

SUMMARY
The hallmarks of cerebellar ataxia are incoordination and movement variability. The cerebellum not only controls motor coordination but also plays a role in cognition and mood. After establishing cerebellar ataxia on examination, determining the underlying cause is necessary. The causes of cerebellar ataxia are extensive, and systematic workup includes laboratory studies and imaging. In some cases, genetic testing often leads to a specific cause for cerebellar ataxia. Acquired and reversible causes should be pursued for disease-modifying treatment. Cerebellar ataxia is rapidly expanding, and new therapies are actively being tested, bringing hope for patients with cerebellar ataxia.
Keywords: Cerebellar ataxia, Spinocerebellar ataxia, Multiple system atrophy, Ataxia, Genetics
INTRODUCTION
Movement precision is critical to voluntary motor execution. Cerebellar ataxia leads to impairment of movement precision, making simple tasks such as eating, dressing, and walking difficult. The causes of cerebellar ataxia can be diverse and thus pose a diagnostic challenge. Therefore, it is important to recognize signs and symptoms, perform a comprehensive workup, and institute a treatment plan for patients with cerebellar ataxia.
The cerebellum is primarily known for control of voluntary movement coordination, balance maintenance, and eye movements. Cerebellar ataxia is a manifestation of cerebellar damage or degeneration and is typified by specific motor impairments. Clinical manifestations of cerebellar ataxia include speech impairment, limb incoordination, gait instability, and eye movement abnormalities. In addition to motor control, the cerebellum is also involved in cognition.1–4 Although the cognitive symptoms associated with cerebellar ataxia are not as widely recognized as motor symptoms, they are still disabling to patients and should be evaluated along with the motor impairments.
The causes of cerebellar ataxia are vast. Age of onset, disease course, associated clinical symptoms, as well as laboratory and imaging data provide clues to construct a differential diagnosis, which can include vascular, infectious, inflammatory, paraneoplastic, neoplastic, hereditary, toxic, and degenerative etiologies. Given the diverse causes, diagnostic workup and management can be challenging. This review aims to provide guidance for the diagnosis and management of cerebellar ataxia with a special emphasis on phenomenology, complmented by video clips. The authors highlight the unique features of certain forms of cerebellar ataxia and provide a stepwise diagnostic workup diagram. They also discuss symptomatic and emerging disease-modifying therapies for cerebellar ataxia.
CEREBELLAR ANATOMY
The first step of classic neurology is localization of neurologic deficits. Understanding cerebellar anatomy is critical for assessing ataxia. The cerebellum is composed of the left and right lateral hemispheres connected by the vermis. Outflow fibers from the ipsilateral deep cerebellar nuclei course through the superior cerebellar peduncles to the contralateral thalamus and subsequent contralateral motor cortex. Because the contralateral motor cortex controls ipsilateral limb movements, damages to the ipsilateral cerebellum will result in ipsilateral limb ataxia. The vermis is a midline structure important for truncal control. The flocculonodular lobe receives inputs from vestibular nuclei and regulates posture and eye movements. Discrete lesions in a particular cerebellar area result in ataxia of the corresponding body parts,4,5 whereas degenerative diseases often involve broad regions of the cerebellum and thus ataxia symptoms are more diffuse.
COGNITION AND MOOD IN CEREBELLAR ATAXIA
The role of the cerebellum in cognition and mood is equally as significant as its role in motor coordination. The cerebellar posterior lobe has extensive neural connections to the limbic and paralimbic areas, prefrontal, temporal, parietal cerebral cortex, and cingulate gyrus.6–9 Posterior lobe degeneration is linked to cognitive dysfunction in executive and visuospatial domains and personality changes of affect flattening, disinhibition, and impulsivity.10–12 Depression is common among inherited and degenerative ataxia syndromes and are independent of disease severity.13,14 Collectively, these impairments are known as cerebellar cognitive affective syndrome.10 Recognizing and screening cerebellar ataxia patients for these cognitive and psychiatric changes is important for quality of life.
