

Abstract
Background
Clear cell renal cell carcinoma (ccRCC) is the most lethal renal cancer. An overwhelming increase of patients experience tumor progression and unfavorable prognosis. However, the molecular events underlying ccRCC tumorigenesis and metastasis remain unclear. Therefore, uncovering the underlying mechanisms will pave the way for developing novel therapeutic targets for ccRCC. In this study, we sought to investigate the role of mitofusin‐2 (MFN2) in supressing ccRCC tumorigenesis and metastasis.
Methods
The expression pattern and clinical significance of MFN2 in ccRCC were analyzed by using the Cancer Genome Atlas datasets and samples from our independent ccRCC cohort. Both in vitro and in vivo experiments, including cell proliferation, xenograft mouse models and transgenic mouse model, were used to determine the role of MFN2 in regulating the malignant behaviors of ccRCC. RNA‐sequencing, mass spectrum analysis, co‐immunoprecipitation, bio‐layer interferometry and immunofluorescence were employed to elucidate the molecular mechanisms for the tumor‐supressing role of MFN2.
Results
we reported a tumor‐suppressing pathway in ccRCC, characterized by mitochondria‐dependent inactivation of epidermal growth factor receptor (EGFR) signaling. This process was mediated by the outer mitochondrial membrane (OMM) protein MFN2. MFN2 was down‐regulated in ccRCC and associated with favorable prognosis of ccRCC patients. in vivo and in vitro assays demonstrated that MFN2 inhibited ccRCC tumor growth and metastasis by suppressing the EGFR signaling pathway. In a kidney‐specific knockout mouse model, loss of MFN2 led to EGFR pathway activation and malignant lesions in kidney. Mechanistically, MFN2 preferably binded small GTPase Rab21 in its GTP‐loading form, which was colocalized with endocytosed EGFR in ccRCC cells. Through this EGFR‐Rab21‐MFN2 interaction, endocytosed EGFR was docked to mitochondria and subsequently dephosphorylated by the OMM‐residing tyrosine‐protein phosphatase receptor type J (PTPRJ).
Conclusions
Our findings uncover an important non‐canonical mitochondria‐dependent pathway regulating EGFR signaling by the Rab21‐MFN2‐PTPRJ axis, which contributes to the development of novel therapeutic strategies for ccRCC.
Keywords: ccRCC, EGFR signaling pathway, MFN2, PTPRJ, Rab21
Abbreviations
- ATCC
the American Type Culture Collection
- AUC
area under curve
- BLI
bio‐layer interferometry
- BSA
bovine serum albumin
- ccRCC
clear cell renal cell carcinoma
- Co‐IP
co‐immunoprecipitation
- CPTAC
the Clinical Proteomic Tumor Analysis Consortium
- DAPI
4,6‐diamidino‐2‐phenylindole
- DDM
1.2% n‐dodecyl‐b‐D‐maltoside
- DFS
disease‐free survival
- DMEM
Dulbecco's Modified Eagle Medium
- EGFR
epidermal growth factor receptor
- FBS
fetal bovine serum
- GST
glutathione S‐transferase
- HK‐2
immortalized renal epithelial cell
- LC‐MS
liquid Chromatography‐Mass Spectrometry
- MAPK
mitogen‐activated protein kinase
- MFN1
mitofusin‐1
- MFN2
mitofusin‐2
- mTOR
mechanistic target of rapamycin
- OMM
outer mitochondrial membrane
- ORF
open reading frame
- OS
overall survival
- PBS
phosphate‐buffered saline
- PDX
Patient‐derived xenograft
- PI3K
phosphatidylinositol 3‐kinase
- PMSF
phenylmethanesulfonylfluoride
- PTPRJ
tyrosine‐protein phosphatase receptor type J. mito‐PTPRJ, OMM‐anchoring form of PTPRJ
- qRT‐PCR
quantitative real‐time polymerase chain reaction
- RCC
Renal cell carcinoma
- RIPA
Radio immunoprecipitation assay
- RNA‐seq
RNA‐sequencing
- RTK
receptor tyrosine kinases
- SDS‐PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- shRNA
short hairpin RNA
- SIM
illumination microscopy
- siRNA
small interfering RNA
- STAT3
signal transducer and activator of transcription 3
- SYSUCC
Sun Yat‐sen University Cancer Center
- TCGA
the Cancer Genome Atlas
- TCGA‐KIRC
the Cancer Genome Atlas kidney renal clear cell carcinoma
- VEGF
vascular endothelial growth factor
- α‐SMA
alpha smooth muscle actin
1. BACKGROUND
Renal cell carcinoma (RCC), which originates from renal tubule epithelial cells, is a common type of kidney cancer, accounting for 3%‐5% of all human malignancies [1]. Over the past 35 years, the incidence and mortality of RCC have been continuously growing in China [2]. Of all RCC cases, approximately 70%‐80% are clear cell RCC (ccRCC) [3]. In recent years, targeted therapies have been developed against specific pathways, such as vascular endothelial growth factor (VEGF), mechanistic target of rapamycin (mTOR) and epidermal growth factor receptor (EGFR) [4, 5]. Sunitinib, a multi‐kinase inhibitor, is used as a first‐line drug for RCC [6, 7, 8]. A randomized phase III clinical trial revealed that lapatinib, an EGFR/receptor tyrosine‐protein kinase ErbB‐2 dual kinase inhibitor, prolonged the overall survival (OS) of advanced‐stage RCC [9]. Furthermore, cells derived from metastatic ccRCC tumors showed significantly higher EGFR signature activity than those from paired primary tumors. Given these clinical advances, most patients with ccRCC eventually experienced tumor progression [10]. Understanding the molecular mechanisms underlying the initiation and development of ccRCC is urgently needed to develop novel therapeutic strategies.
The EGFR signaling pathway is frequently overactivated in ccRCC and plays a critical role in the malignancy of ccRCC [11, 12, 13, 14]. Epidermal growth factor (EGF) is essential for the rapid proliferation of many human RCC cell lines and can be repressed by inhibiting EGFR [15, 16]. Upon binding of growth factors, EGFR becomes dimerized and autophosphorylated, and subsequently activates downstream signaling networks [17, 18]. As an attenuating mechanism, ligand binding induces internalization of EGFR via endocytosis [19]. Endocytosed EGFR targets various organelles for recycling and degradation [20, 21], which is mediated by multiple Rab GTPases [22, 23]. Recently, EGFR was reported to traffic to mitochondria and regulate multiple cellular events, such as cellular ATP production, migration, apoptosis, and drug resistance [24, 25, 26]. Compared to other well‐studied intracellular delivery processes, little is known about how EGFR is delivered to mitochondria. Moreover, current studies regarding the crosstalk between EGFR and mitochondria are mainly focused on the effect of EGFR on mitochondria. Whether mitochondria in turn regulate EGFR signaling activity remains unknown.
Mitochondria dynamically undergo fission and fusion [27]. Mitofusin‐2 (MFN2), located on the outer mitochondrial membrane (OMM), mediates the fusion of OMM. Aberrant MFN2 function causes neuropathy and cardiomyopathy [28, 29]. MFN2 is also related with various malignancies, as recent studies reported that MFN2 was down‐regulated in tumor samples and served as a tumor suppressor [30, 31, 32]. Although the mitochondrial fusogenic activity of MFN2 is thought to be important in these events, the exact mechanism of how MFN2 confers its anti‐tumor function remains to be further understood.
Our group has a long‐standing interest in studying the structure and function of MFN2 [33, 34]. Function of MFN2 in the initiation and progression of kidney cancer remains unclear and warrants further investigation. In this study, we constructed kidney‐specific Mfn2‐knockout mouse models to prove that MFN2 is a key tumor suppressor in ccRCC. By a combination of bioinformatical, biochemical, and imaging approaches, we aimed to uncover the mechanism of how MFN2 induces ccRCC progression. Finally, preclinical study on a PDX mouse model was conducted to reveal the therapeutic potential of increasing MFN2 expression against ccRCC.
2. MATERIALS AND METHODS
2.1. Patients and clinical samples
ccRCC tissues and peri‐tumor tissues were obtained from patients who underwent surgery at the First Affiliated Hospital, Sun Yat‐sen University (FAH‐SYSU; Guangzhou, Guangdong, China) in 2021 for RNA or protein analysis. Paraffin‐embedded tumor samples obtained from 150 patients pathologically diagnosed with primary ccRCC between 2013 and 2015 at Sun Yat‐sen University Cancer Center (SYSUCC; Guangzhou, Guangdong, China) were used for Kaplan–Meier survival analysis. All patients had provided written consent, and all related experimental procedures were performed with the approval of the Internal Review and Ethics Boards of SYSUCC (Approval No. 202201071013000560775z) and of FAH‐SYSU (Approval No. [2021]144). The study complied with all relevant ethical regulations for research involving human participants.
2.2. Plamids and siRNA
The recombinant MFN2 overexpression plasmids for stable transfection were generated by cloning the full‐length open reading frame (ORF) of the human MFN2 gene (NM_001127660.2) into the PLVX‐N1 vector (#632152, Clontech, Palo Alto, CA, USA). For transient transfection, MFN2 was cloned into the pcDNA3.1 (+) vector (V79020, Invitrogen, Carlsbad, CA, USA) with N‐terminal 3 × Flag‐tag. The ORFs of EGFR and Rab21 were cloned into the pSin‐EF2‐puro vector (a gift from prof. Tiebang Kang, SYSUCC) with a C‐terminal HA tag. The cDNA of OMM‐anchoring form of PTPRJ (mito‐PTPRJ) was generated by fusing a cDNA fragment of the hydrophobic region (LILAMLAIGVFSLGAFIKIIQLRKNN) from Listeria monocytogenes actin assembly‐inducing protein (ActA) protein [35] with the PTPRJ ORF. All point mutations were generated by site‐directed mutagenesis. Mutant of MFN2(T105M) was used in our previous study [34]. The short hairpin RNA (shRNA) constructs were cloned into the PLKO.1‐puro vector (#8453, Addgene, Cambridge, MA, USA). The small interfering RNA (siRNA) oligos against Rab21 and PTPRJ were commercially synthesized by Gene Pharma (Shanghai, China) and transfected into cells using Lipofectamine RNAiMAX (Invitrogen). The siRNA sequences are as follows: siMFN2 #1, 5’‐GGAAGACAUUGAGUUCCAUUU‐3’; siMFN2 #2, 5’‐GGUUUAUAAGAAUGAGCUGCA‐3’; siRab21 #1, 5’‐GGAACUCUUUCUUGACCUUUGUAAA‐3’; siRab21 #2, 5’‐GAUUCCAUGCAUUGGGUCCAAUUUA‐3’; siPTPRJ #1, 5’‐ACGAGUCGUCAUCUAACUAUA‐3’; siPTPRJ #2, 5’‐CCGAUACAAUGCCACCGUUUA‐3’.
2.3. Cell culture
Human renal cancer cell lines (786‐O, A‐498 and Caki‐1), immortalized renal epithelial cell line (HK‐2) and 293T cells were originally obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). 786‐O and A‐498 cells were cultured in RPMI‐1640 medium (Gibco, Carlsbad, CA, USA), HK‐2 cells were cultured in keratinocyte serum‐free medium (Gibco), 293T cells were cultured in Dulbecco's Modified Eagle Medium (DMEM; Gibco), and Caki‐1 cells were cultured in McCoy's 5A Medium (Gibco). The media were all supplemented with 10% fetal bovine serum (FBS; Gibco). Cells were cultured in a 5% CO2 humidified incubator at 37°C. MFN2‐knockdown 786‐O or HK‐2 cells were transfected with wild‐type (WT) MFN2 or mutant MFN2 (T105M) lentivirus to construct stable expression cells. The SF9 insect cell line was a gift from Professor Ping Yin (Huazhong Agricultural University, Wuhan, Hubei, China). SF9 cells were grown in Sf‐900 II SFM medium (Invitrogen) at 28°C. All cell lines were tested negative for mycoplasma contamination with MycoBlue Mycoplasma Detector (D101, Vazyme, Nanjing, Jiangsu, China) according to the manufacturer's instructions.
2.4. Cell proliferation assays
For cell proliferation assay, ccRCC cells were seeded in 96‐well plates (1,500 cells/well) in fresh medium. At each time point, 10 μL of Cell Counting Kit‐8 solution (CCK‐8; Dojindo, Tokyo, Japan) was added directly to each well, followed by incubation for 3 h at 37°C. For drug treatment, cells were seeded in 96‐well plates (1,500 cells/well) with 100 μL culture medium, then treated with 2 μmol/L gefitinib (MedChemExpress, Princeton, NJ, USA), lenvatinib (MedChemExpress) or cabozantinib (MedChemExpress) for 72 h, followed by adding CCK‐8 solution. The optical density was measured at 450 nm (OD450) by Microplate Reader (BioTek, Vermont, USA). Each experiment was performed in triplicate.
In the colony formation assay, cells were plated in 6‐well plates (500 cells/well) in 2 mL of fresh medium and cultured for 10 days. Visible colonies were stained with 0.5% crystal violet, and colonies were counted with ImageJ software (National Institutes of Health, Bethesda, MD, USA).
2.5. Transwell assays
For the transwell migration assay, 4 × 104 cells in 200 μL serum‐free DMEM were placed into the upper chamber of a 24‐well transwell insert (pore size, 8.0 μm; Corning Falcon, New York, NY, USA), and the lower chamber was filled with 700 μL cell culture medium supplemented with 10% FBS. For the invasion assay, the chamber membrane was pre‐coated with Matrigel at 37°C for 1 h. After incubation for 12 h, the migrated and invaded cells in the lower filters were fixed in methanol, stained with 0.5% crystal violet, and counted under a microscope (Nikon, Tokyo, Japan). For drug treatment, cells were treated with 2 μmol/L gefitinib, lenvatinib, or cabozantinib (all from MedChemExpress) for 24 h.
2.6. Immunohistochemical (IHC) staining
Paraffin‐embedded tumor samples and mouse kidney tissues were processed into 4 μm sections, and the slides were dried at 65°C for 30 min before deparaffinization in xylene and rehydration in a graded ethanol series. Antigen retrieval was performed with the high‐pressure method for 3 min in 0.01 mol/L citrate buffer (pH 6.0) and subsequently treated with 3% hydrogen peroxide to quench the endogenous peroxidase activity. The slides were incubated with specific primary antibodies overnight at 4°C. Primary antibodies included MFN2 (1:100; ab218162, Abcam, Cambridge, MA, USA), Ki67 (1:200; ab15580, Abcam), Rab21 (1:200; sc‐81917, Santa Cruz Biotechnology, Dallas, TX, USA), p‐EGFR (1:200; #3777, Cell Signaling Technology, Danvers, MA, USA), p‐AKT (1:100; #3777, Cell Signaling Technology), alpha smooth muscle actin (α‐SMA; 1:2,000; ab232784, Abcam), carbonic anhydrase IX (CA9; 1:400, PA1‐16592, Invitrogen) and CD3 (1:200; ab16669, Abcam). For quantification, the slides were assessed according to the intensity and area of the staining by two independent pathologists. The staining intensity was scored as 0 for no staining, 1 for weak staining, 2 for medium staining, and 3 for strong staining. The proportion of positive cells was scored as follows: 0 for <5%, 1 for 5%–25%, 2 for 26%–50%, 3 for 51%–75%, and grade 4 for >75%. The staining index (SI) = staining intensity × proportion of positive cells.
