

Abbreviations
- PPGL
pheochromocytoma and paraganglioma
- TCA
tricarboxylic acid
- OGDH
2‐oxoglutarate dehydrogenase
- OGDHc
oxoglutarate dehydrogenase complex
- SucCoA
succinyl‐coenzyme A
- PTM
post‐translational modification
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- DLD
dihydrolipoamide dehydrogenase
- NADH
nicotinamide adenine dinucleotide reduced
- RNA‐Seq
RNA sequencing
- LCL
lymphoblastoid cell line
- SDH
succinate dehydrogenase
- NES
normalized enrichment scores
- DLST
dihydrolipoamide S‐succinyltransferase
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- KO
knockout
- WT
wild‐type
Dear Editor,
Of all human tumors, pheochromocytomas and paragangliomas (PPGLs) have the highest heritability rate. Over 15% of PPGLs harbor mutations in genes encoding tricarboxylic acid (TCA) cycle‐related enzymes that cause oncometabolite accumulation and drive tumorigenesis via metabolic adaptation to hypoxia and global hypermethylation [1]. The dihydrolipoamide S‐succinyltransferase (DLST) gene was recently described as a new PPGL susceptibility gene [2]. DLST is a component of the 2‐oxoglutarate dehydrogenase (OGDH) complex (OGDHc) that catalyzes the conversion of alpha‐ketoglutarate to succinyl‐coenzyme A (SucCoA) in the TCA cycle. It also plays an understudied role in protein succinylation, a highly conserved post‐translational modification (PTM) involving succinyl group transfer from SucCoA to protein lysine residues [3]. Succinylation causes major chemical and structural changes to proteins and has been linked to the development of diseases, including cancer [4, 5]. Here, we further characterized DLST‐mutated PPGLs, explored the molecular mechanisms underlying their tumorigenesis, and examined the impact of DLST mutations on the succinylome.
Transcriptome analysis was performed for PPGLs carrying the recurrent DLST‐p.G374E mutation and a PPGL carrying the DLST‐p.Y422C variant, whose deleterious effect was supported by a comprehensive study (Supplementary Figure S1). Hierarchical clustering revealed that DLST‐mutated tumors showed a pseudohypoxic transcriptional profile (Figure 1A), supported by the expression of canonical hypoxia‐related genes (Supplementary Figure S2). Conversely, unsupervised clustering of DNA methylation data separated DLST‐mutated tumors from PPGLs with mutations of other TCA cycle‐related genes (Figure 1B), suggesting that DLST‐mutated tumors lacked a hypermethylated phenotype. Thus, we investigated alternative tumorigenic mechanisms.
FIGURE 1.
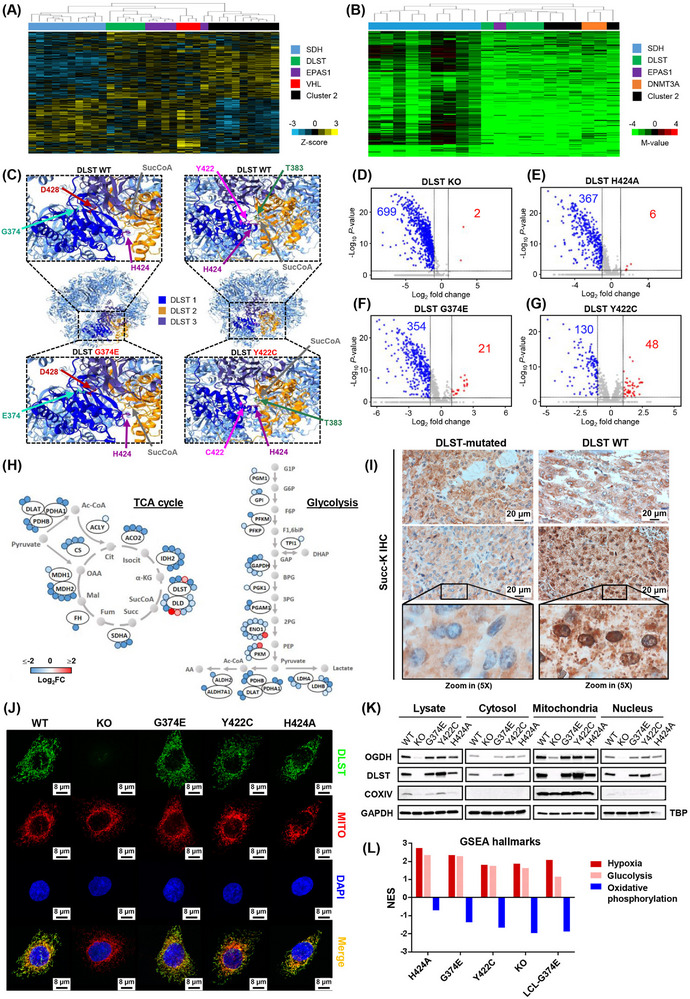
PPGL‐causing DLST mutations remodel the cellular succinylome and up‐regulate the hypoxic pathway. (A) Unsupervised clustering of transcriptomic data (z‐scores) for genes expressed differentially in PPGLs (Burnichon et al. 2011) grouped DLST‐mutated tumors (shown in green) with cases featuring SDH/VHL/EPAS1 (in blue, red, and purple, respectively) mutations (Cluster 1) and separately from Cluster 2 PPGLs (tumors carrying mutations in kinase signaling‐related genes; in