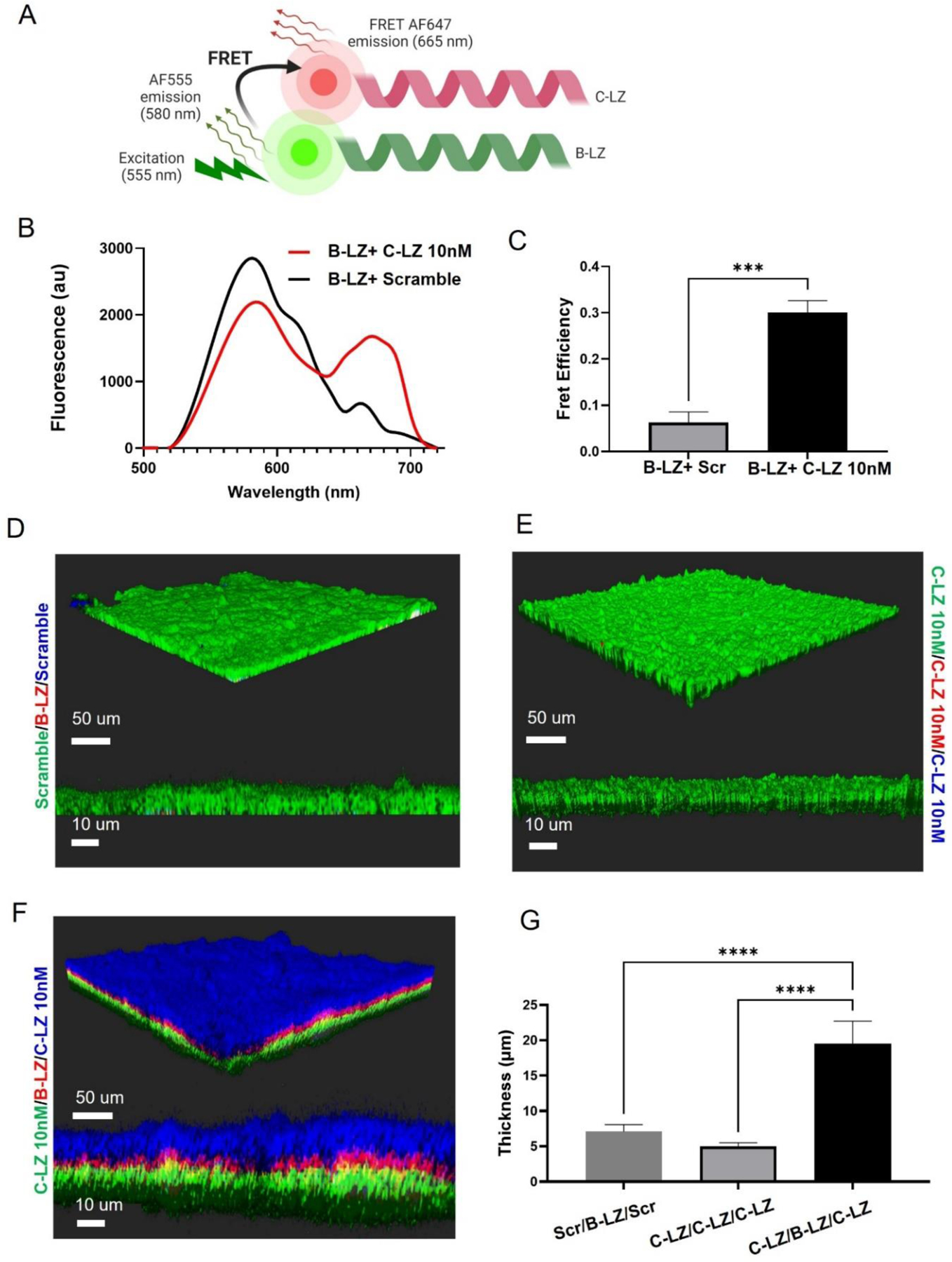
Leucine zipper decoration facilitated crosslinking (A)Schematic of FRET. When bound and excited, AlexaFluor555 labeled B-LZ transfers energy to AlexaFluor647 labeled C-LZ for subsequent emission. (B) FRET spectra of heterodimerizing pairs vs scramble control (C) FRET efficiency (D) A monolayer of scramble-decorated cells (DiO lipid dye = green) was seeded and then incubated with a layer of B-LZ (DiI =red) and an additional layer of MSCs decorated with a scrambled peptide (blue= DiD). (E) A monolayer of C-LZ (DiO) followed by 2 additional layers of C-LZ (DiI & DiD). (F) A monolayer of C-LZ (green=DiO) followed by B-LZ decorated cells (red = DiI lipid dye) and then an additional layer of C-LZ cells (blue=DiD). (G) The average thickness of three-dimensional, layer-by-layer cocultures. Data are presented as mean ± SD with *p <0.05, **p<0.01, ****p<0.0001 by one-way ANOVA followed by Tukey’s post-test, n=3.
ZipperCells Maintain Normal Phenotype
A functional concern of the ZipperCell system was whether they would maintain their “stemness” upon surface decoration. To assess the MSC phenotype, we performed RT-PCR. A panel of positive phenotypic MSC markers such as CD29 & CD90 and negative phenotypic markers (CD14 & CD19) was tested, using GAPDH as a housekeeping gene. We found that all genes were consistent. Upon further investigation, we also found that the ZipperCells maintained their capacity to differentiate into osteoblasts and adipocytes. This indicated their ability to retain their phenotype associated with regenerative wound healing, which is needed at the infarct site. We found no statistical difference between ZipperCells and unmodified MSCs 7 days after surface decoration when examining relative fold change in gene expression (Fig. 4A). This was further confirmed by assessing MSC-specific surface markers via flow cytometry, where both unmodified MSCs and ZipperCells showed comparable levels of CD90, CD29, and CD105 (Fig. S3). Additionally, upon adipogenic and osteogenic differentiation induction, both unmodified MSCs and ZipperCells were able to maintain their capacity to differentiate with no statistical difference in Alizarin Red or Oil Red staining (Fig. 4B-E).
Figure 4.
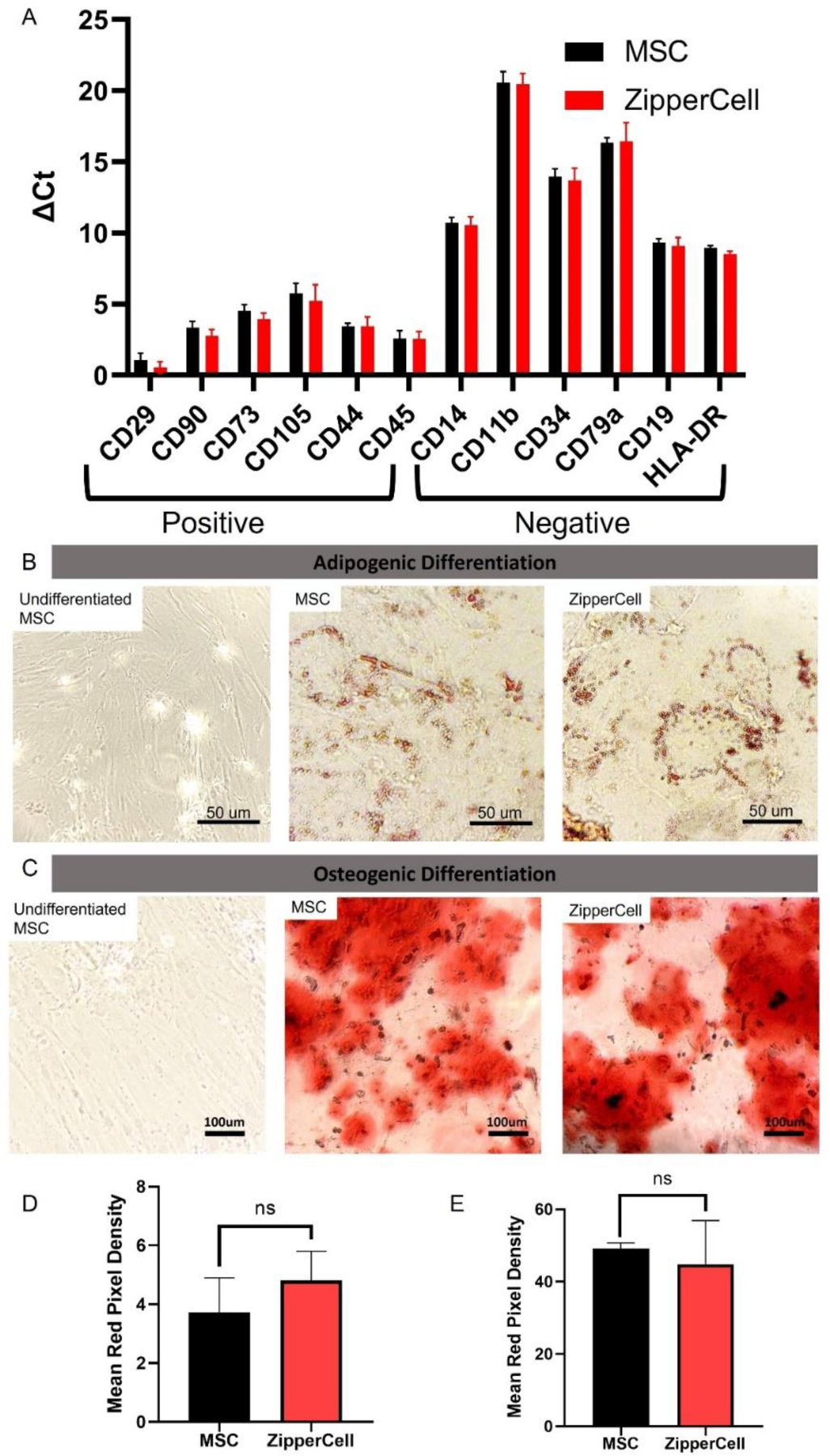
Leucine Zippers maintain normal phenotype in vitro (A) RT-qPCR of phenotypic MSC marker total RNA expression from unmodified MSCs and ZipperCells seven days after surface decoration. ZipperCells retained their capacity to differentiate into (B) osteoblasts (calcium deposits stained with Alizarin Red S) and (C) adipocytes (lipid droplets stained with Oil Red O). (D) Quantification of Oil Red Stain. (E) Quantification of Alizarin Red Stain. Differentiation was performed for 21 days and then characterized. Data are presented as mean ± SD with *p <0.05, **p<0.01, ****p<0.0001 by one-way ANOVA followed by Tukey’s post-test, n=3.