2.7. RNA isolation and quantitative real‐time polymerase chain reaction (qRT‐PCR)
Total RNA was extracted from cell lines and tissue specimens using Trizol reagent (Invitrogen). First‐strand cDNA was synthesized using the HiScript II 1st Strand cDNA Synthesis Kit (Vazyme) with 1 μg of total RNA following the manufacturer's instructions. The cDNA samples used for gene expression analysis were diluted by 1:20. qRT‐PCR was performed on a CFX96 real‐time PCR detection system (Bio‐Rad, Hercules, CA, USA), and a HiScript II One Step qRT‐PCR SYBR Green Kit (Vazyme) was used for gene detection. Tubulin was used as an internal control to calculate the relative mRNA levels. Primers for MFN2 were as follows: forward, 5’‐CACATGGAGCGTTGTACCAG‐3’; reverse, 5’‐TTGAGCACCTCCTTAGCAGAC‐3’. Primers for Tubulin were as follows: forward, 5’‐TGGACTCTGTTCGCTCAGGT‐3’; reverse, 5’‐TGCCTCCTTCCGTACCACAT‐3’. qRT‐PCR was performed according to the manufacturer's instructions. Breifly, the cycling reaction was as follows: initial denaturation at 95˚C for 30 s, followed by 40 cycles of denaturation at 95˚C for 10 s and annealing at 60˚C for 30 s.
2.8. Subcellular fraction isolation
For mitochondria isolation, cells were harvested and washed with phosphate‐buffered saline (PBS), and then resuspended in isolation buffer containing 20 mmol/L 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonicaci (HEPES, pH 7.5; Sigma, St Louis, MO, USA), 70 mmol/L sucrose, 210 mmol/L mannitol, 0.5 mmol/L ethylene diamine tetraacetic acid (EDTA, Sigma), 1 mg/mL bovine serum albumin (BSA, Sigma), and 1 mmol/L phenylmethanesulfonylfluoride (PMSF, Sigma). The cells were lysed on ice using a Dounce homogenizer (Sigma), and the homogenate was centrifuged twice at 1,000 × g for 10 min at 4°C. The supernatant was further centrifuged at 10,000 × g for 20 min at 4°C to obtain the crude mitochondrial fraction. To prepare pure mitochondrial fractions, the isolated crude mitochondrial fractions were centrifuged at 100,000 × g for 30 min at 4°C with a 30% Percoll gradient in isolation buffer. The mitochondria were washed and collected for experiments.
Plasma membrane proteins were isolated using the Plasma Membrane Protein Extraction Kit (ab65400, Abcam). Briefly, cells were harvested, washed twice with ice‐cold PBS, and resuspended in homogenization buffer. The cells were lysed using a Dounce homogenizer, and the homogenate was centrifuged twice at 1,000 × g for 10 min at 4°C. The supernatant was further centrifuged at 10,000 × g for 30 min at 4°C to obtain the cytosolic fraction and pellets. The pellets were further purified using upper and lower phase solution according to the manufacturer's (Abcam) instructions.
To detect the distribution of PTPRJ on mitochondria, mitochondria isolated from 786‐O cells were treated with 10 μg/mL proteinase K (Gibco) with or without 1% Triton X‐100 (Anatrace, Maumee, OH, USA) for 30 min at 4°C, and the reaction was terminated by the addition of PMSF.
2.9. Co‐immunoprecipitation (Co‐IP) and Western blotting
Cells were lysed in ice‐cold lysis buffer (50 mmol/L Tris‐HCl [pH 7.5], 150 mmol/L NaCl, 1% Triton X‐100, 1 mmol/L EDTA) with 1 × protease inhibitor cocktail (C0001, TargetMol, Boston, MA, USA) for 30 min. Heterologously expressed MFN2‐Flag and EGFR‐HA were subjected to anti‐Flag G1 affinity resin (L00432, GenScript, Nanjing, Jiangsu, China) or anti‐HA Magnetic Beads (#88836, Thermo‐Fisher Scientific, Waltham, MA, USA). The precipitated proteins were eluted by 400 μg/mL Flag peptide (RP10586, GenScript) or 1 mg/mL HA peptide (TP1276, TargetMol). For co‐IP of endogenous MFN2, cell lysates were incubated with anti‐MFN2 antibody (1‒2 μg) overnight at 4°C. Subsequently, the pre‐cleared protein A/G magnetic beads (#26162, Thermo‐Fisher Scientific) were added to capture the protein complexes. In both cases, the beads were extensively washed at least five times with lysis buffer to remove non‐specifically associated proteins. Eluted proteins were boiled for 10 min in 1 × sodium dodecyl sulphate (SDS) loading buffer and then analyzed by Western blotting. Protein samples were loaded and separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) and transferred to a 0.45 μm polyvinylidene difluoride (PVDF) membrane (Merck Millipore, Billerica, MA, USA). After blocking, proteins were immunoblotted with the indicated antibodies.
2.10. Protein expression and purification
The full‐length human MFN2 was cloned into pFastBac1 vector (Invitrogen) with an N‐terminal 3 × Flag tag. The recombinant MFN2 was expressed in SF9 insect cells using the Bac‐to‐Bac baculovirus system (Invitrogen). Briefly, bacmid DNAs were generated in DH10Bac cells (Alpalife, Shenzhen, Guangdong, China), and the resulting baculoviruses were generated and amplified in SF9 cells. After baculovirus infection, cells were cultured at 28°C for 48 h before harvesting. To obtain proteins for bio‐layer interferometry (BLI) assays, the truncated human MFN2 (MFN2IM) [34] was cloned into a modified pET28 vector (pSKB) with an N‐terminal 6 × His‐tag; full‐length human Rab21 carried a glutathione S‐transferase (GST)‐tag. Transformed E. coli Rosetta (DE3) cells (Alpalife) were cultured at 37°C and induced with 0.1 mmol/L isopropyl‐1‐thio‐β‐d‐galactopyranoside (IPTG, Sigma) at OD600. Cells were cultured for 20 h at 18°C before harvesting.
For purification of full‐length MFN2, cells were harvested by centrifugation at 800 × g for 20 min at 4°C and washed with PBS, then resuspended in the buffer containing 20 mmol/L HEPES (pH 7.5), 70 mmol/L sucrose, 210 mmol/L mannitol, 0.5 mmol/L EDTA, 1 mg/mL BSA, and 1 mmol/L PMSF. The cells were homogenized on ice using a Dounce homogenizer for 80 cycles, and the homogenate was centrifuged twice at 1,000 × g for 10 min at 4°C. The supernatant was further centrifuged at 10,000 × g for 20 min at 4°C to obtain crude mitochondria. MFN2 was subsequently extracted from the crude mitochondria by 1.2% n‐dodecyl‐b‐D‐maltoside (DDM, Anatrace) in lysis buffer containing 20 mmol/L HEPES (pH 7.5), 500 mmol/L NaCl, 1 mmol/L EDTA, and 1 × protease inhibitor cocktail for 2 h at 4°C. The extraction was centrifuged at 40,000 × g for 1 h at 4°C to remove the insoluble component. The supernatant was incubated with anti‐Flag G1 affinity resin at 4°C for 2 h and then washed three times with 10 column volumes of lysis buffer plus 0.1% DDM. The protein was eluted with lysis buffer containing 1 mmol/L dithiothreitol (DTT; Gibco), 0.1% DDM, and 400 μg/mL Flag peptide. The Flag‐tagged MFN2 was used for further assay.
For the internal truncated version of MFN2 (MFN2IM, used in BLI experiment) [34] and Rab21 purification, E. coli cells were lysed in buffer containing 50 mmol/L HEPES (pH 7.5), 300 mmol/L NaCl, and 2 mmol/L MgCl2 with a cell disruptor (JNBIO, Guangzhou, Guangdong, China). The insoluble fractions were removed by centrifugation at 40,000 × g for 1 h at 4°C. His‐tagged MFN2IM was purified as described previously [34]. For purification of GST‐tagged Rab21, the protein was applied to a GST column (GE Healthcare, Pittsburgh, PA, USA) pre‐equilibrated with lysis buffer, and eluted with lysis buffer containing extra 15 mmol/L reduced glutathione (GSH).
2.11. BLI assay
The BLI assay was conducted on an Octet RED96 (ForteBio, Menlo Park, CA, USA). Anti‐His biosensors (ForteBio) were pre‐equilibrated with buffer containing 50 mmol/L HEPES (pH 7.5), 300 mmol/L NaCl, 2 mmol/L MgCl2, and 2 mmol/L DTT for 10 min before each assay. To determine the dissociation constant (Kd), at least three concentrations of recombinant MFN2IM were added to obtain the binding data. The global fitting algorithm of the Octet Data Analysis software (ForteBio) was used to analyze the data. The results were presented in Graphpad Prism 8.0 (La Jolla, CA, USA).
2.12. Pull‐down assay and liquid chromatography‐mass spectrometry (LC‐MS/MS) analysis
293T or 786‐O cells were lysed with radio immunoprecipitation assay (RIPA) buffer containing 1 × protease inhibitor cocktail. Flag affinity resins with or without Flag‐tagged MFN2 coating (MFN2‐beads) were incubated with cell lysates at 4°C overnight. Beads were washed five times with RIPA buffer and eluted with 400 μg/mL Flag peptide. The eluted protein samples were subjected to SDS‐PAGE or MS analysis. For the Rab21 pull‐down assay, HA magnetic beads (Gibco) were used.
2.13. Immunofluorescence (IF) staining
Paraffin‐embedded tumor samples were continuously sectioned at a thickness of 4 μm. Antigen retrieval was performed using 0.01 mol/L citrate buffer (pH 6.0) in a pressure cooker for 3 min, and subsequently treated with 3% hydrogen peroxide for 5 min. Non‐specific binding was then blocked with 5% BSA for 25 min at room temperature. Cells (2 × 104) for IF staining were seeded on cover slides and fixed with 4% paraformaldehyde for 15 min at room temperature, permeabilized with 0.1% Triton X‐100, and blocked in PBS with 2% BSA for 1 h at room temperature. Subsequently, the samples were incubated with appropriate primary antibodies at 4°C overnight and subjected to secondary antibodies conjugated with Alexa Fluor (Invitrogen) for 1 h at room temperature. Primary antibodies for IF staining included MFN2 (1:200; #11925, Cell Signaling Technology), Rab21 (1:100; sc‐81917, Santa Cruz Biotechnology), EGFR (1:100; #4267, Cell Signaling Technology) and PTPRJ (1:100; sc‐376794, Santa Cruz Biotechnology). Nuclei were counterstained with 4,6‐diamidino‐2‐phenylindole (DAPI; D3571, Invitrogen). For confocal microscopy, confocal images were acquired using a confocal laser‐scanning microscope (FV1000, Olympus, Tokyo, Japan) with a 100 × 1.45 numerical aperture (NA) oil immersion objective (Olympus). For structured illumination microscopy (SIM), super‐resolution images were taken with a Nikon N‐SIM system using a 100 × 1.49 NA oil immersion objective (Nikon). Images were captured with Nikon Imaging Software (NIS)‐Elements and reconstructed using slice reconstruction function. All IF experiments were performed three times with two replica and similar results were acquired.
2.14. Image analysis
The mitochondrial structure was reconstructed using Imaris 9.0 Microscopy Image Analysis Software (https://imaris.oxinst.com/). Mitochondrial contact of Rab21 and EGFR was defined as those with Rab21 or EGFR dots overlapping with mitochondria. The percentage of Rab21 or EGFR in contact with mitochondria was quantified as the area of Rab21 or EGFR dots that formed contact with mitochondria divided by the area of total dots in the view field. The colocalization of Rab21 and EGFR was calculated as Mander's overlap coefficients using the ImageJ software.
2.15. RNA‐sequencing (RNA‐seq)
Total RNA was extracted from 786‐O cells using a Trizol reagent kit. The mRNA was enriched by removing ribosomal RNA, digested and reversely transcribed into second‐strand cDNA, and purified using the QiaQuick PCR extraction kit (QIAGEN, Hilden, Germany). End‐paired segments were subjected to Illumina sequencing adapters (Illumina, San Diego, CA, USA). Next, products were selected by size and amplification. The constructed RNA‐seq libraries were sequenced by the Lianchuan Corporation (Hangzhou, Zhejiang, China). The raw sequencing reads were mapped to the reference genome using TopHat2 RNASeq alignment software (http://ccb.jhu.edu/software/tophat) and quantified by HTSeq (https://pypi.org/project/HTSeq/) to generate read counts for every gene. DESeq2 was used to estimate gene expression levels and identify differentially expressed genes (DEGs). The Benjamini‐Hochberg method was used to estimate the false discovery rate (FDR). DEGs were filtered using a minimum log2‐transformed fold change of 1 and a maximum FDR value of 0.05. Pathway analysis was conducted using the Wikipathways [36] of the WEB‐based Gene Set Analysis Toolkit (WebGestalt) (http://www.webgest‐alt.org/).
2.16. Xenograft mouse model
Male BALB/c nude mice (4 weeks old) were purchased from GemPharmatech (Nanjing, Jiangsu, China) and housed under specific pathogen‐free (SPF) conditions. Mice were randomly allocated into the indicated experiment groups (n = 5 per group). All animal experiments were approved by the Institutional Review Boards and Animal Care and Use Committees of SYSUCC (Approval No. L025501202107050). For the xenograft tumor growth assay, 1 × 107 786‐O or Caki‐1 cells in 150 μL PBS were subcutaneously injected into the dorsal flank of nude mice. Two weeks after inoculation, tumor size was measured every week. Seven weeks later, the mice were euthanized by CO2, and the tumor weights and volumes were calculated. The tumor volume was determined by the formula 0.5 × length × width2. The tumors were excised and fixed for further analysis. For gelfitinib treatment, when tumor volume reached about 4 mm in diameter (day 0), mice were randomly assigned to the following two treatments (n = 5 per group): vehicle and gelfitinib (50 mg/kg daily for 4 weeks with a 2‐week interval). For the pulmonary metastasis assays, 5 × 106 cells were injected into the tail veins of nude mice. Eight weeks later, the mice were euthanized by CO2, and lung nodules were counted and analyzed by hematoxylin and eosin (H&E) staining. When the tumor diameter reached over 20 mm or tumor burden exceeded 10% of the mouse body weight, the mice would be treated humanely with excessive carbon dioxide inhalation.