black), evidencing their pseudohypoxic nature. (B) Hierarchical clustering using DNA methylation data from a list of probes found differentially methylated in SDHB‐ or FH‐mutated tumors showing a CpG island methylator phenotype (Ricketts CJ et al. PLoS One. 2022;17(12):e0278108), separated DLST‐mutated tumors (green) from hypermethylated tumors carrying mutations in SDH genes (blue). (C) Structure of DLST's homooligomeric 24‐mer. Three DLST monomers are highlighted in dark blue, yellow and purple. The upper panels show the structure of DLST WT, while the bottom left and bottom right panels show the p.G374E and p.Y422C mutants, respectively. SucCoa denotes Succinyl‐CoA (only the succinyl group is shown). (D‐G) Volcano plots showing the number of differentially succinylated sites (Log2FC < ‐1 or > 1, P < 0.05) in DLST KO (D), DLST H424A (E), DLST G374E (F), and DLST Y422C (G) cells when compared to DLST WT cells. Blue, red and grey dots represent differentially hyposuccinylated sites, differentially hypersuccinylated sites, and non‐significantly succinylated sites and/or with a Log2FC between ‐1 and 1, respectively. (H) Representation of sites exhibiting significant differential succinylation (colored circles) in TCAc enzymes (left panel) and the Glycolysis/Gluconeogenesis pathway (right panel) when comparing DLST G374E and WT DLST cells. Both pathways exhibit global hyposuccinylation (blue color). (I) Anti‐succinyllysine immunohistochemistry images of PPGLs harboring the DLST mutations p.Y422C (left top) and p.G374E (left bottom) and known mutations in other susceptibility genes as controls (right). The granular pattern and perinuclear staining observed in the images on the right suggest mitochondrial staining, whereas cells from DLST‐mutated PPGLs exhibit more homogeneous cytoplasmic staining. The tumor harboring the p.Y422C DLST mutation (top left) has an intermediate pattern with fewer granules and some perinuclear staining, in accordance with proteomic succinylation data obtained using our cell model. (J) Immunofluorescence analysis of the different cell lines showing that DLST (green) colocalizes with the Mitotracker dye (red, MITO in the figure) and is thus predominantly localized in the mitochondria regardless of its mutations in all cells other than DLST KO cells lacking the DLST protein. Nuclei are stained in blue with DAPI. (K) Immunoblotting assay showing that DLST is present in the cytosol and nucleus compartments as well as the mitochondria‐enriched fraction (named mitochondria in the figure) regardless of PPGL‐causing mutations. OGDH levels are significantly reduced in DLST KO cells lacking DLST. GAPDH and TBP (nucleus) were used as loading controls, and COXIV as a mitochondrial marker. (L) Representation of normalized enrichment scores (NES) relative to DLST WT cells for the GSEA Hallmark gene sets of Hypoxia, Glycolysis and Oxidative phosphorylation in cells harboring DLST alterations (from left to right: DLST H424A, DLST G374E, DLST Y422C, DLST KO and LCL‐G374E).