Furthermore, cells were also tested for their ability to differentiate into cardiomyocyte-like cells. 14 Days after the induction of 5-azacytidine mediated differentiation, cells were fixed and fluorescently stained for GATA4 and Troponin I or lysed for total RNA assessment using RT-qPCR (Fig. S4). Again, both groups of cells were able to maintain their ability to differentiate. Lastly, cells were tested for potential secretion of paracrine factors, as these have been widely implicated in many MSC-mediated wound healing processes and angiogenic potentials [21,22]. Unmodified MSCs and ZipperCells were tested for both vascular endothelial growth factor (VEGF) and epidermal growth factor (EGF) secretion (Fig. S5). MSCs and ZipperCells exhibited biologically relevant levels of both factors, with no statistical differences between groups.
Leucine Zipper Decorated Cells Accumulate after ischemia/reperfusion
After the characterization of ZipperCell crosslinking in vitro, we sought to assess the cardiac accumulation of the ZipperCell system in vivo. We began by examining the biodistribution of DiR-labeled stem cells after systemic administration via tail vein injection in a mouse model of myocardial infarction (MI) (ischemia/reperfusion). Twenty-four hours after surgery, 500,000 cells were injected into mice every 12 hours for a total of three injections. Twelve hours after the final injection, organs were collected, and their DiR fluorescence was imaged ex vivo, using the IVIS Spectrum in vivo imaging system (Fig. S2). Although the majority of both unmodified MSCs and ZipperCells were found in the liver, spleen, and lungs (Fig. 5C), quantification of the fluorescence in the hearts revealed 486±126% higher levels of ZipperCells compared to unmodified MSCs 72 hours post-infarction (Fig. 5A & 5B). Representative sections were taken within the border zone and infarct area, which was defined as the visible infarct area and the 2 mm area encircling the infarct area (Fig. 5D)[23,24]. Upon immunohistochemical staining, we found 9.97±.24-fold greater deposition of ZipperCells within the infarct and border zone of hearts compared to unmodified MSCs (Fig. 5E & Fig.S6A), likely as a result of leaky vasculature and enhanced permeability after MI.
Figure 5.
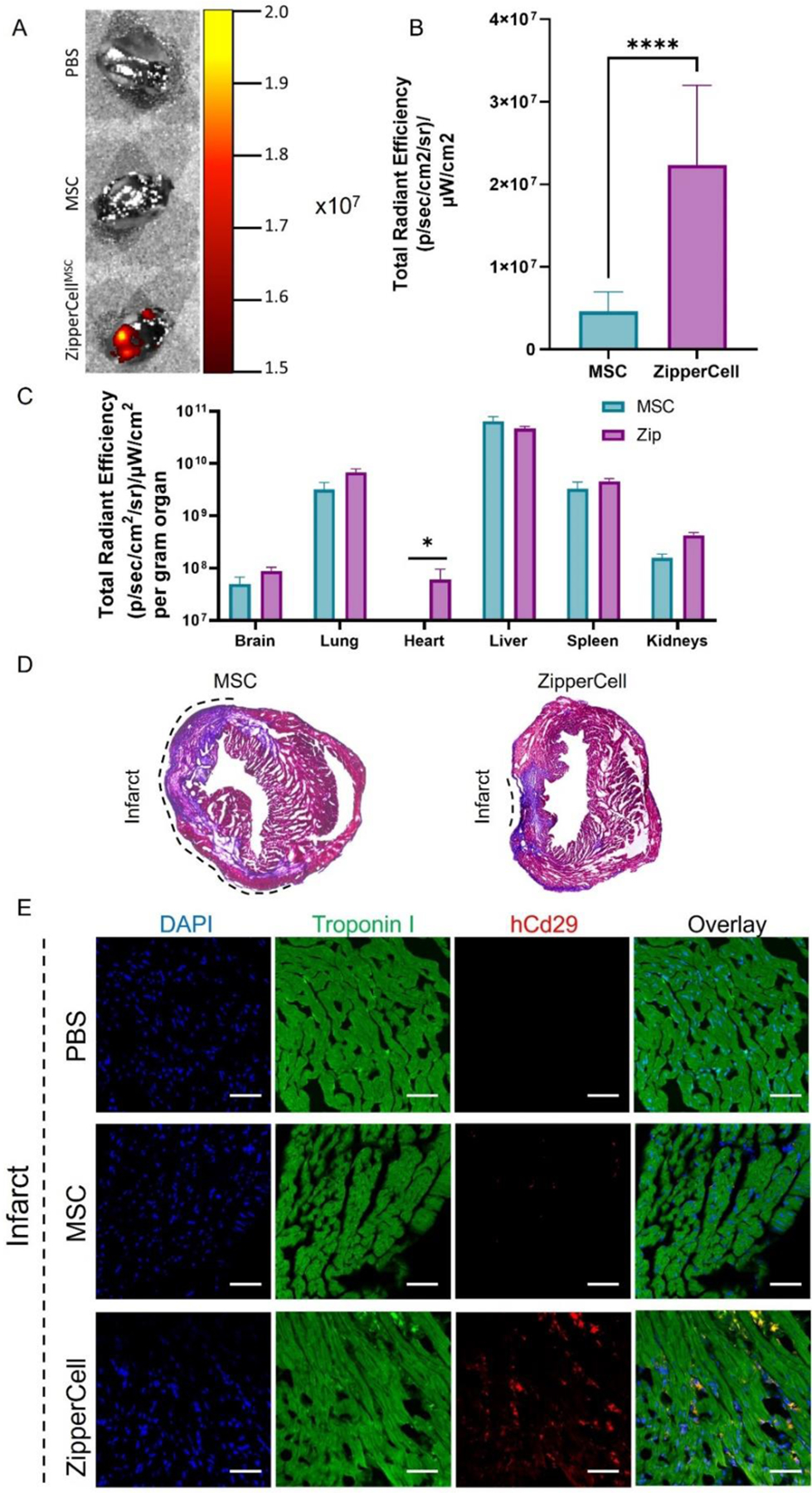
Biodistribution of MSCs in mice with MI. (A)Representative IVIS images using the IVIS Spectrum in vivo imaging system show DiR-labeled MSC deposition in the infarcted heart. (B) Quantification of DiR fluorescence at the infarcted heart. (C) Quantification of fluorescence in major organs. (D) Masson’s Trichrome staining of whole heart sections. (E) Immunofluorescent staining of cryosections of the infarcted heart of mice treated with PBS, unmodified MSCs, and ZipperCells. Cryosections were stained for Troponin I (green), human CD29 (red), and nuclei (DAPI, blue). Scale bar = 50 µm. Data are presented as mean ± standard deviation. Data are presented as mean ± SD with *p <0.05, **p<0.01, ****p<0.0001 by one-way ANOVA followed by Tukey’s post-test, n=7.
To further assess what portion of the retained stem cells was viable, TUNEL staining was performed. TUNEL staining showed that there was a higher number of apoptotic cells within the PBS control group compared to both the MSC and ZipperCells treated groups (Fig. S6B). However, we determined that 66.7±5.7% of resident unmodified MSCs had TUNEL positive nuclei while only 15.33±1.2% of ZipperCells contained TUNEL positive nuclei within the infarct zone, which correlates to ~750 viable MSC and ~9,222 viable ZipperCells at the infarct site, respectively (Fig. S6C).
Long-Term Retention of ZipperCells
After demonstrating that almost 500% more ZipperCells were retained at the infarct site 72 hours after ischemia/reperfusion when compared to unmodified MSCs, we aimed to investigate the potential for retention at longer time points. We found that 10 days after the first injection, 78±11% of the 72-hour ZipperCell population fluorescent signal was still detectable in the mouse hearts via ex vivo IVIS imaging, a ~6-fold improvement over unmodified MSCs (Fig. 6A-B. This suggests that the fold retention of ZipperCells over unmodified MSCs increases over time.
Figure 6.