2.17. Patient‐derived xenograft (PDX) model
ccRCC specimens were obtained intraoperatively from patients at the FAH‐SYSU. Our study was implemented with the informed consent of patients and approved by the Institution Review Board. In addition, all animal experiments were reviewed and approved by the ethics boards and Animal Care and Use Committees of Sun Yat‐sen University (Approval No. L025501202107050). Briefly, freshly sectioned ccRCC tissues were transported in RPMI‐1640 on ice and minced finely into fragments of 1‐2 mm3, mixed with Matrigel, and implanted subcutaneously into the right flank of 6‐week‐old male immunocompromised NSG (NOD/ShiLtJGpt‐Prkdcem26Cd52Il2rgem26Cd22/Gpt) mice (GemPharmatech). When the tumor volume reached 50‐100 mm3, 5 × 108 MFN2 overexpression or control lentiviral particles were injected intratumorally for 2 consecutive days. Tumor growth was monitored every 12 days. On the 36th day, mice were euthanized by CO2, and tumors were collected for further experiments. Tumor volumes were calculated.
2.18. Transgenic mouse models
Cdh16‐Cre mice were purchased from Cyagen (Suzhou, Jiangsu, China), Mfn2 flox/flox mice were generated by GemPharmatech, and Vhl flox/flox mice were obtained from Shanghai Model Organisms (Shanghai, China). All mice were maintained under SPF conditions at the Laboratory Animal Resource Center of Sun Yat‐sen University. Eight‐week‐old male Cdh16‐Cre mice were crossed with 8‐week‐old female Mfn2 flox/flox mice to obtain Cdh16‐Cre/Mfn2 flox/flox mice as homozygous kidney‐specific conditional Mfn2 knockout (Mfn2 CKO) mice. Vhl flox/flox mice were crossed with Mfn2 CKO mice to generate Cdh16‐Cre/Vhl flox/flox (Vhl CKO) and Cdh16‐Cre/Vhl flox/flox /Mfn2 flox/flox (VhlMfn2 CKO) mice. The genotypes of transgenic mice were identified by PCR amplification of target DNA from mouse tails and agarose gel electrophoresis using the Animal Genomic DNA Quick Extraction Kit for PCR Analysis (D0065S, Beyotime, Shanghai, China). Primers for Cdh16‐Cre transgenic genotyping were forward (5’‐GCAGATCTGGCTCTCCAAAG‐3’) and reverse (5’‐AGGCAAATTTTGGTGTACGG‐3’). Primers for floxed Mfn2 knockout allele genotyping were 5’loxP forward (5’‐GGCAGCTTTTATTCTGGCCTCAGA‐3’) and 3’ loxP reverse (5’‐TCAGGAGAGAGAGGTAGGAGGGTCTCT‐3’). Vhl‐flox allele primers were 5’loxP forward (5’‐TAGTCCCCAGCATAACAAACACC‐3’) and 3’ loxP reverse (5’‐TGCAACTGCCCAAAATTCTCAAAG‐3’).
For primary culture of renal tubule cells, the kidney cortex collected from Mfn2 CKO mice were digested with 1 mg/mL Liberase TM (Roche, Indianapolis, IN, USA) and 1 μmol/L DNase I (Takara, Tokyo, Japan) for 30 min at 37°C and filtered through the 100‐μm mesh to isolate cells. Cells were cultured in RPMI‐1640 containing 10% FBS, 20 ng/mL EGF, and 1% penicillin/streptomycin (Gibco) in a 5% CO2 humidified incubator at 37°C. Cells were starved and stimulated with 100 ng/mL EGF for 30 min before Western blotting analysis.
Blood urea nitrogen (BUN) and creatinine were measured by the Urea (BUN) Quick Test Strips (MAS008, Sigma) or the Creatinine Assay Kit (MAK080, Sigma) as manufacturer's suggestions. For flow cytometry, kidney lesions or cortex from healthy mice were digested and prepared into single‐cell suspension. Cells were incubated with antibodies for 30 min. Antibodies included CD45 (1:200; #103126, Biolegend, San Diego, CA, USA), CD11b (1:200; #101206, Biolegend), CD11c (1:200; #117308, Biolegend), and F4/80 (1:200; #123110, Biolegend). The samples were determined by the Beckman CytoFLEX Flow cytometer (Beckman Coulter, Brea, CA, USA), and data were analyzed by FlowJo V10 (FlowJo LLC, Ashland, OR, USA).
2.19. Identification of apoptosis
Apoptosis of HK‐2 and 786‐O cells were detected by an apoptosis detection kit (MultiSciences, Hangzhou, Zhejiang, China). Cells were stained with annexin V and propidine iodide (PI) and determined by flow cytometry (Beckman Coulter). Data were analyzed by FlowJo V10 (FlowJo LLC).
2.20. Analyses of TCGA data
The clinical data and RNA‐seq data of the Cancer Genome Atlas kidney renal clear cell carcinoma (TCGA‐KIRC) cohort were downloaded from Firebrowse (http://firebrowse.org/). The mRNA expression data of primary and metastatic ccRCC samples were downloaded from the Gene Expression Omnibus (GEO) database (GSE73121) [37]. Normalized protein expression data of TCGA‐KIRC samples were download from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) data portal (https://pdc.cancer.gov/pdc/browse). R package DESeq2 (version 1.30.1) was used to calculate the fold change and adjusted P values for the mRNA expression of MFN2 between ccRCC samples and adjacent normal tissues. R packages DESeq2 (version 1.30.1) was used to identify the DEGs between normal and tumor tissues as well as the DEGs between MFN2‐high (>50%) and ‐low (≤50%) expression tumor tissues. The R packages Seurat (version 4.0.1) was used to calculate the DEGs between primary and metastatic ccRCC PDX tissues. R packages pROC (version 1.18.0) was used for area under curve (AUC) evaluation of the predictive prognosis value of candidate genes. The heatmap was plotted by the pheatmap package (version 1.0.12) with the gene expression values normalized by the rows, and the chord graph was drawn with ggplot2 package (version 3.0.0).
2.21. Statistical analysis
Statistical tests were performed as indicated in the figure legends and text. All data are presented as mean ± standard deviation (SD), with a minimum of three biologically independent experiments for significance. Data were analyzed using GraphPad Prism 8.0 software (GraphPad Software, San Diego, CA, USA). Unpaired and two‐tailed student's t‐tests were used for the intergroup comparisons and one‐way analysis of variance (ANOVA) for comparisons among multiple groups unless otherwise stated. Survival analysis was performed using the Kaplan‐Meier method, and the log‐rank test was used for comparisons. OS was defined as the interval from being diagnosed to death of any cause; disease‐free survival (DFS) was defined as the interval from being diagnosed to recurrence or death. Tumor cells or colonies were counted using ImageJ software.
3. RESULTS
3.1. Identification of MFN2 as a key regulator of tumor progression and a favorable prognosis marker in ccRCC
To screen key regulators of tumorigenesis and metastasis of ccRCC, we analyzed gene expression in two ccRCC cohorts from TCGA‐KIRC and GSE73121 datasets, respectively. Totally, 3,604 up‐regulated and 2,704 down‐regulated genes were found in TCGA‐KIRC tumor tissues (Figure 1A), while 579 up‐regulated and 1,341 down‐regulated genes were found in metastatic ccRCC samples (Supplementary Figure S1A). To identify the tumor suppressors that inhibit the initiation and development of ccRCC, we overlapped the down‐regulated genes between TCGA‐KIRC and GSE73121 datasets, and 354 genes were noted (Figure 1B). Candidate genes were subjected to AUC analysis to evaluate their prognostic values using the TCGA‐KIRC database (Figure 1C). Among these genes, MFN2 was significantly down‐regulated in ccRCC tumor tissues, especially in metastatic ccRCC (Figure 1D, Supplementary Figure S1B). While reported as a tumor suppressor in various tumors [30, 31], its role in ccRCC has not yet been illustrated. Subsequent analysis of the CPTAC database indicated that MFN2 protein level was significantly decreased in ccRCC (Supplementary Figure S1C). The down‐regulation of MFN2 in ccRCC samples of patients from FAH‐SYSU was subsequently validated by Western blotting analysis (Figure 1E), IHC and IF staining (Figure 1F‐G), as well as qPCR (Supplementary Figure S1D). Further analysis of the TCGA‐KIRC data revealed that MFN2 expression was associated with clinical stage (Figure 1H), tumor stage (Figure 1I), metastasis status (Figure 1J) and histological grade (Supplementary Figure S1E). Next, we examined MFN2 expression levels in a panel of human ccRCC cells (786‐O, A‐498, and Caki‐1) and in HK‐2 cells. MFN2 expression levels were significantly lower in tumor cells than in normal cells (Figure 1K‐L). These results suggested that reduced MFN2 expression was a prominent feature of ccRCC.
FIGURE 1.
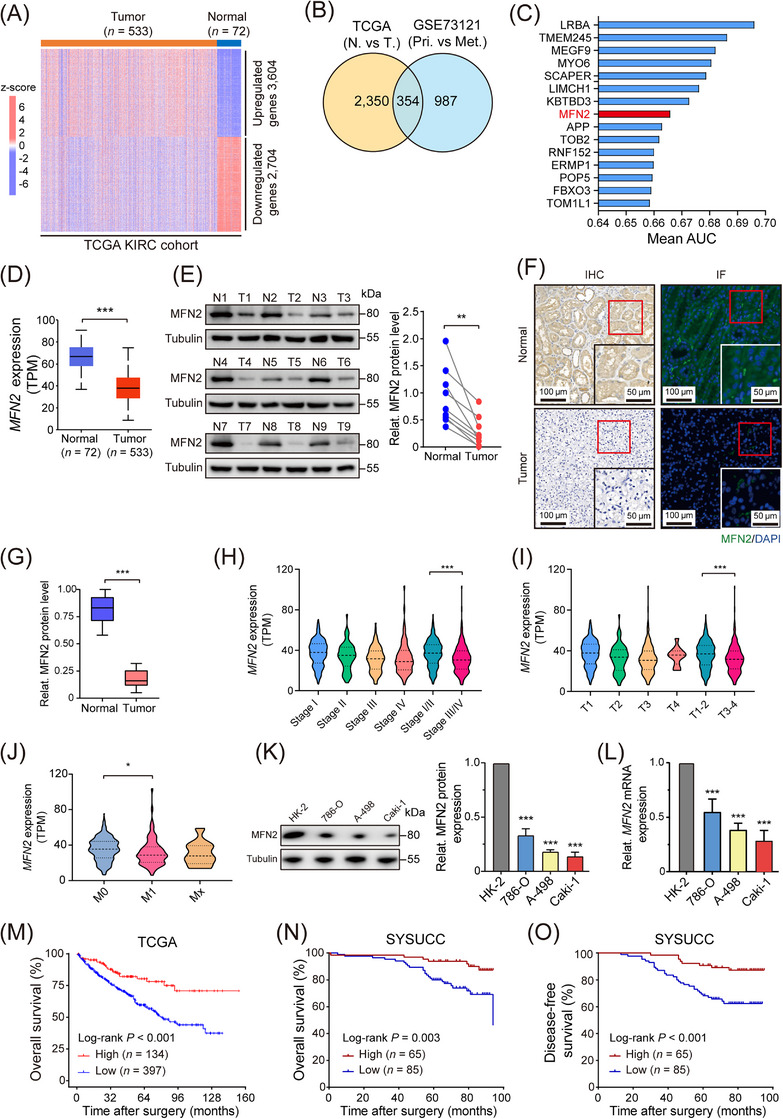
MFN2 is down‐regulated in human ccRCC and associated with favorable prognosis. (A) Heat map showing differentially expressed genes between normal and tumor tissues in TCGA‐KIRC cohort. (B) Strategy to identify candidate genes involved in tumorigenesis and metastasis of ccRCC. (C) AUC evaluation of the prognostic value of identified candidate genes based on TCGA‐KIRC database. (D) MFN2 mRNA expression levels in normal and tumor tissues from TCGA‐KIRC database. (E) Representative images of MFN2 protein levels in nine pairs of human ccRCC tumor tissues and adjacent normal tissues. Tubulin was used as a loading control. The statistical analysis is shown in the right panel. (F) Representative images of IHC staining (left panels) and IF staining (right panels) for MFN2 (green) and DAPI (blue) in human ccRCC tumor and normal tissues. (G) Quantification of MFN2 protein levels in human ccRCC tumor and normal tissues from IHC staining of Figure 1F (n = 10). (H‐J) MFN2 mRNA expression levels in different grades and stages in TCGA‐KIRC database: clinical stage (H), tumor stage (I) and metastasis status (J) according to the 8th edition of AJCC TNM Staging. (K) Western blotting image (left panel) and statistical analysis (right panel) of MFN2 protein expression detected in immortalized renal epithelial cell line HK‐2 and ccRCC cell lines (786‐O, A‐498, Caki‐1). Tubulin was used as a loading control. (L) Relative MFN2 mRNA expression of HK‐2 and ccRCC cells. Tubulin was used as a loading control. (M) OS of ccRCC patients with high (n = 134) or low (n = 397) expression of MFN2 in TCGA‐KIRC cohort. (N‐O) OS (N) and DFS (O) of ccRCC patients with high (n = 65) or low (n = 85) expression of MFN2 in SYSUCC cohort, determined by MFN2 IHC staining, Data are presented as means ± SD. *P < 0.05, ** P < 0.01, *** P < 0.001, by Student's t test (D, E, G, H, I, J, K) or log‐rank test (M, N, O).
Abbreviations: TCGA‐KIRC, the Cancer Genome Atlas kidney renal clear cell carcinoma; MFN2, mitofusin‐2; AUC, area under curve; N, normal; T, tumor; Pri, primary tumor; Met, metastatic tumor; M, metastasis; TPM, transcripts Per Kilobase of exon model per Million mapped reads; IHC, immunohistochemistry; IF, immunofluorescence; DAPI, 4,6‐diamidino‐2‐phenylindole; Relat., relative; SYSUCC, Sun Yat‐sen University Cancer Center; OS, overall survival; DFS, disease‐free survival.
To assess the clinical prognostic value of MFN2 in ccRCC, we analyzed the corresponding prognosis data in TCGA‐KIRC cohort, which indicated that higher expression of MFN2 was associated with better patient survival (Figure 1M). Mitofusin‐1 (MFN1), the MFN2 homolog also possessing mitochondrial fusogenic activity, showed less degree of down‐regulation in tumor tissues and no prognostic potential in TCGA‐KIRC cohort (Supplementary Figure S1F‐G). Consistent with TCGA results, we found that high MFN2 expression was associated with a favorable OS and DFS in an independent ccRCC cohort from SYSUCC (Figure 1N‐O, Supplementary Figure S1H). Collectively, these results indicated that MFN2 was a favorable prognostic marker for patients with ccRCC.
