

Abstract
Clonal hematopoiesis of indeterminate potential (CHIP) is a premalignant expansion of mutated hematopoietic stem cells. As CHIP-associated mutations are known to alter the development and function of myeloid cells, we hypothesized that CHIP may also be associated with the risk of Alzheimer’s disease (AD), a disease in which brain-resident myeloid cells are thought to have a major role. To perform association tests between CHIP and AD dementia, we analyzed blood DNA sequencing data from 1,362 individuals with AD and 4,368 individuals without AD. Individuals with CHIP had a lower risk of AD dementia (meta-analysis odds ratio (OR) = 0.64, P = 3.8 × 10−5), and Mendelian randomization analyses supported a potential causal association. We observed that the same mutations found in blood were also detected in microglia-enriched fraction of the brain in seven of eight CHIP carriers. Single-nucleus chromatin accessibility profiling of brain-derived nuclei in six CHIP carriers revealed that the mutated cells comprised a large proportion of the microglial pool in the samples examined. While additional studies are required to validate the mechanistic findings, these results suggest that CHIP may have a role in attenuating the risk of AD.
Subject terms: Immunology, Genetic association study
Clonal hematopoiesis of indeterminate potential (CHIP) was found to be associated with a protective effect from Alzheimer’s disease (AD) in large population-based cohorts.
Main
Clonal hematopoiesis of indeterminate potential (CHIP) is an age-associated expansion of hematopoietic stem cells (HSCs) found in 10–30% of those older than 70 (refs. 1–4). It most commonly occurs due to truncating or loss-of-function mutations in transcriptional regulators such as DNMT3A, TET2 and ASXL1 and can be detected by sequencing of DNA from peripheral blood or bone marrow cells5. As these are also founding mutations for hematological neoplasms such as acute myeloid leukemia, it is unsurprising that CHIP associates with a higher risk of developing these cancers1,2,6,7. However, CHIP also associates with an increased risk of atherosclerotic cardiovascular disease and death8–10. This link is believed to be causal as mice that are deficient for Tet2 or Dnmt3a in hematopoietic cells develop more severe cardiovascular phenotypes, possibly due to altered gene expression in mutant macrophages, which favors the more rapid progression of the lesions8,11,12.
Alzheimer’s disease (AD) remains a leading cause of morbidity and mortality in the elderly, but therapies that can effectively slow or halt its progression are lacking. Genome-wide association studies (GWAS) have implicated functional alterations of microglia (MG), the macrophage-like hematopoietic cells in the brain, as a major driver of AD risk13. Because CHIP-associated mutations influence the function of myeloid cells8,11,14, we tested whether CHIP was associated with the risk of AD.
Results
Association between CHIP and AD dementia
To test the association between CHIP and incident AD dementia, we used data from the Framingham Heart Study (FHS) and the Cardiovascular Health Study (CHS), which are two cohorts within the Trans-omics for Precision Medicine (TOPMed) project15. CHIP variants (Supplementary Table 1) were identified from blood-derived whole-genome sequencing (WGS) data as previously described9. AD dementia was diagnosed when participants met the criteria of the National Institute of Neurological and Communicative Disorders and Stroke (NINCDS) and the AD and Related Disorders Association (ADRDA) for definite, probable or possible AD.
After excluding those with coronary heart disease, stroke or prior dementia, there were 2,437 participants in FHS, of whom 92 developed incident AD dementia, and 743 participants in CHS, of whom 166 developed incident AD dementia (Supplementary Table 2). Participants in CHS were substantially older on average and a higher proportion was female compared to participants in FHS, which contributed to a higher rate of AD dementia in CHS compared to FHS (22.3% versus 3.8%) in the follow-up period (Supplementary Table 2). Contrary to our expectations, the presence of CHIP was associated with a lower subdistribution hazard ratio (SHR) for incident AD dementia in fully adjusted competing risks regression (CRR) models (SHR = 0.69, P = 0.13 in CHS; SHR = 0.51, P = 0.068 in FHS; SHR = 0.63, P = 0.024 in a fixed-effects meta-analysis of the two cohorts), while the effects of age, sex and APOE genotype were as expected based on prior studies (Fig. 1a). APOE genotype was strongly associated with AD dementia risk in those without CHIP age 60 years or older (P = 8.1 × 10−8 by log-rank test), but not in CHIP carriers of the same age (P = 0.42 by log-rank test) (Fig. 1b), possibly due to smaller sample size. The inclusion of MG-associated germline polymorphisms from AD GWAS did not attenuate the effect of CHIP in these models (Supplementary Table 3).
Fig. 1. Association of CHIP and AD in longitudinal cohorts.

a, Effect of CHIP on the risk of incident AD in CHS and FHS. SHR, CI95 and two-sided Wald P values were calculated for each covariate from CRR models, which included age at blood draw, sex and APOE genotype as covariates. The measure of center is the SHR and the lengths of the lines represent the CI95 for the SHR. Results from CHS and FHS were then meta-analyzed using a fixed-effects model for the two cohorts, the center of the diamonds represents the SHR and the lengths represent the CI95 for the SHR. People in FHS, n = 2,437 and people in CHS, n = 743. b, Kaplan–Meier curve showing AD-free probability in CHIP noncarriers (left) and carriers (right), stratified by APOE genotype. Analysis was restricted to those in FHS and CHS older than 60 years at the time of blood draw, and the results of a two-sided log-rank test are shown. People for non-CHIP carriers, n = 1,800 and people for CHIP carriers, n = 246. CI95, 95% confidence interval.
Next, we sought to replicate the association between CHIP and AD dementia in Alzheimer’s Disease Sequencing Project (ADSP), a case-control study with similar criteria for AD diagnosis as TOPMed. Due to a selection process that targeted cases with minimal risk as predicted by known risk factors (age, sex and APOE genotype) and targeted controls with the least probability of conversion to AD by age 85 years16, only APOE ε3ε3 cases and controls were well matched with respect to age and sex (Methods). Therefore, we focused our analysis on the 1,104 individuals with AD dementia and 1,446 individuals without AD dementia who had the APOE ε3ε3 genotype (Supplementary Table 4). The sequencing depth in ADSP WES samples (~80×) was higher than that for TOPMed WGS samples (~38×), which resulted in greater sensitivity to detect smaller clones (Extended Data Fig. 1). Clone size, which is approximated by the variant allele fraction (VAF), has previously been shown to be an important predictor of risk for blood cancer1,17 and cardiovascular outcomes8,9. To directly compare the outcomes of ADSP to TOPMed, both cohorts would need to have a similar distribution of VAF in CHIP carriers. Therefore, we limited the definition of CHIP carriers to those with VAF > 0.08 in ADSP—a cutoff that was empirically chosen because it resulted in a VAF distribution that was nearly identical to TOPMed (Extended Data Fig. 1). We found that CHIP with VAF > 0.08 was associated with lower risk of AD dementia in ADSP using logistic regression (odds ratio (OR) = 0.66, P = 5.5 × 10−4; Fig. 2a and Supplementary Table 5). In contrast, having VAF ≤ 0.08 had no association with AD dementia (OR = 1.25, P = 0.23; Supplementary Table 6). Higher VAF was also significantly associated with protection from AD dementia when modeled as a continuous variable (Supplementary Table 6). The effect of CHIP remained significant when using a VAF cutoff of >0.02 (OR = 0.79, P = 0.024; Supplementary Table 5). A meta-analysis of ADSP, CHS and FHS showed that CHIP carriers had a significantly lower risk of AD dementia (OR = 0.64, P = 3.8 × 10−5; Fig. 2b).
Extended Data Fig. 1. The VAF distribution in TOPMed and ADSP.

Density plots of VAF from all CHIP carriers in ADSP (left) or in CHIP carriers with VAF greater than 0.08 (right) compared to VAF distribution from TOPMed CHIP carriers (red) in each plot.
Fig. 2. CHIP is associated with protection from AD dementia and AD neuropathologic change (ADNC).

a. Effect of CHIP on the risk of AD in those with APOE ε3ε3 genotype from the ADSP. OR, CI95 and two-sided Wald P value were calculated from a logistic regression model that also included age at the time of blood draw and sex as covariates (Supplementary Table 5 for full regression results). People, n = 2,550. b, Fixed-effect meta-analysis for risk of AD in CHIP carriers using logistic regression in ADSP, FHS and CHS with two-sided Wald P value shown. c, MR for risk of AD based on genetic risk of CHIP using the weighted median estimator on summary statistics from Schwartzentruber et al.18 AD GWAS and GWAX, Finngen AD GWAS and Gr@ACE AD GWAS, which were meta-analyzed using a fixed-effects model. OR, CI95 and two-sided Wald P values for risk of AD per 1 log-odds increase in genetic risk of CHIP are shown. The GWAX analysis was one-sample MR (both CHIP and AD effect estimates were derived from UK Biobank data), while the other studies were two-sample MR. People, n = 692,045. d, Effect of CHIP on the risk of increased CERAD neuritic plaque score in ADSP participants without a dementia diagnosis. OR and CI95 were calculated from an ordinal logistic regression model, which included age at autopsy, APOE genotype, sex and CHIP status as covariates. Two-sided P values were calculated by comparing the t-statistic for each covariate against a standard normal distribution. Full regression results are in Supplementary Table 9. People, n = 427. e, Effect of CHIP on the risk of increased Braak stage in ADSP participants without a dementia diagnosis. OR and CI95 were calculated from an ordinal logistic regression model, which included age at autopsy, APOE genotype, sex and CHIP status as covariates. Two-sided P values were calculated by comparing the t-statistic for each covariate against a standard normal distribution. Full regression results are in Supplementary Table 9. People, n = 454. For all forest plots, the measure of center is the OR and the lines represent the CI95 for the OR.
Mendelian randomization analyses of CHIP and AD
MR is a form of causal inference in which inherited genetic polymorphisms known to influence the risk of a particular exposure or trait (in this case CHIP) are assessed for an association with a disease or trait (in this case AD). One- and two-sample MR using 24 CHIP-associated polymorphisms18 as the instruments (Supplementary Table 7) found that higher genetic risk of CHIP was associated with reduced odds of AD using the weighted median estimator (OR = 0.90 per 1 log-odds increase in risk of CHIP, P = 3.3 × 10−4, Fig. 2c). Consistent results were obtained with other MR methods (Supplementary Table 8 and Extended Data Fig. 2). To rule out reverse causation (that having AD causally reduces the risk of CHIP), we also performed two-sample MR using 36 polymorphisms for risk of AD as the instruments18 and CHIP as the outcome, which found no evidence of a causal effect in this direction (OR = 0.97 per 1 log-odds increase in risk of AD, P = 0.26 using the weighted median estimator).
Extended Data Fig. 2. Scatterplot of effect sizes from the Mendelian randomization study.

a) Regression line for the IVW model is shown. The variants shown are from previously published genome-wide association studies (GWAS) of CHIP as described in Methods and listed in Supplementary Table 7. The measure of center is the beta estimate for each variant and the lines represent the 95 percent confidence interval for the beta estimate. All values were taken from published summary statistics data from CHIP or AD GWAS. b) Regression lines for each model are shown using the same data as in A).