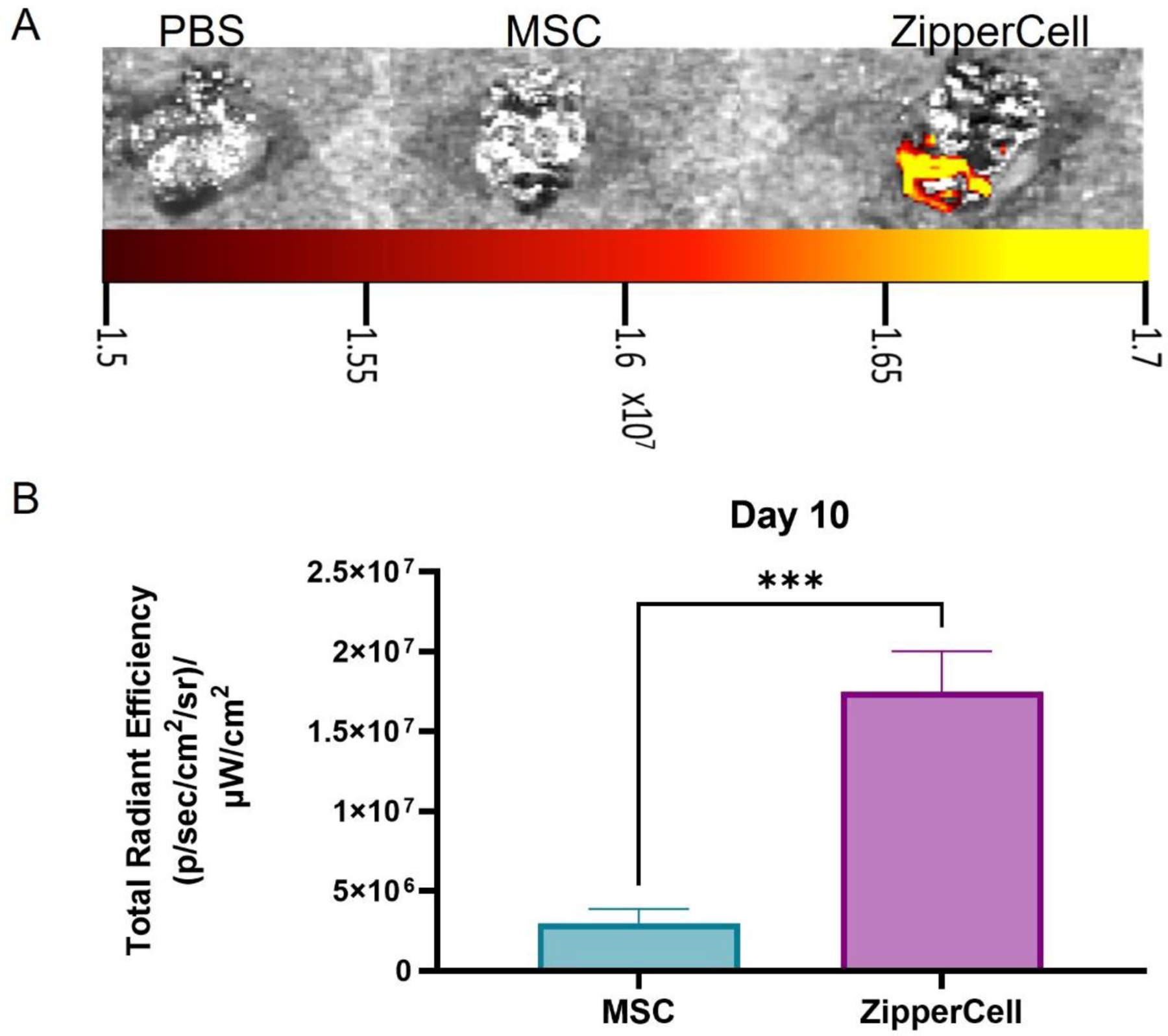
Long-term leucine zipper expression and cellular retention at the infarct site. (A) Representative IVIS representative images of DiR-labeled MSCs ten days after the initial injection. (B) Quantitation of fluorescent cell signal from hearts ten days after the initial injection. Data are presented as mean ± SD with *p <0.05, **p<0.01, ****p<0.0001 by one-way ANOVA followed by Tukey’s post-test, n=3.
Competitive Leucine Zippers Facilitate Targeted Disassembly
It is critically important to have the “disassembly-on-demand” feature when using multi-dose crosslinking, to ensure personalized clinical safety by design. To test if the scaffold and cell clusters can be disassembled, we took advantage of competitive ligand binding.
We hypothesized that lower affinity leucine zipper pairs can be knocked off by dosing with a competing high-affinity leucine zipper, leading to disassembly of the system. This built-in safety mechanism may allow for rapid removal in the case of potential toxicity or immunogenicity. To test this in vitro, we seeded a monolayer of B-LZ leucine zipper ZipperCells stained with DiO (green). Then we independently incubated with DiI (red) fluorescently labeled populations of C-LZ 80nM affinity ZipperCells (Fig. 7A). Following a 3-hour incubation, wells were then treated with 100x fold excess of free C-LZ 10nM peptide for an additional 3 hours (Fig. 7B). We observed that after C-LZ 10nM incubation effectively removed the majority of the C-LZ 80 nM signal (Fig. 7C). Finally, to determine the in vivo effects of competition-mediated disassembly, the C-LZ 80 nM affinity leucine zipper pair was dosed based on the previously described schedule. Twenty-four hours after the final injection, each mouse was dosed with one mg of free C-LZ 10 nM. Twelve hours later, mice were euthanized, and their organs were imaged (Fig. 7D). We observed a 68.8±10% decrease in the DiR signal at the infarct of mice treated with the highest affinity leucine zipper (Fig. 7E).
Figure 7.
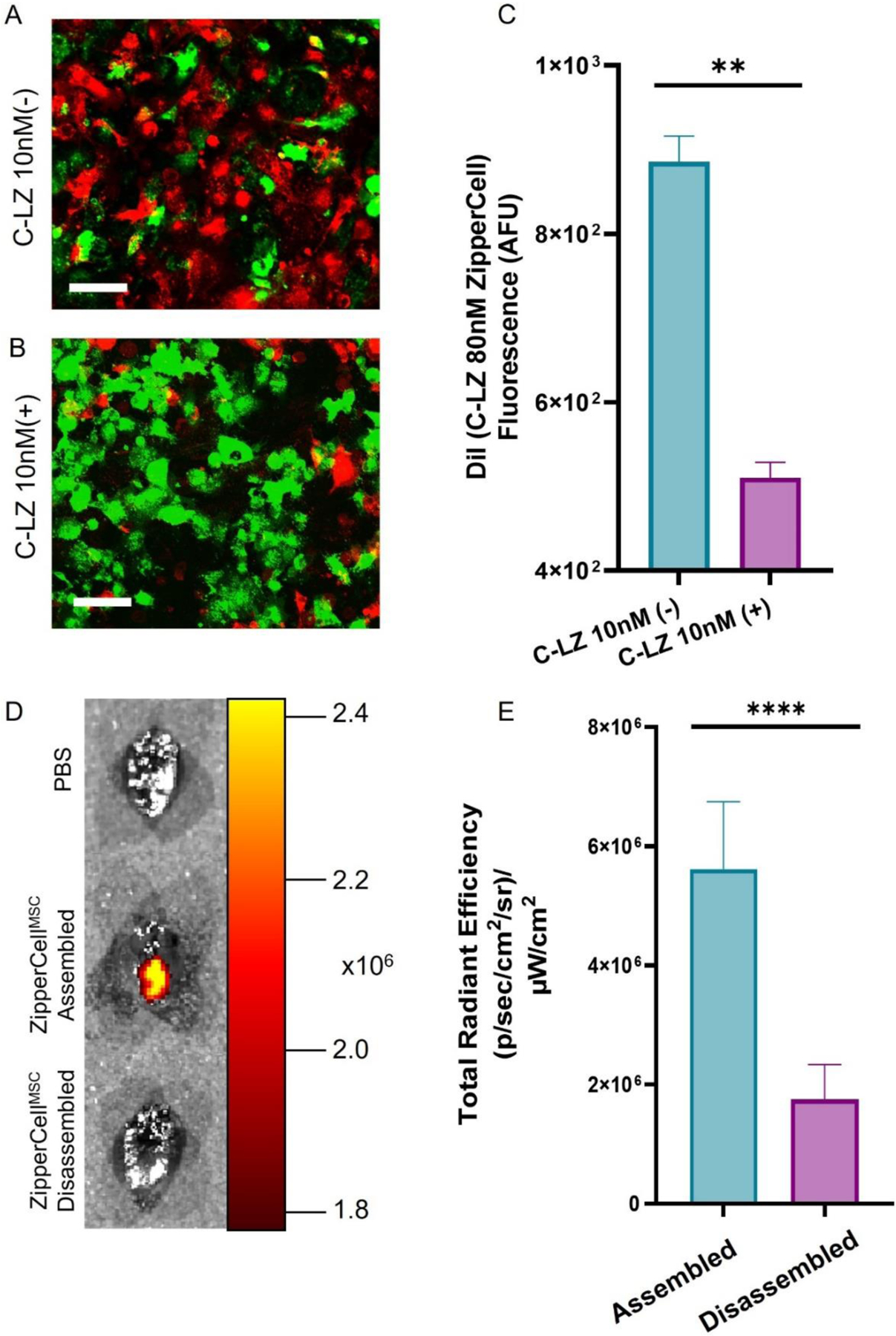
Disassembly of accumulated ZipperCells in vitro and in vivo via competition. Coculture of the monolayer of DiO B-LZ & DiI C-LZ 80 nM without (A) and with (B) the addition of 100x C-LZ 10 nM affinity zipper. (C) Quantitation of DiI signal with and without the C-LZ 10 nM affinity competition. (D) Representative IVIS images were acquired of the C-LZ 80 nM affinity ZipperCell pair followed by a 1 µg dose of C-LZ 10 nM affinity leucine zipper. (E) Quantification of DiR fluorescence at the infarcted heart. Scale Bar = 50 µm. Data are presented as mean ± SD with *p <0.05, **p<0.01, ****p<0.0001 by one way ANOVA followed by Tukey’s post-test, n=3
ZipperCells are Non-Immunogenic and Maintain Cytokine Expression Levels
Next, to verify that the long-term accumulation and retention of ZipperCells would not cause toxicity, anti-drug antibody assays were performed. ELISA assays using either C-LZ 10 nM (Fig. 8A) or B-LZ (Fig. 8B) as the coated antigen, were used to detect total IgG content 6 weeks after injection. We found very low antibody titers in all injected mice, signaling a lack of immune response. As additional mediators of systemic inflammatory responses, we also wanted to investigate the impact of multiple ZipperCells administrations on cytokine profiles. To complete this, serum samples were collected from mice the day before surgery (D0) and 4 hours after the second injection (40 hours post-MI). Serum levels of 40 cytokines were then tested using the Proteome Profiler Cytokine Array, Panel A. We observed that induction of MI overall increased cytokine expression in all mice, including the PBS control. Both groups of cell-treated mice showed similar profiles of pro-inflammatory cytokines, but ZipperCell treated mice mediated robust upregulation of several increased anti-inflammatory and pleiotropic cytokines (Fig. 8C), likely due to enhanced accumulation. After the second dose, we found a statistically significant increase in multiple anti-inflammatory factors such as G-CSF, IL-4, IL-6, and SDF-1in ZipperCell treated mice compared to unmodified MSC-treated mice, which have been shown to reprogram recruited macrophages to an anti-inflammatory phenotype [25–27]
Figure 8.