3.2. MFN2 loss promoted EGFR signaling and accelerated ccRCC growth and metastasis
To explore the potential role of MFN2 in ccRCC development, we knocked down MFN2 in cells with high MFN2 expression (HK‐2, 786‐O cells) and overexpressed MFN2 in cells with low MFN2 expression (Caki‐1, A‐498 cells) (Supplementary Figure S2A). MFN2 depletion promoted the proliferation, migration and invasion of 786‐O and HK‐2 cells, whereas exogenous overexpression of MFN2 in Caki‐1 and A‐498 cells led to opposite effects (Supplementary Figure S2B‐D). However, MFN2 depletion did not affect the survival of HK‐2 and 786‐O cells (Supplementary Figure S2E). To further elucidate the exact role of MFN2 in suppressing ccRCC initiation and progression, we analyzed the transcriptome profiles from TCGA‐KIRC cohort based on MFN2 expression levels using WikiPathway. We found that MFN2 was closely related to EGFR and its downstream signaling pathways, including the RAS‐mitogen‐activated protein kinase (MAPK), phosphatidylinositol 3‐kinase (PI3K)‐AKT‐mTOR, and Janus‐family tyrosine kinase (JAK)‐signal transducer and activator of transcription 3 (STAT3) pathways (Figure 2A). To confirm this, we performed transcriptome profiling in MFN2‐knockdown and control 786‐O cells, and observed significant difference between the two groups (Supplementary Figure S3A). RNA‐seq results showed that depletion of MFN2 in 786‐O cells led to up‐regulation of genes functionally enriched in EGFR and related downstream pathways (Figure 2B), which is consistent with the TCGA data. These results were further validated by Western blotting. Knockdown of MFN2 in 786‐O cells increased the phosphorylation level of EGFR (p‐EGFR), as well as those of the downstream targets STAT3 (p‐STAT3), AKT (p‐AKT), and ERK (p‐ERK), while MFN2 overexpression decreased the phosphorylation levels of these proteins in Caki‐1 cells (Figure 2C). We then found that the protein level of MFN2 was negatively correlated with that of p‐EGFR in human ccRCC samples (Figure 2D, Supplementary Figure S3B).
FIGURE 2.
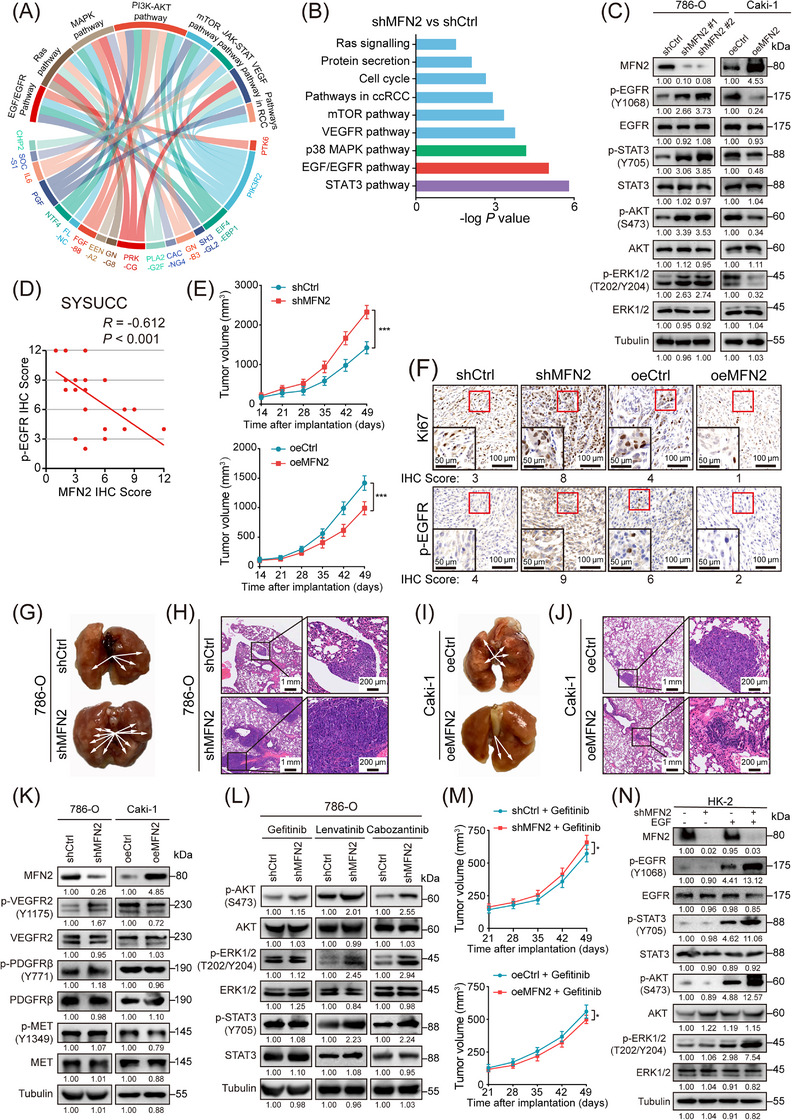
MFN2 inhibits the proliferation and metastasis of ccRCC via EGFR signaling. (A) Chord graph representing 17 differentially expressed genes between MFN2 high and low expression groups in TCGA‐KIRC cohort and the association of these genes to the corresponding oncogenic pathways. (B) Pathway analysis showed the significantly altered signaling pathways upon knockdown of MFN2 in 786‐O cells. (C) Western blotting analysis of EGFR pathway proteins in MFN2‐knockdown 786‐O cells and MFN2‐overexpressed Caki‐1 cells using the indicated antibodies. (D) Correlation analysis between MFN2 and p‐EGFR protein levels in human ccRCC samples (n = 30). (E) Growth curves of tumors from BALB/c nude mice subcutaneously implanted with MFN2‐knockdown and control 786‐O cells (upper panel) or MFN2‐overexpressing and control Caki‐1 cells (lower panel) (5 mice/group). Tumor volumes were measured at the indicated times. (F) Representative IHC staining images of Ki67 and p‐EGFR in MFN2 knockdown 786‐O tumors or MFN2 overexpression Caki‐1 tumors. IHC Scores are shown at the bottom. (G‐H) Representative images of lungs with metastatic nodules (G) and H&E staining (H) of metastatic lesions in the lungs of BALB/c nude mice after tail vein injection with MFN2‐knockdown and control 786‐O cells (n = 5 per group). Arrows indicate the metastatic colonies in the lung tissues. (I‐J) Representative images of lungs with metastatic nodules (I) and H&E staining (J) of metastatic lesions in the lungs of BALB/c nude mice after tail vein injection with MFN2‐overexpression and control Caki‐1 cells (n = 5 per group). Arrows indicate the metastatic colonies in the lung tissues. (K) Western blotting analysis of indicated RTKs (VEGFR2, PDGFRβ, c‐MET) in MFN2 knockdown 786‐O cells or MFN2 overexpression Caki‐1 cells using the indicated antibodies. (L) Western blotting analysis of indicated proteins in MFN2 knockdown 786‐O cells treated with indicated RTK inhibitors. (M) Growth curves of tumors from BALB/c nude mice subcutaneously implanted with MFN2 knockdown and control 786‐O cells (upper panel) or MFN2 overexpression and control Caki‐1 cells (lower panel) treated with Gefitinib (50 mg/kg, 5 mice per group). Tumor volumes were measured at the indicated times. (N) Western blotting assay of EGFR pathway proteins in MFN2 knockdown HK‐2 cells with or without EGF stimulation (100 ng/mL) using the indicated antibodies, Data are presented as means ± SD. ns, not significant. * P < 0.05, *** P < 0.001, by one‐way ANOVA (E, M) or Person's correlation analysis (D).
Abbreviations: oeCtrl, empty overexpression control; oeMFN2, overexpression of MFN2; shCtrl, negative control shRNA; shRNA, short hairpin RNA; IHC, immunohistochemistry; SYSUCC, Sun Yat‐sen University Cancer Center; VEGFR, vascular endothelial growth factor receptor; PDGFR, platelet‐derived growth factor receptor; MET, mesenchymal epithelial transition factor. p‐EGFR, phosphorylated epidermal growth factor receptor; p‐AKT, phosphorylated protein kinase B; p‐ERK1/2, phosphorylated extracellular signal‐regulated kinase 1/2; H&E, hematoxylin and eosin; RTK, receptor tyrosine kinase.
We then used a subcutaneous xenograft model of nude mice to investigate the effect of MFN2 on ccRCC tumor growth in vivo. Consistent with the in vitro results, knockdown of MFN2 in 786‐O tumors showed accelerated growth (Figure 2E, Supplementary Figure S3C), while overexpression of MFN2 in Caki‐1 tumors showed decelerated growth (Figure 2E, Supplementary Figure S3D). In line with the differences in tumor growth rate, the positive ratio of Ki67, a cell division marker, as well as the p‐EGFR level, were increased in 786‐O tumors with MFN2 knockdown, but decreased in Caki‐1 tumors with MFN2 overexpression (Figure 2F). In addition, pulmonary metastasis model was employed to explore the role of MFN2 in ccRCC tumor metastasis in vivo. Compared to the control groups, a greater number of metastatic nodules were detected in the group with MFN2 knockdown (Figure 2G‐H, Supplementary Figure S3E), and fewer metastatic nodules were found in the group with MFN2 overexpression (Figure 2I‐J, Supplementary Figure S3F). Taken together, these results showed that MFN2 inhibited ccRCC cell growth and metastasis in vitro and in vivo. Collectively, MFN2 was important for both neoplastic transformation and metastatic progression in renal cancer.
3.3. MFN2 suppressed ccRCC growth and metastasis through EGFR signaling pathway
Multiple receptor tyrosine kinase (RTK) signaling pathways, especially vascular endothelial growth factor receptor (VEGFR), platelet‐derived growth factor receptor (PDGFR) and EGFR pathways, are activated in ccRCC [38]. We sought to identify whether MFN2 regulates ccRCC growth and metastasis mainly through EGFR signaling. We screened multiple RTKs and found that compared with the effect on EGFR (Figure 2C), MFN2 overexpression only slightly inhibited VEGFR2 phosphorylation and did not significantly alter the phosphorylation of PDGFRβ and MET (Figure 2K). Gefitinib is a small molecule that inhibits ATP binding within the tyrosine‐kinase domain of EGFR, thus completely blocking EGF‐stimulated EGFR autophosphorylation on cell surface and subsequent signal transduction from activated EGFR [39]. Gefitinib, but not lenvatinib (an inhibitor of VEGFR, fibroblast growth factor receptor (FGFR), PDGFR, c‐kit and RET) or cabozantinib (an inhibitor of VEGFR, MET, c‐kit and Axl), inhibited the elevated levels of p‐AKT, p‐ERK and p‐STAT3 as well as the malignant phenotypes induced by MFN2 knockdown (Figure 2L, Supplementary Figure S4A‐C). MFN2 knockdown‐induced differences in in vivo tumor growth could be largely alleviated by gefitinib treatment (Figure 2M). Importantly, the effects of MFN2 knockdown on p‐EGFR and its downstream targets (p‐STAT3, p‐AKT and p‐ERK), as well as on the phenotypes of cell proliferation, migration and invasion, were dependent on EGF stimulation (Figure 2N, Supplementary Figure S4D‐I). These findings demonstrated that MFN2 restricted EGF‐induced EGFR activation and repressed ccRCC growth and metastasis in an EGFR signaling‐dependent manner.
3.4. Mice with kidney‐specific Mfn2‐knockout developed malignant lesions and were characterized by increased EGFR signaling activity
To test whether MFN2 loss contributes to renal carcinogenesis, we generated kidney‐specific Mfn2‐knockout mice (Cdh16‐Cre/Mfn2 flox/flox, hereby denoted as Mfn2 CKO) by crossbreeding Mfn2 flox/flox and Cdh16‐Cre transgenic mice [40] (Figure 3A). The Cdh16‐Cre promoter drove expression throughout the kidney epithelium, with the highest expression levels in the collecting ducts and more distal nephron segments [41]. Homozygous Mfn2 CKO mice were verified by genotyping and IHC staining (Supplementary Figure S5A‐B). Kidney functions, which were indicated by blood urea nitrogen (BUN) and creatinine levels, were unaffected in Mfn2 CKO mice (Supplementary Figure S5C). Mfn2 CKO mice were born at expected Mendelian ratios and did not appear sickly at birth. At 12 weeks of age, Mfn2 CKO mice had grossly normal kidneys similar to the controls, but the kidneys of 3 of 8 Mfn2 CKO mice at the age of 40 weeks developed typical malignant lesions (Figure 3B‐C). Compared with kidneys of control mice which exhibited organized tubule structures, kidneys of Mfn2 CKO mice displayed significant solid dysplasia, typical clear cytoplasm and a spectrum of cystic lesions (Figure 3D). Tubular lesions developed from irregular luminal contour, high cellularity and multiple layering of the epithelium to thorough structural abnormality surrounded by fibroblastic cells; dilated tubules, glomerular cysts, and vascular dysplasia were also observed in malignant regions (Supplementary Figure S5D). IHC analysis with anti‐Ki67 antibody indicated enhanced proliferation of renal tubular epithelial cells upon Mfn2 deletion (Supplementary Figure S5E). All pathological lesions showed strong immunoreactivity for pan‐cytokeratin (CK) (a marker for RCC) and c‐Myc (a marker for neoplastic lesions), and weak or median immunostaining for α‐SMA (a marker for myofibroblasts), indicating they were likely neoplastic lesions (Supplementary Figure S5F). Moreover, the Mfn2 CKO kidney lesions were infiltrated by CD3‐positive T lymphocytes (Supplementary Figure S6A) and myeloid cells including CD11b‐positive, F4/80‐positive and CD11c‐positive cells (Supplementary Figure S6B‐C), indicating an active inflammatory response.
FIGURE 3.
