

Re‐analysis of next‐generation sequencing (NGS) data can help to resolve undiagnosed cases. 1 We report on two patients with dystonia to illustrate how stringent NGS‐variant filtering criteria can lead to missed diagnoses in the context of variants lying in a homopolymeric nucleotide tract; at the same time, relaxing bioinformatic‐filter settings can yield false‐positive pathogenic variant‐hits in the same homopolymeric stretch, further complicating the NGS‐based diagnostic process.
Patient‐1, a 16‐year‐old boy of Czech origin, presented with repetitive attacks of dystonic spasms (distal‐limb predominance), lasting a few seconds. These abnormal movements were triggered by voluntary action and showed response to carbamazepine treatment. Patient‐2 was a 10‐year‐old girl with unremarkable family history who manifested isolated lower‐limb dystonia since age 6. Examination revealed bilateral inward rotation of the legs and twisting movements of the toes during longer walks.
Both patients underwent NGS studies in a program applying whole‐exome sequencing (WES) as a first‐tier molecular test. 2 However, data analyses implementing standard minor‐allele frequency (MAF) filters (<0.001 for heterozygous variants) 2 could not identify any (likely) pathogenic alterations. Motivated by the recent observation of dystonia‐causal mutations with higher‐than‐expected population frequencies, 3 , 4 we re‐evaluated NGS datasets with MAF cutoffs of 1%. This led to identification of described pathogenic PRRT2 variants situated in a homopolymeric stretch in patient‐1 (NM_145239.3:c.649dup) and patient‐2 (NM_145239.3:c.649del) (Fig. 1A,B). The variants were investigated with Sanger‐sequencing, which verified c.649dup (patient‐1), but demonstrated c.649del (patient‐2) to be an erroneous mutation call (Fig. 1C–E). Segregation analysis detected c.649dup in patients‐1's father who had epilepsy during childhood. There was no indication of allelic dropout in patient‐2. Re‐analysis of parent–child trio‐WES data identified no other relevant candidate variants for patient‐2. Homopolymeric stretches represent strings of identical nucleotides that are spread across the genome and highly mutable. These tracts are prone to slipped strand mispairing which consequently leads to polymerase‐induced errors such as insertions or deletions (indels) of one or more nucleotides creating a new genomic variant. 5 The same can occur under laboratory PCR approaches, representing standard steps in targeted short‐read NGS experiments. Examinations of WES outputs indicate that PCR amplifications can introduce a substantial number of artificial indels, especially in low‐complexity regions such as homopolymers. 6 , 7 The homopolymeric cytosine tract in PRRT2's exon 2 (NM_145239.3:c.641–649) has recently been highlighted as a pitfall in NGS studies. 8 Both c.649dup and c.649del have repeatedly been reported as causative for PRRT2‐related movement disorders. 9 Nevertheless, these indels were filtered out upon primary WES‐data analyses because of their high MAFs in controls (gnomAD‐v2.1.1: c.649dup:0.003; c.649del:0.004). These MAFs, inconsistent with the rarity of PRRT2‐associated diseases, are explainable by sequencing artifacts in PRRT2's nucleotide‐tract c.641–649. 8 From an analytic‐technical point‐of‐view, two conclusions emerge: (i) NGS‐based diagnostics of phenotypes linked to variants in homopolymeric stretches should be done with expanded MAF‐filter settings; and (ii) pathogenic‐variant hits identified in such homopolymers should be treated with caution and prompt secondary validation. From a clinical‐practical point‐of‐view, the following three points deserve consideration: (i) Moving from single‐gene tests to WES has substantially increased the diagnostic yield, but the method comes with greater challenges in data analysis. Although the pretest probability for a PRRT2 alteration was high for patient‐1, unbiased sequencing strategies are now considered the method of choice for studying dystonic disorders due to genetic heterogeneity and impracticability of exon‐by‐exon Sanger approaches. 2 Nevertheless, we highlight that phenomenology plays a huge role in guiding genetic testing, and one may argue that a targeted screen for known PRRT2 mutations may have been another straightforward way to diagnosis in patient‐1; (ii) evaluation of the significance of NGS variants relies heavily on the phenotypic information provided by ordering clinicians. For patient‐2, a phenotype description of childhood‐onset limb dystonia with fluctuating intensity was considered during WES‐data interpretation. Our knowledge of genotype–phenotype correlations expands steadily and we are not yet able to decipher all associations between detected variants and disease expressions. Notably, there have been reports describing exercise‐related longer‐lasting leg stiffness, resembling features of patient‐2, in association to mutant PRRT2. 10 Therefore, the PRRT2 c.649del variant‐call was prioritized as the potential cause of patient‐2's clinical manifestation. This stresses the need for delivery of precise phenotype catalogs to genetic laboratories and interdisciplinary exchange. In the case of patient‐2, a genomic board involving geneticists and neuropediatric experts was instrumental in determining that the patient's clinical picture was more atypical for PRRT2‐related disease; and (iii) phenotypic expansions are discovered at a fast pace, and clinicians are facing many uncommon presentations of well‐defined inherited conditions. In view of the NGS‐identified c.649del variant‐call, neuropediatricians considered the possibility of an atypical manifestation of PRRT2‐related disease for patient‐2, but simultaneously initiated Sanger‐sequencing characterized c.649del as a false‐positive NGS hit, so that in the end no erroneous diagnosis was made. Our observations highlight the need for consideration of analytic pitfalls in WES data analyses, precise genotypic‐phenotypic correlations, and multidisciplinary team discussions to ensure accurate diagnostic output from NGS studies.