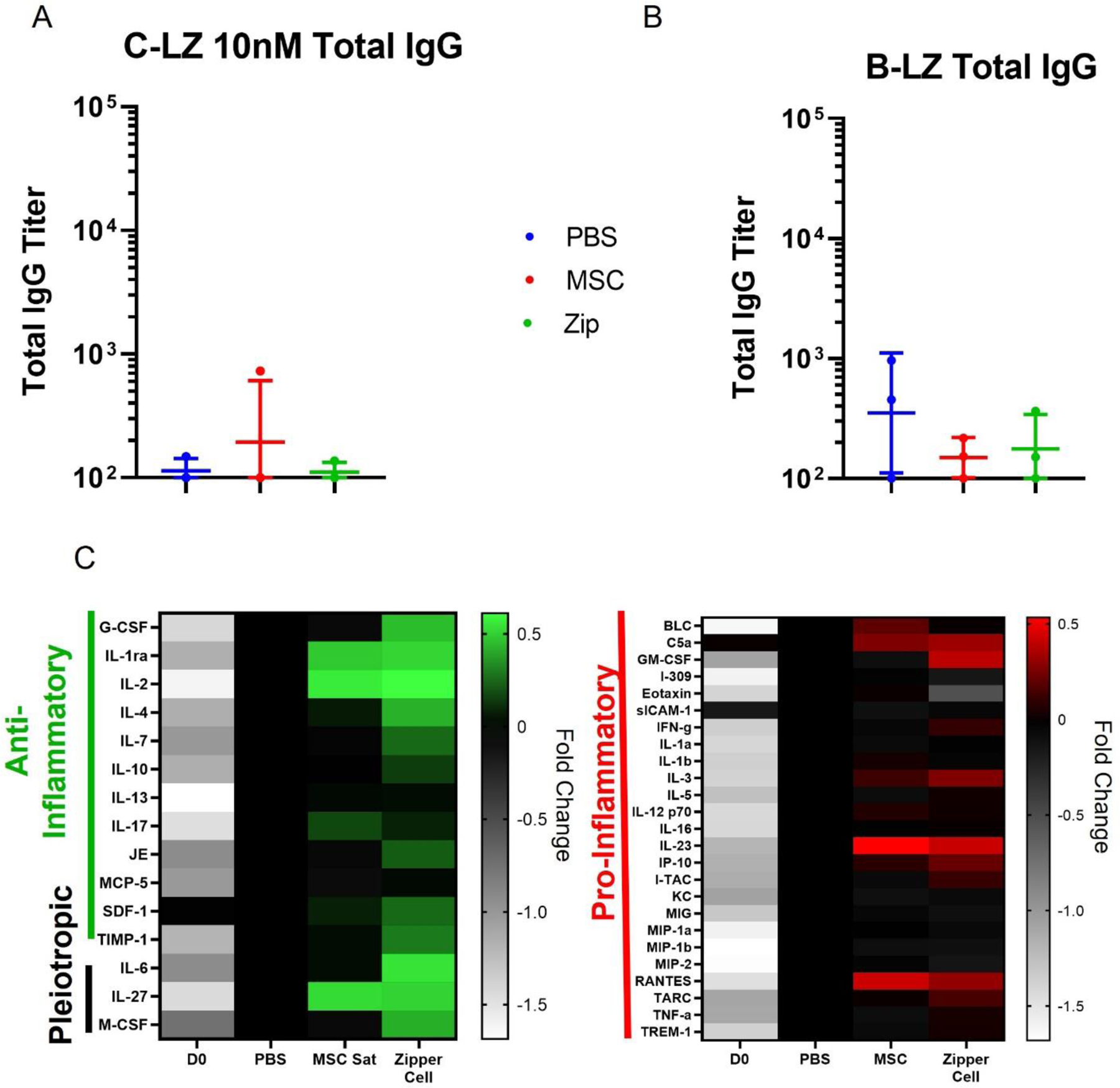
ZipperCells Are non-immunogenic. (A) Total IgG detection against C-LZ 10nM in serum. (B) Total IgG detection against B-LZ. in serum. (C) Relative cytokine expression levels of mice the day before surgery (D0) and four hours after the 2nd injection (40 hours post-MI) of PBS, unmodified MSCs, or ZipperCells. Data are presented as mean ± SD with *p <0.05, **p<0.01, ****p<0.0001 by one-way ANOVA followed by Tukey’s post-test, n=3.
To further confirm biocompatibility and the effect of maximal off-target organ retention, a set of healthy mice were injected with PBS, unmodified MSCs, or ZipperCells, as previously described. We hypothesized that without inflammatory signaling or leaky vasculature from myocardial infarction, homing to the heart would be reduced and maximum retention would be observed in the major clearance organs such as the liver and spleen[28–30]. We found that in healthy mice targeting to the heart was negligible (Fig. S7A & B), showing an active homing-dependent mechanism. Furthermore, we examined the ratio of cell accumulation in infarcted hearts vs healthy hearts and found significantly less accumulation in healthy hearts (Fig. S8A & B). When examining the ratios of % injected dose per gram of organ vs % injected dose per gram of heart in healthy mice, we observed that for both MSCs and ZipperCells, we saw similar levels of off-target accumulation, again supporting a conclusion that within healthy mice, homing to the heart is limited (Fig. S8C & D). As shown in Fig. S8E & F, ratios of off-target organs vs infarcted heart accumulation are substantially lower in mice treated with ZipperCells compared to mice treated with unmodified MSCs. This provides additional evidence that there is an injury-specific signaling required for accumulation of ZipperCells and MSCs into the infarcted heart.
Additionally, pathological analysis of H&E staining in the healthy mice liver, lung, and spleen tissues showed no noticeable differences between any groups and no detectable legions (Fig. S9A). Furthermore, CD45+ staining, a marker of immune cell infiltration, showed no signal in the liver or lung of any samples and minimal signal in the spleens of both cell-treated groups (Fig. S9B). Overall, this data demonstrates a lack of significant immune response and a biocompatible delivery system.
3. Discussion
Mesenchymal stem cells are exemplary mediators of wound healing. Their ability to self-renew, immunomodulate, and differentiate into a variety of cell types makes them ideal tools for regenerative medicine, and their ability to reduce infarct size, reduce fibrosis, and promote new vascularization at the wound site makes them particularly suited for treating myocardial infarctions [5,31,32]. MSCs have been shown to mediate cardioprotection by inhibiting inflammatory transcription factors, such as NF-kB[33], secreting protective paracrine factors[34], and activating of toll-like receptors[35]. Studies have also shown that the secretome of injected MSCs may aid in their cardioprotection by depleting the infarct proteomic landscape of proteins controlling cell death, apoptosis, and inflammation[33,34]. Despite these successes, there are still no FDA-approved MSC treatments and an overwhelming consensus within the field is that poor accumulation and retention are primary hurdles to clinical translation [36,37].
Here, we have engineered a system of self-assembling MSCs with multiple flexible features and fine-tune ability for enhanced accumulation and retention. An essential component of the system is the layer-by-layer dosing strategy. Inspiration for this aspect was drawn from recent studies showing that repeat dosing of MSCs is well-tolerated and further decreases infarct size and improves cardiac function [38–40]. Our approach allows several waves of cellular delivery in a non-invasive manner through circulation. Unlike conventional delivery strategies, which do not allow the subsequent accumulation of cells or carriers after saturation of the target infarct site, each dose of cells in our platform serves as an additional capturing surface for the next dose of cells. This dramatically amplifies the targetable surface area for additional waves of cell attachment. Additionally, the extensive multivalent crosslinking helps resist venous washout, allowing cells to persist long term.
While unmodified MSCs showed only 0.15±.07% retention of the injected dose at the infarct site, which is in line with previously published results [41–43], ZipperCells showed ~ 500% improvement at the infarct site compared to unmodified MSCs. This improved retention is further widened by day 10, where ZipperCells demonstrate a ~ 600% improvement over unmodified MSCs.
Another key feature of the ZipperCell system is its non-invasive, yet targeted, systemic administration. We take advantage of the natural ability of MSCs to migrate to the site of infarct and promote cardiac repair. Previous studies have shown that following a myocardial infarction, chemokines such as CXCL12 are secreted and overexpressed at the site of injury [44,45]. As a result, MSCs expressing chemokine receptors, including CXCR4, migrate to the site of injury via chemoattraction. This was shown by our preferential retention in the ischemic myocardium region by both unmodified MSCs and ZipperCells. This was also validated by a lack of cardiac accumulation in healthy mice; without inflammatory signaling or leaky vasculature caused by injury, homing was significantly reduced. Future studies assessing this feature of the platform in other cardiac failure models, such as chronic ischemic cardiomyopathy, would be warranted as well.
In clinical settings of MI, it is standard practice to establish intravenous access in at least one peripheral vein and maintain this access for the duration of the hospital stay to administer any IV therapies. As such, ZipperCell administration would be painless and unobtrusive for the patient. The average length of hospital stay after MI in the United States is 3 days and longer in other countries [14], so ZipperCell infusion is not expected to prolong the stay. Moreover, several clinical studies have shown that patients tolerate multiple MSC injections with no adverse effects.
Recent strategies for enhancing cellular retention have largely focused on shielding cells from venous washout. Encapsulating materials, such as injectable hydrogels, are of great interest as they provide protective structural support and can be comprised of a range of materials. Scaffolds made of materials such as injectable fibrin hydrogels[46] or polymer fibers[47] have garnered particular preclinical interest. While these materials may be engineered to incorporate additional therapeutic elements and tunability, this also requires extensive material characterization of factors such as stiffness, porosity, and adhesion [48]. Furthermore, they are often plagued by low cellular or payload capacity [49,50]. The inclusion of additional materials widens the likelihood of toxicity and immunogenicity as well as the alteration of cellular function and engraftment in vivo [51]. Other approaches such as cell sheets and cardiac patches attempt to overcome poor retention through the delivery of cells with established extracellular matrices [52,53]. Recently, a four-dimensional GelMA patch was engineered to account for biomechanical and physicochemical properties, in which cell engraftment was significantly improved [54]. While strategies like this have advanced the field with insight into biomimetic design, they are also bound by extensive microfabrication equipment costs and lengthy culture protocols. In contrast, our simplified approach circumvents these challenges and allows ZipperCells to form their own scaffold-like network in situ, making them more potent and biocompatible.