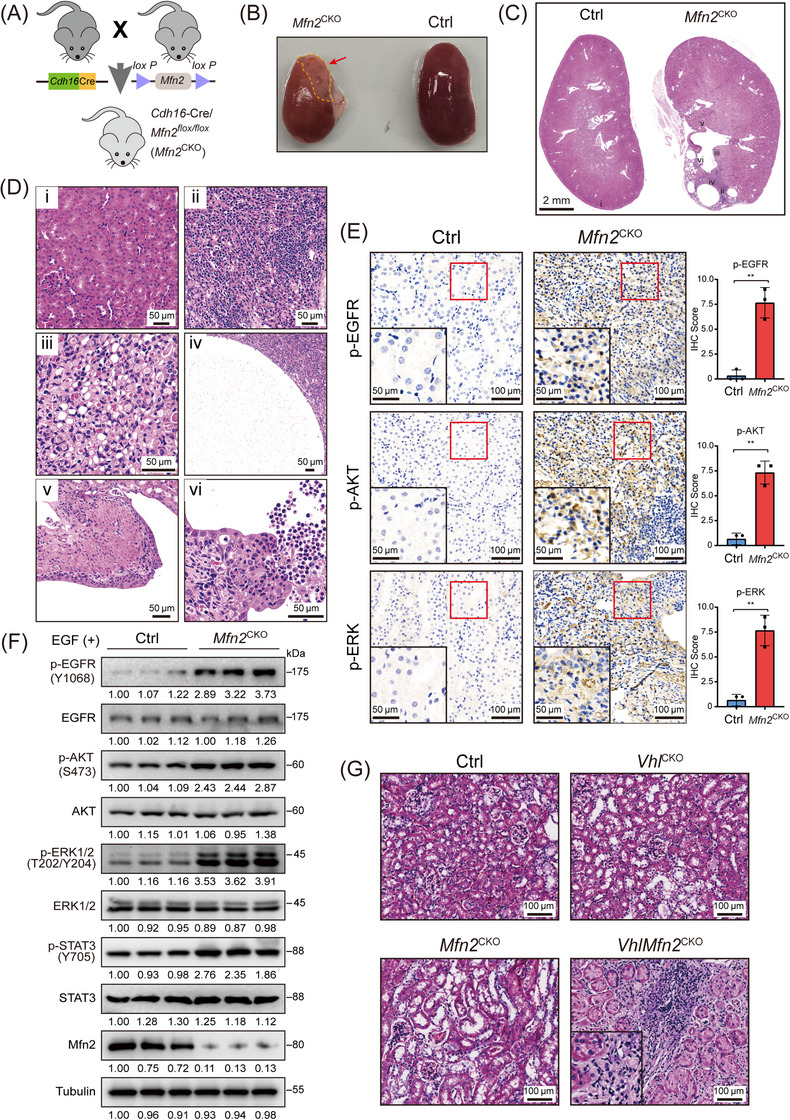
Cdh16‐Cre/Mfn2 flox/flox mice develops malignant lesions and exhibits EGFR signaling pathway activation. (A) Experimental scheme for generating the Mfn2 CKO mice. (B) Representative images of kidneys of Mfn2 CKO and control Mfn2 flox/flox mice taken at 40 weeks of age. (C) Representative H&E staining of kidney sections of 40‐week‐old control and Mfn2 CKO mice. (D) Representative H&E staining of kidney sections. (i) H&E staining of control mice display normal kidney morphology. (ii‐vi) H&E staining of kidney sections of Mfn2 CKO mice shows solid dysplasia (ii), clear cytoplasm (iii), a simple cyst lined by a single epithelial layer (iv), a cyst showing atypical epithelial cell proliferation (multi‐layered) (v) and a cyst showing extensive proliferation of epithelial cells into the cyst lumen (vi). (E) Representative IHC staining images of p‐EGFR, p‐AKT and p‐ERK in kidney sections of control and Mfn2 CKO mice (n = 3 per group). IHC scores are shown. (F) Western blotting analysis of indicated proteins in EGF‐treated primary renal tubular epithelial cells isolated from control and Mfn2 CKO mice. (G) Representative H&E staining of kidney sections from Ctrl, Vhl CKO, Mfn2 CKO and VhlMfn2 CKO mice.
Abbreviations: Mfn2 CKO, Cdh16‐Cre/Mfn2 flox/flox mice; CKO, conditional knockout; Ctrl, control Mfn2 flox/flox mice; H&E, hematoxylin and eosin; EGF, epidermal growth factor; Vhl, Von Hippel‐Lindau Tumor Suppressor; Vhl CKO, Cdh16‐CreVhl flox/flox mouse; VhlMfn2 CKO, Cdh16‐Cre/Vhl flox/flox /Mfn2 flox/flox mice.
We then investigated whether MFN2 deletion in kidney epithelial cells affected EGFR signaling activity. IHC analysis of kidney sections showed increased levels of p‐EGFR, p‐AKT and p‐ERK in Mfn2 CKO mice (Figure 3E). To further study the effect of MFN2 on kidney epithelial cells, we isolated renal tubule cells from kidneys of Mfn2 CKO and control mice. Western blotting analysis showed that MFN2 loss significantly increased EGF‐induced p‐EGFR, p‐AKT, p‐ERK and p‐STAT3 levels in cultured renal tubule cells compared to their counterparts in the control groups (Figure 3F). These transgenic mouse data demonstrated that Mfn2 deletion in kidney epithelial cells resulted in elevated EGFR signaling, which may cause malignancy of renal epithelium and serve as an oncogenic driver for kidney cancer.
To determine whether loss of MFN2 in the kidney accelerates development of ccRCC, we crossed Vhl flox/flox mice with Mfn2 CKO mice to generate Cdh16‐CreVhl flox/flox (Vhl CKO) and Cdh16‐Cre/Vhl flox/flox /Mfn2 flox/flox (VhlMfn2 CKO) mice (Supplementary Figure S7A). No renal abnormality was observed for aged control, Vhl CKO, or Mfn2 flox/flox mice. In contrast, 2 of 8 VhlMfn2 CKO mice developed renal lesions by 20 weeks of age (Figure 3G, Supplementary Figure S7B). Importantly, all renal lesions in double‐mutant mice showed immunoreactivity to carbonic anhydrase 9 (CA9, a marker of ccRCC and VHL inactivation) [42] and increased proliferation (Supplementary Figure S7C), indicating that combined loss of Vhl and Mfn2 in the kidney accelerated the development of renal neoplasia resembling ccRCC.
3.5. MFN2 interacted with Rab21 and tethered Rab21 to mitochondria
Given the above results, we were curious about whether the function of MFN2 in regulating EGFR signaling was dependent on its mitochondrial fusogenic activity. To verify this, we compared the p‐EGFR‐regulatory activity and biological function of a previously reported fusion‐incompetent MFN2 mutant, MFN2(T105M) [34], with wild‐type (WT) MFN2. Overexpression of MFN2(T105M) in MFN2‐knockdown cells reduced the p‐EGFR level as well as cell proliferation, migration and invasion with a similar efficiency as WT MFN2 upon EGF stimulation (Supplementary Figure S8A‐D), suggesting that the mitochondrial fusogenic activity of MFN2 was dispensable for its regulatory function in the EGFR signaling pathway in ccRCC cells.
To further elucidate the molecular mechanisms of MFN2 in regulating the EGFR signaling, Flag‐tagged MFN2 proteins purified from insect cells were subjected to pull‐down assays with lysates from 786‐O or 293T cells. Silver staining of the resulting SDS‐PAGE gel revealed a prominent band at ∼25 kDa (Figure 4A). Subsequent LC‐MS/MS identified several Rab proteins, among which Rab5A, Rab21, and Rab5C were the most enriched (Figure 4B, Supplementary Figure S8E). Co‐IP assays confirmed the interaction between MFN2 and Rab21 in 786‐O cells (Figure 4C‐D), but not Rab5A or Rab5C (Supplementary Figure S8F). BLI assays showed that purified Rab21 bound with a truncated version of MFN2 containing the GTPase domain and helical domain 1 (HD1) [34] with a dissociation constant (Kd) of 0.2 μmol/L (Figure 4E). Moreover, the interaction between MFN2 and Rab21 was not affected by MFN2 mutations that disrupted the GTPase and mitochondrial fusogenic activities (Supplementary Figure S8G), which was in agreement with the results that MFN2(T105M) restored the p‐EGFR levels and cell phenotypes in MFN2‐knockdown cells.
FIGURE 4.
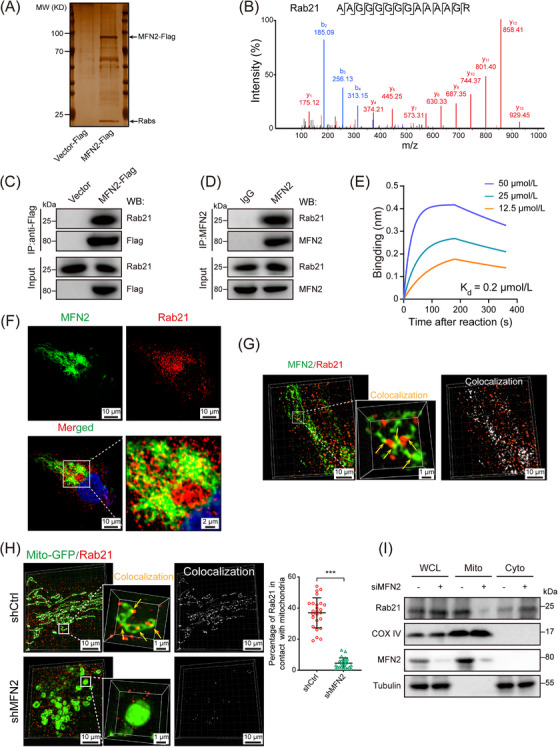
MFN2 interacts with Rab21 in ccRCC cells. (A) Silver staining showed that MFN2‐Flag was immunoprecipitated with Rab proteins. (B) Mass spectrometry identified Rab21‐unique 14‐aa peptide sequences in 786‐O cells. (C) Co‐IP of endogenous Rab21 with overexpressed MFN2‐Flag. Whole cell lysates were prepared from 786‐O cells stably expressing MFN2‐Flag. Co‐IP and Western blotting were performed with the indicated antibodies. (D) Co‐IP of endogenous Rab21 with endogenous MFN2. Whole cell lysates were prepared from 786‐O cells. Co‐IP and Western blotting were performed with the indicated antibodies and IgG was used as a control. (E) BLI assay showed that MFN2IM bound to Rab21 in vitro. Three MFN2 IM concentrations were used. (F) Immunofluorescence staining showing the co‐localization of endogenous MFN2 (green) and Rab21 (red) in 786‐O cells. (G) Representative 3D‐SIM images of MFN2 colocalization with Rab21 (yellow arrows) in 786‐O cells immunofluorescent stained for Rab21 and MFN2. (H) Representative 3D‐SIM images of mitochondrial localization of Rab21 (yellow arrows) in 786‐O cells stably transfected with mito‐GFP. Quantification of Rab21 in contact with mitochondria is shown in the right panel (n = 25 fields). (I) Mitochondrial localization of Rab21 is MFN2‐dependent. Whole cell lysates, mitochondria fractions, and cytoplasm were prepared from 786‐O cells treated with MFN2 or control siRNA and subjected to Western blotting, Data are presented as means ± SD. *** P < 0.001, by Student's t test (H).
Abbreviations: MW, molecular weight; Rabs, Ras related proteins; Co‐IP, co‐immunoprecipitation; WCL, whole cell lysates; Mito, mitochondria; Cyto, cytoplasm. BLI, bio‐layer interferometry; MFN2IM, truncated human MFN2; GFP, Green fluorescent protein; 3D‐SIM, three‐dimensional structured illumination microscopy; COX IV, cytochrome c oxidase subunit IV.
Rab21 is a small GTPase involved in endosome trafficking control [43] and has been reported to attenuate EGFR signaling transduction [44], but the underlying molecular mechanism remains unclear. Therefore, we focused on Rab21 to explore whether it was involved in the regulation of EGFR signaling by MFN2. IF co‐staining with MFN2 and Rab21 antibodies showed that Rab21 exhibited an apparent cytoplasm distribution and co‐localized with MFN2 in ccRCC cells (Figure 4F, Supplementary Figure S8H). Such co‐localization was further confirmed by 3D‐structured SIM analysis (Figure 4G). Further analysis revealed that a portion of cellular Rab21 was in close contact with mitochondria, which was diminished upon MFN2 depletion (Figure 4H, Supplementary Figure S8I). Moreover, we compared Rab21 levels between the mitochondrial and cytoplasmic fractions of 786‐O cells and found that MFN2 depletion led to significantly decreased levels of mitochondrial‐located Rab21 (Figure 4I). Thus, our results indicated that Rab21 can be driven in close proximity to mitochondria by physically interacting with MFN2.
3.6. Rab21 repressed ccRCC cell proliferation and migration in a MFN2‐dependent manner
By analyzing TCGA data and animal samples, we found that Rab21 has relatively high expression in ccRCC tumor tissues compared to other cancer types, and Rab21 also exhibited higher expression in normal kidney tissues than in other organs (Supplementary Figure S9A‐B), implying a critical role of Rab21 in ccRCC. Interestingly, high expression level of Rab21 was related with favorable patient survival in the TCGA‐KIRC cohort (Figure 5A), but with poor patient survival of many other cancer types (data not shown) in TCGA database. Consistent with TCGA data, we found that Rab21 was associated with prolonged OS and DFS in our independent ccRCC cohort from SYSUCC (Figure 5B‐C, Supplementary Figure S9C). Furthermore, in both TCGA‐KIRC and SYSUCC ccRCC cohort, patients with simultaneous high expression of MFN2 and Rab21 had optimal survival (Figure 5D‐E).
FIGURE 5.