SYMPTOMS OF CEREBELLAR DISEASE
Recognition of the core symptoms and signs is the first step in approaching patients with suspected cerebellar ataxia. The most common and often first presenting symptom of cerebellar ataxia is gait imbalance.15 Patients may describe unexpected loss of balance, easily tripping or bumping into objects, and finally, progressive falls. Ataxic gait may be described as appearing intoxicated from alcohol. Other ancillary information regarding gait imbalance will further ascertain a diagnosis of cerebellar ataxia. Specifically, gait changes (veering, intoxicated, or shortened cadence), the circumstances surrounding the fall (occurring primarily in the dark or with the eyes closed, foot dragging, sudden balance loss “out of nowhere”), and the progressive nature of falls are important diagnostic clues. Patients with cerebellar ataxia are more likely to have veering or changing gait direction and difficulty turning. Falling with eyes closed or in the dark suggests sensory neuropathy.
Appendicular ataxia manifests as imprecise movements in the hands or legs. Patients complain of “clumsiness,” uncoordinated handwriting, or knocking over or dropping objects due to miscalculation of object location or weight. Eye and speech complaints are also common in patients with cerebellar ataxia. Patients may notice “jumpy” eyes or burry vision, corresponding to nystagmus. Double vision is not common in pure cerebellar disease unless late stages of spinocerebellar ataxia (SCA) types 2 and 3 cause partial ophthalmoplegia. Speech may be slurred or reported as more difficult to understand. Lastly, patients may describe vertigo, a sensation the environment or oneself is moving. Oftentimes, patients experience lightheadedness without frank vertigo. Cerebellar vertigo is central in origin and thus typically does not worsen with head movements and is less severe than peripheral vertigo, which is often associated with nausea and vomiting.
EXAMINING PATIENTS WITH CEREBELLAR ATAXIA
Evaluation should begin while observing the patient in the waiting room and walking to the examination room. Observation of speech, eye movements, and abnormal movements continues before direct examination. How much of the history is provided by the patient versus a friend or family member can also help clinicians gauge severity of speech disorder or cognitive and emotional impairment. This spontaneous information provides a valuable glimpse of how ataxia symptoms may affect patients’ lives.
Eye Examination
The eye examination should first begin in primary position. Instruct the patient to fixate on a far target, such as a painting on the wall. The examiner is looking for square wave jerks (SWJ), which means saccadic intrusions in the horizontal plane that interrupt visual fixation on a target. Some healthy adults may exhibit SWJ; conversely, larger amplitude SWJ are seen in certain cerebellar diseases, such as Friedreich ataxia. SWJ may be observed in atypical parkinsonism with cerebellar involvement, including progressive supranuclear gaze palsy (PSP) and multiple system atrophy (MSA).16,17
Next, the patient should gaze at and follow the examiner’s finger in vertical and horizontal directions to look for nystagmus or bring out ocular flutter or opsoclonus. Inquire about diplopia during smooth pursuit. Nystagmus is involuntary, repetitive eye movements that occur in a to-and-fro manner.18 Nystagmus of a cerebellar origin involves fast and slow phases and can be directional or multidirectional. Nystagmus is the most common eye finding in cerebellar ataxia. Ocular flutter is irregular, oscillatory horizontal/unidirectional bursts of saccadic eye movements, typically induced by blinking or voluntary gaze.18 Opsoclonus is similar to ocular flutter but saccadic oscillations are multidirectional.18 Both are at a higher frequency than nystagmus and are seen in infectious/parainfectious, immune-mediated, or paraneoplastic cerebellar ataxia.19
Lastly, the examiner should test saccades to assess speed and accuracy of eye movements. To test saccadic movement, have the patient direct gaze between an examiner’s finger then switch gaze quickly to the examiner’s nose on command. Slow saccades are seen in SCA2 and SCA320,21; however, SCA2 patients particularly have profoundly slow saccades. Hypometric saccades occur when eye movements fall short of reaching the intended gaze target. The eyes then make one or more corrective saccades to reach the target. Hypermetric saccades occur when eye movements pass the intended gaze target then correct back to the target. Patients with cerebellar ataxia may have co-existence of hypometric and hypermetric saccades with hypometric in one direction and hypermetric saccades in another direction. Oculomotor apraxia is a loss of voluntary eye movement control and presents as difficulty initiating a saccade. Eye blinks or head thrust are used to maneuver gaze in a particular direction. Examples of eye findings associated with cerebellar ataxia are shown in Video 1, and eye findings commonly associated with specific cerebellar ataxia syndromes are listed in Table 1.