A unique component of the ZipperCell system is the intrinsic safety-by-design feature. Unlike traditional therapeutic depots, ZipperCell disassembly or removal can be performed without surgery, which could cause additional acute stress to the patient and increase the possibility of morbidity [55,56]. Our disassembly mechanism uses a simple intravenous injection to remove the majority of all cells at the target site. The ability to rapidly and non-invasively remove the therapeutic depot is especially important in the case that patients begin to show adverse effects or toxicity and need to terminate the treatment immediately. Furthermore, conventional therapeutic depots such as the aforementioned hydrogels and patches cannot be easily modified, making it difficult to adjust drug regimens in response to the evolving disease state, which is becoming ever more important to optimize outcomes in the personalized medical era. While our results show that ZipperCells are well-tolerated and do not induce significant immune responses, this mechanism adds to the overall potential clinical translation of our system.
Future studies are required to test the therapeutic effect of enhanced cellular retention on cardiac repair. While we exploited the fact that mesenchymal stem cells are naturally able to migrate to sites of inflammation, the incorporation of a targeting ligand on the cell surface would likely further enhance accumulation at the site. Additionally, while many studies have attributed secretome factors as mediators of cardio-regeneration[57,58], future studies exploring the local secretome release could aid in further understanding this mechanism of action. The use of alternative therapeutic carriers such as nanoparticles, microparticles, and mixed systems would be warranted as well and further underscore the wide utility of our newly engineered system. Furthermore, optimization of dosing intervals would be beneficial.
4. Conclusion
In conclusion, here we report the first-of-its-kind, non-invasive, self-assembling cell depot for significantly enhanced retention and accumulation at the infarct site. This novel cell delivery strategy amplifies the available binding area in a layer-by-layer fashion. Additionally. this method ensures minimal invasiveness for not only initial delivery but also for removal, as needed. In this study, we demonstrate that the cell surface decoration of leucine zippers does not interfere with the MSC phenotype or the capacity to differentiate can facilitate cellular crosslinking to retain cells at the infarct site effectively. Conceptually and technically, this platform can serve as a proof-of-concept for a variety of non-invasive, in situ forming depots. The proposed research will also lay the foundation for a better understanding of the biocompatibility and immunogenicity of leucine zipper-based therapeutics in combination with stem cell therapy.
5. Experimental Section
Cell Culture:
Human Bone-marrow derived MSCs were obtained from the American Type Culture Collection (ATCC PCS-500-012) and cultured with DMEM/F12 medium supplemented with 10% FBS (Gibco™, Gaithersburg, MD). Cells were maintained in hypoxic conditions (5% CO2, 5% O2) for 24 hours prior to studies.
Protein Production and Purification:
The customized leucine zipper sequences were cloned into a pET21a vector (Novagen, Burlington, MA) and expressed in BL-21 (DE3) cells (Lucigen, Middleton, WI). Bacterial cultures were grown overnight at 37°C in 2x YT (Fisher Scientific, Waltham, MA) medium and ampicillin. For large-scale production, a 10L 2x YT culture was inoculated with 1% of the overnight culture and grown to a high density at 37°C for 8 hours. Protein expression was induced with 20g/L alpha lactose. After overnight incubation at 24°C, the cells were harvested 16 h post-induction by centrifugation. The cell pellet was resuspended in lysis buffer containing 50 mM Tris, 100 mM KCl, 1mM DTT, 10% glycerol, and 100µg/ml lysozyme, pH 8.0. The cell suspension was sonicated and then centrifuged at 17,000 x g for 40 minutes at 4°C, after which Ni-NTA Resin (Gbiosciences, St. Louis, MO) was added to the solution. The beads were incubated with the solution and the protein was isolated by immobilized metal affinity chromatography (IMAC). Endotoxin removal was performed using Pierce High-Capacity Endotoxin Removal Resin (Thermofisher) and validated using the Pierce LAL Chromogenic Endotoxin Kit Quantitation Kit (Thermofisher).
Enzyme-linked immunosorbent assay (ELISA):
Nunc MaxiSorp 96-well plates were coated with Leucine Zippers at 2.5 µg/mL and incubated overnight at 4°C. The plate was thoroughly washed with 0.1% TBS-Tween (TBST). The wells were washed with 0.1% PBST and then blocked with BSA blocking buffer for 1 h at room temperature. Then incubated with 3.3 μM complimentary leucine zipper at 37°C for 2 hours before washing. Washes were followed by the addition of 100 μL of Anti-FLAG horseradish peroxidase (HRP)-linked monoclonal antibody (mAb, #A8592, Sigma Aldrich, St. Louis, MO) for 1 h at room temperature with rocking. The wells were washed with 0.1% PBST followed by the addition of TMB-ELISA substrate. After 10 minute incubation, 2 M H2SO4 was added to stop the reaction. The absorbance was measured at 450 nm and 570 nm with a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA).
Protein-Cell Conjugation:
Cells were trypsinized, washed, and resuspended in HBSS. At a concentration of 1.0E6 cells/ml cells were incubated with 2.5µM DiR dye and incubated with sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (Sulfo-SMCC). at a final concentration of 24 μg/ml for 30 minutes at 37°C.To reduce protein for conjugation, proteins were incubated with 1mM tris(2-carboxyethyl)phosphine (TCEP) at room temperature for 20 minutes. After washing cells via centrifugation, the protein was added to cells and incubated at room temperature for 30 minutes before final washing and resuspension.
Fluorescent Protein Labeling:
For general protein labeling, protein solutions were labeled with fluorescein isothiocyanate. For Förster resonance energy transfer (FRET), proteins were labeled using N-hydroxysuccinimide ester amine chemistry with Alexa Fluor™ 555 C2 Maleimide (Invitrogen) for the base leucine zipper or AF657 NHS Ester (Invitrogen) for complementary leucine zippers. Protein samples were first reduced using 5mM TCEP. Then proteins were mixed with 10-fold molar excess dye and incubated at room temperature for 1 hour. After labeling, the free dye was removed using a PD-10 column. Labeling efficiency (1:1 ratio) was ensured by measuring the absorbance at 555 nm (AF555, ε = 158,000 M−1 c−1) or 650 nm (AF647, ε = 270,000 M−1 c−1) in PBS for the dye and 280 nm in PBS for the protein.
Computational Modeling:
Computational modeling was used to predict the number of leucine zippers required per cell, to allow for stable network formation by overcoming the shear forces due to blood flow. The model simulated the hierarchical microvasculature in mice [59] with an inlet flow rate of 0.3 ml/minute [60] at the arterial side and −120 mmHg pressure at the venous side.[61] At the central infarct region, the permeability of the leaky microvessels was governed by the Kozeny-Carman permeability model [62,63](cell radius (rc) = 10 μm and porosity (ε) = 40% [62]). Within each layer, the shear forces (
| (1) |
where 𝜏 is shear stress) were assumed to act obliquely on each cell.[64] The force required to dissociate the bound proteins
| (2) |
was determined as a function of binding affinity (KD) and the length of the crosslinked chain between the cells
| (3) |
where lz is zipper length (7.5 nm), lc is the crosslinker length (0.7 nm) and lsp is the average cell surface protein length (2.2 nm). The number of proteins required on the surface of each cell was
| (4) |
where
| (5) |
is the fraction of the cell surface area participating in dimerization.[64] The number of bonds (Nb) needed to overcome the viscous shear forces was, therefore,
| (6) |
For zipper dimerization at 200 nM, 80 nM, and 10 nM binding affinity (KD), the number of zippers (Np) required was calculated as
| (7) |
where the required on the surface of each cell (fsa) was approximated by
| (8) |
Protein Dilution Study:
ZipperCells were generated as described above. Cells were seeded at a density of 10,000 cells per well of a 96 well plate in triplicate. The first time point was recorded 12 hours after plating. Cells were washed and fixed with 4% paraformaldehyde (PFA) for 10 minutes. Cells were then incubated with 100 μl of 1ug/ml Hoechst 3342 to normalize via cell number. Next, cells were treated with blocking buffer (1% BSA in PBS) for 1 h at 37°C. Wells were incubated with diluted Anti-Flag HRP antibody for 30 minutes at 37°C and washed with PBS. Finally, cells were incubated with 3,3′,5,5′-Tetramethylbenzidine (TMB) for 15 minutes before measuring absorbance at 650 nm on the plate reader. Hoechst fluorescence (361/486 nm) signal was also measured for quantification of cell number. This was repeated daily for 10 days.
Cell Viability Assay (MTS):
ZipperCells were prepared as described above and plated at a density of 10,000 cells per well of a 96 well plate in triplicate along with unmodified MSCs. 3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) reagent (MTS Reagent Powder, Promega Corporation Cat. # G1111) was prepared according to the manufacturer’s instruction. Wells were incubated for 12 hours before recording the first time point. MTS solution was added to each well at a final concentration of 0.33 mg/ml. Cells were incubated for 2 hours at 37°C before measuring absorbance at 490 nm.