Association between CHIP and ADNC
The hallmark neuropathological features of AD, regional accumulation of β-amyloid plaques and tau neurofibrillary tangles, can also be found in some people without a clinical diagnosis of dementia. A neuritic plaque density score developed by the Consortium to Establish a Registry for AD (CERAD)19 and Braak stage for neurofibrillary tangle distribution20 are commonly used to assess these changes at brain autopsy, with a higher score indicative of more extensive accumulation of pathologic features. A subset of participants in ADSP had a brain autopsy performed after death, which allowed us to test whether CHIP was associated with ADNC in those without dementia (Supplementary Table 9a,b). Here the presence of CHIP was associated with having a lower CERAD score (OR = 0.50, P = 3.2 × 10−3) and Braak stage (OR = 0.56, P = 0.015) using ordinal logistic regression after adjusting for age at death, sex and APOE genotype (Fig. 2d,e and Supplementary Table 9c,d).
Stratified associations between CHIP and AD
APOE genotype is the strongest genetic risk factor for AD21, with APOE ε2 conferring protection from disease and APOE ε4 conferring higher risk, compared to the APOE ε3 allele (Fig. 1a). In CHS and FHS, the decrement in risk for AD dementia associated with CHIP was of a similar magnitude among people with the APOE ε3ε3 genotype or with an APOE ε4 allele, but not seen in those with APOE ε2ε2 or APOE ε2ε3 genotypes (Fig. 3a and Supplementary Tables 10a–c). In ADSP participants without dementia, ADNC was lower in CHIP carriers compared to noncarriers with the APOE ε3ε3 genotype or with an APOE ε4 allele, but this effect was seen not in those with APOE ε2ε2 or APOE ε2ε3 genotypes (Fig. 3b). Regression models that examined the interaction of CHIP with APOE genotype showed a consistent direction of effect as the stratified models for both AD and ADNC, although the individual interaction terms did not reach statistical significance (Supplementary Tables 9e and 10d). While these results are suggestive, they require confirmation in larger cohorts.
Fig. 3. Associations of CHIP to AD by APOE genotype and mutated driver gene.
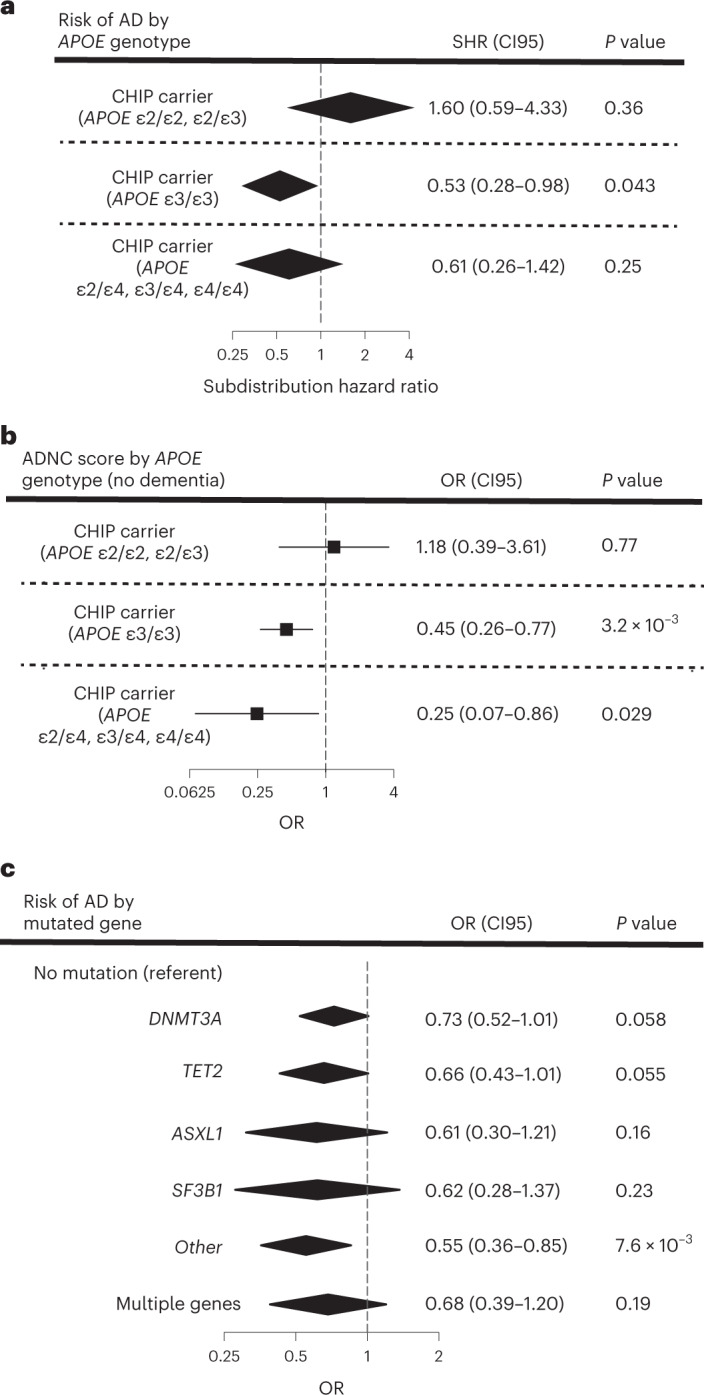
a, Effect of CHIP on AD dementia risk in participants from CHS and FHS stratified by APOE genotype. Participants were binned into those with neutral (APOE ε3ε3), low-risk (APOE ε2ε2 and ε2ε3) and high-risk (any APOE ε4 allele) groups. SHR, 95% confidence intervals (CI95) and two-sided Wald P values were calculated for each covariate (age at the time of blood draw for sequencing, sex and CHIP carrier status) from CRR models, and results from FHS and CHS were then meta-analyzed using a fixed-effects model (Supplementary Table 10 for full regression results). Adjustments for multiple comparisons were not performed. People, n = 3,180. b, Effect of CHIP on ADNC score in cognitively intact participants from ADSP stratified by APOE genotype. Participants were binned into those with neutral (APOE ε3ε3), low-risk (APOE ε2ε2 and ε2ε3) and high-risk (any APOE ε4 allele) groups. ORs, CI95 and two-sided Wald P values were calculated for a six-point composite score of CERAD and Braak stages and analyzed using an ordinal logistic regression model with age at death, sex and CHIP carrier status as covariates (Supplementary Table 10 for full regression results). Adjustments for multiple comparisons were not performed. People, n = 422. c, Effect of mutated CHIP gene on AD in participants from CHS, FHS and ADSP. OR, CI95 and two-sided Wald P values were calculated for each covariate (age at the time of blood draw for sequencing, sex, cohort and APOE genotype) from logistic regression models, and results from the TOPMed cohorts and ADSP were then meta-analyzed using a fixed-effects model (Supplementary Table 12 for full regression results). Adjustments for multiple comparisons were not performed. People, n = 5,730. For all forest plots, the measure of center is the SHR or OR and the lines represent the CI95 for the SHR or OR.
In analyses stratified by sex, there was a similar degree of protection for both male and female CHIP carriers (Supplementary Table 11). We also assessed whether the risk of AD dementia varied based on the specific mutated gene. Of the most commonly mutated genes in CHIP, all were associated with protection from AD dementia to a similar degree (Fig. 3c and Supplementary Table 12).
Detection of CHIP variants in the human brain
We wondered whether cells bearing CHIP-associated mutations could be found in the brain, a finding that would support a causal association between CHIP and AD risk. We first obtained brain DNA-derived WES data from 1,776 persons in ADSP and assessed for the presence of CHIP-associated variants. Similar to a prior study22, we found mutations consistent with CHIP in 17 brain samples (Fig. 4a and Supplementary Table 13), although paired blood samples were not available for this set.
Fig. 4. CHIP variants can be found in the MG-enriched fraction of the brain.

a, Barplot of putative CHIP mutations identified from whole exome sequencing of brain DNA from 1,775 persons in ADSP. Details about the variants identified are in Supplementary Table 13. b, Schematic of experimental workflow. Autopsy samples from the occipital cortex, cerebellum and putamen were digested to prepare single nuclei suspensions. Nuclei were then stained and sorted using antibodies to c-MAF+ (a marker of myeloid cells) and NeuN+ (a marker of neuronal cells), followed by amplicon sequencing for CHIP variants. c, Barplot of the VAF of the CHIP variants from eight donors (ACT1 to ACT8). For each sample, the VAF in the blood and in the brain c-MAF+ NeuN− population is shown. The occipital cortex was available for all eight donors. A bar for cerebellum or putamen is shown if available, otherwise, NA in the corresponding color designates lack of an available sample (purple for cerebellum and red for putamen). The CHIP mutations present in each participant are reported in the box on the right of the barplot. Details about the samples and the VAF from sorted populations are shown in Supplementary Table 14.
To test definitively whether blood-derived cells from CHIP carriers were present in the brain, we obtained tissue samples from the occipital lobe, and in some cases putamen or cerebellum, of eight donors from the ACT cohort who were known to have CHIP from blood exome sequencing, as well as four donors without CHIP (Supplementary Table 14). All persons were in their 80s and 10 of 12 were without dementia and had no/low ADNC at the time of death. The eight CHIP carriers had mutations in DNMT3A, TET2, ASXL1, SF3B1 and GNB1 with the highest frequency in DNMT3A (four of eight) and TET2 (three of eight) (Supplementary Table 14). In addition, two of the eight harbored two different CHIP mutations. We digested the frozen brain tissue and isolated intact nuclei, from which we extracted DNA for amplicon sequencing. We detected the same mutations that were present in blood in six of eight unfractionated brains with VAF ranging from 0.004 to 0.02 (Extended Data Fig. 3 and Supplementary Table 14).
Extended Data Fig. 3. Sorting of nuclei from brains of CHIP carriers.

Flow cytometry gating strategy for nuclei sorting for the 8 brain samples using c-MAF and NeuN markers. The arrow points to the c-MAF+ NeuN− sorted population and the VAF is indicated for each sample. The brain regions are abbreviated as Ce for Cerebellum, OC for Occipital cortex, and P for putamen.
We hypothesized that the presence of CHIP mutations represented blood cells, likely myeloid, that had engrafted in the brain. To test this hypothesis, we devised a flow cytometric strategy to enrich myeloid cells and deplete other blood cell types. Because the tissue was not viably cryopreserved, isolation of cells based on the expression of membrane antigens was not possible. Instead, we stained nuclei for the neuronal-specific transcription factor NeuN (encoded by the RBFOX3 gene) and c-MAF (encoded by the MAF gene), a transcription factor highly expressed in mononuclear phagocytes, including monocytes, macrophages, dendritic cells and MG, as well as some neurons and nonhematopoietic glial cells, but not granulocytes or most lymphocytes. We then sorted four populations based on the presence or absence of these markers (Fig. 4b). The CHIP somatic variants were found in the NeuN− c-MAF+ population in seven out of eight brains, with a VAF that ranged from 0.02 to 0.28 (representing 4% to 56% of NeuN− c-MAF+ nuclei). In contrast, CHIP somatic variants were not detected in the NeuN+ c-MAF− neuronal population and were absent or at low levels in the other two populations (Fig. 4c and Extended Data Fig. 3), effectively excluding granulocytes and lymphocytes as the source of the mutations. To determine whether the observed VAF was reproducible across replicates, we repeated the VAF measurement in the NeuN− c-MAF+ gate for four mutations and obtained concordant results (Extended Data Fig. 4a).
Extended Data Fig. 4. Correlation of repeated VAF measurements from brain.

