

List of abbreviations
- AML
acute myeloid leukemia
- CDK
cyclin dependent kinase
- CHD
clonal hematopoietic disorder
- CNA
copy number alteration
- EDTA
ethylenediamine tetraacetic acid
- G0
Gap 0
- GSI
γ‐secretase inhibitor
- HSC
hematopoietic stem cell
- IHC
immunohistochemistry
- KO
knock‐out
- LPE
low protein expression
- MCH
mean corpuscular hemoglobin
- MCV
mean corpuscular volume
- MDS
myelodysplastic syndromes
- MEF
mouse embryo fibroblast
- MPO
myeloperoxidase
- RBC
red blood cell
- RBPJ
recombinant binding protein‐J
- WT
wild type
1.
Dear Editor,
The tumor suppressor gene CDKN1B, encoding for the p27Kip1 (p27) protein, defines the smallest region of deletion on chromosome 12p13 described in clonal hematopoietic disorders (CHDs) [1]. Among them, myelodysplastic syndromes (MDSs) display typical onset in the elderly and an indolent behavior that may evolve in acute myeloid leukemia (AML) [2]. Recent evidences support a model of parallel clonal evolution at the stem or progenitor cell level and implicate the need of more preclinical, translational and clinical research to identify better ways to timely target clones that may evolve toward malignancy [3, 4]. In mice, p27 plays a key role in regulating number, expansion and self‐renewal of hematopoietic stem and progenitor cells and the growth of lymphoid organs [5, 6, 7]. Yet, its role in adult mice and in the onset/progression of CHDs has not been studied in depth, although a fusion construct of mVenus‐p27K‐, acting as biomarker of G0 phase of the cell cycle, has been used to discriminate dormant and active hematopoietic stem cells (HSCs) in 48 weeks‐old mice [8]. Here, we filled this gap by studying adult/elderly (i.e. one‐year‐old, 1Y) mice.
As observed in young animals, 1Y Cdkn1b knock‐out (Cdkn1bKO) mice maintained a significant increase in body and lymphoid organs weight, particularly in the spleen, compared to wild type (WT) littermates (Supplementary Figure S1A‐B, detailed methods are provided in Supplementary Materials). Splenocytes from 1Y Cdkn1bKO mice showed a decrease in the G1 population (Supplementary Figure S1C), with a highly significant reduction in CD45R+CD19+ B‐lymphocytes, and no differences in the number of CD3+ and CD4+CD8+ T cells (Figure 1A‐B, Supplementary Figure S1D‐E). A decrease in CD45R+CD19+ B cells with a slight increase in CD3+ T cells was already observed in young mice (8‐week‐old, 8W) (Supplementary Figure S2A‐B).
FIGURE 1.
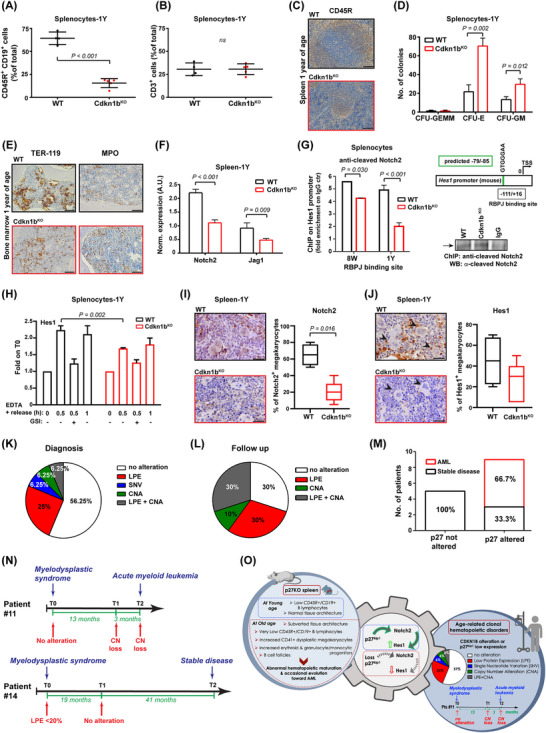
Cdkn1bKO mice developed an MDS linked to Notch2 pathway alteration. (A‐B) Graph reports the CD45R+ CD19+ (A) and CD3 (B) positive splenocytes, extracted from 1Y WT or Cdkn1bKO mice, analyzed by FACS and expressed as mean ± SD. The percentage of CD45R+ CD19+ cells is strongly impaired in 1Y Cdkn1bKO spleen. Each dot corresponds to one mouse. (C) Representative images of spleen sections from 1Y WT (n = 5, upper panel) and Cdkn1bKO (n = 16, lower panel) mice, stained for B cells marker CD45R through IHC. Pictures shows a profound alteration in spleen architecture of Cdkn1bKO mice. Scale bars, 100 μm. (D) Graph reports the quantification of CFU derived from 1Y WT and Cdkn1bKO splenocytes. Numbers of CFU‐E, CFU‐GM, and CFU‐GEMM progenitor cell‐derived colonies were assessed. A higher number of CFU‐E and CFU‐GM colonies was generated by 1Y Cdkn1bKO splenocytes compared to WT counterpart. Results represent the mean ± SD of n = 3 independent experiments, assayed in triplicate. (E) Representative IHC of bone marrow sections