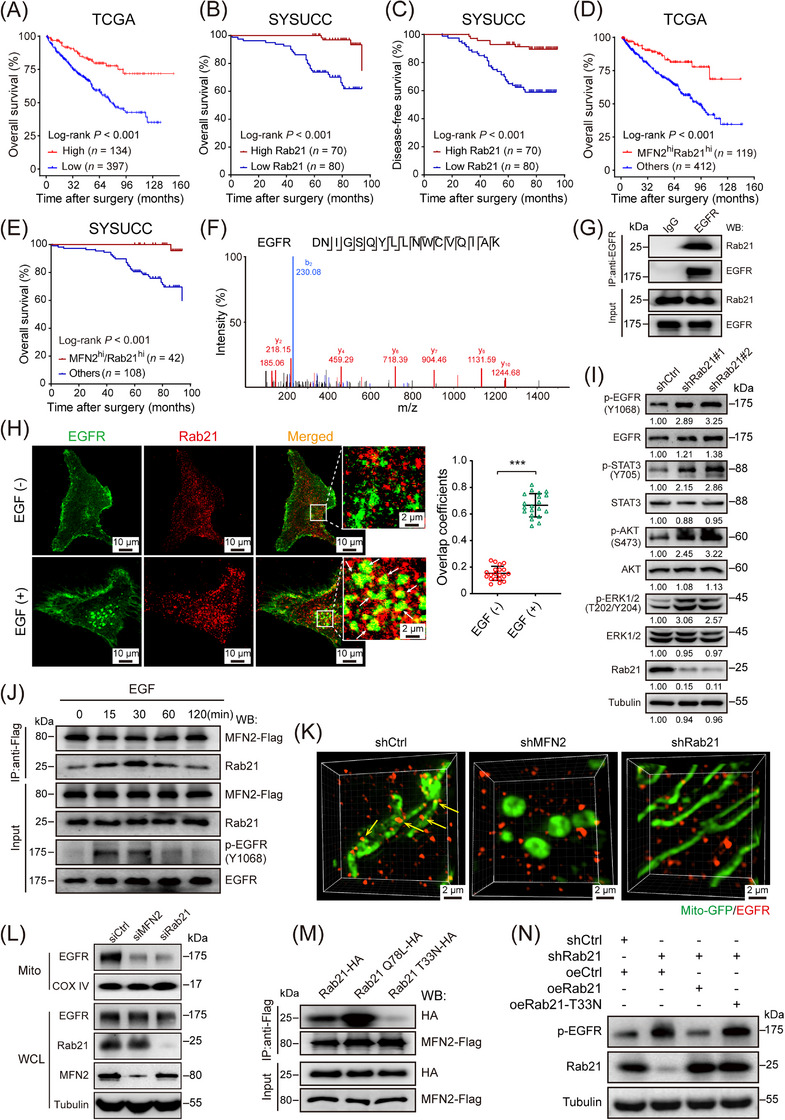
Rab21 interacts with EGFR and delivers EGFR to mitochondria in ccRCC cells. (A) OS of ccRCC patients with high (n = 134) or low (n = 397) expression of Rab21 in TCGA‐KIRC cohort. (B‐C) OS (B) and DFS (C) of ccRCC patients with high (n = 70) or low (n = 80) expression of Rab21 in SYSUCC cohort. (D) OS of ccRCC patients, with regard to the simultaneous high expression of MFN2 and Rab21 (n = 119), in TCGA‐KIRC cohort. (E) OS of ccRCC patients, with regard to the simultaneous high expression of MFN2 and Rab21 (n = 42), in SYSUCC cohort. (F) LC‐MS identified an EGFR‐unique 17‐aa peptide sequences in 786‐O cells. (G) Co‐IP of endogenous EGFR from 786‐O cells indicated that EGFR couples with Rab21. IgG was used as a control. (H) Representative immunofluorescence staining showing the co‐localization of EGFR and Rab21 in 786‐O cells upon EGF stimulation (left panel). EGF (‐): cells not treated with EGF; EGF (+): cells treated with 100 ng/mL EGF for 20 min. Statistical quantification of the co‐localization between MFN2 and Rab21 is shown in right panel. (I) Western blotting analysis of EGFR pathway proteins in Rab21‐knockdown 786‐O cells using the indicated antibodies. (J) MFN2 dynamically interacts with Rab21 in 786‐O cells. 786‐O cells were transfected with MFN2‐Flag, treated with EGF (100 ng/mL) for the indicated time points and total cell lysates were prepared. Co‐IP and the Western blotting were performed with the indicated antibodies. The interaction between MFN2 and Rab21 changed with prolonged EGF stimulation; the strongest interaction was at 30 min before declining over time. (K) Representative 3D‐SIM images of mitochondrial (mito‐GFP) localization of EGFR in the indicated 786‐O cells. (L) Mitochondrial localization of EGFR is MFN2 and Rab21‐dependent. Whole cell lysates and mitochondria were prepared from 786‐O cells with MFN2 or Rab21 silencing and then subjected to Western blotting analysis. (M) Co‐IP of MFN2‐Flag with WT Rab21, Rab21(Q78L) mutant, or Rab21(T33N) mutant indicated that MFN2 strongly interacts with the constant GTP‐loading variant Rab21(Q78L). (N) Western blotting assay of p‐EGFR levels in Rab21‐knockdown 786‐O cells with wild‐type or Rab21 mutant (T33N) reconstitution. Data are presented as means ± SD. *** P < 0.001, by Student's t test (H) or log‐rank test (A, B, C, D, E).
Abbreviations: OS, overall survival; DFS, disease‐free survival; hi, high; ccRCC, clear cell renal cell carcinoma; TCGA‐KIRC, the Cancer Genome Atlas kidney renal clear cell carcinoma; shCtrl, negative control shRNA; siCtrl, negative control siRNA; oeCtrl, empty overexpression control. LC‐MS, liquid chromatography‐mass spectrometry; WCL, whole cell lysates; Mito, mitochondria; WT, wild‐type; EGF, epidermal growth factor; IP, immunoprecipitation.
We further investigated the role of Rab21 in ccRCC tumorigenesis and metastasis. Knockdown of Rab21 significantly promoted the proliferation, migration and invasion capacities of 786‐O and HK‐2 cells (Supplementary Figure S9D‐F). However, these effects were similar to that of simultaneous MFN2 and Rab21‐knockdown (Supplementary Figure S9D‐F), suggesting that the tumor‐suppressive activity of Rab21 was largely dependent on MFN2.
3.7. Rab21 docked EGFR to mitochondria by cooperating with MFN2 to mediate EGFR signaling
We next sought to elucidate how MFN2‐Rab21 interaction works to suppress the progression of ccRCC. Interactome analysis revealed that EGFR was a strong candidate for the binding partner of Rab21 in 786‐O cells (Figure 5F, Supplementary Figure S9G). The association between endogenous MFN2 and Rab21 was confirmed by IP assays (Figure 5G). IF staining indicated that the co‐localization of Rab21 with EGFR was promoted upon EGF stimulation (Figure 5H). Similar to MFN2 (Figure 2C), knockdown of Rab21 increased p‐EGFR, p‐STAT3, p‐AKT and p‐ERK levels in 786‐O cells (Figure 5I). In human ccRCC samples, Rab21 protein levels were also negatively correlated with those of p‐EGFR (Supplementary Figure S9H). Furthermore, co‐IP assays indicated that the interaction between MFN2 and Rab21 underwent dynamic change during EGF stimulation, and p‐EGFR levels were decreased along with the enhancement of MFN2‐Rab21 interaction (Figure 5J). Given the potentials of Rab21 in binding with both MFN2 and EGFR, we wondered whether EGFR could be delivered to mitochondria via MFN2‐Rab21 interaction. Using 3D‐SIM analysis, we observed that EGFR and mitochondria formed abundant contact sites in 786‐O cells, and the number of these sites were significantly reduced when MFN2 or Rab21 was knocked down (Figure 5K, Supplementary Figure S9I), suggesting that MFN2 and Rab21 were both required for mitochondria juxtaposition of EGFR. In line with this, Western blotting showed that EGFR was present in the isolated mitochondrial fraction, and mitochondrial juxtaposition of EGFR was dramatically decreased with the silencing of MFN2 or Rab21 (Figure 5L). Taken together, these findings demonstrated that Rab21‐MFN2 interaction was essential for the mitochondrial delivery of EGFR.
As a Ras‐like GTPase, the activation status of Rab21 was determined by its binding with different guanine nucleotides [22]. To understand whether the GTP binding status of Rab21 affects its interaction with MFN2, we performed co‐IP assays and found that MFN2 preferably interacted with the constant GTP‐loading variant Rab21(Q78L), but barely the GDP‐binding variant Rab21(T33N) (Figure 5M). Consistent with the weak interaction between MFN2 and Rab21(T33N), overexpression of Rab21(T33N) failed to efficiently reduce p‐EGFR level (Figure 5N), or to inhibit proliferation, migration and invasion of 786‐O cells with depleted endogenous Rab21 (Supplementary Figure S9J‐L). Taken together, these results further supported that Rab21 acted as a bridge to tether EGFR to mitochondria through interacting with MFN2.
3.8. PTPRJ dephosphorylated mitochondria‐juxtaposed EGFR to inactivate the EGFR signaling pathway
To investigate the underlying mechanism of how the EGFR signaling pathway was inactivated through MFN2‐Rab21 interaction, we performed proteomic analysis on mitochondria extracted from 786‐O cells to look for potential EGFR‐targeting phosphatases, and identified tyrosine‐protein phosphatase receptor type J (PTPRJ) (Figure 6A), which was previously reported to be involved in the dephosphorylation of EGFR [45]. To test whether PTPRJ was responsible for the dephosphorylation of mitochondria‐juxtaposed EGFR, we first checked the subcellular localization of this phosphatase, which has been unclear in human cells. IF staining showed that in 786‐O cells, PTPRJ had a pervasive distribution, with apparent overlapping to mitochondria (Figure 6B). As a further verification, we isolated plasma membrane, mitochondria, and cytoplasm fractions from 786‐O cells, and PTPRJ was detected in all three cell fractions (Figure 6C). Further analysis revealed that the mitochondrial localization of PTPRJ was independent of EGF stimulation (Supplementary Figure S10A). In consistence with our data, a recent published quantitative human mitochondrial proteomic data also reported mitochondrial localization of PTPRJ [46].
FIGURE 6.
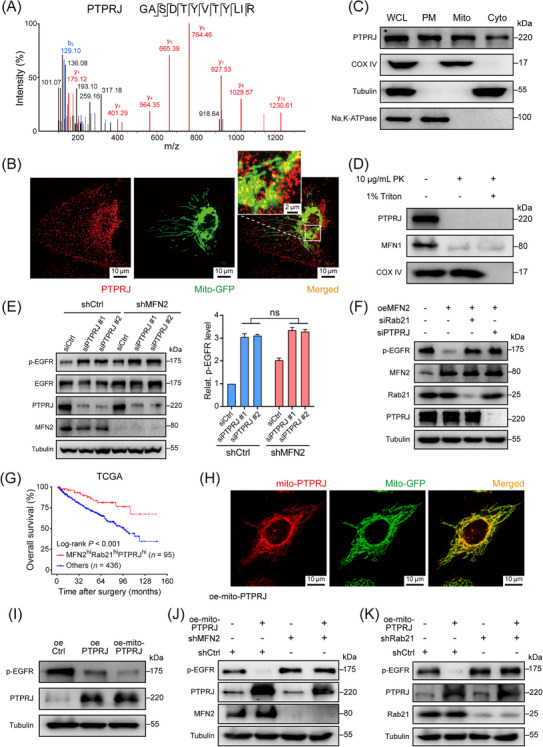
Mitochondria‐located PTPRJ dephosphorylates EGFR and inactivates EGFR signaling transduction. (A) LC‐MS analysis of mitochondria isolated from 786‐O cells identified PTPRJ as a mitochondria‐located tyrosine‐protein phosphatase. (B) Representative immunofluorescence staining image showing the co‐localization of mitochondria (green) and PTPRJ (red) in 786‐O cells stably transfected with mito‐GFP. (C) Mitochondria, cytosol and plasma membrane fractions of 786‐O cells were isolated and PTPRJ were detected by Western blotting. COX IV, tubulin, and Na, K‐ATPase were used as markers for mitochondria, cytoplasm, and plasma membrane, respectively. (D) Western blotting showing the distribution of PTPRJ on mitochondria. MFN1 and COX IV were used as markers of mitochondrial outer membrane and mitochondrial inner membrane. (E) Representative western blot image (left panel) and statistical analysis (right panel) of p‐EGFR and total EGFR protein levels using the indicated antibodies in MFN2 knockdown and control 786‐O cells transfected with PTPRJ siRNAs or control siRNA. (F) Western blotting showing p‐EGFR levels in MFN2‐overexpressing or control Caki‐1 cells with Rab21 or PTPRJ silencing. (G) OS of ccRCC patients, with regard to the simultaneous high expression of MFN2, Rab21 and PTPRJ (n = 95) in TCGA‐KIRC cohort. (H) Representative immunofluorescence staining image showing the co‐localization of mitochondria (green) and mito‐PTPRJ (red) in 786‐O cells stably transfected with mito‐GFP. (I) Western blotting showing p‐EGFR levels in 786‐O cells with PTPRJ or mito‐PTPRJ overexpression. (J) Western blotting showing p‐EGFR levels in MFN2‐knockdown or control 786‐O cells with mito‐PTPRJ overexpression. (K) Western blotting showing p‐EGFR levels in Rab21‐knockdown or control 786‐O cells with mito‐PTPRJ overexpression. Data are presented as mean ± SD. ns, not significant, by Student's t test (E) or log‐rank test (G).
Abbreviations: LC‐MS, liquid chromatography‐mass spectrometry; PTPRJ, tyrosine‐protein phosphatase receptor type J; hi, high; Mito, mitochondria; GFP, Green fluorescent protein; COX IV, cytochrome c oxidase subunit IV; Cyto, cytoplasm; PM, plasma membrane; WCL, whole cell lysate; PK, proteinase K; oe‐mito‐PTPRJ, overexpression of OMM‐anchoring form of PTPRJ; siCtrl, negative control siRNA; shCtrl, negative control shRNA; oeCtrl, empty overexpression control. OS, overall survival; Relat., relative; TCGA, the Cancer Genome Atlas.
To clarify the exact distribution of PTPRJ on mitochondria, we extracted mitochondria from 786‐O cells. The extracted mitochondria were treated with proteinase K in the presence or absence of Triton X‐100, a detergent deforming the OMM but not the inner mitochondrial membrane. Degradation of PTPRJ was detected (Figure 6D), suggesting that PTPRJ was localized to the OMM. Moreover, PTPRJ expression was associated with better prognosis of ccRCC patients in both TCGA‐KIRC and SYSUCC cohort (Supplementary Figure S10B‐C), and PTPRJ protein levels were negatively correlated with p‐EGFR levels in human ccRCC samples (Supplementary Figure S10D). Similar to MFN2 and Rab21, PTPRJ silencing promoted the malignant phenotypes of 786‐O and HK‐2 cells (Supplementary Figure S10E‐H).
To verify the contributions of PTPRJ to EGFR signaling, we knocked down PTPRJ in MFN2‐depleted and control 786‐O cells. PTPRJ silencing led to significantly elevated p‐EGFR levels in both groups, and the increase of the p‐EGFR level in MFN2‐depleted cells was not as dramatic as that in control cells (Figure 6E). Additionally, in MFN2‐overexpressing Caki‐1 cells, knockdown of Rab21 or PTPRJ by siRNAs dramatically promoted the p‐EGFR level as well as cell proliferation, migration and invasion capacities (Figure 6F, Supplementary Figure S10I‐K). Moreover, we found that simultaneous high expression of MFN2, Rab21 and PTPRJ could be a robust biomarker which was tightly associated with the best outcome of the patients in TCGA‐KIRC and our SYSUCC cohort (Figure 6G, Supplementary Figure S10L). All these results suggested that PTPRJ may coordinate with MFN2 and Rab21 to attenuate EGFR signaling.
In cells, protein phosphatases usually have a self‐inhibitory effect until they are activated by certain events, including translocation to a certain organelle [47]. To check whether the mitochondrial localization of PTPRJ affects its activity on dephosphorylating endocytosed EGFR, we constructed a mito‐PTPRJ [35], and the mitochondrial localization of this modified PTPRJ was confirmed by IF (Figure 6H, Supplementary Figure S10M). Interestingly, 786‐O cells overexpressing mito‐PTPRJ exhibited more prominent decrease in p‐EGFR levels than cells overexpressing WT PTPRJ (Figure 6I), suggesting that mitochondrial localization promotes dephosphorylation activity of PTPRJ. Moreover, the effect of mito‐PTPRJ on EGFR dephosphorylation was abolished by the depletion of either MFN2 or Rab21 (Figure 6J‐K), reconfirming that MFN2 and Rab21 is essential for the delivery of EGFR to mitochondria. Taken together, our data demonstrated a Rab21‐MFN2‐PTPRJ axis‐mediated EGFR processing pipeline in ccRCC. Endocytosed, Rab21‐associated EGFR was docked to mitochondria through Rab21‐MFN2 interaction, where it was dephosphorylated by activated, mitochondria‐localized PTPRJ, eventually resulting in attenuated EGFR oncogenic signaling transduction (Figure 7).
FIGURE 7.