Table 1.
Specific eye findings seen in cerebellar ataxia syndromes
| Square Wave Jerks | Friedreich Ataxia |
|---|---|
| Slowed saccades | SCA2 SCA3 SCA7 |
| Downbeating nystagmus | SCA6 Multiple system atrophy |
| Pigmentary retinal macular degeneration | SCA7 |
| Ophthalmoparesis | SCA2 SCA3 SCA7 Kearns-Sayre syndrome Miller Fisher syndrome |
| Oculomotor apraxia | Ataxia with oculomotor ataxia types 1 & 2 Ataxia telangiectasia |
| Supranuclear ophthalmoplegia | Niemann-Pick type C |
| Retinitis pigmentosa | Refsum disease Abetalipoproteinemia Kearns-Sayre syndrome Neuropathy, ataxia, & retinitis pigmentosa |
Abbreviation: SCA, spinocerebellar ataxia.
Speech Examination
Speech examination begins while listening to the patient give the history. The most common presenting cerebellar speech impairments are dysarthria and scanning speech. Dysarthria is an abnormal articulation of speech phonemes. Ataxic dysarthria is due to uncoordinated, antagonistic muscle movements resulting in imprecise speech. Scanning speech has variability in speed, power, syllabic stress, and coordination of muscle movements.22 It is also described as a staccato speech where words or syllables are interrupted by variable pauses. For example, when saying, “Today is a sunny day,” a patient may say, “Today is a” pause “sunny day,” or “Today is a sun” pause “-ny day.” Consonants may be overemphasized or vowels underemphasized or distorted. Another key feature of cerebellar speech is slow speed, possibly a compensatory response to imprecision. Examples of speech impairment in cerebellar ataxia can be listened to in Video 2.
Appendicular Examination
Appendicular ataxia is evaluated by a series of assessments centered around the core clinical feature of appendicular ataxia: dysmetria. Dysmetria is inaccurate reaching of a target by the hands, arms, or legs. The ability to predict the accurate distance and speed needed to coordinate muscle movements is the exact function of the cerebellum and is consequently impaired in cerebellar ataxia. Two methods used to assess dysmetria are finger chase and heel-shin slide. Finger chase is performed by the examiner quickly moving his or her finger in a random fashion through space. The patient rapidly mirrors the movements exactly. The degree of dysmetria is estimated by the difference the patient’s finger deviates from the examiner’s finger. Leg dysmetria is tested by performing heel-shin slide in the lying down position with shoes and socks off and no obstructive clothing below the knee. The patient places a heel onto the contralateral knee and accurately and slowly slides the heel down the shin. The heel is then first rested on the table or bed, then repositioned back to the knee to perform successive movements. Thus, the patient is only instructed to slide down the shin but not up the shin. Grading severity is based on how well the heel maintains contact and the number of times the heel slides off the shin.
Dysdiadochokinesia is irregular and inaccurate rapid alternating synchronous movements. Because one major cerebellar function is control of movement rhythm, cerebellar ataxia manifests as abnormal rapid alternating movements. To perform rapid alternating hand movements, the patient alternates between pronating and supinating the hand as accurately as possible on the ipsilateral thigh. The palmar aspect of the hand will alternate touching the thigh with the dorsal aspect of the hand. To find further subtle cerebellar abnormalities, instruct the patient to perform the movements as fast as possible.