stained for the immature erythroid marker TER‐119 and myeloid precursor MPO, from 1Y WT (n = 5, upper panel) and Cdkn1bKO (n = 5, lower panel) mice. Disarrangement of TER‐119+ erythroid islands and non paratrabecular distribution of MPO+ myelocytes is present in Cdkn1bKO sections. Scale bars, 50 μm. (F) qRT‐PCR analysis of Notch2 receptor and Jagged‐2 ligand in spleen collected from 1Y WT and Cdkn1bKO mice. A decreased expression of Notch2 and Jagged‐2 was found in Cdkn1bKO spleen. Results (mean ± SD) derive from the use of n = 6 samples/genotype, performed in duplicate, normalized and expressed in A.U. (G) ChIP of WT and Cdkn1bKO splenocytes from 8W and 1Y mice (pool of 3 samples/genotype/age), as indicated, treated with EDTA 10 mmol/L for 20 minutes. The binding of anti‐cleaved Notch2 antibody to the Hes1 promoter (RBPJ binding site) was evaluated. Data are expressed as fold enrichment over the value of ChIP performed with control IgG. On the right, schematic representation of amplified mouse Hes1 promoter region containing the predicted RBPJ binding sequence (‐79/‐85, in green) (upper panel) and WB reporting representative chromatin IP in WT and Cdkn1bKO splenocytes, using anti‐cleaved Notch2 antibody or control IgG (lower panel). Notch2 binds significantly less to the Hes1 promoter in Cdkn1bKO splenocytes, compared to WT ones. (H) qRT‐PCR analysis of Notch target gene Hes1, in WT or Cdkn1bKO splenocytes treated with EDTA 10 mmol/L for the indicated times and pre‐treated or not with GSI 20 mmol/L for 2 hours. After EDTA stimulation, Cdkn1bKO splenocytes display a decreased transcription of Hes1. Results (mean ± SD) derive from the use of n = 3 splenocytes populations/genotype. (I‐J) IHC analysis and quantification of Notch2 (I) and Hes1 (J) positive megakaryocytes in spleen from 1Y WT (upper panels) or Cdkn1bKO mice (lower panels). Images shows an impaired expression of Notch2 and Hes1 in Cdkn1bKO spleen. Scale bars, 30 μm. In J, arrowheads point to megakaryocytes. (K) Pie chart reports the percentage of patients affected by MDS (n = 32, see Supplementary Table S1) that, at diagnosis, show low p27 protein expression evaluated in bone marrow sections via IHC (LPE), CDKN1B single nucleotide variant (SNV) or copy number alteration (CNA). The presence of SNV in CDKN1B sequence was assessed through NGS analysis, the CNA through ddPCR. At diagnosis, more than 40% of MDS patients results to be altered for p27. (L) Same as in (K), but referred to the follow‐up of 10 out of 18 patients that showed no alteration of p27 at diagnosis. Graph indicates that 70% of MDS patients acquire p27 alterations during follow up. (M) Graph reports the number and percentage of MDS patients that evolved in AML, stratified according to p27 status. When p27 protein or gene is altered, most of patients (66,7%) evolved in AML. (N) Timelines of two representative MDS patients. Alteration in p27 protein or gene at diagnosis (T0) and follow up (T1, T2) are indicated. Patient #11 acquired CDKN1B copy number loss during follow up and evolved in AML. Patient #14 presented no alteration in p27 protein or gene during follow up and maintained a stable disease. (O) Schematic representation of this work. Loss or dysregulation of p27 is associated with development and progression of clonal hematopoietic disorders in human and mice likely, through the regulation of Notch2 signaling. In all graphs, statistical significance was calculated by Student's t‐test or Mann–Whitney, as appropriate, and expressed by P < 0.05.