FRET Assay to Measure Heterodimerization:
The fluorescence emission of N-terminally AF647 labeled leucine zippers was measured alone and, when mixed with C-terminally Alexa Fluor 555-labeled partners, in triplicate. Samples (100 μL) were mixed at 100 nM concentration of each protein in PBS pH 7.4, 5 mM TCEP, allowed to incubate for 2 h at 37 °C, and then equilibrated for 1 h at room temperature. Samples were excited at 555 nm, and emission spectra were monitored from 400 nm to 750 nm at 25 °C. Samples were assayed in 96-well black plates (Corning) using a Synergy H1 plate reader (Winooski, VT). FRET efficiency was calculated using the following formula[67]:
| (9) |
Reverse transcription-polymerase chain reaction (RT-PCR) of Phenotypic Markers:
ZipperCells and unmodified MSCs were plated at a density of 35,000 cells/cm2. Seven days later, the gene expression profiles of unmodified MSCs and ZipperCells were quantified by real-time polymerase chain reaction (qPCR). Briefly, harvested cells were fixed in TRIzol, and RNA was extracted from the homogenized cell lysate through a series of rinse, elution, and centrifugation steps. The RNA samples were then reverse transcribed into cDNA using ProtoScript II First Strand cDNA Synthesis Kit reagents (New England Biolabs, Ipswich, MA) following the manufacturer’s instructions. In the differentiation studies, the gene expression of interest was determined using iTaq Universal SYBR Green Supermix (Hercules, MA). Five positive MSC phenotypic markers and five negative MSC phenotypic markers were examined[68,69]. The fluorescent signals were amplified and detected using a QuantStudio 3 sequence detector (Applied Biosystems). The cycle threshold (Ct) value for each sample was averaged from triplicates. A ΔCt approach was used where the fluorescent signals were normalized to the corresponding housekeeping gene (glyceraldehyde-3-phosphate dehydrogenase (GAPDH)).
In vitro MSC Differentiation:
Cells were plated at a density of 5 × 103 cells/cm2 in a 12-well plate. When the cells were 60% confluent, differentiation was initiated by replacing the complete growth medium with the osteogenic, adipogenic [70], or cardiomyogenic medium.[68] The osteogenic medium was composed of DMEM High Glucose supplemented with 10% FBS, 1% P/S, 100 nM of dexamethasone, 50 μM of ascorbic acid, and 10 mM of sodium β-glycerophosphate. The adipogenic medium was composed of DMEM High Glucose supplemented with 10% FBS, 1% P/S, 1 µM dexamethasone, 1 µM insulin, and 200 µM indomethacin. The cardiomyogenic medium was composed of α-MEM supplemented with 10% FBS, 1% P/S, and 10 μM 5- Azacytidine. In parallel, the control undifferentiated MSCs were grown in standard complete medium. For osteogenic and adipogenic differentiation, cells were grown for 21 days at 37° C in a humidified 5% CO2 atmosphere. For cardiomyogenic differentiation, cells were grown in differentiation medium for 24 hours before changing to a standard complete medium for an additional 13 days at 37° C in a humidified 5% CO2 atmosphere. The medium for all cells was changed every 3 days.
Osteogenic Staining:
After 21 days, cells were washed with PBS before fixing them with 4% PFA for 15 minutes. Fixative was removed and cells were washed 3x with Deionized (DI water). 1 ml of 1% aqueous Alizarin Red solution (GFS Chemicals) was added to each well and incubated with gentle rocking for 45 minutes. Dye was removed and cells were washed 3x with DI water before imaging using brightfield microscopy.
Adipogenic Staining:
After 21 days, cells were washed with PBS before fixing them with 4% PFA for 15 minutes. Fixative was removed and cells were washed 3x with PBS. 1ml of 0.5% Oil Red solution (Sigma-Aldrich) was added to each well and incubated for 5 minutes at room temperature. Dye was removed and cells were washed 3x with DI water before imaging using brightfield microscopy.
Cardiomyogenic Staining:
After 14 days, cells were washed with PBS before fixing them with 4% PFA for 15 minutes. Fixative was removed and cells were washed 3x with PBS. Cells were then stained with either Troponin I (Thermo Fisher, Waltham, MA, Cat # BS-0799R) or anti- GATA4 (Proteintech, Rosemont, IL, Cat # 19530-1-AP). This was followed by secondary antibody staining with anti-rabbit IgG Alexa488 (Invitrogen, Cat # A-11008). Cells were washed 3x with DI water and DAPI mounted using Fluoromount-G, with DAPI (Cat# 00-4959-52, Invitrogen) before imaging using confocal microscopy.
Animals:
Female C57BL/6 mice were used (10-12 weeks) for the in vivo studies. All animal procedures were approved by the University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee (IACUC). All methods and experiments were performed in accordance with the U.S National Institutes of Health Guide for Care and Use of Laboratory Animals. Humane care and treatment of animals were ensured.
Mouse Model of Ischemia/Reperfusion (I/R):
All surgeries were performed in the McAllister Heart Institute (MHI) Cardiovascular Physiology and Phenotyping Core. Briefly, mice were anesthetized with isoflurane. A small incision was made under the mandible to visualize the trachea before intubation with a 20-gauge blunt needle and ventilation. A left lateral thoracotomy will expose the heart, and the left coronary artery (LCA) was identified and temporarily occluded with a 7–0 nylon suture for 40 minutes. Reperfusion was confirmed by electrocardiogram (ECG). The thorax was closed in layers (ribs, muscles, and skin)[71]. Mice were provided with analgesics and monitored per protocol.
Biodistribution of MSCs:
For biodistribution studies, 500,000 1,1’-Dioctadecyl-3,3,3’,3’-Tetramethylindotricarbocyanine Iodide (DiR)-labeled ZipperCells or unmodified MSC, or PBS was administered via tail vein injection into C57BL/6 mice 24 hours after inducing MI. A total of three injections were administered every twelve hours. Twelve hours after the final injection (60 hours post-MI), mice were sacrificed and organs including brain, lung, heart, liver, spleen, and kidneys were collected and weighed. Fluorescent biodistribution was analyzed using the IVIS Spectrum in vivo imaging system (PerkinElmer, Waltham, MA). Average region of interest (ROI) signals were calculated using Living Image 4.5.2 software (PerkinElmer). The data is presented as total radiant efficiency/g organ.
Histological Assessment of MSCS in the Infarcted Heart:
For histological studies (n=3), 500,000 1,1’-Dioctadecyl-3,3,3’,3’-Tetramethylindotricarbocyanine Iodide (DiR)-labeled ZipperCells or unmodified MSC, or PBS was administered via tail vein injection into C57BL/6 mice 24 hours after inducing MI. A total of three injections were administered every twelve hours. Twelve hours after the final injection (60 hours post-MI), mice were sacrificed, and hearts were collected. The left ventricle was briefly perfused with potassium chloride (30 mM) to arrest the heart in diastole and harvested mouse hearts were embedded in OCT. The infarct was cryosectioned in 10 μm sections.
(1). Immunohistochemistry of MSCS in the Infarcted Heart:
Heart cryosections were incubated with anti-Troponin I (Cat # bs-0799R, Bioss USA) and APC anti hCD29 (Cat # 102901C0, AAT Bioquest) overnight at 4°C, then stained with a secondary antibody for Troponin I (Cat# A10042, Invitrogen). Sections were then washed 3x with PBST, followed by DAPI mounting using Fluoromount-G, with DAPI (Cat# 00-4959-52, Invitrogen). Images were taken with an Olympus FV3000RS and analyzed using ImageJ.
(2). Masson’s Trichrome Staining:
Masson’s staining was performed using a kit (Sigma-Aldrich, Burlington, MA). After cryosectioning and immediate fixation in 95% ethanol, sections were immersed in Bouin’s Solution overnight at room temperature. Sections were incubated in working Weigert’s Iron Hematoxylin Solution to stain nuclei, Biebrich Scarlet-Acid Fuchsin to identify the cytoplasm and muscle fibers, and Aniline Blue Solution to stain collagen fibers. Whole heart images were taken with the Nikon Eclipse Ti2 microscope.
Cytokine Release Assays:
The Proteome Profiler Mouse Cytokine Array A, (ARY006, R&D Systems) was used to quantify the 40 mouse proteins (cytokines, chemokines, and growth factors) from serum collected the day before surgery, and four hours after the 2nd injection from mice injected with PBS, unmodified MSCs, and ZipperCells. Serum was diluted and mixed with a cocktail of biotinylated detection antibodies, according to the manufacturer’s instructions. The sample/antibody mixture was then incubated with the array membrane overnight at 4°C. The membranes were washed and incubated with streptavidin–horseradish peroxidase followed by chemiluminescent detection. The array data were quantitated to generate a protein profile and the results are presented as the average signal (pixel density) of the pairs of duplicate spots representing each cytokine or chemokine analyzed using MATLAB. The data presented are from three biological samples per group.
Detection of Antibody Responses (ELISA):
Mice were injected with B-LZ & C-LZ 10 nM ZipperCells, or PBS, as previously described. After 6 weeks, blood was collected. The serum was obtained by centrifugation of blood for 20 minutes at 1500 x g. Nunc MaxiSorp plates were coated with B-LZ or C-LZ 10nM at 1 μg/ml and incubated overnight at 4°C. The plates were thoroughly washed 3x with 0.1% PBS-Tween (PBST), and incubated with ELISA blocking buffer (3% (w/v) instant dry milk (Food Lion) in PBS) for 1 h at RT. The wells were again washed 3x with 0.1% PBST. Serum was added to the top wells at a 1:100 dilution in ELISA blocking buffer and then serially diluted 3-fold followed by a 2 h incubation at RT. Plates were washed again 3x with 0.1% PBST. Washes were followed by the addition of 100 μL of Goat Anti-Mouse IgG Fc-HRP (Cat# 1033-05, SouthernBiotech) for 1 h at room temperature. The wells were washed 5x with 0.1% PBST followed by the addition of the TMB-ELISA substrate. After a 10 minute incubation, 50 μl of 2 N H2SO4 was added to stop the reaction. The absorbance was measured at 450 nm and 570 nm with a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA).
Flow Cytometry of Cell Surface Markers:
The following antibodies were used for flow cytometric immunophenotyping of human, BM-MSCs: Anti-Human CD90 FITC (PeproTech, Cranbury, NJ), Anti-Human CD105 PE (Peptrotech), Anti-Human CD29 APC (AAT Bioquest, Sunnyvale, CA), Anti Human CD3 APC Isotype Control (Biolegend, San Diego, CA), Anti-Human Cd192 PE Isotype Control (Miltenyi, Gaithersburg, MD), and Anti-Human Foxp3 FITC Isotype Control (Biolegend, San Diego, CA). Samples were analyzed on the Attune NxT V6 flow cytometer (Thermofisher) with minimum cell counts of 10,000 cells per sample. An FSC/SSC threshold of 25.0 was set to exclude debris. FITC-conjugated mAbs were excited with the 488 (50 mW) laser under the 503 long-pass mirror under the 530/30 BL1 detector. PE-conjugated mAbs were excited with a 561 laser (50 mW) and detected under the 577 long-pass mirror in the 585/16 YL1 detector. APC-conjugated mAbs were excited under the 637 laser (100 mW) under the 654 long-pass mirror in the 670/14 RL1 detector. Cell solution volumes were adjusted to 1mL and run at a speed of 500 µL/min.