Several types of tremors are present in cerebellar ataxia. Intention tremor occurs when tremor amplitude increases as nearing a target and is the most common type of tremor in cerebellar ataxia.23 Intention tremor is observed in finger-nose-finger testing performed by having the patient extend the index finger and alternate between touching the examiner’s finger and the patient’s nose or chin (depending on tremor severity). Note the examiner’s finger should be fixed during finger-nose-finger testing. Another form of kinetic tremor, postural tremor, can also be present.23,24 Assess postural tremor by observing the patient maintaining their arms in an outstretched or wing-beating position. Lastly, look inside the patient’s mouth to assess for palatal tremor seen in progressive ataxia associated with hypertrophic olivary degeneration. The examples of appendicular examinations for cerebellar ataxia can be viewed in Videos 3 and 4.
Sitting Examination
Truncal ataxia manifests as incoordination of the trunk, shoulders, and hips. Truncal sway can be observed in the seated position, without back support, and the arms outstretched. In severe cases, patients may not be able to sit unsupported. Truncal titubation means increased shaking of the body when standing or sitting, which is common in patients with cerebellar ataxia.
Stance and Gait Examination
Stance is evaluated by observing the patient stand in the natural position, with the feet together and with the feet situated in tandem/heel-to-toe. Titubation and truncal sway are seen in sitting and standing positions in cerebellar ataxia. Ataxia also manifests as inability to maintain balance, needing to catch oneself by holding onto an object or side-stepping while walking. If ataxia is suspected, the examiner should remain close to the patient to ensure patient safety. Sensory ataxia, ataxia due to erroneous sensory inputs, worsens with the eyes closed. Additional examination maneuvers that can bring out subtle ataxic signs include having patients stand on one foot at a time or hop on one foot. Patients with cerebellar ataxia will lose balance.
Gait is evaluated by watching the patient comfortably and safely walking with turns. Turning requires more coordination than natural walking; therefore, patients with cerebellar ataxia may find turning more difficult. Sequentially, tandem walking can further bring out ataxia. Ataxic gait is typically wide based to compensate for imbalance and staggering. Tandem walking forces the patient to walk on a very narrow base, amplifying ataxia. Ataxic patients attempting to walk in tandem may have large amplitude sway or side-stepping to maintain balance. Although wide-based gait is commonly associated with ataxia, the core feature of cerebellar gait is stride variability, which is an inability to walk with consistent cadence or direction throughout. The examiner cannot predict where the next step will occur due to variation in stride length, cadence, or speed. Stride variability should still be observed in patients who exhibit wide-based gait. Lastly, some patients may have co-existing spastic gait. Spastic gait is a stiff leg walk with foot dragging and circumduction of the leg. A good example is autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). The examples of ataxic stance and gait can be viewed in Videos 5 and 6.
OTHER MOVEMENT DISORDER EXAMINATION CONSIDERATIONS
Once ataxia is established by cerebellar-specific examination as discussed earlier, looking for other signs of movement disorders is important. These signs include parkinsonism, dystonia, and myoclonus. Parkinsonism is defined by bradykinesia, hypokinesia, and rigidity. Parkinsonism is present in MSA, PSP, SCA2, SCA3, and SCA17. Rest tremor is uncommon but may be seen in MSA and SCA2. Dystonia is sustained or intermittent muscle contractions that results in abnormal posturing, twisting, or repetitive movements. Facial dystonia and anterocollis (forward neck flexion) are common in MSA. Although dystonia is the prominent feature of Wilson disease, an autosomal recessive disorder caused by excessive copper deposition, some cases can also present with ataxia. Dystonia can be seen in autosomal dominant ataxias, including SCA3 and SCA8, and in autosomal recessive ataxias, including abetalipoproteinemia, ataxia telangiectasia, Niemann-Pick type C (NPC), and polymerase gamma gene-related disorder (POLG) ataxia. Chorea is involuntary, random, unsustained, dancelike movements. In the autosomal dominant ataxias, chorea is typically accompanied by dementia in dentatorubral-pallidoluysian atrophy and SCA17. In the autosomal recessive ataxias, chorea is seen in ataxia telangiectasia, abetalipoproteinemia, ataxia with oculomotor apraxia (AOA) types 1 and 2, and POLG ataxia. Myoclonus is a fast, involuntary muscle jerk frequently present in mitochondrial disorders and progressive myoclonic epilepsies.