Abbreviations: WT, wild‐type; KO, knock‐out; 1Y, 1‐year old; FACS, fluorescence‐activated cell sorting; SD, standard deviation; IHC, immunohistochemistry; CFU, colony forming unit; CFU‐E, colony forming unit‐erythroid; CFU‐GM, colony forming unit‐granulocyte monocyte; CFU‐GEMM, colony forming units‐granulocyte, erythrocyte, monocyte, megakaryocyte; MPO, myeloperoxidase; qRT‐PCR, quantitative reverse transcription polymerase chain reaction; A.U., arbitrary units; ChIP, chromatin immunoprecipitation; 8W, 8‐weeks old; EDTA, ethylenediamine tetraacetic acid; RBPJ, recombining binding protein suppressor of hairless; IgG, immunoglobulin G; WB, western blot; IP, immunoprecipitation; GSI, g‐secretase inhibitor; MDS, myelodysplastic syndrome; LPE, low protein expression; SNV, single nucleotide variant; CNA, copy number alteration; NGS, next generation sequencing; ddPCR, droplet digital polymerase chain reaction; AML, acute myeloid leukemia; CHD, clonal hematopoietic disorder.
Spleens from 1Y Cdkn1bKO mice displayed a profound architectural subversion, due to a reduction of B‐cell follicles (CD45R+) in the white pulp and an abnormal expansion of the red pulp, infiltrated by bigger, immature cells displaying blue nuclei, fine chromatin, inconspicuous nucleoli and scant cytoplasm (Figure 1C, Supplementary Figure S2C‐D). Immunohistochemistry (IHC) showed an increased presence of CD41+ megakaryocytes with markedly dysplastic mono/hypo‐lobulated or bizarre segmentation of nuclei and a tendency to form clusters (Supplementary Figure S2D‐E). In colony‐forming‐unit assays, 1Y Cdkn1bKO splenocytes formed more and bigger colonies of the erythroid and granulocyte/monocyte lineages (Figure 1D, Supplementary Figure S2F). Flow cytometry excluded alterations in other populations including natural killers, neutrophils, and monocytes, and in the CD34+c‐KIT+ subpopulation (Supplementary Figure S3A), excluding the presence of nonlymphoid leukemia underlying the splenic subversion.
Further analyses of 1Y Cdkn1bKO bone marrows showed atypical localization of immature precursors with disordered erythropoiesis (disarrangement of TER119+ erythroid islands) and myeloperoxidase positive (MPO+) myelocytes, with a non‐paratrabecular involvement (Figure 1E). Peripheral blood cell count showed a decreased hematocrit, due to a hyper‐chromic macrocytic anemia (low red blood cell count [RBC], high mean corpuscular volume [MCV], and high mean corpuscular hemoglobin [MCH]), thrombocytopenia, lymphocytopenia associated with neutrophilia, together with the presence of scattering erythroblasts and Howell‐jolly bodies within erythrocytes in peripheral blood smear (Supplementary Figure S3B‐C). Liver from Cdkn1bKO (n = 2/5) but not WT mice (n = 0/5) had scattered foci of hematopoiesis (Supplementary Figure S3D). Altogether, these findings were consistent with an abnormal maturation of hematopoietic cells and suggested that Cdkn1bKO mice developed an age‐dependent CHD, highly reminiscent of MDS [9]. Interestingly, 1/5 mice showed a progression of this phenotype, characterized by liver necrosis and diffuse bone marrow infiltration by immature MPO+ cells, consistent with leukemic evolution (Supplementary Figure S3E).
Notch2 plays a unique role in the spleen by regulating the marginal zone B‐cell differentiation, in human and mice [10]. Moreover, Notch2 mutations can drive clonal evolution of human MDS [3]. Expression of Notch2 and its ligand Jag1 was reduced in spleens from 1Y, but not 8W Cdkn1bKO mice (Figure 1F, Supplementary Figure S4A‐F). Yet, spleens from both 1Y and 8W Cdkn1bKO mice displayed a reduction in Notch2 target gene, Hes1, known to mediate the repressive role of Notch toward myelomonocytic‐specific differentiation in mice [11] (Supplementary Figure S4E‐F), indicating that a possible Notch2 signaling impairment was operating in Cdkn1bKO mice, well before the appearance of a pathological phenotype. Accordingly, in both 8W and 1Y Cdkn1bKO splenocytes, Notch2 bound significantly less to the Hes1 promoter (Figure 1G), suggesting that p27 could positively regulate Notch2 transcriptional activity. To test this hypothesis, we stimulated Notch activity using EDTA (Supplementary Figure S5A). We observed that the induction of Hes1 expression was significantly impaired in Cdkn1bKO compared to WT splenocytes, especially after 30 minutes (Figure 1H), and specifically blocked by treatment with γ‐Secretase inhibitors (GSI). Similar results were obtained by analyzing the expression of another Notch target gene, Hey1 (Supplementary Figure S5B). IHC staining of the spleens confirmed that Notch2 and Hes1 proteins expression was decreased in Cdkn1bKO compared to WT mice (Figure 1I‐J, Supplementary Figure S5C). Of note, dysplastic megakaryocytes in Cdkn1bKO spleens were negative for both Hes1 and Notch2 expression (Figure 1I‐J).