Abbreviations: PPGL, pheochromocytoma and paraganglioma; SucCoA, succinyl coenzyme A; TCAc, tricarboxylic acid cycle; DAPI, 4’,6‐diamidino‐2‐phenylindole, dihydrochloride; NES, normalized enrichment score; GSEA, gene set enrichment analysis; LCL, lymphoblastoid cell line; Succ‐K IHC, succinylated lysine immunohistochemistry; Log2FC, Log2 fold change.
Structural modeling of the recently described homooligomeric DLST 24‐mer [6] showed that glycine 374 is physically close to aspartic acid 428 of a nearby DLST monomer (Figure 1C, left panel). The DLST‐p.G374E mutation substitutes this glycine with a glutamic acid, potentially introducing a repulsive interaction between the two anionic side chains that could alter the oligomer's structure. Conversely, the DLST‐p.Y422C mutation could disrupt a likely interaction between tyrosine 422 and threonine 383 from an adjacent DLST monomer (Figure 1C, right panel), altering the substrate pocket's three‐dimensional conformation. DLST mutations identified in PPGL patients thus likely alter the homooligomeric DLST assembly and hence the native structure of the OGDHc.
To determine whether DLST alterations may affect the cellular succinylome, mass spectrometry was applied to previously generated DLST knockout (KO) H838 cell lines into which DLST constructs, including the wild‐type (WT), the two aforementioned PPGL‐causing mutants, and the catalytically dead p.H424A mutant [2] were stably introduced (Supplementary Figure S3A‐B). DLST‐KO cells exhibited dramatically reduced succinylation levels (Figure 1D, Supplementary Table S1) that were not caused by reduced protein abundance (Supplementary Figure S3C). The number of hyposuccinylated lysines differed between proteins, ranging from 1 to 20 (Supplementary Figure S3D). Moreover, hyposuccinylated proteins were involved in multiple pathways and mapped to all cellular compartments (Supplementary Figure S3E, Supplementary Table S2), indicating a widespread down‐regulation of succinylation. These findings enable us to experimentally link the absence of DLST to a profound decrease in this PTM. Although succinylation was once believed to be a pH‐dependent non‐enzymatic reaction regulated by donor (SucCoA) concentration, Gibson et al. [7] found that succinylation efficiency increased in the presence of α‐ketoglutarate and the OGDHc, suggesting that OGDHc could catalyze the succinylation of proteins. Our results support the role of DLST (and hence the OGDHc) as a major regulator of protein succinylation.
Compared to DLST‐WT cells, those expressing DLST‐p.G374E and DLST‐p.H424A exhibited global hyposuccinylation, while cells expressing DLST‐p.Y422C exhibited significant but more limited hyposuccinylation (Figure 1E‐G, Supplementary Table S3). Some proteins with multiple down‐regulated succinylation sites in DLST G374E cells were involved in the metabolic Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, such as the TCA cycle and glycolysis/gluconeogenesis (Figure 1H). Overall, cells carrying PPGL‐causing DLST mutations exhibited hyposuccinylation of mitochondrial proteins, which was particularly pronounced in G374E‐carrying cells (Supplementary Figure S4A). Changes in mitochondrial succinylation were validated in PPGL tumors using anti‐succinyllysine immunohistochemistry: DLST‐mutated tumors exhibited diffuse cytoplasmic staining, whereas PPGLs with mutations in other susceptibility genes had granular and perinuclear succinylation patterns (Figure 1I). These results suggest that DLST mutations remodeled the cellular succinylome, causing mitochondrial hyposuccinylation and potentially altering the function of key proteins in PPGL development. Numerous metabolic enzymes are regulated by succinylation, either modulating their activity, degradation, or ability to interact with other proteins. For example, desuccinylation of the succinate dehydrogenase complex flavoprotein subunit A was reported to inhibit its enzymatic activity and promote clear cell renal cell carcinoma cell proliferation in vitro, and GLS desuccinylation protected it from ubiquitin‐mediated degradation, promoting breast tumorigenesis [8]. Overall, histone succinylation was reduced by DLST‐KO and was not fully restored to WT levels upon reintroducing DLST mutants (Supplementary Figure S4B, Supplementary Table S4). The molecular consequences of OGDHc‐mediated histone succinylation have been described [4], but further studies are needed to unravel the implications of hyposuccinylation of specific histone residues.