GENERAL NEUROLOGIC EXAMINATION CONSIDERATIONS
Cerebellar disease can affect executive, visuospatial, language, and emotional processing, and these should be screened for in office. The cerebellar cognitive affective/Schmahmann syndrome scale (CCAS-Scale) was devised specifically for patients with cerebellar ataxia.25 This easy-to-administer scale assesses verbal fluency, executive function, memory, visuospatial, and affect domains.
Additional eye examinations can add to the diagnosis, as delineated in Table 1.21 Fundoscopic examination showing papilledema can suggest optic neuritis due to demyelinating or infectious processes. Relative afferent pupillary defect also suggests a demyelinating process. Pigmentary retinal macular degeneration can be found in SCA7, whereas retinitis pigmentosa is associated with Refsum disease, abetalipoproteinemia, and Kearns-Sayre syndrome.
Muscle strength examination is not only important for determining cerebellar ataxia cause but also uncovers contributions of weakness to the clinical presentation. Myopathies typically affect proximal muscles first and are associated with mitochondrial disorders, which often have cerebellar involvement. Distal weakness can be seen in myopathy, neuropathy, or motor neuron disease. Friedreich ataxia is a good example of ataxia associated with distal weakness.
Detailed sensory examination is critical when evaluating a patient for ataxia to distinguish mainly between cerebellar ataxia and sensory ataxia. Sensory ataxia results from loss of proprioceptive input. Testing vibratory sensation and joint position sense can assess large fiber sensory nerve function. Loss of visual input by closing the eyes can worsen sensory ataxia, whereas it has only minor effects on cerebellar ataxia. Thus, in sensory ataxia, imbalance occurs more at nighttime when it is dark or with the eyes closed; this can be further tested by Romberg sign where the patient stands with feet together and eyes closed. Of course, if the patient falls or steps to the side with the feet together when the eyes are opened, testing with the eyes closed would not yield additional information. Sensory loss associated with polyneuropathy can be seen in many of the autosomal recessive ataxias, mitochondrial neuropathies, and paraneoplastic disorders.
OTHER EXAMINATION CONSIDERATIONS
Examination of the skin and extremities can yield important clinical information. Cholesterol depositions in tendons (ie, xanthomas) are seen in cerebrotendinous xanthomatosis as subcutaneous nodules or skin papules.26 Telangiectasias seen in ataxia telangiectasias first appear in the eyes around age 3 to 6 years, then spread to involve the face. Pes cavus and scoliosis are common in Friedreich ataxia. NPC may have hepatosplenomegaly.
CLINICAL RATING SCALES TO TRACK ATAXIA SEVERITY
Clinical ratings scales are important for standardizing evaluation and tracking disease severity; this is particularly important in clinical trials. The scale for the assessment and rating of ataxia (SARA) is a 40-point widely used rating scale for ataxia (Table 2).27 SARA evaluates speech, limb ataxia, tremor, stance, and gait using defined objective measures. SARA is the most widely used ataxia scale and has been validated in the natural history studies for SCAs in both North America28 and Europe.29 Eye findings are not included and are better evaluated by the more comprehensive 100-point International Cooperative Ataxia Rating Scale (ICARS),30 which evaluates eye movements at rest, with pursuit, for nystagmus and saccadic dysmetria. Alternatively, the brief ataxia rating scale is a more concise ICARS.31
Table 2.