Like splenocytes, mouse embryonic fibroblasts (MEF) preferentially express Notch2 and represent a useful model to study Notch signaling (Supplementary Figure S5D). Notch2 basal expression and its nuclear translocation was not affected by p27 expression, but Notch‐dependent Hes1 transcription was largely impaired in Cdkn1bKO, as observed using EDTA and GSI treatments (Supplementary Figure S6A‐B). Accordingly, EDTA‐induced activity of an exogenously transfected recombinant binding protein‐J (RBPJ)‐responsive reporter was strongly reduced in Cdkn1bKO compared to Cdkn1bWT cells (Supplementary Figure S6C‐D). Reintroduction of p27 in different Cdkn1bKO models effectively rescued EDTA‐stimulated RBPJ promoter activity, in a dose‐dependent manner (Supplementary Figure S6E‐F). Interestingly, p27 increased the EDTA‐induced RBPJ activity, independently from its ability to bind and inhibit cyclin/cyclin dependent kinase (CDK) complexes (Supplementary Figure S6G).
To evaluate the possible relevance of our findings in human disease, we assessed the levels of p27 protein expression, the copy number alteration (CNA) and mutational status of CDKN1B in a panel of bone marrow samples from patients with CHD (Supplementary Table S1). Over 40% (14/32) of patients presented a p27 alterations, due to CDKN1B gene inactivation via copy number alteration (CNA) or mutation and/or p27 low protein expression (LPE), generally associated with worse clinical parameters (Figure 1K, Supplementary Table S1‐2). We could retrieve follow‐up samples from 10 of the 18 patients who did not display baseline CDKN1B alterations. Of these patients, four acquired CNA and six LPE during their follow‐up (Figure 1L). A significantly higher proportion of patients displaying p27 alterations evolved from MDS to AML (Figure 1M), as also evidenced from the analyses of patients’ clinical histories (Figure 1N, Supplementary Figure S7).
Our findings are supported by other studies, describing Notch pathway having a tumor suppressive function in myeloid leukemia, both in human and in mice [3, 11] and defining CDKN1B gene as the smallest region of deletions on chromosome 12p13, in MDS and AML [1]. In our study, the extent of p27 alteration largely exceeds the percentage of CDKN1B and 12p deletion observed in MDS human samples. This is due, in part, to the fact that here we put together LPE, CNA and mutations, and, in part, by the relatively low number of analyzed samples, certainly representing a possible limitation. In silico analyses further supported our results, showing that CDKN1B and, in part, NOTCH2 expression progressively decreases when passing from normal bone marrow to MDS and to AML. Further, NOTCH2 and CDKN1B genomic alterations were mutually exclusive in human MDS samples (Supplementary Figure S8A‐C).
Although a causal relationship is not formally demonstrated, our data, collected both in human samples and in mice, suggest that loss of p27 may contribute to the pathogenesis of CHD by suppressing Notch2‐Hes1 transcriptional activity (Figure 1O) and that CDKN1B and p27 protein alterations in MDS may have been underestimated in previous studies. Overall, our work provides new knowledge on the molecular basis underlying CHD onset and evolution and might open the way to the design of novel diagnostic and therapeutic tools for a better management of these patients.
DECLARATIONS
AUTHOR CONTRIBUTIONS
Ilenia Segatto, Gian Luca Rampioni Vinciguerra, Barbara Belletti, and Gustavo Baldassarre: conceptualization. Ilenia Segatto, Gian Luca Rampioni Vinciguerra, Ilenia Pellarin, Alessandra Dall'Acqua, Stefania Berton, Francesca Citron, Sara D'Andrea, Giorgia Mungo, Davide Viotto, and Lorena Musco: methodology. Ilenia Segatto, Gian Luca Rampioni Vinciguerra, Vincenzo Canzonieri, Valter Gattei, Andrea Vecchione, Barbara Belletti, and Gustavo Baldassarre: data curation and analysis. Arianna Di Napoli, Maria Antonietta Aloe Spiriti, and Andrea Vecchione: clinical research support. Ilenia Segatto, Gian Luca Rampioni Vinciguerra, Barbara Belletti, and Gustavo Baldassarre: writing‐original draft preparation, visualization, writing‐reviewing, and editing. Barbara Belletti and Gustavo Baldassarre: funding acquisition. All authors read and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The authors have no conflict of interests.