a) c-MAF+ NeuN− nuclei were sorted from the samples shown and VAF for the CHIP variants was assessed. A repeat sample was sorted to determine the variability in the VAF of sorted populations. b) DNA from unsorted occipital cortex nuclei was obtained for the samples shown. VAF of the CHIP variants was determined from two independent sequencing libraries generated for each sample to assess the variability in the VAF across sequencing runs. The y = x line is drawn. R2, coefficient, and y-intercept values calculated from linear regression are shown.
Single-nucleus chromatin accessibility profiling of human brains
MG are believed to have little contribution from marrow-derived cells in adulthood23, so it would be surprising to find the mutations in this compartment. To better understand the identity of the mutated myeloid cells in the brains of CHIP carriers, we performed single nucleus ATAC-sequencing (snATAC-seq) on brain samples from the four ACT donors without CHIP (ACT9-12), as well as the six donors with detectable CHIP mutations in the unsorted brain (ACT2-7). All ten donors were free of AD and had no/low ADNC (Supplementary Table 14). We analyzed the occipital cortex from all donors to assess the cell state in a single brain region across all samples. We also analyzed the cerebellum and putamen for the ACT6 donor to determine if the infiltration of mutant cells varied by brain region within a single donor. snATAC-seq was performed on unsorted nuclei for each sample, as well as sorted NeuN− c-MAF+ nuclei for ACT2, ACT6 and ACT9.
In total, we recovered high-quality snATAC-seq profiles for 94,367 nuclei (Extended Data Fig. 5). We then aggregated our data with previously published snATAC-seq data from ten samples from the adult human brain,24 as well as from circulating immune cells25 (additional 93,387 nuclei in total). We identified 14 clusters encompassing the major brain and hematopoietic cell types (Fig. 5a,b), including one cluster that contained previously described MG, as well as myeloid cells from each of our samples (C6; Fig. 5b, Extended Data Fig. 6 and Supplementary Table 15). Small numbers of cells in our brain samples clustered with circulating immune cells, predominantly T cells, and were found in all unsorted brains (mean = 7.2% of total hematopoietic cells, range 3.7–12.5%). There was no enrichment of nonmicroglial hematopoietic cells in the CHIP carriers (mean = 6.5%) compared to controls (mean = 8.7%; Fig. 5c and Supplementary Table 15). For nuclei within C6 grouped by sample, accessible chromatin at MG marker genes (TMEM119, P2RY12, SALL1 and APOE) in each of our samples was visually and quantitatively similar to the reference MG but distinct from profiles of blood monocytes or dendritic cells (Fig. 5d and Extended Data Fig. 7). Furthermore, the C6 tracks in all brain samples had low accessibility at ANPEP (CD13), FOSB, VIM and S100A10, in contrast to monocytes or DCs. To further investigate heterogeneity within C6, we reclustered cells from our samples within C6 (Extended Data Fig. 8). We identified three subclusters, all of which exhibited accessibility profiles consistent with MG, but we did not observe consistent differences in subcluster proportions between CHIP and non-CHIP samples (Extended Data Fig. 8). In total, these results indicate that the cells in C6 exhibit aggregate similarity to MG and are distinct from monocytes (C7) and dendritic cells (C8). Furthermore, the C6 cells in CHIP carriers were similar to C6 cells in noncarriers in several different analyses.
Extended Data Fig. 5. High-quality snATAC-seq libraries from ACT brain samples.

Quality control metrics for each nucleus in the indicated scATAC-seq sample. Aggregated Corces 2020 samples and Satpathy 2019 samples are also included. Top: Enrichment of fragments in transcription start sites (TSS). Center: number of fragments. Bottom: fraction of reads in peaks (FRIP) for each nucleus. Unless otherwise indicated, the samples are derived from occipital cortex (OC) and are unsorted. The brain regions are abbreviated as Ce for Cerebellum, OC for Occipital cortex, and P for putamen. The n for each sample is the number of nuclei analyzed in a single experiment for each sample, which is listed in Supplementary Table 15 under column ‘TOTAL’.
Fig. 5. snATAC-seq of brain samples from CHIP carriers reveals that the mutated cells are similar to MG and comprise a large proportion of the microglial pool.

a, snATAC-seq profiles of 187,754 cells from our dataset, the Corces 2020 adult human brain dataset and the Satpathy 2019 human blood dataset. Each dot represents the snATAC-seq profile of one cell and is colored by its assigned cluster. b, snATAC-seq profiles of all cells colored by which donor it originated from. Samples from Corces 2020 and Satpathy 2019 are aggregated and shown in gray and dark gray, respectively. c, Cluster composition of hematopoietic cells in each sample in this study. d, Pseudo-bulk tracks for selected gene loci. The top eight tracks show snATAC-seq coverage of cells from the indicated sample (or aggregated Corces 2020 samples) within cluster C6–MG. Clusters C7–monocyte and C8–DC are also included for visual reference. e, Calculation of the proportion of MG bearing a CHIP mutation in each sample. Error bars depict the simulated 95% confidence interval for percent mutant MG in each sample using n = 106 random samples (see Methods for details). f, Correspondence between the fraction of mutant blood cells and mutant MG for each donor (n = 6). Pearson product-moment correlation coefficient and two-sided P value calculated from a t-statistic based on the correlation coefficient are shown. g, Fraction of MG relative to the total glial pool in occipital cortex samples from CHIP and non-CHIP donors, computed from frequencies of the indicated clusters. Two-sided Wald P value = 0.042 by quasibinomial regression of the cell counts of MG, oligodendrocytes and astrocytes. n = 10 donors. Box plot shows median, first quartile and third quartile. Box plot whiskers show minimum and maximum values, which are capped at a distance of 1.5× (interquartile range) away from the box. The colors in d–g represent the sample name as depicted in the box under Fig. 4b. Detailed information for each donor is available in Supplementary Table 14. Samples are unsorted and are taken from the occipital cortex unless otherwise indicated. Ce, cerebellum; P, putamen.
Extended Data Fig. 6. Marker genes and additional data for scATAC-seq clusters.

a) snATAC-seq profiles of all nuclei colored by accessibility of indicated marker genes (quantified using GeneScore). b) snATAC-seq profiles of all nuclei colored by which study they originated from (left), sort status (center), or brain region (right). Brain regions are abbreviated as Ce for Cerebellum, OC for Occipital cortex, and P for putamen.
Extended Data Fig. 7. Quantification of gene accessibility in microglia subsets.

a) For each gene, accessibility in each single nucleus is plotted (quantified via ArchR GeneScore). The n for each brain sample is the number of nuclei in the microglia cluster (C6) analyzed in a single experiment for each sample, which is listed in Supplementary Table 15. n = 7,982 nuclei for C7-monocyte and n = 2,369 nuclei for C8-DC. b) Fold change and statistical significance when comparing all pairs of groups for SALL1 (left) and ANPEP (right). (A-B) Within the microglia cluster (C6), cells are split by donor and brain region. Aggregated Corces 2020 microglia, as well as C7-Monocyte and C8-DC clusters, are included for reference. Statistical significance and fold change was computed for all pairs of groups for all GeneScores using the Wilcoxon test. * denotes FDR < 0.05.
Extended Data Fig. 8. Sub-clustering of cells in our samples within cluster C6-MG.

(a) snATAC-seq profiles of each nucleus colored by which donor it originated from. (b) snATAC-seq profiles colored by sub-cluster (left) and sub-cluster composition of each sample (right). (c) snATAC-seq profiles of nuclei colored by brain region (top) or sort status (bottom). (d) Quantification of accessibility in each single nucleus for selected marker genes (ArchR GeneScore). Nuclei from our samples are grouped by sub-cluster, and microglia from the Corces 2020 reference dataset, as well as C7 and C8 from Fig. 4, are included for reference.
We next used the snATAC-seq data to estimate the proportion of mutated MG in each sample. We determined that the percentage of MG ranged from 7.6% to 25% in the sorted samples, representing an 11.5-fold to 34.5-fold enrichment for MG in the occipital cortex and cerebellum (Fig. 5e and Supplementary Table 16). However, this indicated that there were still large numbers of contaminating non-MG cells in the NeuN− c-MAF+ gate. To better estimate the percentage of mutated MG, we first assessed the VAF for the CHIP variant in each unsorted brain sample. We confirmed that VAF estimates from unsorted nuclei were consistent across replicates of the same sample (Extended Data Fig. 4b). We also determined that the VAF estimates were unlikely to be inflated by artifactual variant reads because amplicon sequencing of a brain sample from a donor without CHIP did not detect any mutant alleles (Supplementary Table 14). Because these are heterozygous mutations, multiplying the VAF by two gives an estimate of the percentage of mutated cells in the sample. We conservatively assumed that all nonmicroglial hematopoietic cells were mutant and subtracted the number of these cells from the total mutant cell count (Supplementary Table 15 and Methods). Having excluded the contribution of all nonmicroglial hematopoietic cells, we reasoned that we could divide the number of remaining mutant cells by the total number of MG in each unsorted sample to estimate the percentage of mutant MG. Overall, we observed a range of mutant MG from 30% to 95% in the six CHIP samples (Fig. 5e and Supplementary Table 16). For the ACT6 donor, 68%, 38% and 47% of putamen, cerebellar and occipital cortex MG harbored the TET2 mutation, respectively, suggesting that multiple brain regions in CHIP carriers are likely to be infiltrated by marrow-derived myeloid cells that resemble MG. We observed a very strong correlation between the proportion of mutated cells in peripheral blood and in occipital cortex MG from the six CHIP donors (R2 = 96%, Fig. 5f). This suggests a linear relationship between the size of the mutant clone in the periphery and infiltration of the brain and may contribute to the observed association between higher peripheral blood VAF and protection from AD dementia (Supplementary Table 6).
Finally, as CHIP mutations are known to increase the fitness of HSCs and myeloid progenitors5, we looked for evidence of enhanced fitness of mutant MG in these samples. The mean proportion of MG amongst all glia was 5.1% in the four control occipital cortex samples (Fig. 5g), but this proportion was increased to 8.2% in the six occipital cortex samples from CHIP carriers (P = 0.042). If confirmed in larger studies, these results may suggest a possible role for CHIP mutations in increasing MG number via enhanced homing, survival or proliferation of marrow-derived MG or their precursors in the brain.
Discussion
We provide here several lines of evidence supporting the role of mutant, marrow-derived cells in protecting against the risk of AD. First, CHIP is associated with protection from AD dementia in multiple cohorts, an effect that could not be attributed to survival bias. Second, MR analyses were consistent with a causal role for CHIP in reducing AD risk. Third, CHIP was associated with lower levels of neuritic plaques and neurofibrillary tangles in those without dementia, indicating a possible modulating effect of CHIP on the underlying pathophysiology of AD. Fourth, consistent with this hypothesis, we also provided preliminary evidence of substantial infiltration of the brain by marrow-derived mutant cells, which adopted a microglial-like phenotype. The degree of protection from AD dementia seen in CHIP carriers is similar to carrying an APOE ε2 allele, which is the most protective common inherited variant for AD26.