Scale of the Assessment and Rating of Ataxia distribution of score27
| Assessment | Score Rating |
|---|---|
| Gait | 0–8 0—Normal 1—Slight difficulties seen with tandem only 2—Abnormal gait, cannot perform tandem 3—Staggering gait but walks without support 4—Marked staggering, touching walls intermittently 5—Severe staggering, support of walking stick/cane or light one arm support needed 6—Can walk >10 m but needs walker or person 7—Can walk <10 m with walker or person 8—Cannot walk even with support |
| Stance | 0–6 0—Normal 1—Cannot stand in tandem for >10 s, no sway in other positions 2—Can stand with feet together for >10 s but has sway 3—Can stand in natural position only >10 s 4—Can stand in natural position only >10 s with intermittent support 5—Can stand in natural position only >10 s with constant arm support 6—Cannot stand in natural position >10 s even with constant support |
| Sitting | 0–4 0—Normal 1—Intermittent sway with sitting >10 s 2—Constant sway with sitting >10 s 3—Intermittent support needed with sitting >10 s 4—Unable to sit >10 s without continuous support |
| Speech disturbance | 0–6 0—Normal 1—Slight speech impairment 2—Noticeably impaired speech but easily understood 3—Occasional words difficult to understand 4—Many words difficult to understand 5—Only single words understood 6—Cannot understand speech/anarthria |
| Finger chase | Right 0–4; Left 0–4 0—Normal 1—Dysmetria <5 cm 2—Dysmetria <15 cm 3—Dysmetria >15 cm 4—Cannot perform 5 consecutive movements |
| Finger-nose-finger test | Right 0–4; Left 0–4 0—Normal 1—Tremor amplitude <2 cm 2—Tremor amplitude <5 cm 3—Tremor amplitude >5 cm 4—Unable to perform 5 movements |
| Rapid alternating movements | Right 0–4; Left 0–4 0—Normal 1—Slight irregularities, can perform 10 alternations <10 s 2—Clear irregularities, can perform 10 alternations <10 s 3—Many irregularities, interruptions, and undistinguishable movements, performs alterations >10 s 4—Cannot perform 10 alternating movements |
| Heel-shin slide | Right 0–4; Left 0–4 0—Normal 1—Slightly abnormal, contact with shin maintained 2—Clearly abnormal, can go off shin ≤3 times 3—Severely abnormal, goes off shin ≥4 times 4—Cannot perform |
| Total: 40 |
Appendicular examination, including finger chase, finger-nose-finger, rapid alternating movements, and heel-shin slide, are averaged scores of the left and right sides.
EVALUATION OF ATAXIA CAUSE BASED ON PRESENTATION
Once ataxia is established based on examination, the clinician should determine the cause. Chronicity of disease, age of onset, and other notable clinical features can indicate differential diagnosis and direct investigatory evaluation.
Chronicity
Chronicity of disease can be separated into acute (hours to days), subacute (weeks), chronic (months to years) (Fig. 1), or episodic (discrete intervals of worsening) presentations. The ataxia causes of various chronicity are detailed in Table 3.32–34 Acute presentations of ataxia include vascular, demyelinating, and infectious/parainfectious. Varicella is one of the most common causes of acute infectious ataxia, and cerebellar disease is the most frequent neurologic manifestation. As varicella causes a pancerebellar syndrome, it presents with impaired gait, dysarthria, dysmetria, and tremor.35 Subacute presentations include immune-mediated, paraneoplastic, neoplastic, and some infectious/parainfectious causes. Chronic causes of ataxia include degenerative, hereditary, idiopathic, and structural lesions such as slow-growing neoplasms. Toxic/metabolic causes of cerebellar ataxia span from acute to chronic depending on the dose and exposure.
Fig. 1.