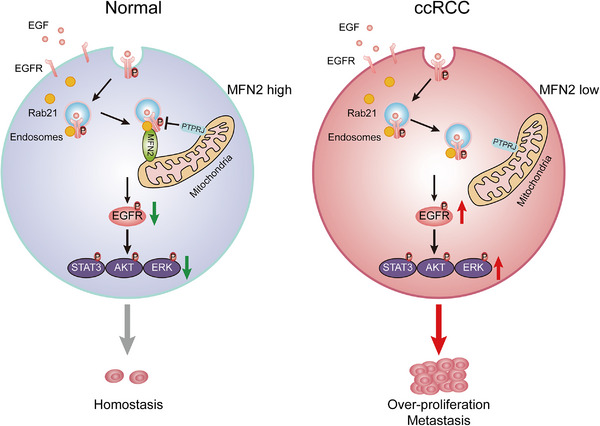
Schematic diagram showing the mechanism of EGFR signaling attenuation mediated by Rab21‐MFN2‐PTPRJ axis. In normal kidney epithelial cells, the interaction between MFN2 and Rab21 facilitates the mitochondria juxtaposition of internalized EGFR, where EGFR is dephosphorylated by PTPRJ, thus, attenuating the downstream signal transduction. In ccRCC cells, MFN2 deficiency leads to reduced EGFR dephosphorylation by PTPRJ in the OMM, thereby causing enhanced downstream signaling.
Abbreviations: CCRCC, clear cell renal cell carcinoma; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; P, Phosphorylation; STAT3, signal transducer and activator of transcription; AKT, protein kinase B; ERK, extracellular signal‐regulated kinase; OMM, outer mitochondrial membrane.
3.9. Therapeutic potential of boosting MFN2 in preclinical ccRCC PDX mouse models
The above results uncovered the non‐canonical tumor‐suppressor function of MFN2 in ccRCC, raising the intriguing possibility that restoring MFN2 expression may be beneficial to ccRCC patients. To better mimic the histopathologic characteristics of ccRCC, we utilized a preclinical model, in which immunocompromised NCG mice were implanted with ccRCC PDX. We injected MFN2 overexpression recombinant lentivirus or control lentivirus into tumors of PDX models (Figure 8A). Compared with the control group, intra‐tumoral injection of MFN2 overexpression lentivirus significantly suppressed tumor growth (Figure 8B‐D). Ki67 staining indicated that the proliferation of tumor cells was notably decreased upon MFN2 overexpression (Figure 8E‐F). Western blotting analysis revealed that restoring MFN2 expression dramatically inhibited the activity of EGFR signaling pathway (Figure 8G). Collectively, these findings provided convincing evidence that enhancing MFN2 expression may be a promising clinical therapeutic strategy against ccRCC.
FIGURE 8.
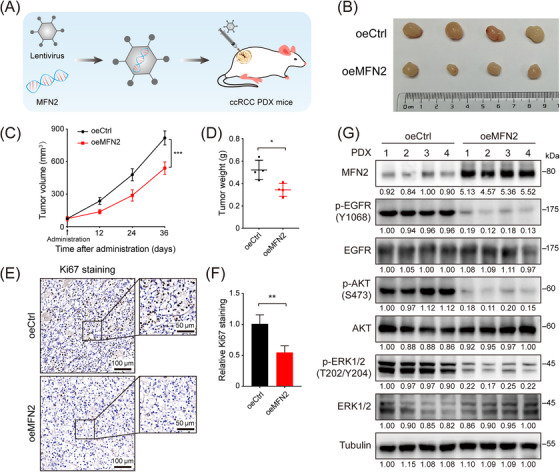
Therapeutic potential of restoring MFN2 expression in ccRCC PDX mouse models. (A) Diagram of MFN2 overexpression recombinant lentivirus intratumorally injected into tumors of ccRCC PDX mice. (B‐D) Tumor image (B), tumor volume (C) and tumor weight (D) of ccRCC PDX tumors treated with MFN2 overexpression or control lentivirus (n = 4 per group). (E‐F) Representative IHC staining images (E) and statistical analysis (F) of Ki67 in ccRCC PDX tumors treated with MFN2 overexpression or control lentivirus. (G) Western blotting of EGFR pathway proteins in ccRCC PDX tumors treated with MFN2 overexpression or control lentivirus using the indicated antibodies. Data are presented as means ± SD. * P < 0.05, ** P < 0.01, *** P < 0.001, by one‐way ANOVA (C) or Student's t test (D, F).
Abbreviations: PDX, Patient‐derived xenograft; oeCtrl, empty overexpression control; oeMFN2, overexpression of MFN2; p‐EGFR, phosphorylated epidermal growth factor receptor; p‐AKT, phosphorylated protein kinase B; p‐ERK1/2, phosphorylated extracellular signal‐regulated kinase 1/2.
4. DISCUSSION
ccRCC is the most common renal malignancy. However, the molecular events underlying ccRCC progression remain unclear. It is urgently needed to uncover the underlying mechanisms and to identify novel therapeutic targets. In this study, we identified MFN2, which suppressed ccRCC tumor growth and metastasis in vivo and in vitro, as a critical tumor suppressor in ccRCC. MFN2 attenuated EGFR signaling activity through the Rab21‐MFN2‐PTPRJ axis. By interacting with both MFN2 and EGFR, Rab21 docked endocytosed EGFR to mitochondrial outer membrane, where EGFR was dephosphorylated by PTPRJ. Here, we identified a novel non‐canonical function of MFN2 in inhibiting ccRCC progression, which might be a potential target against ccRCC.
The molecular mechanisms underlying the tumorigenesis and metastasis of ccRCC remain elusive. Mutations in the gene encoding von Hippel‐Lindau disease tumor suppressor (VHL), which present in a large proportion of ccRCC patients, is widely considered as a central process for the pathogenesis of ccRCC [48, 49]. However, given the fact that VHL mutations are not conclusive for the tumorigenesis of ccRCC, to identify new driving factors is still necessary [50]. Here, we identified MFN2 as a key regulator of tumor initiation and progression in ccRCC. MFN2 was down‐regulated in ccRCC tumor tissues, and high expression of MFN2 was associated with favorable prognosis for patients. Defects in MFN2 are closely related to the onset of many neuropathies and metabolic diseases, which are characterized by disrupted mitochondrial fusion and a reduction in the oxidative phosphorylation (OXPHOS) capacity of energy‐consuming‐sensitive cells, such as cardiomyocytes and neurons [51, 52, 53]. As tumor cells are less dependent on OXPHOS and they actively go on cell division that requires mitochondrial fragmentation, it is reasonable to conclude that the mitochondrial fusogenic activity of MFN2 endows cells with a common tumor suppressive potential. By introducing fusion‐deficient mutations, however, we showed that MFN2 also served as a docking molecule that facilitated the mitochondria juxtaposition of EGFR to suppress ccRCC in a fusogenic activity‐independent manner. Thus, we uncover a noncanonical tumor‐suppressive function of MFN2 in ccRCC, which is marked by a novel mechanism for mitochondria‐dependent dephosphorylation of EGFR.
EGFR signaling is indispensable for cellular renewal and homeostasis under physiological conditions. Activation of EGFR signaling has been implicated in acute kidney injury and the progression of chronic kidney disease [54], as well as human epithelial malignancies, including RCC [14, 55, 56]. Using in vitro and in vivo functional experiments as well as transgenic kidney‐specific Mfn2‐knockout mouse models, we found that MFN2 suppressed ccRCC initiation and progression by inhibiting EGFR signaling. To avoid overactivation, EGFR is subjected to ligand‐induced endocytosis for degradation or recycling [19, 20, 57]. The endocytic trafficking of EGFR is regulated by multiple Rab proteins, and among them Rab5 is well studied [58]. Rab21, a member of the Rab5 subfamily [59], is abundant in the kidney. Rab21 is predominantly localized in the early endocytic pathway and regulates early endosome dynamics [43, 60]. So far, the role of Rab21 in tumor is not clearly understood. It was reported that Rab21 participates in β1‐integrin and FAK signaling, and is a poor prognostic factor in breast cancer [61, 62]. Here, our study demonstrated a different role of Rab21 as a favorable prognostic factor in ccRCC. We speculated that the integrin signaling might play a minor role in ccRCC, as compared to the over‐activated EGFR signaling pathway. Thus, with significantly altered MFN2 expression level, Rab21 tended to participate more in mitochondria‐dependent inactivation of EGFR in ccRCC, thereby becoming a favorable prognosis factor.
Intracellular organelles frequently communicate and integrate their activities, including signal transduction, lipid metabolism and biogenesis [63, 64]. In particular, mitochondria physically and functionally interact with the endoplasmic reticulum and endosomes [63, 64, 65]. Mitochondrion‐endosome contact is crucial for lipid/ion transfer and mitochondria quality control [65]. Upon EGF stimulation, EGFR is internalized into endosomes, which are further matured into multivesicular endosomes and finally destined to lysosomes for degradation [20]. Although the pathway taken by endocytosed EGFR to other organelles (such as lysosomes and Golgi apparatus) has been extensively studied, the mechanism for the trafficking of EGFR‐containing endosomes to mitochondrial remains poorly understood. Here, we found that MFN2‐Rab21 interaction is vital for docking EGFR to mitochondria. Previous studies mainly focused on the impact of oncogenic signaling on mitochondrial dynamics [30, 66‐68], in which mitochondria are generally considered downstream effectors of tumor progression. Nevertheless, our results demonstrated that mitochondria might act as upstream regulators of oncogenic signaling, and defined mitochondria as a primordial tumor‐suppressing force to limit EGFR pathway in kidney epithelial cells.
Protein tyrosine phosphatases are critical inhibitors of tumor initiation and progression [69, 70]. PTPRJ, a phosphatase localized mainly on the plasma membrane, has been reported to decrease EGFR phosphorylation and downstream signaling [45] and serves as a tumor suppressor [71, 72]. We found that patients with high expression of PTPRJ have prolonged survival, especially for those with simultaneous high expression of MFN2, Rab21 and PTPRJ, in the TCGA‐KIRC cohort. Organelle‐localized protein tyrosine phosphatase has been reported to dephosphorylate EGFR and down‐regulate EGFR signaling [34]. By careful cell biology and biochemical assays, we pinpointed the OMM distribution of PTPRJ. PTPRJ is likely to be activated by OMM localization, and thus becomes a key hub for the dephosphorylation of EGFR that was docked to mitochondria through MFN2‐Rab21 interaction. Our study uncovered a novel mitochondria‐localized protein tyrosine phosphatase and further elucidated its role in the tumorigenesis and metastasis of ccRCC. Further studies are needed to explore the cellular functions and detailed mechanisms for mitochondria‐localized phosphatases in dephosphorylating signaling molecules of various pathways.
There are some limitations in our present study. First of all, the molecular mechanism contributed to the down‐regulation of MFN2 in ccRCC is still elusive and further studies are warranted. Moreover, whether MFN2 is involved in the resistance to RTK inhibitors (TKIs), such as sunitinib, axitinib and cabozantinib, remains unclear. It is urgently needed to explore the roles of MFN2 in TKI resistance and validate whether boosting MFN2 combined with TKI treatment will have synergistic effects.
5. CONCLUSIONS
Our study reveals a mitochondria‐dependent Rab21‐MFN2‐PTPRJ axis meditating EGFR signal trunsduction to suppresss ccRCC progression, suggesting MFN2 as a potential therapeutic molecule for RCC.
DECLARATIONS
AUTHOR CONTRIBUTIONS
SG and JY conceived the project, SG, DW and JL supervised the project. LL, YP and JY performed experiments. QW performed BLI analysis. JF and BY helped with protein purification. DW, TK, JY and JL analysed the data. SG, LL and JY wrote the manuscript. All authors discussed and approved the manuscript.
CONFLICTS OF INTEREST STATEMENT
The authors declare that they have no competing interests.
CONSENT FOR PUBLICATION
Not applicable.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Experiments regarding clinical samples were performed with the approval of the Internal Review and Ethics Boards of SYSUCC (Approval No. 202201071013000560775z) and of FAH‐SYSU (Approval No. [2021]144). All clinical samples were collected from patients with written informed consent for the use of tissues and clinical data. All animal experiments were approved by the Institutional Review Boards and Animal Care and Use Committees of SYSUCC (Approval No. L025501202107050).
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGEMENTS
We acknowledge the participants who generously gave their help on the study. This study was supported by the National Key R&D Program of China (2018YFA0508300 to SG), National Natural Science Foundation of China (82173098 and 31722016 to SG; 81725016 and 81872094 to JL), and Natural Science Foundation of Guangdong Province (2019TX05Y598 to SG).
Luo L, Wei D, Pan Y, Wang Q‐X, Feng J‐X, Yu B, et al. MFN2 suppresses clear cell renal cell carcinoma progression by modulating mitochondria‐dependent dephosphorylation of EGFR. Cancer Communications. 2023;43:808–833. 10.1002/cac2.12428
Contributor Information
Junhang Luo, Email: luojunh@mail.sysu.edu.cn.
Jiefeng Yang, Email: yangjf28@mail.sysu.edu.cn.
Song Gao, Email: gaosong@sysucc.org.cn.
DATA AVAILABILITY STATEMENT
The raw RNA sequencing data are available at the Genome Sequence Archive (GSA) database (accession number HRA002229). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE33 partner repository with the dataset identifiers PXD033112 for the interactome of MFN2, PXD033104 for the interactome of Rab21, and PXD033099 for the mitochondrial proteome of 786‐O cells. The key raw data have been deposited into the Research Data Deposit (www.researchdata.org.cn), with the approval number of 2302230001.