One of the more surprising findings in our study is the high proportion of mutant MG-like cells observed in the brains of CHIP carriers. This was demonstrated by amplicon sequencing of nuclei from both sorted and unsorted brain fractions, although the estimated fraction of mutant cells is likely more reliable from the unsorted brain. Although there is some uncertainty in the estimates due to known or unknown causes of measurement error, we found a very strong correlation between the mutant cell fraction in blood and MG, arguing against imprecision in the estimates due to stochastic variation from sample preparation or sequencing. We were also able to rule out contamination by circulating blood cells as an explanation for this finding, as ~88% to 96% of the hematopoietic cells in the brain samples clustered as MG and were distinct from other immune cells. Nonetheless, our study has limitations. We did not find differences in chromatin accessibility between the MG from CHIP carriers compared to noncarriers that could explain the protection from AD. This may be due to limited sample size, mutational heterogeneity or the fact that we only assessed MG from people without AD or significant ADNC. It is possible that differences in microglial phenotypes due to CHIP are not evident at steady-state, but only after perturbation, as has been observed in other contexts8,11,27,28. Therefore, while the data presented here are suggestive, we cannot currently conclude that the mechanism of protection from AD seen in CHIP carriers is due to these infiltrating cells. Identifying biologically meaningful differences between mutant and wild-type MG would strengthen the hypothesis that mutant cells residing in the brain have a causal role in protection from AD. Functional studies of mutant MG are likely to be informative, as are technologies that are able to obtain both genotype and transcriptome data from single cells within the same sample29,30.
There is a growing body of evidence from mice that bone marrow-derived myeloid cells can enter the brain during homeostasis31,32. Our study demonstrates that this may be true in humans as well. Marrow-derived cells may have an ameliorative effect on neurodegenerative phenotypes in mice33,34, possibly due to their superior phagocytic capacity35. GWAS have strongly implicated microglial genes involved in ligand recognition and phagocytosis, such as APOE, TREM2, CD33 and the MS4A cluster, in the biology of AD36. We hypothesize that reduced phagocytic capacity of the endogenous microglial system during aging elevates the risk of AD, but that this is rescued in CHIP carriers, possibly due to the influx of peripherally derived myeloid cells into the brain parenchyma, which are able to outcompete endogenous MG. The observation that the effect sizes were similar with several different driver genes suggests that there could be shared mechanisms leading to protection from AD across several different mutations. For example, there may be convergent gene expression patterns in innate immune cells or a common effect of the mutations on MG survival or proliferation, similar to what has been observed in other contexts12,37. Along these lines, we provide preliminary evidence of a quantitative increase in the fraction of MG in the brains of CHIP carriers. We predict that more detailed characterization of phenotypic differences between mutant and wild-type MG may provide insights into slowing the progression of AD.
Methods
Study population
FHS
The FHS is a single-site, prospective and population-based study that has followed participants from the town of Framingham, MA to investigate risk factors for cardiovascular diseases. The population of Framingham was almost entirely white at the beginning of the study. The FHS comprises three generations of participants. The first generation (Original cohort/Gen1) (ref. 38), followed since 1948, enrolled 5,209 male and female participants who comprised two-thirds of the adult population then residing in Framingham, MA, USA. Survivors continue to receive biennial examinations. The second generation (Offspring cohort/Gen2) (ref. 39), followed since 1971, comprised 5,124 offspring of Gen1 and spouses of the offspring (including 3,514 biological offspring) who attended examinations every 4–8 years. The third generation (Gen3) (ref. 40), enrolled in 2002, included 4,095 children from the largest offspring families who attended three examinations 4 years apart. All cohorts continue under active surveillance for cardiovascular events, stroke and dementia. The FHS was approved by the Institutional Review Board of the Boston University Medical Center. All study participants provided written informed consent at each examination.
A total of 4,195 samples were sequenced as part of the TOPMed project Freeze 6 release as previously described9. The selection of participants for sequencing was mostly a random selection of those with available DNA but also included some related individuals for family studies. After the exclusion of participants with coronary heart disease, ischemic stroke or with missing information on age at blood draw or AD diagnosis, a total of 2,437 persons remained for this analysis. Adjudication for Alzheimer’s phenotypes was done by a committee41, comprising at least two neurologists and a neuropsychologist. Multiple types of information were used to evaluate participants with suspected dementia, including neurologic and neuropsychological assessments, a telephone interview with a family member or caregiver, medical records, imaging studies and autopsy data when available. AD was diagnosed when participants met the criteria of the NINCDS and the ADRDA for definite, probable or possible AD.
CHS
The CHS is a prospective, multi-ethnic, longitudinal study of risk factors for coronary heart disease and stroke in people aged 65 and older. A total of 2,840 samples were sequenced as part of the TOPMed project as previously described9. The samples selected for WGS as part of TOPMed were heavily oversampled for cardiovascular disease cases. After exclusion of participants with coronary heart disease or stroke, or with missing information on age at blood draw or AD diagnosis, a total of 743 persons remained for this analysis. AD was diagnosed as probable and possible following the NINCDS–ADRDA criteria in 1997–98 and 2002–03 (ref. 42). All CHS participants provided informed consent, and the study was approved by the Institutional Review Board of the University of Washington.
Each study in TOPMed received institutional certification before deposition in dbGaP, which certified that all relevant institutional ethics committees approved the individual studies and that the genomic and phenotypic data submission was compliant with all relevant ethical regulations. Secondary analysis of the dbGaP data in this manuscript was approved by the Stanford University Institutional Review Board, and this work is compliant with all relevant ethical regulations.
ADSP
The ADSP is a collaborative effort of the National Institutes of Aging, the NHGRI and the Alzheimer’s community to understand the genetic basis of AD16. The whole exome sequencing (WES) set of ADSP was a case-control design where cases met NINCDS–ADRDA criteria for possible, probable or definite AD had documented age at onset or age at death and APOE genotyping. A case-control selection strategy was chosen that targeted cases with minimal risk as predicted by known risk factors (age, sex and APOE) and targeted controls with the least probability of conversion to AD by age 85 years. A total of 5,096 cases and 4,965 controls from 24 cohorts were chosen for WES. This selection strategy was chosen to maximize the power to detect germline variant associations. Written informed consent was obtained from all human participants in each of the studies that contributed to ADSP in our analysis, which are listed below along with the name of the institutional review board that approved the study. Secondary analysis of the dbGaP data in this manuscript was approved by the Partners Healthcare and Stanford University Institutional Review Boards, and this work is compliant with all relevant ethical regulations.
ACT—University of Washington IRB
National Institute on Aging ADC—39 centers contributed to this data and IRBs at each institution approved the study
CHAP—Rush University Medical Center IRB
EFIGA—Columbia University IRB
NIA-LOAD—Columbia University IRB
MAP—Rush University Medical Center IRB
NCRAD—Indiana University IRB
ROS—Rush University Medical Center IRB
TARC—Baylor College of Medicine, Texas Tech University Health Sciences Center, University of North Texas Health Science Center, The University of Texas Health Sciences Center at San Antonio, The University of Texas Southwestern Medical Center and Texas A & M Health Science Center IRBs
Variant calling and annotation
WGS of TOPMed samples15 and WES of ADSP samples15 were previously performed. FASTQ files were aligned to reference genome hg38 for TOPMed WGS and hg19 for ADSP WES and the resulting BAM files were passed through Mutect/Indelocator (ADSP) or Mutect2 (TOPMed) pipelines to identify putative variants15. The Mutect/Mutect2 pipelines were run at default settings in Tumor-Normal mode, with one person from each cohort known not to have CHIP used as the ‘normal’ sample. These algorithms excluded variants that had characteristics of common artifacts, such as oxoguanine artifact, end-of-read artifact and PCR artifact (strand bias). Common polymorphisms present in germline databases were also excluded. Rare error modes were excluded by using a Panel of Normals compiled from 1,000 persons without CHIP in the same sequencing centers. Output from the Mutect/Mutect2 pipelines were then annotated for known CHIP variants in 73 genes from a curated whitelist (Supplementary Table 1).
Statistical analysis plan
TOPMed
We wished to test for an association of AD dementia to CHIP. We hypothesized that CHIP carriers would have an increased risk of AD dementia based on prior data that CHIP carriers have more inflammation in innate immune cells11 and that enhanced inflammasome activation was associated with worsened AD phenotypes in mice43.
For the discovery set, we used the two cohorts in TOPMed with available data on incident AD diagnoses, FHS and CHS. The CHS sample was heavily oversampled for those with cardiovascular diseases, especially coronary heart disease (CHD) and stroke (1,838 of 2,840 participants had these conditions). CHIP is known to be associated with atherosclerotic cardiovascular disease11. As systemic atherosclerosis is a risk factor for vascular dementia, which can mimic AD dementia symptoms, we wished to exclude anyone with these conditions to prevent confounding. To do this, we excluded anyone with an event type of myocardial infarction (MI), stroke, angioplasty, coronary artery bypass surgery, silent MI or death due to coronary heart disease using CHS event codes 1, 3, 7, 8, 10 and 11 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/variable.cgi?study_id=phs000287.v2.p1&phv=100824&phd=2793&pha=3548&pht=1466&phvf=&phdf=&phaf=&phtf=&dssp=1&consent=&temp=1) and excluded those selected on the basis of CHD, stroke or ‘other’ sampling group codes 3, 4, 5 and 6 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/variable.cgi?study_id=phs001368.v2.p2&phv=377683&phd=8024&pha=&pht=7957&phvf=&phdf=&phaf=&phtf=&dssp=1&consent=&temp=1). For FHS, we also excluded anyone with codes for coronary heart disease or ischemic stroke. For both cohorts, we also excluded anyone with prior dementia. The final study sample for CHS was 743 people, 491 of these were female. The median age was 72 years at the time of blood draw for WGS. There were 123 people with ε2ε2 or ε2ε3 genotype, 26 people with ε2ε4 genotype, 424 people with ε3ε4 genotype, 162 people with ε3ε4 genotype, and 8 people with ε4ε4 genotype at APOE. The final study sample for FHS was 2,437 people, 1,385 of these were female. The median age was 61 years at the time of blood draw for WGS. There were 310 people with ε2ε2 or ε2ε3 genotype, 46 people with ε2ε4 genotype, 1,584 people with ε3ε3 genotype, 458 people with ε3ε4 genotype and 39 people with ε4ε4 genotype at APOE.
FHS and CHS are both prospective studies with information on incident AD diagnosis. We, therefore, used time-to-event regression models to test for an association of CHIP to incident AD dementia in both cohorts. To exclude confounding due to survivorship bias, we performed the analysis using CRR, with death as the competing risk. After excluding those without information on AD diagnosis, there were 2,437 persons in FHS and 620 persons in CHS. Other variables included in these models were age at blood draw used for sequencing, APOE genotype, and sex for both cohorts and in addition, self-reported race and study site for CHS. For FHS, some participants were selected as part of family studies, which could potentially lead to biased estimates in the regression models due to correlated genetic or environmental factors. To control for this possibility, we also included family as a cluster variable in the CRR model for FHS using the R package crrSC (https://cran.r-project.org/web/packages/crrSC/index.html). The results from both cohorts were then meta-analyzed using a fixed-effects meta-analysis. The R packages meta (https://cran.r-project.org/web/packages/meta/index.html) and cmprsk (https://cran.r-project.org/web/packages/cmprsk/index.html) were used to perform the meta-analysis and CRR, respectively. For both risk of AD and death, visual examination of a plot of the Schoenfeld residuals revealed that the proportional hazards assumption was met for each covariate and visual examination of a plot of the Martingale residuals for age revealed that the linearity assumption was met.