FUNDING INFORMATION
This research was supported by CRO Ricerca Corrente core grant (linea 1) of Ministero della Salute, Associazione Italiana Ricerca sul Cancro (AIRC) IG to Gustavo Baldassarre (#16865) and Barbara Belletti (#20061), and Associazione Italiana Ricerca sul Cancro (AIRC) fellowship to Ilenia Segatto (#18171) and Francesca Citron (#20902).
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Human specimens from patients were collected after signed written informed consent was obtained, in accordance with recognized ethical guidelines and following approval by the Institutional Review Board of University of Rome “La Sapienza” Santo Andrea Hospital (Rome, Italy) and Ethical Committee (Prot. CE n. 168/03). All studies were performed in compliance with the 1975 Declaration of Helsinki, as revised in 1983.
CONSENT FOR PUBLICATION
Not applicable.
Supporting information
Supporting information
ACKNOWLEDGEMENTS
We like to thank all patients who have donated their tissue and made our research possible, Mr Roberto Cirombella for his valuable technical support, and all members of the SCICC lab for critical discussion of the data.
Open access funding provided by BIBLIOSAN.
Contributor Information
Barbara Belletti, Email: bbelletti@cro.it.
Gustavo Baldassarre, Email: gbaldassarre@cro.it.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request. All sequencing data are publicly available (BioProject ID: PRJNA943150).
REFERENCES
- 1. Sato Y, Suto Y, Pietenpol J, Golub TR, Gilliland DG, Davis EM, et al. TEL and KIP1 define the smallest region of deletions on 12p13 in hematopoietic malignancies. Blood. 1995;86(4):1525–33. [PubMed] [Google Scholar]
- 2. Kennedy AL, Shimamura A. Genetic predisposition to MDS: clinical features and clonal evolution. Blood. 2019;133(10):1071–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen J, Kao YR, Sun D, Todorova TI, Reynolds D, Narayanagari SR, et al. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat Med. 2019;25(1):103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Deininger MWN, Tyner JW. Solary E. Turning the tide in myelodysplastic/myeloproliferative neoplasms. Nat Rev Cancer. 2017;17(7):425–40. [DOI] [PubMed] [Google Scholar]
- 5. Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, et al. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)‐deficient mice. Cell. 1996;85(5):733–44. [DOI] [PubMed] [Google Scholar]
- 6. Cheng T, Rodrigues N, Dombkowski D, Stier S, Scadden DT. Stem cell repopulation efficiency but not pool size is governed by p27(kip1). Nat Med. 2000;6(11):1235–40. [DOI] [PubMed] [Google Scholar]
- 7. Berton S, Pellizzari I, Fabris L, D'Andrea S, Segatto I, Canzonieri V, et al. Genetic characterization of p27(kip1) and stathmin in controlling cell proliferation in vivo. Cell Cycle. 2014;13(19):3100–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fukushima T, Tanaka Y, Hamey FK, Chang CH, Oki T, Asada S, et al. Discrimination of Dormant and Active Hematopoietic Stem Cells by G(0) Marker Reveals Dormancy Regulation by Cytoplasmic Calcium. Cell Rep. 2019;29(12):4144–58 e7. [DOI] [PubMed] [Google Scholar]
- 9. Kogan SC, Ward JM, Anver MR, Berman JJ, Brayton C, Cardiff RD, et al. Bethesda proposals for classification of nonlymphoid hematopoietic neoplasms in mice. Blood. 2002;100(1):238–45. [DOI] [PubMed] [Google Scholar]
- 10. Lobry C, Oh P, Mansour MR, Look AT, Aifantis I. Notch signaling: switching an oncogene to a tumor suppressor. Blood. 2014;123(16):2451–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Klinakis A, Lobry C, Abdel‐Wahab O, Oh P, Haeno H, Buonamici S, et al. A novel tumour‐suppressor function for the Notch pathway in myeloid leukaemia. Nature. 2011;473(7346):230–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. All sequencing data are publicly available (BioProject ID: PRJNA943150).