REFERENCES
- 1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33. [DOI] [PubMed] [Google Scholar]
- 2. Qiu H, Cao S, Xu R. Cancer incidence, mortality, and burden in China: a time‐trend analysis and comparison with the United States and United Kingdom based on the global epidemiological data released in 2020. Cancer Commun (Lond). 2021;41(10):1037–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Capitanio U, Montorsi F. Renal cancer. Lancet. 2016;387(10021):894–906. [DOI] [PubMed] [Google Scholar]
- 4. Choueiri TK, Kaelin WG, Jr . Targeting the HIF2‐VEGF axis in renal cell carcinoma. Nat Med. 2020;26(10):1519–30. [DOI] [PubMed] [Google Scholar]
- 5. Sequist LV, Han JY, Ahn MJ, Cho BC, Yu H, Kim SW, et al. Osimertinib plus savolitinib in patients with EGFR mutation‐positive, MET‐amplified, non‐small‐cell lung cancer after progression on EGFR tyrosine kinase inhibitors: interim results from a multicentre, open‐label, phase 1b study. Lancet Oncol. 2020;21(3):373–86. [DOI] [PubMed] [Google Scholar]
- 6. Motzer RJ, Rini BI, Mcdermott DF, Aren Frontera O, Hammers HJ, Carducci MA, et al. Nivolumab plus ipilimumab versus sunitinib in first‐line treatment for advanced renal cell carcinoma: extended follow‐up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol. 2019;20(10):1370–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mcdermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI, Escudier B, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med. 2018;24(6):749–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cella D, Grunwald V, Escudier B, Hammers HJ, George S, Nathan P, et al. Patient‐reported outcomes of patients with advanced renal cell carcinoma treated with nivolumab plus ipilimumab versus sunitinib (CheckMate 214): a randomised, phase 3 trial. Lancet Oncol. 2019;20(2):297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ravaud A, Hawkins R, Gardner JP, Von Der Maase H, Zantl N, Harper P, et al. Lapatinib versus hormone therapy in patients with advanced renal cell carcinoma: a randomized phase III clinical trial. J Clin Oncol. 2008;26(14):2285–91. [DOI] [PubMed] [Google Scholar]
- 10. Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, et al. Renal cell carcinoma. Nat Rev Dis Primers. 2017;3:17009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Franovic A, Gunaratnam L, Smith K, Robert I, Patten D, Lee S. Translational up‐regulation of the EGFR by tumor hypoxia provides a nonmutational explanation for its overexpression in human cancer. Proc Natl Acad Sci U S A. 2007;104(32):13092–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kassouf W. Editorial comment on: Von Hippel‐Lindau tumor suppressor gene loss in renal cell carcinoma promotes oncogenic epidermal growth factor receptor signaling via Akt‐1 and MEK‐1. Eur Urol. 2008;54(4):853–4. [DOI] [PubMed] [Google Scholar]
- 13. Wang A, Bao Y, Wu Z, Zhao T, Wang D, Shi J, et al. Long noncoding RNA EGFR‐AS1 promotes cell growth and metastasis via affecting HuR mediated mRNA stability of EGFR in renal cancer. Cell Death Dis. 2019;10(3):154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Feng ZH, Fang Y, Zhao LY, Lu J, Wang YQ, Chen ZH, et al. RIN1 promotes renal cell carcinoma malignancy by activating EGFR signaling through Rab25. Cancer Sci. 2017;108(8):1620–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Massari F, Ciccarese C, Santoni M, Brunelli M, Piva F, Modena A, et al. Metabolic alterations in renal cell carcinoma. Cancer Treat Rev. 2015;41(9):767–76. [DOI] [PubMed] [Google Scholar]
- 16. Schoffski P, Dumez H, Clement P, Hoeben A, Prenen H, Wolter P, et al. Emerging role of tyrosine kinase inhibitors in the treatment of advanced renal cell cancer: a review. Ann Oncol. 2006;17(8):1185–96. [DOI] [PubMed] [Google Scholar]
- 17. Tomas A, Futter CE, Eden ER. EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol. 2014;24(1):26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yarden Y, Shilo BZ. SnapShot: EGFR signaling pathway. Cell. 2007;131(5):1018. [DOI] [PubMed] [Google Scholar]
- 19. Ceresa BP, Schmid SL. Regulation of signal transduction by endocytosis. Curr Opin Cell Biol. 2000;12(2):204–10. [DOI] [PubMed] [Google Scholar]
- 20. Dobrowolski R, De Robertis EM. Endocytic control of growth factor signalling: multivesicular bodies as signalling organelles. Nat Rev Mol Cell Biol. 2011;13(1):53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goh LK, Sorkin A. Endocytosis of receptor tyrosine kinases. Cold Spring Harb Perspect Biol. 2013;5(5):a017459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10(8):513–25. [DOI] [PubMed] [Google Scholar]
- 23. Wei D, Zhan W, Gao Y, Huang L, Gong R, Wang W, et al. RAB31 marks and controls an ESCRT‐independent exosome pathway. Cell Res. 2021;31(2):157–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Boerner JL, Demory ML, Silva C, Parsons SJ. Phosphorylation of Y845 on the epidermal growth factor receptor mediates binding to the mitochondrial protein cytochrome c oxidase subunit II. Mol Cell Biol. 2004;24(16):7059–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Demory ML, Boerner JL, Davidson R, Faust W, Miyake T, Lee I, et al. Epidermal growth factor receptor translocation to the mitochondria: regulation and effect. J Biol Chem. 2009;284(52):36592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yue X, Song W, Zhang W, Chen L, Xi Z, Xin Z, et al. Mitochondrially localized EGFR is subjected to autophagic regulation and implicated in cell survival. Autophagy. 2008;4(5):641–9. [DOI] [PubMed] [Google Scholar]
- 27. Youle RJ, Van Der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337(6098):1062–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Archer SL. Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N Engl J Med. 2013;369(23):2236–51. [DOI] [PubMed] [Google Scholar]
- 29. Gao S, Hu J. Mitochondrial Fusion: The Machineries In and Out. Trends Cell Biol. 2021;31(1):62–74. [DOI] [PubMed] [Google Scholar]
- 30. Qi M, Dai D, Liu J, Li Z, Liang P, Wang Y, et al. AIM2 promotes the development of non‐small cell lung cancer by modulating mitochondrial dynamics. Oncogene. 2020;39(13):2707–23. [DOI] [PubMed] [Google Scholar]
- 31. Hernandez‐Alvarez MI, Sebastian D, Vives S, Ivanova S, Bartoccioni P, Kakimoto P, et al. Deficient Endoplasmic Reticulum‐Mitochondrial Phosphatidylserine Transfer Causes Liver Disease. Cell. 2019;177(4):881‐95 e17. [DOI] [PubMed] [Google Scholar]
- 32. Schrepfer E, Scorrano L. Mitofusins, from Mitochondria to Metabolism. Mol Cell. 2016;61(5):683–94. [DOI] [PubMed] [Google Scholar]
- 33. Cao YL, Meng S, Chen Y, Feng JX, Gu DD, Yu B, et al. MFN1 structures reveal nucleotide‐triggered dimerization critical for mitochondrial fusion. Nature. 2017;542(7641):372–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li YJ, Cao YL, Feng JX, Qi Y, Meng S, Yang JF, et al. Structural insights of human mitofusin‐2 into mitochondrial fusion and CMT2A onset. Nat Commun. 2019;10(1):4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang J, Fonovic M, Suyama K, Bogyo M, Scott MP. Rab35 controls actin bundling by recruiting fascin as an effector protein. Science. 2009;325(5945):1250–4. [DOI] [PubMed] [Google Scholar]
- 36. Martens M, Ammar A, Riutta A, Waagmeester A, Slenter DN, Hanspers K, et al. WikiPathways: connecting communities. Nucleic Acids Res. 2021;49(D1):D613‐D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim KT, Lee HW, Lee HO, Song HJ, Jeong Da E, Shin S, et al. Application of single‐cell RNA sequencing in optimizing a combinatorial therapeutic strategy in metastatic renal cell carcinoma. Genome Biol. 2016;17:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brugarolas J. Renal‐cell carcinoma–molecular pathways and therapies. N Engl J Med. 2007;356(2):185–7. [DOI] [PubMed] [Google Scholar]
- 39. Herbst RS, Fukuoka M, Baselga J. Gefitinib–a novel targeted approach to treating cancer. Nat Rev Cancer. 2004;4(12):956–65. [DOI] [PubMed] [Google Scholar]
- 40. Qiu C, Huang S, Park J, Park Y, Ko YA, Seasock MJ, et al. Renal compartment‐specific genetic variation analyses identify new pathways in chronic kidney disease. Nat Med. 2018;24(11):1721–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Carter P, Schnell U, Chaney C, Tong B, Pan X, Ye J, et al. Deletion of Lats1/2 in adult kidney epithelia leads to renal cell carcinoma. J Clin Invest. 2021;131(11): e144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tostain J, Li G, Gentil‐Perret A, Gigante M. Carbonic anhydrase 9 in clear cell renal cell carcinoma: a marker for diagnosis, prognosis and treatment. Eur J Cancer. 2010;46(18):3141–8. [DOI] [PubMed] [Google Scholar]
- 43. Eden ER, White IJ, Tsapara A, Futter CE. Membrane contacts between endosomes and ER provide sites for PTP1B‐epidermal growth factor receptor interaction. Nat Cell Biol. 2010;12(3):267–72. [DOI] [PubMed] [Google Scholar]
- 44. Yang X, Zhang Y, Li S, Liu C, Jin Z, Wang Y, et al. Rab21 attenuates EGF‐mediated MAPK signaling through enhancing EGFR internalization and degradation. Biochem Biophys Res Commun. 2012;421(4):651–7. [DOI] [PubMed] [Google Scholar]
- 45. Tarcic G, Boguslavsky SK, Wakim J, Kiuchi T, Liu A, Reinitz F, et al. An unbiased screen identifies DEP‐1 tumor suppressor as a phosphatase controlling EGFR endocytosis. Curr Biol. 2009;19(21):1788–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Morgenstern M, Peikert CD, Lubbert P, Suppanz I, Klemm C, Alka O, et al. Quantitative high‐confidence human mitochondrial proteome and its dynamics in cellular context. Cell Metab. 2021;33(12):2464‐83 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mattila E, Pellinen T, Nevo J, Vuoriluoto K, Arjonen A, Ivaska J. Negative regulation of EGFR signalling through integrin‐alpha1beta1‐mediated activation of protein tyrosine phosphatase TCPTP. Nat Cell Biol. 2005;7(1):78–85. [DOI] [PubMed] [Google Scholar]
- 48. De Cubas AA, Rathmell WK. Epigenetic modifiers: activities in renal cell carcinoma. Nat Rev Urol. 2018;15(10):599–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yang J, Luo L, Zhao C, Li X, Wang Z, Zeng Z, et al. A Positive Feedback Loop between Inactive VHL‐Triggered Histone Lactylation and PDGFRbeta Signaling Drives Clear Cell Renal Cell Carcinoma Progression. Int J Biol Sci. 2022;18(8):3470–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kaelin WG, Jr . The von Hippel‐Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8(11):865–73. [DOI] [PubMed] [Google Scholar]
- 51. Zuchner S, Mersiyanova IV, Muglia M, Bissar‐Tadmouri N, Rochelle J, Dadali EL, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot‐Marie‐Tooth neuropathy type 2A. Nat Genet. 2004;36(5):449–51. [DOI] [PubMed] [Google Scholar]
- 52. Lim KL, Ng XH, Grace LG, Yao TP. Mitochondrial dynamics and Parkinson's disease: focus on parkin. Antioxid Redox Signal. 2012;16(9):935–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Su B, Wang X, Bonda D, Perry G, Smith M, Zhu X. Abnormal mitochondrial dynamics–a novel therapeutic target for Alzheimer's disease? Mol Neurobiol. 2010;41(2‐3):87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tang J, Liu N, Zhuang S. Role of epidermal growth factor receptor in acute and chronic kidney injury. Kidney Int. 2013;83(5):804–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu C, Liu L, Wang K, Li XF, Ge LY, Ma RZ, et al. VHL‐HIF‐2alpha axis‐induced SMYD3 upregulation drives renal cell carcinoma progression via direct trans‐activation of EGFR. Oncogene. 2020;39(21):4286–98. [DOI] [PubMed] [Google Scholar]
- 56. Morris ZS, Mcclatchey AI. Aberrant epithelial morphology and persistent epidermal growth factor receptor signaling in a mouse model of renal carcinoma. Proc Natl Acad Sci U S A. 2009;106(24):9767–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Carpenter G. The EGF receptor: a nexus for trafficking and signaling. BioEssays: news and reviews in molecular, cellular and developmental biology. 2000;22(8):697–707. [DOI] [PubMed] [Google Scholar]
- 58. Miaczynska M. Effects of membrane trafficking on signaling by receptor tyrosine kinases. Cold Spring Harb Perspect Biol. 2013;5(11):a009035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jean S, Kiger AA. Coordination between RAB GTPase and phosphoinositide regulation and functions. Nat Rev Mol Cell Biol. 2012;13(7):463–70. [DOI] [PubMed] [Google Scholar]
- 60. Simpson JC, Griffiths G, Wessling‐Resnick M, Fransen JA, Bennett H, Jones AT. A role for the small GTPase Rab21 in the early endocytic pathway. J Cell Sci. 2004;117(Pt 26):6297–311. [DOI] [PubMed] [Google Scholar]
- 61. Alanko J, Mai A, Jacquemet G, Schauer K, Kaukonen R, Saari M, et al. Integrin endosomal signalling suppresses anoikis. Nat Cell Biol. 2015;17(11):1412–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Moreno‐Layseca P, Jantti NZ, Godbole R, Sommer C, Jacquemet G, Al‐Akhrass H, et al. Cargo‐specific recruitment in clathrin‐ and dynamin‐independent endocytosis. Nat Cell Biol. 2021;23(10):1073–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Burte F, Carelli V, Chinnery PF, Yu‐Wai‐Man P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol. 2015;11(1):11–24. [DOI] [PubMed] [Google Scholar]
- 64. Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12(9):823–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Das A, Nag S, Mason AB, Barroso MM. Endosome‐mitochondria interactions are modulated by iron release from transferrin. J Cell Biol. 2016;214(7):831–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, et al. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK‐driven tumor growth. Mol Cell. 2015;57(3):537–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li T, Han J, Jia L, Hu X, Chen L, Wang Y. PKM2 coordinates glycolysis with mitochondrial fusion and oxidative phosphorylation. Protein Cell. 2019;10(8):583–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yu Y, Peng XD, Qian XJ, Zhang KM, Huang X, Chen YH, et al. Fis1 phosphorylation by Met promotes mitochondrial fission and hepatocellular carcinoma metastasis. Signal Transduct Target Ther. 2021;6(1):401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sun T, Aceto N, Meerbrey KL, Kessler JD, Zhou C, Migliaccio I, et al. Activation of multiple proto‐oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell. 2011;144(5):703–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Julien SG, Dube N, Read M, Penney J, Paquet M, Han Y, et al. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2‐induced mammary tumorigenesis and protects from lung metastasis. Nat Genet. 2007;39(3):338–46. [DOI] [PubMed] [Google Scholar]
- 71. Ruivenkamp C, Hermsen M, Postma C, Klous A, Baak J, Meijer G, et al. LOH of PTPRJ occurs early in colorectal cancer and is associated with chromosomal loss of 18q12‐21. Oncogene. 2003;22(22):3472–4. [DOI] [PubMed] [Google Scholar]
- 72. Balavenkatraman KK, Jandt E, Friedrich K, Kautenburger T, Pool‐Zobel BL, Ostman A, et al. DEP‐1 protein tyrosine phosphatase inhibits proliferation and migration of colon carcinoma cells and is upregulated by protective nutrients. Oncogene. 2006;25(47):6319–24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The raw RNA sequencing data are available at the Genome Sequence Archive (GSA) database (accession number HRA002229). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE33 partner repository with the dataset identifiers PXD033112 for the interactome of MFN2, PXD033104 for the interactome of Rab21, and PXD033099 for the mitochondrial proteome of 786‐O cells. The key raw data have been deposited into the Research Data Deposit (www.researchdata.org.cn), with the approval number of 2302230001.