ADSP
Having demonstrated a surprising inverse association between CHIP and AD dementia in the discovery set, we wished to replicate the finding. For this, we used the ADSP data. As described above, the selection strategy for ADSP was chosen to maximize the power to detect germline variant associations. As CHIP is strongly associated with age, and to a lesser extent sex, this study design can lead to selection biases that confound a CHIP analysis. These biases resulted in imbalanced distributions for cases and controls based on these variables. This is illustrated by the distribution of AD cases and controls amongst APOE ε4 carriers, where AD cases were strongly enriched for younger females and controls were preferentially older males. This age and sex imbalance did not occur for APOE ε3/ε3 carriers, presumably because the overall neutral effect of ε3/ε3 genotype led to relaxed selection based on age and sex. Furthermore, 1,776 samples were sequenced from the brain, not blood, DNA. We limited our CHIP/AD association analysis to those samples where DNA was derived from blood and where the age at blood draw used for sequencing was known. In most cases, the AD diagnosis was made before the blood draw; however, the diagnosis was usually within 5 years of the time of blood sampling for both prevalent and incident cases. To assess whether there was an association between CHIP and APOE genotype, which could confound our results, we determined the prevalence of APOE genotypes stratified by CHIP status in TOPMed. Among 4,276 people without CHIP in TOPMed, 2,688 (62.9%) were APOE ε3ε3, 600 (14.0%) were ε2ε2 or ε2ε3 and 988 (23.1%) were ε2e4, ε3ε4 or ε4ε4. Among 485 people with CHIP in TOPMed, 321 (66.2%) were APOE e3e3, 59 (12.2%) were ε2ε2 or ε2ε3, and 105 (21.6%) were ε2ε4, ε3ε4 or ε4ε4. Thus, CHIP is not associated with APOE genotype.
A second major difference between ADSP and TOPMed is the use of higher-depth WES in ADSP, compared to lower-depth WGS in TOPMed. To perform a power calculation for the replication study in ADSP, we had to ensure the VAF was comparable between ADSP and TOPMed for two reasons. First, the sensitivity to detect CHIP is linked to the sequencing depth, therefore the prevalence of CHIP was higher in ADSP. Second, the associations for previously studied health outcomes related to CHIP are dependent on clone size, with small clones having less of an effect size. We empirically determined that a cutoff of VAF at 0.08 gave a nearly identical VAF distribution for CHIP clones in ADSP as compared to TOPMed (Extended Data Fig. 1).
After excluding those without blood DNA or known age at blood draw and further limiting to APOE ε3ε3 carriers, we had 1,104 AD cases and 1,446 controls who were well matched by age. The median age was 81 years at the time of blood draw for WES and there were 1,458 females. We then used the powerMediation (https://cran.r-project.org/web/packages/powerMediation/index.html) package in R to perform a power calculation for varying effect sizes of CHIP at an alpha of 0.1. For an OR of 0.6 (similar to the hazard ratio for CHIP obtained from TOPMed), the power was 1. For an OR of 0.8, the power was 0.96. For an OR of 0.9, the power was 0.50. Thus, we were well-powered for the replication analysis in ADSP. We used logistic regression to assess the association between CHIP and AD in ADSP, with age at blood draw and sex as other explanatory variables in the model. For privacy concerns, those age 90 or older did not have an exact age available on dbGaP, and were considered to be age 90 for the purposes of this analysis.
We further assessed whether smaller clones were associated with AD dementia in two ways. First, we performed a logistic regression for AD where CHIP status was modeled as a three-factor variable (no CHIP, CHIP with VAF ≤ 0.08 or CHIP with VAF > 0.08). Second, we performed a logistic regression for AD dementia where VAF was included as a continuous variable using only CHIP carriers.
A fixed-effect meta-analysis for the risk of AD in CHS, FHS and ADSP was performed using logistic regression models for each cohort with age at blood draw, sex, APOE genotype and CHIP carrier status as covariates.
We wished to test whether CHIP status was associated with AD-related pathologic changes in people without clinical dementia symptoms. For a subset of participants in ADSP who died and donated their brains for research, a neuritic plaque score based on the CERAD criteria and Braak stage was assessed. For this analysis, we used all APOE genotypes and limited the analysis to those with available age at autopsy. Information on CERAD score was obtained from the NACC/ADRC and the ACT cohort. Information on Braak stage was available from NACC/ADRC, ACT, FHS and the GD cohorts. For all cohorts, anyone with a clinical dementia diagnosis was excluded. For NACC/ADRC and ACT, we also excluded anyone with mild cognitive impairment. A total of 427 persons had CERAD neuritic plaque scores and 454 persons had Braak stages available for this analysis after these exclusions. We performed ordinal logistic regression for CERAD score (0–3) and Braak stage (grouped as 0/I/II (1), III/IV (2) and V/VI (3)) using the PoLR function in the MASS package in R (https://cran.r-project.org/web/packages/MASS/index.html). Explanatory variables included CHIP, age at death, sex and APOE genotype. The t values from the ordinal logistic model were used to calculate P values for each of the covariates using a standard normal distribution. For the analysis of CHIP and ADNC stratified by APOE genotype, we created a composite score of CERAD amyloid burden and Braak tau spread by adding the two scores together, yielding a total score that ranged from 1 to 6. The composite score was then analyzed using ordinal logistic regression adjusted for age at death, sex and CHIP status.
Sex-specific analyses
We assessed whether there were sex-specific effects for CHIP by performing analyses for males and females separately in CHS, FHS and ADSP. Sex was self-reported in these cohorts. We used logistic regression models with AD as the outcome variable and age at blood draw, CHIP status, APOE genotype (for CHS and FHS), race (for CHS) and study site (for CHS) as explanatory variables. Family was used as a cluster variable in FHS. Results were then meta-analyzed using fixed-effects models.
Mendelian randomization
To perform two-sample MR, we used the 27 independent genome-wide significant hits from ref. 44 as the instrumental variables for CHIP exposure (Supplementary Table 7) and summary statistics from several AD association studies for the outcome. Of the 27 CHIP GWAS loci, 24 were available in the AD GWAS summary statistics from ref. 18, which provided summary statistics from a large AD GWAS/GWAX meta-analysis. The AD GWAS included ~22,000 AD cases and ~42,000 controls from ref. 45, while the GWAX contained ~53,000 AD-by-proxy cases and ~378,000 controls from UK Biobank. We further obtained summary statistics from an AD GWAS from Finngen release 5 (case definition: Dementia in AD F5) comprising 2,191 cases and 209,487 controls and for an AD GWAS from Gr@ACE46 comprising 4,210 AD cases and 3,289 controls. The β values and standard errors for the 24 variants from the CHIP and AD GWAS summary statistics were then used to perform weighted median MR, inverse-variance weighted MR using a multiplicative random effects model, mode-based estimation MR and MR-Egger MR using the MendelianRandomization package (https://cran.r-project.org/web/packages/MendelianRandomization/index.html). We also performed analyses using MR-PRESSO (https://github.com/rondolab/MR-PRESSO) and MR-RAPS (https://github.com/qingyuanzhao/mr.raps). Weighted median MR was chosen as the primary analysis due to its ability to provide robust estimates under several scenarios; for example, the estimate would be reliable even if up to 50% of the instrumental variable weight was invalid due to horizontal pleiotropy47 (https://wellcomeopenresearch.org/articles/4-186/v2). The results from each GWAS or GWAX were meta-analyzed using fixed-effects meta-analysis in the meta package in R.
The assumptions for the validity of MR analyses are relevance, independence and exclusion restriction of the independent variables (IVs)48. We explain why the particular IVs chosen for our study satisfy these assumptions below.
Relevance: We selected as our instrumental variables 24 independent variants recently identified as associated with CHIP in a large GWAS at P < 5 × 10−8 (Supplementary Table 7). Given the genome-wide significance threshold for inclusion, we consider these to be strong instruments. We estimated the narrow-sense heritability (h2) for CHIP to be 9.3% using the sum of the scaled marginal effects of these 24 variants. CHIP has moderate heritability relative to other traits (https://nealelab.github.io/UKBB_ldsc/index.html).
Independence: This assumption presupposes that the CHIP IVs are not associated with unmeasured confounders. While this cannot be assessed directly, the detection of associations between IVs and other measured covariates might suggest violations of this assumption. Well-established risk factors for AD that were included as covariates in our observational study were female sex and APOE genotype. Clearly, our selected germline IVs are independent of sex and other unlinked genetic variants. See also below for an analysis of the association between the genetic risk of CHIP and hypertension, another potential confounder that could violate the independence assumption. Finally, we obtained similar effect estimates in all of the cohorts we examined, arguing against cohort-specific confounding.
Exclusion restriction: A final assumption is that the CHIP IVs influence AD risk only through CHIP and not in other ways (no horizontal pleiotropy). Like the independence assumption, this is difficult to test directly. To address this, we used several different approaches. First, we used MR methods that are robust to pleiotropy including weighted median (primary analysis), weighted mode, MR-RAPS and MR-PRESSO (Supplementary Table 8). All methods provided similar effect estimates. We further used the MR-PRESSO global test49 to assess for horizontal pleiotropy and found no significant P values in the four studies used in the MR meta-analysis (UKB GWAX P = 0.07, UKB GWAS P = 0.20, FinnGen P = 0.84 and GR@ACE P = 0.91, all P values unadjusted for multiple comparisons). Second, we assessed whether any of the proximal genes to CHIP IVs had plausible connections to AD via other mechanisms. None of these genes have an established biological link to AD, but some are thought to be relevant to cancers generally, including solid tumors (TERT, PARP1, ATM, TP53, BCL2L1 and SETBP1). This is relevant because previous observational studies have found inverse associations between cancer and AD, but the mechanism for this is unknown50. It is unlikely that tumors of solid organs could directly influence the biology of AD. We speculate that the association between cancer and AD could instead be due to carriers of solid tumors having enrichment of genetic variants that also increase the risk of CHIP, which is an area for future investigation. Along these lines, a recent paper found that the genetic liability of CHIP was associated with an increased risk of lung, prostate, ovarian, oral and endometrial cancers7. Finally, we noticed that there were multiple shared loci in the CHIP GWAS and a recent GWAS for hypertension51. As mid-life hypertension has been reported to be associated with an increased risk of AD in observational studies52, we wondered whether the association between CHIP and AD was mediated by hypertension. We, therefore, performed MR using CHIP as the exposure and systolic blood pressure as the outcome. We found that there was a significant effect in some methods, but not others (IVW, effect = 0.144, P = 0.62; weighted median, effect = 0.666, P = 6.3 × 10−5; MR-PRESSO, effect = 0.144, P = 0.63; MR-RAPS, effect = 0.173, P = 0.014). However, the directionality of effect in all models indicated that increased genetic risk of CHIP was associated with higher blood pressure. Given that higher blood pressure is a risk factor for AD, this does not explain the inverse association between CHIP and AD seen in our MR analysis and observational study.
To assess the reverse association, we used 36 genome-wide significant variants for AD risk from Schwartzentruber et al.18 as the instrumental variables and the summary statistics from the CHIP GWAS from ref. 9 in two-sample weighted median MR analysis.
Nuclei isolation from human postmortem brain tissue
ACT is a longitudinal, community-based observational study of brain aging in participants older than 65 randomly sampled from the Group Health Cooperative (now Kaiser Permanente Washington), a health management organization in King County, Washington. A subset of participants in the study donate their brains for research upon death, and a comprehensive neuropathological exam is performed to assess for AD and related neurodegenerative disease pathologies53. For decedents with postmortem intervals of less than 8 h, a rapid autopsy is performed in which numerous samples from multiple brain regions are taken from one hemisphere and flash-frozen in liquid nitrogen. Consent for brain donation was obtained from each donor and the study was approved by the University of Washington Institutional Review Board and by the Kaiser Permanente Washington Institutional Review Board.