Histological Assessment of MSCS in Healthy Mice:
For histological studies (n=3), 500,000 1,1’-Dioctadecyl-3,3,3’,3’-Tetramethylindotricarbocyanine Iodide (DiR)-labeled ZipperCells or unmodified MSC, or PBS was administered via tail vein injection into C57BL/6 mice. A total of three injections were administered every twelve hours. Twelve hours after the final injection (60 hours post-MI), mice were sacrificed, and liver, spleens, and lungs were collected. Tissues were fixed in formalin for two days, dehydrated, and then cleared before paraffin embedding. Tissues were then cut into 7.5 μm sections. Sections were deparaffinized and rehydrated for subsequent staining.
(1). Hematoxylin & Eosin Staining:
H & E staining was performed using a kit (Biovision, Milpitas, CA). Sections were incubated in Hematoxylin to stain nuclei, Eosin to identify the cytoplasm, and bluing reagent for color development. Sections were dehydrated and cleared before mounting in Permount (Fisher Chemical, Waltham, MA). Images were taken with the Nikon Eclipse Ti2 microscope.
(2). Immunohistochemistry of Off Target Organs:
After deparaffinization and rehydration, heat-induced epitope retrieval was performed in citrate buffer at pH 6 for 20 minutes. Samples were then blocked in a peroxidase blocking solution for 5 minutes at room temperature. Liver, lung, and spleen sections were then incubated with anti-mouse CD45 (Tonbo Biosciences, San Diego, CA, Cat # 70-0451-U100) at a 1:200 dilution for 30 minutes at room temperature. This was followed by secondary antibody staining with a goat anti-rat HRP secondary antibody (Tonbo Biosciences, San Diego, CA, Cat # 72-8104-M001). Substrate development was performed with a DAB HRP substrate (Vector Labs, Newark, CA, Cat # SK-4105). Samples were then counterstained with hematoxylin and bluing reagent (Biovision, Milpitas, CA) before mounting with Permount. Images were taken with the Nikon Eclipse Ti2 microscope.
Cytokine Release Assays:
The Proteome Profiler Mouse Cytokine Array A, (ARY006, R&D Systems) was used to quantify the 40 mouse proteins (cytokines, chemokines, and growth factors) from serum collected the day before surgery, and four hours after the 2nd injection from mice injected with PBS, unmodified MSCs, and ZipperCells. Serum was diluted and mixed with a cocktail of biotinylated detection antibodies, according to the manufacturer’s instructions. The sample/antibody mixture was then incubated with the array membrane overnight at 4°C. The membranes were washed and incubated with streptavidin–horseradish peroxidase followed by chemiluminescent detection. The array data were quantitated to generate a protein profile and the results are presented as the average signal (pixel density) of the pairs of duplicate spots representing each cytokine or chemokine analyzed using MATLAB. The data presented are from three biological samples per group.
TUNEL Assessment of Apoptosis:
60 hours post-MI the hearts were collected, and the infarct region was cryosectioned into 10µm sections. After fixation in 4% paraformaldehyde for 15 min, sections were permeabilized with 0.1% Triton X-100 in PBS for 10 minutes at room temperature. Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) of DNA strand breaks were stained using the Alexa Fluor 488 Click-iT™ Plus TUNEL Assay Kit for In Situ Apoptosis Detection (Thermo Fisher, Waltham, MA) according to the manufacturer protocol. This was followed by the previously described CD29 APC staining to detect mesenchymal stem cells. Following mounting, sections were visualized with an Olympus FV3000RS confocal microscope. To determine the percentage of apoptotic cells, TUNEL positive cells and CD29 positive cells were counted using the ImageJ software (version 1.53s, NIH).
Paracrine Secretion (ELISA):
Cells were plated at a density of 5 × 103 cells/cm2 in a 6-well plate. When the cells were 60% confluent, they were switched to low serum media. After 72 hours, the conditioned media was collected. Conditioned media was then spun at 500g for 10 minutes and then 2000g for 15 minutes to remove possible cell debris and apoptotic bodies. The resulting cleared conditioned media were then used in VEGF and EGF ELISA kits (Peprotech, Cat # 900-TM10 & 900-M05) according to manufacturer protocols.
Statistical Analysis:
All the quantitative data were presented as mean± SD. The mean values are based on at least three replicates. Comparisons between groups were analyzed using one-way ANOVA with a post hoc Tukey test (*p<0.05, **p<0.01, ***p<0.001). All statistical analyses were performed using Prism 7 (GraphPad Software, La Jolla, CA). The error bars represent standard deviations (SD).
Supplementary Material
Acknowledgments
The authors acknowledge Dr. Rani Sellers (UNC Department of Pathology and Laboratory Medicine) for the pathological assessment of healthy tissue samples. We acknowledge funding through the National Institutes of Health (R01HL161456, R01EB023262 and R01AI147497) and the Eshelman Institute of Innovation. Parts of figures 1 and 4 were created using Biorender.com. Confocal images were acquired at the UNC Neuroscience Microscopy Core 593 (RRID:SCR_019060), which is supported, in part, by funding from the NIH-NINDS 594 Neuroscience Center Support Grant P30 NS045892 and the NIH-NICHD Intellectual and Developmental Disabilities Research Center Support Grant P50 HD103573. IVIS images were acquired at the UNC Biomedical Research Center. We also acknowledge funding from the Pharmaceutical Research and Manufacturers of America (PhRMA) Foundation for N.Jasiewicz.
Contributor Information
Natalie E. Jasiewicz, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy. University of North Carolina, Chapel Hill, NC 27599, USA
Kuo-Ching Mei, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy. University of North Carolina, Chapel Hill, NC 27599, USA.
Hannah M. Oh, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy. University of North Carolina, Chapel Hill, NC 27599, USA
Parth Chansoria, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy. University of North Carolina, Chapel Hill, NC 27599, USA.
Dylan A. Hendy, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy. University of North Carolina, Chapel Hill, NC 27599, USA
Emily, E. Bonacquisti, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy. University of North Carolina, Chapel Hill, NC 27599, USA
Eric M. Bachelder, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy. University of North Carolina, Chapel Hill, NC 27599, USA
Kristy M. Ainslie, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy. University of North Carolina, Chapel Hill, NC 27599, USA
Haifeng Yin, McAllister Heart Institute, University of North Carolina, Chapel Hill, NC 27599, USA.
Li Qian, McAllister Heart Institute, University of North Carolina, Chapel Hill, NC 27599, USA; Department of Pathology and Laboratory Medicine, University of North Carolina, Chapel Hill, NC 27599, USA.
Brian C. Jensen, McAllister Heart Institute, University of North Carolina, Chapel Hill, NC 27599, USA; Department of Medicine, Division of Cardiology, University of North Carolina, Chapel Hill, NC 27599, USA
Juliane Nguyen, Division of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy. University of North Carolina, Chapel Hill, NC 27599, USA.