FIG. 1.
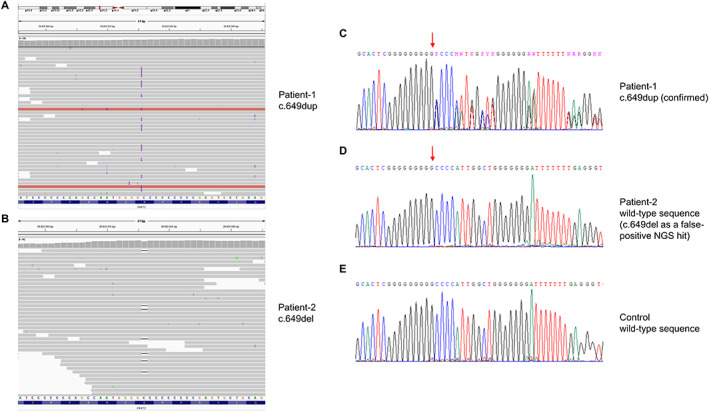
Whole‐exome and Sanger sequencing results for PRRT2 variants in a homopolymeric region. Patient‐derived exome data visualized with the Integrative Genomics Viewer show a heterozygous duplication in patient‐1 (A) and a heterozygous deletion in patient‐2 (B). Sanger electopherogram confirms the presence of the duplication (C), whereas the deletion is not found by Sanger analysis (D). A control is also shown (E); red arrows indicate the affected position in PRRT2.
Author Roles
(1) Research project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript: A. Writing of the first draft, B. Review and Critique.
I.D.: 1A, 1C, 3A.
E.G.: 1A, 1C, 3B.
M.B.: 1C, 3B.
R.B.: 1C, 3B.
I.P.: 1C, 3B.
A.B.: 1C, 3B.
J.W.: 1B, 3B.
R.J.: 1B, 3B.
K.V.: 1B, 3B.
M.Z.: 1A, 1B, 1C, 3B.
Disclosures
Ethical Compliance Statement: Genetic studies and de‐identified reporting of clinical and molecular data were performed in accordance with respective ethics review board approvals (Technical University of Munich, Munich, Germany). Written informed consent was obtained from all participants of this study (including parents in case of minors). We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: This study was funded by in‐house institutional funding of Technical University of Munich, Munich, Germany. JW and MZ receive research support from the German Research Foundation (DFG 458949627; WI 1820/14‐1; ZE 1213/2‐1). There are no conflicts of interest to report.
Financial Disclosures for the Previous 12 Months: The authors declare that there are no additional disclosures to report.
Acknowledgments
We thank the patients for their generous participation. Open Access funding enabled and organized by Projekt DEAL.
References
- 1. Liu P, Meng L, Normand EA, et al. Reanalysis of clinical exome sequencing data. N Engl J Med 2019;380(25):2478–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zech M, Jech R, Boesch S, et al. Monogenic variants in dystonia: an exome‐wide sequencing study. Lancet Neurol 2020;19(11):908–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Skorvanek M, Rektorova I, Mandemakers W, et al. WARS2 mutations cause dopa‐responsive early‐onset parkinsonism and progressive myoclonus ataxia. Parkinsonism Relat Disord 2022;94:54–61. [DOI] [PubMed] [Google Scholar]
- 4. Indelicato E, Boesch S, Baumgartner M, Plecko B, Winkelmann J, Zech M. Confirmation of a causal role for SHQ1 variants in early Infantile‐Onset recessive dystonia. Mov Disord 2022;38:355–357. [DOI] [PubMed] [Google Scholar]
- 5. Montes‐Moreno S, Routbort MJ, Lohman EJ, et al. Clinical molecular testing for ASXL1 c.1934dupG p.Gly646fs mutation in hematologic neoplasms in the NGS era. PLoS One 2018;13(9):e0204218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fang H, Wu Y, Narzisi G, et al. Reducing INDEL calling errors in whole genome and exome sequencing data. Genome Med 2014;6(10):89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stoler N, Nekrutenko A. Sequencing error profiles of Illumina sequencing instruments. NAR Genom Bioinform 2021;3(1):lqab019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Corominas J, Smeekens SP, Nelen MR, Yntema HG, Kamsteeg EJ, Pfundt R, Gilissen C. Clinical exome sequencing‐mistakes and caveats. Hum Mutat 2022;43(8):1041–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Landolfi A, Barone P, Erro R. The Spectrum of PRRT2‐Associated Disorders: Update on clinical features and pathophysiology. Front Neurol 2021;12:629747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marano M, Motolese F, Consoli F, De Luca A, Di Lazzaro V. Paroxysmal dyskinesias in a PRRT2 mutation carrier. Tremor Other Hyperkinet Mov 2018;8:616. [DOI] [PMC free article] [PubMed] [Google Scholar]