For this analysis, we obtained occipital cortex samples from 12 ACT brain donors (eight CHIP carriers and four noncarriers). Three of these also had a frozen sample from the cerebellum available, and two had a frozen sample from the putamen. The eight CHIP carriers represented all donors known to have CHIP and with autopsy specimens available. The four without CHIP represented a random selection of the study cohort. Of the 12 donors, two had AD dementia, seven were female, ten were APOE ε3ε3, two were APOE ε3ε4 and the median age at autopsy was 90.5 years. For nuclei isolation, we performed and adapted the protocol from ref. 54. Briefly, around 250 mg of frozen postmortem brain tissue was thawed in 5 ml lysis buffer and transferred to a douncer placed on ice. After 20–30 strokes, the homogenized tissue was transferred to a clear 50 ml ultracentrifuge tube and the volume was adjusted to 12 ml. 21 ml of sucrose buffer was added to the bottom of the clear ultracentrifuge tube, to create a concentration gradient with the homogenized tissue solution on top of the sucrose buffer. The tubes were placed in buckets in a SW32Ti swinging rotor (Beckton Dickinson). The samples were ultracentrifuged at 107163g for 2.5 h at 4 °C. The supernatant was removed and 500 μl of 1X PBS was added to the pellet and incubated for 20 min on ice. The nuclei were then resuspended and transferred into a microcentrifuge tube. The nuclei were counted using trypan blue dilution and then centrifuged at 500g for 5 min. The lysis buffer comprised 0.32 M sucrose, 5 mM CaCl2, 3 mM Mg(acetate)2, 0.1 mM EDTA, 10 mM Tris–HCl pH8, 1 mM DTT, 0.1%Triton X-100 in H2O. The sucrose buffer comprised 1.8 M sucrose, 3 mM Mg(acetate)2, 1 mM DTT, 10 mM Tris–HCl, pH8 in H2O.
Immunostaining and sorting of the nuclei
The nuclei were resuspended at a concentration of 200,000 cells in 50 μl of 0.5% BSA in 1× PBS solution and stained for 45 min with Anti-NeuN Antibody Alexa Fluor 488(EMD Millipore) at a concentration of 1:400, and Anti-Maf antibody PE (BD biosciences) at a concentration of 1:50. The nuclei were then washed and strained using a 40 µm strainer. The sorting was done on an Aria II sorter using a 100 µm nozzle (data collected using FACSDIVA version 8.0.1 or earlier, BD Pharmingen). The nuclei were collected in 0.5% BSA in 1× PBS solution and centrifuged at 500g for 5 min. Flow cytometry analyses were performed using FlowJo vlO from BD Biosciences.
DNA extraction, amplification and sequencing
DNA was extracted from the nuclei using the Qiagen QIAmp DNA micro kit. DNA concentration was measured using the Qubit fluorometer. PCR was performed to amplify the region surrounding the mutation of interest (around 300 bp) using the Phusion high-fidelity master mix (New England Biolabs). The amplified DNA was purified using the Qiagen QIAquick PCR purification kit according to the manufacturer’s recommendations. Libraries were generated from the pooled amplicons using the Celero DNA-seq library kit (NuGEN). Sequencing of the libraries was performed using MiSeq Nano v2 kits. Sequencing reads were aligned with BWA (http://bio-bwa.sourceforge.net), and variant calling and annotation were done with Varscan (http://varscan.sourceforge.net) and Annovar (https://annovar.openbioinformatics.org/en/latest/).
To confirm the reproducibility of the VAF measurements, DNA from unsorted brain nuclei or sorted Maf+ NeuN− was used for library prep and sequencing in a second replicate. To rule out sequencing errors or other technical artifacts that could be artificially inflating the mutant allele fraction, we also performed amplicon sequencing using the same primers on a negative control brain sample from a donor known to not be a CHIP carrier. Primer sequences can be found in Supplementary Table 17. The VAFs of the unsorted brain and the sorted subsets were obtained from the same tissue prep for each sample, and the results can be found in Supplementary Table 14. For some samples, the same tissue prep was also used for snATAC-seq. However, in some cases, the tissue prep used for snATAC-seq was from a different piece of tissue from the same donor. In these cases, the VAFs were determined again using the same tissue prep as used for snATAC-seq, and the VAFs can be found in Supplementary Table 16.
Single-nucleus ATAC-sequencing
Sample processing
After nuclei isolation as described above, samples were transposed, single cells were barcoded using 10X Genomics GEMs (Gel Bead in-EMulsions) and libraries were prepared for sequencing according to the commercially available 10X Chromium Next GEM Single Cell ATAC Library & Gel Bead Kit v1.1. Paired-end sequencing was performed on an Illumina HiSeq 2500. We performed single-nucleus ATAC-sequencing (snATAC-seq) on occipital cortex samples from all six donors with detected CHIP mutations in the brain, as well as the four donors without CHIP. We also performed snATAC-seq on putamen and cerebellum samples from donor ACT6.
Reference datasets
To aid in the interpretation of cell types from our snATAC-seq data, we also included two previously published datasets24. For the Corces et al. brain dataset, original fastq files of all ten snATAC samples (available under GEO accession no. GSE147672) were aligned to the hg19 reference genome and then processed as described above55. For the Satpathy et al. hematopoiesis dataset, we downloaded fragments files for the seven samples most relevant to our study, focusing on dendritic cells and monocytes24. Accession numbers and sample names of these are as follows:
GSM3722015_PBMC_Rep1_fragments.tsv.gz
GSM3722076_PBMC_Rep2_fragments.tsv.gz
GSM3722075_PBMC_Rep3_fragments.tsv.gz
GSM3722077_PBMC_Rep4_fragments.tsv.gz
GSM3722039_Dendritic_all_cells_fragments.tsv.gz
GSM3722026_Dendritic_Cells_fragments-Reformat.tsv.gz
GSM3722027_Monocytes_fragments.tsv.gz
Analysis pipeline
Fastq files were trimmed, deduplicated, filtered and aligned using the 10X cellranger-atac count pipeline, yielding a file of high-quality ATAC-seq fragments for all cells per sample. Reference genome hg19 was used for compatibility with the hematopoiesis reference dataset (described in ‘Reference datasets’)25. The fragments file for each sample, including the reference samples, was then loaded into ArchR for downstream analysis55.
Nuclei quality control and clustering were performed using the standard ArchR pipeline. Briefly, barcodes were called nuclei based on fragments per barcode and enrichment of fragments in transcription start sites (TSS) genome-wide. Doublets were removed in two steps. First, for each sample, doublets were predicted and removed based on similarity to computationally simulated doublets. Second, after initial clustering, we identified a cluster of putative doublets that was weakly enriched for markers of multiple cell types. This cluster was removed and the remaining nuclei were reprocessed and reclustered. The TileMatrix and GeneScoreMatrix were computed using default settings. For the GeneScoreMatrix, imputation was performed using the ArchR implementation of MAGIC to aid visualization of the sparse ATAC-seq signals in single nuclei. Dimensionality reduction and clustering were performed using the TileMatrix, which tiles the genome into 500 bp windows. Although Harmony batch correction is implemented and part of the standard workflow in ArchR, we did not use any batch correction to ensure that any biological differences between the samples would be preserved. After clustering, reproducible peaks were determined for each cluster individually to ensure that cell type-specific peaks were retained. Reproducible peaks for each cluster were merged into a set of disjoint, fixed width (500 bp) peaks, which were used to create the cell by peak matrix. ATAC-seq pseudo-bulk tracks for selected groups of nuclei were exported from ArchR using the ‘getGroupBW’ function. All tracks were identically normalized using ReadsInTSS, which corrects for variation in sequencing depth and also nuclei quality between different groups of nuclei. Specific regions in the genome were visualized using the Integrative Genomics Viewer (https://software.broadinstitute.org/software/igv/).
To evaluate the fraction of mutated MG (as shown in Fig. 5d and Supplementary Table 16), we first determined the number of MG and nonmicroglial hematopoietic cells in each brain sample. We assumed that all nonmicroglial hematopoietic cells were mutant, therefore we subtracted out the expected contribution of mutant alleles from these nonmicroglial cells to calculate an adjusted VAF for each sample where mutant alleles could only be contributed by MG. We then divided the adjusted percent mutant cells in each sample by the percent MG in each sample to calculate the percent mutant MG. An example calculation using the ACT3 sample is provided below.
vaf = 0.035 # VAF of the DNMT3A mutation from ACT3 unsorted brain
non_mg = 64 # Number of monocyte+DC+T-cell+B-cell in the sample
mg = 801 # Number of MG in the sample
total = 11,762 # Total number of nuclei identified in snATAC-seq
# We assume that only hematopoietic cells can carry the CHIP variant.
# We calculate the total number of mutant MG by
# multiplying the total*VAF*2 (gives total mutant cell burden) and
# subtracting all the nonmicroglial hematopoietic cells (assuming they
# are all mutant), which in this case is calculated to be 759.
mut_mg = round(total*vaf*2-non_mg)
# We calculate the proportion of mutant MG by dividing the
# number of mutant MG calculated above by the total number of
# MG. The value is 94.8%. Without accounting for the non
# microglial hematopoietic cells, the value would be vaf*total*2/mg,
# or 103%.
prop_mut_mg = mut_mg/mg
To model the expected distribution of the percentages of mutant MG based on this calculation, we simulated a binomial model for both the distribution of VAF obtained from amplicon sequencing and percentage of MG in unsorted brain nuclei. For this simulation, we assumed 200,000 haploid genomes (representing 100,000 starting nuclei, which is the minimum number used in our studies) and 8,000 nuclei assessed by snATAC-seq (representing approximately the median value in our samples). We varied the VAF based on the expected percent MG ranging from 10% to 90% and varied the percent MG in each sample from 1% to 3%. We then obtained histograms and 95% confidence intervals for each set of input parameters. These simulations indicated that the confidence intervals for the estimate of mutant MG are not overly broad based on expected input parameters. We then estimated bootstrapped confidence intervals for percent mutant MG for each sample using a similar simulation based on input parameters for VAF, percent MG, and a number of cells unique to each sample.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41591-023-02397-2.
Supplementary information
Supplementary Appendix—TOPMed Consortium author list.
Supplementary Tables 1–17.