Immunofluorescence staining showed that all DLST protein variants were mainly localized in the mitochondria regardless of mutations (Figure 1J), though they were also identified in other compartments by immunoblotting (Figure 1K). Of the other OGDHc components, dihydrolipoamide dehydrogenase (DLD) was localized in the mitochondria but was also present in the nucleus, while OGDH was generally found in mitochondria but was absent in DLST‐KO cells (Supplementary Figure S5). The low OGDH levels in DLST‐KO cells were confirmed by immunoblotting (Figure 1K), suggesting that without DLST, OGDHc assembly is impaired and OGDH is degraded, consistent with the high OGDH degradation rates observed in the presence of OGDH mutations causing protein structural instability [9]. This result suggests that DLST plays a key role in OGDHc assembly, supporting the proposed stepwise assembly mechanism in which the stronger OGDH‐DLST interaction forms before DLD is anchored [6]. Immunoblotting revealed that OGDHc assembly was unaffected by PPGL‐causing DLST mutations, but a significant decrease in NADH production demonstrated the mutations’ profound effect on OGDHc activity (Supplementary Figure S6). Overall, these findings showed that DLST mutations might not affect OGDHc assembly or subcellular location but impair its enzymatic activity.
Gene set enrichment analysis of RNA‐sequencing data showed that when compared to DLST‐WT cells, the hallmark gene sets of hypoxia and glycolysis were up‐regulated, while the oxidative phosphorylation set was down‐regulated in all cell lines carrying DLST alterations, including a lymphoblastoid cell line derived from a patient with the DLST‐p.G374E mutation (Figure 1L, Supplementary Figure S7). This link between DLST mutations and hypoxia/glycolysis pathway up‐regulation was confirmed by functional studies showing differences in glucose‐dependent growth, oxygen consumption and ATP production in cells carrying DLST alterations (Supplementary Figure S8). However, DNA methylation profiling revealed no statistically significant differences between the H838 cell lines, indicating that the change in gene expression is not due to altered methylation (data not shown). This is consistent with the pseudohypoxic expression profile and absence of global hypermethylation observed in DLST‐mutated tumors. It has been proposed that the shift from oxidative to glycolytic metabolism observed in esophageal squamous cell carcinoma cells could lead to global hyposuccinylation due to SucCoA depletion [10]. Our results suggest the existence of a feedback loop whereby reduced protein succinylation induces a similar metabolic shift towards a hypoxic state.
In conclusion, we show in this study that global protein succinylation levels depend strongly on DLST, advancing our understanding of this PTM's mechanistic role and highlighting DLST as a promising therapeutic target for treating diseases linked to dysregulated succinylation. Furthermore, we show that DLST mutations found in PPGL patients can remodel the cellular succinylome and cause a transcriptional shift from oxidative phosphorylation to a hypoxic cellular state.
AUTHOR CONTRIBUTIONS
Sara Mellid contributed to experimental design, data acquisition, analysis, interpretation, and manuscript writing. Fernando García and Javier Muñoz contributed to data acquisition, analysis and interpretation. Alberto Díaz‐Talavera and Ángel Mario Martínez‐Montes contributed to bioinformatics and statistical analysis. Juan María Roldán‐Romero, Eduardo Gil, Carlos Valdivia, Scherezade Jiménez, Manuel Pérez‐Martínez, Ana Cerezo, Clara María Santiveri, Manel Esteller, Ramón Campos‐Olivas and Javier Coloma helped conducting the experiments. Bruna Calsina, María Monteagudo, Rocío Letón, María Santos, Javier Lanillos, Natalia Martínez‐Puente, Javier de Nicolás‐Hernández, Emiliano Honrado and Eduardo Caleiras contributed to data analysis and interpretation. Cristina Montero‐Conde, Luis Javier Leandro‐García, Cristina Rodríguez‐Antona and Mercedes Robledo provided suggestions and contributed to critical revision of the manuscript. Alberto Cascón contributed to the conception, experimental design, data interpretation, and critical revision of the manuscript. All authors reviewed and edited the manuscript. All authors read and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
FUNDING INFORMATION
This work was supported by the Instituto de Salud Carlos III (ISCIII) through the “Acción Estratégica en Salud” (AES) (projects PI18/00454 and PI22/01490 to A.C. and PI20/01169 to M.R.), cofounded by the European Regional Development Fund (ERDF). Sara Mellid was supported by the Spanish Ministry of Science, Innovation and Universities “Formación del Profesorado Universitario‐ FPU” fellowship with ID number FPU19/04940. Luis J. Leandro‐García is supported by ‘la Caixa’ Foundation (ID 100010434) under agreement LCF/BQ/PI20/11760011.