References
- [1].Ioacara S, Popescu AC, Tenenbaum J, Dimulescu DR, Popescu MR, Sirbu A, Fica S, Int J Environ Res Public Health 2019. Dec 31;17(1):285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Switaj TL, Christensen S, Brewer DM, Am Fam Physician 2017, 95, 232. [PubMed] [Google Scholar]
- [3].Johansson S, Rosengren A, Young K, Jennings E, BMC Cardiovasc Disord 2017, 17. [DOI] [PMC free article] [PubMed]
- [4].Hall M, Dondo TB, Yan AT, Mamas MA, Timmis AD, Deanfield JE, Jernberg T, Hemingway H, Fox KAA, Gale CP, PLoS Med 2018, 15, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Richardson WJ, Clarke SA, Alexander Quinn T, Holmes JW, Compr Physiol 2015, 5, 1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].TANG J, XIE Q, PAN G, WANG J, WANG M, European Journal of Cardio-Thoracic Surgery 2006, 30, 353. [DOI] [PubMed] [Google Scholar]
- [7].Ayala-Cuellar AP, Kang JH, Jeung EB, Choi KC, Roles of mesenchymal stem cells in tissue regeneration and immunomodulation, Vol. 27, Korean Society of Applied Pharmacology, 2019, pp. 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhu J, Lu K, Zhang N, Zhao Y, Ma Q, Shen J, Lin Y, Xiang P, Tang Y, Hu X, Chen J, Zhu W, Webster KA, Wang J, Yu H, Artif Cells Nanomed Biotechnol 2018. [DOI] [PMC free article] [PubMed]
- [9].Liew LC, Ho BX, Soh BS, Stem Cell Research & Therapy 2020 11:1 2020, 11, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sheng CC, Zhou L, Hao J, Biomed Res Int 2013, 2013, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fukushima S, Varela-Carver A, Coppen SR, Yamahara K, Felkin LE, Lee J, Barton PJR, Terracciano CMN, Yacoub MH, Suzuki K, Circulation 2007, 115, 2254. [DOI] [PubMed] [Google Scholar]
- [12].Zhao ZA, Han X, Lei W, Li J, Yang Z, Wu J, Yao M, Lu XA, He L, Chen Y, Zhou B, Hu S, Circ Res 2018, 123, E21. [DOI] [PubMed] [Google Scholar]
- [13].Baldazzi F, Jørgensen E, Ripa RS, Kastrup J, Eur Heart J 2008, 29, 1819. [DOI] [PubMed] [Google Scholar]
- [14].Malliaras K, Marbán E, Br Med Bull 2011, 98, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Williams AR, Hare JM, Mesenchymal stem cells: Biology, pathophysiology, translational findings, and therapeutic implications for cardiac disease, Vol. 109, NIH Public Access, 2011, pp. 923–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Karantalis V, Hare JM, Use of mesenchymal stem cells for therapy of cardiac disease, Vol. 116, Lippincott Williams and Wilkins, 2015, pp. 1413–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bashor CJ, Helman NC, Yan S, Lim WA, Science (1979) 2008, 319, 1539. [DOI] [PubMed] [Google Scholar]
- [18].Krylov D, Barchi J, Vinson C, J Mol Biol 1998, 279, 959. [DOI] [PubMed] [Google Scholar]
- [19].Krylov D, Mikhailenko I, Vinson C, EMBO J 1994, 13, 2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Reinke AW, Grant RA, Keating AE, J Am Chem Soc 2010, 132, 6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mabotuwana NS, Rech L, Lim Joyce, Hardy SA, Murtha LA, Rainer PP, Andrew, Boyle J, Stem Cell Reviews and Reports 2022 2022, 1, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Pankajakshan D, Agrawal DK, J Biomed Technol Res 2014, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wang J, Lee CJ, Deci MB, Jasiewicz N, Verma A, Canty JM, Nguyen J, Nanomedicine 2020, 27, 102201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kido M, Du L, Sullivan CC, Li X, Deutsch R, Jamieson SW, Thistlethwaite PA, J Am Coll Cardiol 2005, 46, 2116. [DOI] [PubMed] [Google Scholar]
- [25].Zhang Z, Tian H, Yang C, Liu J, Zhang H, Wang J, Hu S, Sun Z, He K, Chen G, J Am Heart Assoc 2020, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shintani Y, Ito T, Fields L, Shiraishi M, Ichihara Y, Sato N, Podaru M, Kainuma S, Tanaka H, Suzuki K, Scientific Reports 2017 7:1 2017, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hofmann U, Frantz S, Transl Pediatr 2018, 7, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Horwitz LD, ; Kaufman Dimitri, Keller MW, Kong Y, Time Course of Coronary Endothelial Healing After Injury Due To Ischemia and Reperfusion, 1994. [DOI] [PubMed]
- [29].Nguyen MM, Carlini AS, Chien MP, Sonnenberg S, Luo C, Braden RL, Osborn KG, Li Y, Gianneschi NC, Christman KL, Advanced Materials 2015, 27, 5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nguyen J, Sievers R, Motion JPM, Kivimäe S, Fang Q, Lee RJ, Mol Pharm 2015, 12, 1150. [DOI] [PubMed] [Google Scholar]
- [31].Timmers L, Lim SK, Arslan F, Armstrong JS, Hoefer IE, Doevendans PA, Piek JJ, El Oakley RM, Choo A, Lee CN, Pasterkamp G, de Kleijn DPV, Stem Cell Res 2007, 1, 129. [DOI] [PubMed] [Google Scholar]
- [32].Freyman T, Polin G, Osman H, Crary J, Lu MM, Cheng L, Palasis M, Wilensky RL, Eur Heart J 2006, 27, 1114. [DOI] [PubMed] [Google Scholar]
- [33].Rogers TB, Pati S, Gaa S, Riley D, Khakoo AY, Patel S, Wardlow RD, Frederick CA, Hall G, He LP, Lederer WJ, J Mol Cell Cardiol 2011, 50, 346. [DOI] [PubMed] [Google Scholar]
- [34].Huang YL, Qiu RF, Mai WY, Kuang J, Cai XY, Dong YG, Hu YZ, bin Song Y, Cai AP, Jiang ZG, J Zhejiang Univ Sci B 2012, 13, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mastri M, Shah Z, McLaughlin T, Greene CJ, Baum L, Suzuki G, Lee T, Am J Physiol Cell Physiol 2012, 303, C1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Levy O, Kuai R, Siren EMJ, Bhere D, Milton Y, Nissar N, de Biasio M, Heinelt M, Reeve B, Abdi R, Alturki M, Fallatah M, Almalik A, Alhasan AH, Shah K, Karp JM, Sci Adv 2020, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tang J-N, Cores J, Huang K, Cui X-L, Luo L, Zhang J-Y, Li T-S, Qian L, Cheng K, Stem Cells Transl Med 2018, 7, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Guo Y, Wysoczynski M, Nong Y, Tomlin A, Zhu X, Gumpert AM, Nasr M, Muthusamy S, Li H, Book M, Khan A, Hong KU, Li Q, Bolli R, Basic Res Cardiol 2017, 112, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tokita Y, Tang XL, Li Q, Wysoczynski M, Hong KU, Nakamura S, Wu WJ, Xie W, Li D, Hunt G, Ou Q, Stowers H, Bolli R, Circ Res 2016, 119, 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tang XL, Nakamura S, Li Q, Wysoczynski M, Gumpert AM, Wu WJ, Hunt G, Stowers H, Ou Q, Bolli R, Journal of the American Heart Association: Cardiovascular and Cerebrovascular Disease 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lee RH, Pulin AA, Seo MJ, Kota DJ, Ylostalo J, Larson BL, Semprun-Prieto L, Delafontaine P, Prockop DJ, Cell Stem Cell 2009, 5, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Luger D, Lipinski MJ, Westman PC, Glover DK, Dimastromatteo J, Frias JC, Albelda MT, Sikora S, Kharazi A, Vertelov G, Waksman R, Epstein SE, Circ Res 2017, 120, 1598. [DOI] [PubMed] [Google Scholar]
- [43].Li SH, Lai TYY, Sun Z, Han M, Moriyama E, Wilson B, Fazel S, Weisel RD, Yau T, Wu JC, Li RK, Journal of Thoracic and Cardiovascular Surgery 2009, 137, 1225. [DOI] [PubMed] [Google Scholar]
- [44].Cavalera M, Frangogiannis NG, Curr Pharm Des 2014, 20, 1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Noels H, Weber C, Koenen RR, Arterioscler Thromb Vasc Biol 2019, 39, 583. [DOI] [PubMed] [Google Scholar]
- [46].Christman KL, Vardanian AJ, Fang Q, Sievers RE, Fok HH, Lee RJ, J Am Coll Cardiol 2004, 44, 654. [DOI] [PubMed] [Google Scholar]
- [47].Kook YM, Hwang S, Kim H, Rhee KJ, Lee K, Koh WG, Scientific Reports 2020 10:1 2020, 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hoeeg C, Dolatshahi-Pirouz A, Follin B, Gels 2021, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wolinsky JB, Colson YL, Grinstaff MW, J Control Release 2012, 159, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ullah M, Liu DD, Thakor AS, iScience 2019, 15, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Caliari SR, Burdick JA, Nature Methods 2016 13:5 2016, 13, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wang CC, Chen CH, Lin WW, Hwang SM, Hsieh PCH, Lai PH, Yeh YC, Chang Y, Sung HW, Cardiovasc Res 2008, 77, 515. [DOI] [PubMed] [Google Scholar]
- [53].Christman KL, Gaetani R, Yin C, Srikumar N, Braden R, Doevendans PA, Sluijter JPG, Cell Transplant 2016, 25, 1653. [DOI] [PubMed] [Google Scholar]
- [54].Cui H, Liu C, Esworthy T, Huang Y, Yu ZX, Zhou X, San H, Lee SJ, Hann SY, Boehm M, Mohiuddin M, Fisher JP, Zhang LG, Sci Adv 2020, 6. [DOI] [PMC free article] [PubMed]
- [55].Freedland KE, Skala JA, Carney RM, Rubin EH, Lustman PJ, Dávila-Román VG, Steinmeyer BC, Hogue CW, Arch Gen Psychiatry 2009, 66, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tully PJ, Baker RA, J Geriatr Cardiol 2012, 9, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Cai M, Shen R, Song L, Lu M, Wang J, Zhao S, Tang Y, Meng X, Li Z, He ZX, Scientific Reports 2016 6:1 2016, 6, 1. [Google Scholar]
- [58].Noiseux N, Gnecchi M, Lopez-Ilasaca M, Zhang L, Solomon SD, Deb A, Dzau VJ, Pratt RE, Molecular Therapy 2006, 14, 840. [DOI] [PubMed] [Google Scholar]
- [59].Feng Y, Wang X, Fan T, Li L, Sun X, Zhang W, Cao M, Liu J, Li J, Huo Y, Front Physiol 2018, 9, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Teng B, Tilley SL, Ledent C, Mustafa SJ, Physiol Rep 2016, 4. [DOI] [PMC free article] [PubMed]
- [61].Janssen B, Debets J, Leenders P, Smits J, Am J Physiol Regul Integr Comp Physiol 2002, 282. [DOI] [PubMed] [Google Scholar]
- [62].Huang ZJ, Tarbell JM, Am J Physiol 1997, 273. [DOI] [PubMed] [Google Scholar]
- [63].Battiato I, Tartakovsky D, Cabrales P, Intaglietta M, 2017 European Conference on Circuit Theory and Design, ECCTD 2017 2017. [Google Scholar]
- [64].Carpentier B, Pierobon P, Hivroz C, Henry N, PLoS One 2009, 4, e4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Moy VT, Florin EL, Gaub HE, Science 1994, 266, 257. [DOI] [PubMed] [Google Scholar]
- [66].Claveria-Gimeno R, Vega S, Abian O, Velazquez-Campoy A, Expert Opin Drug Discov 2017, 12, 363. [DOI] [PubMed] [Google Scholar]
- [67].Thompson KE, Bashor CJ, Lim WA, Keating AE, ACS Synth Biol 2012, 1, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Rojewski MT, Weber BM, Schrezenmeier H, Transfusion Medicine and Hemotherapy 2008, 35, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kwon A, Kim Y, Kim M, Kim J, Choi H, Jekarl DW, Lee S, Kim JM, Shin JC, Park IY, Sci Rep 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Yang YHK, Ogando CR, Wang See C, Chang TY, Barabino GA, Stem Cell Res Ther 2018, 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Li L, Li J, Drum BM, Chen Y, Yin H, Guo X, Luckey SW, Gilbert ML, Stanley McKnight G, Scott JD, Fernando Santana L, Liu Q, Cardiovasc Res 2017, 113, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.