Acknowledgements
We thank M. Miller (National Institute of Aging), K. Faber (Indiana University), A. Kuzma (University of Pennsylvania), W. Lee (Kaiser Permanente) and K.-R. Johnson (Kaiser Permanente) for providing age at blood draw for ADSP participants, A. Schantz (University of Washington) for compiling the neuropathology data from ACT, W. Kukull (University of Washington) and J. Culhane (University of Washington) for compiling neuropathology data from NACC/ADRCs, B. Ebert (Dana Farber Cancer Institute) and R. Corces (Gladstone Institutes) for helpful discussions and T. Mack (Vanderbilt University) for TOPMed germline genotype information. Notably, we thank the studies and participants who provided biological samples and data. S.J. is supported by the Burroughs Wellcome Fund Career Award for Medical Scientists, Fondation Leducq (TNE-18CVD04), the Ludwig Center for Cancer Stem Cell Research at Stanford University, The Phil and Penny Knight Initiative for Brain Resilience at Stanford University and the National Institutes of Health (DP2HL157540). S.J. and H.B. are supported by the National Institutes of Health (R01HL148565). T.J.M. is supported by the National Institutes of Health (AG053959, AG077443). C.D.K. is supported by the Nancy and Buster Alvord Endowment. A.T.S. is supported by the National Institutes of Health K08CA230188 and the Burroughs Wellcome Fund Career Award for Medical Scientists. P.N. is supported by the National Institutes of Health (R01HL142711, R01HL148050, R01HL151283, R01HL148565, R01HL135242, R01HL151152, R01DK125782), Fondation Leducq (TNE-18CVD04) and MGH (Fireman Endowed Chair in Vascular Medicine). C.S is supported by the NIA (R00 AG066849). A.B. is supported by the Burroughs Wellcome Fund Career Award for Medical Scientists (DP5 OD029586). C.L.S is supported by the National Institutes of Health (AG059727), (UH2 NS100605). S.S. is supported by the National Institutes of Health (U01 AG052409). The following are study-specific acknowledgements:
TOPMed
WGS for the TOPMed program was supported by the National Heart, Lung and Blood Institute (NHLBI). Centralized read mapping and genotype calling, along with variant quality metrics and filtering, were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Phenotype harmonization, data management, sample-identity quality control and general study coordination were provided by the TOPMed Data Coordinating Center (R01HL-120393; U01HL-120393; contract HHSN268201800001I). Genome sequencing for ‘NHLBI TOPMed: WGS and Related Phenotypes in the FHS’ (phs000974.v1.p1) was performed at the Broad Institute Genomics Platform (3R01HL092577-06S1, 3U54HG003067-12S2). Genome sequencing for ‘NHLBI TOPMed: WGS Project: CHS’ (phs001369.v2.p2) was performed at the Broad Institute Genomics Platform (3R01HL092577-06S1, 3U54HG003067-12S2) and Baylor University (HHSM268201600033I, 3U54HG003273-12S2, HHSN268201500015C). We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed.
FHS
The FHS acknowledges the support of contracts NO1-HC-25195, HHSN268201500001I and 75N92019D00031 from the National Heart, Lung and Blood Institute and grant supplement R01 HL092577-06S1 for this research. The FHS was also supported by grants from the National Institutes of Aging (AG054076 and AG058589). We also acknowledge the dedication of the FHS study participants, without whom this research would not be possible. Dr. Vasan is supported in part by the Evans Medical Foundation and the Jay and Louis Coffman Endowment from the Department of Medicine, Boston University School of Medicine.
CHS
This research was supported by contracts HHSN268201200036C, HHSN268200800007C, HHSN268201800001C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086 and 75N92021D00006, and grants U01HL080295 and U01HL130114 from the NHLBI, with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided by R01AG023629 from the National Institute on Aging (NIA). A full list of principal CHS investigators and institutions can be found at CHS-NHLBI.org. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ADSP
The ADSP is comprised of two AD genetics consortia and three National Human Genome Research Institute (NHGRI) funded Large Scale Sequencing and Analysis Centers (LSAC). The two AD genetics consortia are the Alzheimer’s Disease Genetics Consortium (ADGC) funded by NIA (U01 AG032984), and the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) funded by NIA (R01 AG033193), the National Heart, Lung, and Blood Institute (NHLBI), other National Institute of Health (NIH) institutes and other foreign governmental and nongovernmental organizations. The Discovery Phase analysis of sequence data is supported through UF1AG047133 (to G. Schellenberg, L. Farrer, M. Pericak-Vance, R. Mayeux and J. Haines); U01AG049505 to S. Seshadri; U01AG049506 to E. Boerwinkle; U01AG049507 to E. Wijsman; and U01AG049508 to A. Goate and the Discovery Extension Phase analysis is supported through U01AG052411 to A. Goate, U01AG052410 to M. Pericak-Vance and U01 AG052409 to S. Seshadri and M. Fornage. Data generation and harmonization in the Follow-up Phases are supported by U54AG052427 (to G. Schellenberg and L. Wang).
The ADGC cohorts include the followings: Adult Changes in Thought (ACT) funded by NIA (U01AG006781 and U19AG066567), the Alzheimer’s Disease Research Centers (ADRCs) including the University of Washington ADRC funded by NIA (P50 AG005136 and P30 AG066509), the Chicago Health and Aging Project (CHAP), the Memory and Aging Project (MAP), Mayo Clinic (MAYO), Mayo Parkinson’s Disease controls, University of Miami, the Multi-Institutional Research in Alzheimer’s Genetic Epidemiology Study (MIRAGE), the National Cell Repository for Alzheimer’s Disease (NCRAD), the National Institute on Aging Late Onset Alzheimer’s Disease Family Study (NIA-LOAD), the Religious Orders Study (ROS), the Texas Alzheimer’s Research and Care Consortium (TARC), Vanderbilt University/Case Western Reserve University, the Washington Heights-Inwood Columbia Aging Project and the Washington University Sequencing Project, the Columbia University Hispanic-Estudio Familiar de Influencia Genetica de Alzheimer (EFIGA), the University of Toronto, and Genetic Differences (GD).
The CHARGE cohorts are supported in part by NHLBI infrastructure grant HL105756 (B. Psaty) and RC2HL102419 (E. Boerwinkle), and the neurology working group is supported by the National Institute on Aging (NIA) R01 grant AG033193 and RF1 AG059421. The CHARGE cohorts participating in the ADSP include the following: Austrian Stroke Prevention Study (ASPS), ASPS-Family study and the Prospective Dementia Registry-Austria (ASPS/PRODEM-Aus), the Atherosclerosis Risk in Communities (ARIC) Study, the CHS, the Erasmus Rucphen Family Study (ERF), the FHS and the Rotterdam Study (RS). ASPS is funded by the Austrian Science Fond (FWF), grant numbers P20545-P05 and P13180, and the Medical University of Graz. The ASPS-Fam is funded by the Austrian Science Fund (FWF) project I904), the EU Joint Programme—Neurodegenerative Disease Research (JPND) in frame of the BRIDGET project (Austria, Ministry of Science) and the Medical University of Graz and the Steiermärkische Krankenanstalten Gesellschaft. PRODEM-Austria is supported by the Austrian Research Promotion Agency (FFG; Project No. 827462) and by the Austrian National Bank (Anniversary Fund, project 15435). ARIC research is carried out as a collaborative study supported by NHLBI contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C and HHSN268201100012C). Neurocognitive data in ARIC is collected by U01 2U01HL096812, 2U01HL096814, 2U01HL096899, 2U01HL096902 and 2U01HL096917 from the NIH (NHLBI, NINDS, NIA and NIDCD), and with previous brain MRI examinations funded by R01-HL70825 from the NHLBI. CHS research was supported by contracts HHSN268201200036C, HHSN268200800007C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083 and N01HC85086 and grants U01HL080295 and U01HL130114 from the NHLBI with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided by R01AG023629, R01AG15928 and R01AG20098 from the NIA. FHS research is supported by NHLBI contracts N01-HC-25195, HHSN268201500001I and 75N9201900031. This study was also supported by additional grants from the NIA (P30 AG066546, R01s AG054076, AG049607 and AG033040 and NINDS (R01 NS017950)). The ERF study as a part of European Special Populations Research Network was supported by European Commission FP6 STRP grant number 018947 (LSHG-CT-2006-01947) and also received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013)/grant agreement HEALTH-F4-2007-201413 by the European Commission under the program ‘Quality of Life and Management of the Living Resources’ of 5th Framework Programme (no. QLG2-CT-2002-01254). High-throughput analysis of the ERF data was supported by a joint grant from the Netherlands Organization for Scientific Research and the Russian Foundation for Basic Research (NWO-RFBR 047.017.043). The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam, the Netherlands Organization for Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII), and the municipality of Rotterdam. Genetic datasets are also supported by the Netherlands Organization of Scientific Research NWO Investments (175.010.2005.011, 911-03-012), the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, the RIDE (014-93-015; RIDE2) and the Netherlands Genomics Initiative/Netherlands Organization for Scientific Research Netherlands Consortium for Healthy Aging, project 050-060-810. All studies are grateful to their participants, faculty and staff. The content of these manuscripts is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the U.S. Department of Health and Human Services.
The four LSACs are as follows: the Human Genome Sequencing Center at the Baylor College of Medicine (U54 HG003273), the Broad Institute Genome Center (U54HG003067), The American Genome Center at the Uniformed Services University of the Health Sciences (U01AG057659) and the Washington University Genome Institute (U54HG003079). Biological samples and associated phenotypic data used in primary data analyses were stored at Study Investigators institutions, and at the NCRAD (U24AG021886) at Indiana University funded by NIA. Associated Phenotypic Data used in primary and secondary data analyses were provided by Study Investigators, the NIA-funded Alzheimer’s Disease Centers (ADCs), and the National Alzheimer’s Coordinating Center (NACC, U01AG016976) and the National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site (U24AG041689) at the University of Pennsylvania, funded by NIA, and at the Database for Genotypes and Phenotypes (dbGaP) funded by NIH. This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Library of Medicine. Contributors to the Genetic Analysis Data included Study Investigators on projects that were individually funded by NIA, and other NIH institutes, and by private U.S. organizations, or foreign governmental or nongovernmental organizations.
NACC
The NACC database is funded by NIA/NIH Grant U24 AG072122. NACC data are contributed by the NIA-funded ADCs—P30 AG019610 (PI E. Reiman), P30 AG013846 (PI N. Kowall), P50 AG008702 (PI S. Small), P50 AG025688 (PI A. Levey), P50 AG047266 (PI T. Golde), P30 AG010133 (PI A. Saykin.), P50 AG005146 (PI M. Albert), P50 AG005134 (PI B. Hyman), P50 AG016574 (PI R. Petersen), P50 AG005138 (PI M. Sano), P30 AG008051 (PI T. Wisniewski), P30 AG013854 (PI R. Vassar), P30 AG008017 (PI J. Kaye), P30 AG010161 (PI D. Bennett), P50 AG047366 (PI V. Henderson), P30 AG010129 (PI C. DeCarli), P50 AG016573 (PI F. LaFerla), P50 AG005131 (PI J. Brewer), P50 AG023501 (PI B. Miller), P30 AG035982 (PI R. Swerdlow), P30 AG028383 (PI L. Van Eldik), P30 AG053760 (PI H. Paulson), P30 AG010124 (PI J. Trojanowski), P50 AG005133 (PI O. Lopez), P50 AG005142 (PI H. Chui), P30 AG012300 (PI R. Rosenberg), P30 AG049638 (PI S. Craft), P50 AG005136 (PI T. Grabowski), P50 AG033514 (PI S. Asthana), P50 AG005681 (PI J. Morris), P50 AG047270 (PI S. Strittmatter).
FinnGen
We want to acknowledge the participants and investigators of FinnGen study.
Extended data
Author contributions
H.B. conceptualized experimental design and performed all research investigations on brain tissue. J.A.B. performed single-cell ATAC-seq experiments and analysis with assistance from Y.Q. and W.Y. M.J. performed analyses and data curation of ADSP data. S.W., L.M., M.C., H.A. and D.N. provided genotyping and variant calling for CHIP from human samples. A.B., C.S., J.C.B., M.F., W.T.L., O.L.L., B.M.P., C.L.S. and S.S. contributed to neurocognitive phenotype curation in CHS and FHS and provided guidance on epidemiological study design. A.G.B., P.N. and J.W. performed CHIP calling in TOPMed. E.B.L., P.K.C. and C.D.K. provided brain tissues and neuropathology data from ACT. A.T.S. provided analysis, resources and supervision for single-cell ATAC-seq. T.J.M. provided supervision, conceptualization and access to resources for studies involving brain autopsy tissue. S.J. conceived the research plan and supervision for all analyses. H.B., J.A.B. and S.J. wrote the original draft with review from all authors.