CONSENT FOR PUBLICATION
Not applicable.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The study was approved by the ethics committee of Instituto de Salud Carlos III (CEI PI54_2016‐v2) and obtained patients’ consent to participate.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors have nothing to report.
DATA AVAILABILITY STATEMENT
Data and materials supporting these findings will be available upon reasonable request. The DNA methylation data are available in the National Center for Biotechnology Information GEO database under the accession numbers GSE210809, GSE111336 and GSE123185. The raw data of the RNA‐seq used in this study are available in GEO under the accession number GSE210808 and in the European Genome‐phenome archive under the accession number EGAS00001006044. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium with the dataset identifier PXD036782.
REFERENCES
- 1. Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23(6):739–52. [DOI] [PubMed] [Google Scholar]
- 2. Remacha L, Pirman D, Mahoney CE, Coloma J, Calsina B, Curras‐Freixes M, et al. Recurrent Germline DLST Mutations in Individuals with Multiple Pheochromocytomas and Paragangliomas. Am J Hum Genet. 2019;104(4):651–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang Z, Tan M, Xie Z, Dai L, Chen Y, Zhao Y. Identification of lysine succinylation as a new post‐translational modification. Nat Chem Biol. 2011;7(1):58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang Y, Guo YR, Liu K, Yin Z, Liu R, Xia Y, et al. KAT2A coupled with the alpha‐KGDH complex acts as a histone H3 succinyltransferase. Nature. 2017;552(7684):273–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhao G, Zhen J, Liu X, Guo J, Li D, Xie J, et al. Protein post‐translational modification by lysine succinylation: Biochemistry, biological implications, and therapeutic opportunities. Genes Dis. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nagy B, Polak M, Ozohanics O, Zambo Z, Szabo E, Hubert A, et al. Structure of the dihydrolipoamide succinyltransferase (E2) component of the human alpha‐ketoglutarate dehydrogenase complex (hKGDHc) revealed by cryo‐EM and cross‐linking mass spectrometry: Implications for the overall hKGDHc structure. Biochim Biophys Acta Gen Subj. 2021;1865(6):129889. [DOI] [PubMed] [Google Scholar]
- 7. Gibson GE, Xu H, Chen HL, Chen W, Denton TT, Zhang S. Alpha‐ketoglutarate dehydrogenase complex‐dependent succinylation of proteins in neurons and neuronal cell lines. J Neurochem. 2015;134(1):86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lu K, Han D. A review of the mechanism of succinylation in cancer. Medicine (Baltimore). 2022;101(45):e31493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Whittle EF, Chilian M, Karimiani EG, Progri H, Buhas D, Kose M, et al. Biallelic variants in OGDH encoding oxoglutarate dehydrogenase lead to a neurodevelopmental disorder characterized by global developmental delay, movement disorder, and metabolic abnormalities. Genet Med. 2022;25(2):100332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guo Z, Pan F, Peng L, Tian S, Jiao J, Liao L, et al. Systematic Proteome and Lysine Succinylome Analysis Reveals Enhanced Cell Migration by Hyposuccinylation in Esophageal Squamous Cell Carcinoma. Mol Cell Proteomics. 2021;20:100053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Data Availability Statement
Data and materials supporting these findings will be available upon reasonable request. The DNA methylation data are available in the National Center for Biotechnology Information GEO database under the accession numbers GSE210809, GSE111336 and GSE123185. The raw data of the RNA‐seq used in this study are available in GEO under the accession number GSE210808 and in the European Genome‐phenome archive under the accession number EGAS00001006044. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium with the dataset identifier PXD036782.