Peer review
Peer review information
Nature Medicine thanks Ricardo D’Oliveira Albanus, Ross Levine and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling editor: Anna Maria Ranzoni, in collaboration with the Nature Medicine team.
Data availability
Individual whole-genome sequencing data and individual-level harmonized phenotypes from TOPMed are available through dbGaP for investigators who follow dbGaP procedures for requesting controlled access data, as detailed at https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/about.html#request-controlled. All whole exome sequencing data and phenotype data from ADSP are available on dbGaP for investigators who apply for access through https://www.niagads.org.
dbGaP accession numbers:
https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000974.v1.p1
https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001368.v2.p2
https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000572.v8.p4
Single-nucleus ATAC-seq data from human brain samples are available in Gene Expression Omnibus under accession GSE192838.
Code availability
Code and additional metadata tables for the snATAC-seq data are available at https://github.com/juliabelk/CHIP_and_AD (10.5281/zenodo.7809346).
Competing interests
A.G.B is a scientific cofounder and is on the scientific advisory board of TenSixteen Bio. A.T.S. is a founder of Immunai and Cartography Biosciences and receives research funding from 10X Genomics, Arsenal Biosciences and Allogene Therapeutics. B.M.P. serves on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson. M.J. reports consulting fees from RA Ventures. P.N. reports grants from Allelica, Amgen, Apple, Boston Scientific, Genentech and Novartis, is a consultant to Allelica, Apple, AstraZeneca, Blackstone Life Sciences, Foresite Labs, HeartFlow, Novartis, Genentech and GV, scientific advisory board membership to Esperion Therapeutics, Preciseli and TenSixteen Bio, is a scientific cofounder of TenSixteen Bio, and spousal employment at Vertex Pharmaceuticals, all unrelated to the present work. S.J. is a consultant to Novartis, Roche Genentech, AVRO Bio and Foresite Labs, reports speaking fees from GSK, and is on the scientific advisory board for Bitterroot Bio. S.J., P.N. and A.G.B. are founders, equity holders and on the scientific advisory board of TenSixteen Bio, unrelated to the present work. The remaining authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Hind Bouzid, Julia A. Belk.
A list of authors and their affiliations appears at the end of the paper.
A full list of members and their affiliations appears in the Supplementary Information.
Contributor Information
Siddhartha Jaiswal, Email: sjaiswal@stanford.edu.
NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium:
Extended data
is available for this paper at 10.1038/s41591-023-02397-2.
Supplementary information
The online version contains supplementary material available at 10.1038/s41591-023-02397-2.
References
- 1.Jaiswal S, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014;371:2488–98. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Genovese G, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014;371:2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie M, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014;20:1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKerrell T, et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 2015;10:1239–1245. doi: 10.1016/j.celrep.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jaiswal S, Elbert BL. Clonal hematopoiesis in human aging and disease. Science. 2019;366:eaan4673. doi: 10.1126/science.aan4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Desai P, et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat. Med. 2018;24:1015–1023. doi: 10.1038/s41591-018-0081-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kar, S. P. et al. Genome-wide analyses of 200,453 individuals yield new insights into the causes and consequences of clonal hematopoiesis. Nat. Genet. 10.1038/s41588-022-01121-z (2022). [DOI] [PMC free article] [PubMed]
- 8.Jaiswal S, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 2017;377:111–121. doi: 10.1056/NEJMoa1701719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bick AG, et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature. 2020;586:763–768. doi: 10.1038/s41586-020-2819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nachun D, et al. Clonal hematopoiesis associated with epigenetic aging and clinical outcomes. Aging Cell. 2021;20:e13366. doi: 10.1111/acel.13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fuster JJ, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–847. doi: 10.1126/science.aag1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sano S, et al. CRISPR-mediated gene editing to assess the roles of Tet2 and Dnmt3a in clonal hematopoiesis and cardiovascular disease. Circ. Res. 2018;123:335–341. doi: 10.1161/CIRCRESAHA.118.313225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nott A, et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science. 2019;366:1134–1139. doi: 10.1126/science.aay0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Q, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature. 2015;525:389–393. doi: 10.1038/nature15252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taliun, D. et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature10.1038/s41586-021-03205-y (2021). [DOI] [PMC free article] [PubMed]
- 16.Beecham GW, et al. The Alzheimer’s Disease Sequencing Project: study design and sample selection. Neurol. Genet. 2017;3:e194. doi: 10.1212/NXG.0000000000000194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abelson S, et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature. 2018;559:400–404. doi: 10.1038/s41586-018-0317-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwartzentruber J, et al. Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer’s disease risk genes. Nat. Genet. 2021;53:392–402. doi: 10.1038/s41588-020-00776-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hyman BT, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol. 2019;15:501–518. doi: 10.1038/s41582-019-0228-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keogh MJ, et al. High prevalence of focal and multi-focal somatic genetic variants in the human brain. Nat. Commun. 2018;9:4257. doi: 10.1038/s41467-018-06331-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014;14:392–404. doi: 10.1038/nri3671. [DOI] [PubMed] [Google Scholar]
- 24.Corces MR, et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet. 2020;52:1158–1168. doi: 10.1038/s41588-020-00721-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Satpathy AT, et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 2019;3:925–936. doi: 10.1038/s41587-019-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serrano-Pozo A, Das S, Hyman BT. APOE and Alzheimer’s disease: advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021;20:68–80. doi: 10.1016/S1474-4422(20)30412-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shull LR, et al. Biological and induction effects of phenobarbital and 3-methylcholanthrene in mink (Mustela vison) Drug Metab. Dispos. 1983;11:441–445. [PubMed] [Google Scholar]
- 28.Nam AS, et al. Somatic mutations and cell identity linked by genotyping of transcriptomes. Nature. 2019;571:355–360. doi: 10.1038/s41586-019-1367-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nam AS, et al. Single-cell multi-omics of human clonal hematopoiesis reveals that DNMT3A R882 mutations perturb early progenitor states through selective hypomethylation. Nat. Genet. 2022;54:1514–1526. doi: 10.1038/s41588-022-01179-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller TE, et al. Mitochondrial variant enrichment from high-throughput single-cell RNA sequencing resolves clonal populations. Nat. Biotechnol. 2022;40:1030–1034. doi: 10.1038/s41587-022-01210-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cugurra A, et al. Skull and vertebral bone marrow are myeloid cell reservoirs for the meninges and CNS parenchyma. Science. 2021;373:eabf7844. doi: 10.1126/science.abf7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herisson F, et al. Direct vascular channels connect skull bone marrow and the brain surface enabling myeloid cell migration. Nat. Neurosci. 2018;21:1209–1217. doi: 10.1038/s41593-018-0213-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 34.El Khoury J, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 35.Lund H, et al. Competitive repopulation of an empty microglial niche yields functionally distinct subsets of microglia-like cells. Nat. Commun. 2018;9:4845. doi: 10.1038/s41467-018-07295-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Efthymiou AG, Goate AM. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 2017;12:43. doi: 10.1186/s13024-017-0184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinstock JS, et al. Aberrant activation of TCL1A promotes stem cell expansion in clonal haematopoiesis. Nature. 2023;616:755–763. doi: 10.1038/s41586-023-05806-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dawber TR, Kannel WB. The Framingham Study: an epidemiological approach to coronary heart disease. Circulation. 1966;34:553–555. doi: 10.1161/01.CIR.34.4.553. [DOI] [PubMed] [Google Scholar]
- 39.Feinleib M, Kannel WB, Garrison RJ, McNamara PM, Castelli WP. The Framingham Offspring Study. Design and preliminary data. Prev. Med. 1975;4:518–525. doi: 10.1016/0091-7435(75)90037-7. [DOI] [PubMed] [Google Scholar]
- 40.Splansky GL, et al. The Third Generation Cohort of the National Heart, Lung, and Blood Institute’s Framingham Heart Study: design, recruitment, and initial examination. Am. J. Epidemiol. 2007;165:1328–1335. doi: 10.1093/aje/kwm021. [DOI] [PubMed] [Google Scholar]
- 41.Seshadri S, et al. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N. Engl. J. Med. 2002;346:476–483. doi: 10.1056/NEJMoa011613. [DOI] [PubMed] [Google Scholar]
- 42.Lopez OL, et al. Evaluation of dementia in the cardiovascular health cognition study. Neuroepidemiology. 2003;22:1–12. doi: 10.1159/000067110. [DOI] [PubMed] [Google Scholar]
- 43.Heneka MT, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kessler, M. D. et al. Common and rare variant associations with clonal haematopoiesis phenotypes Nature10.1038/s41586-022-05448-9 (2022). [DOI] [PMC free article] [PubMed]
- 45.Kunkle BW, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019;51:414–430. doi: 10.1038/s41588-019-0358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moreno-Grau S, et al. Genome-wide association analysis of dementia and its clinical endophenotypes reveal novel loci associated with Alzheimer’s disease and three causality networks: the GR@ACE project. Alzheimers Dement. J. Alzheimers Assoc. 2019;15:1333–1347. doi: 10.1016/j.jalz.2019.06.4950. [DOI] [PubMed] [Google Scholar]
- 47.Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 2016;40:304–314. doi: 10.1002/gepi.21965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Skrivankova VW, et al. Strengthening the reporting of observational studies in epidemiology using Mendelian randomization: the STROBE-MR statement. JAMA. 2021;326:1614–1621. doi: 10.1001/jama.2021.18236. [DOI] [PubMed] [Google Scholar]
- 49.Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018;50:693–698. doi: 10.1038/s41588-018-0099-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ospina-Romero M, et al. Association between Alzheimer disease and cancer with evaluation of study biases: a systematic review and meta-analysis. JAMA Netw. Open. 2020;3:e2025515. doi: 10.1001/jamanetworkopen.2020.25515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Evangelou E, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet. 2018;50:1412–1425. doi: 10.1038/s41588-018-0205-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011;7:137–152. doi: 10.1038/nrneurol.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kukull WA, et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch. Neurol. 2002;59:1737–1746. doi: 10.1001/archneur.59.11.1737. [DOI] [PubMed] [Google Scholar]
- 54.Matevossian A, Akbarian S. Neuronal nuclei isolation from human postmortem brain tissue. J. Vis. Exp. 2008;20:914. doi: 10.3791/914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Granja JM, et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 2021;53:403–411. doi: 10.1038/s41588-021-00790-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Appendix—TOPMed Consortium author list.
Supplementary Tables 1–17.
Data Availability Statement
Individual whole-genome sequencing data and individual-level harmonized phenotypes from TOPMed are available through dbGaP for investigators who follow dbGaP procedures for requesting controlled access data, as detailed at https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/about.html#request-controlled. All whole exome sequencing data and phenotype data from ADSP are available on dbGaP for investigators who apply for access through https://www.niagads.org.
dbGaP accession numbers:
https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000974.v1.p1
https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001368.v2.p2
https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000572.v8.p4
Single-nucleus ATAC-seq data from human brain samples are available in Gene Expression Omnibus under accession GSE192838.
Code and additional metadata tables for the snATAC-seq data are available at https://github.com/juliabelk/CHIP_and_AD (10.5281/zenodo.7809346).