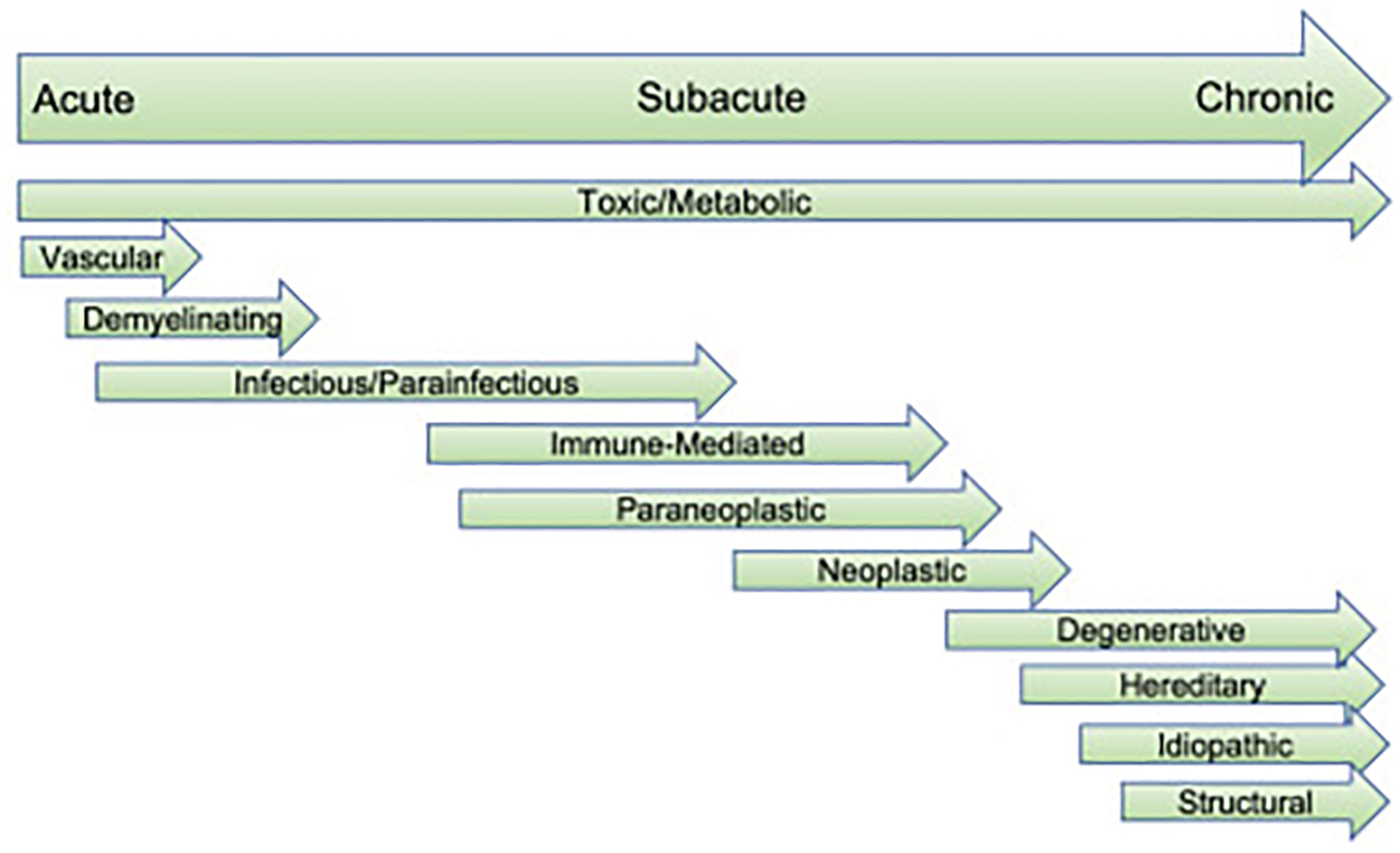
Causes of cerebellar ataxia and acuity of disease presentation.
Table 3.
Disease duration of cerebellar ataxia based on acuity of presentation
| Cerebellar Ataxia Causes Based on Disease Duration | |
|---|---|
| Acute | Vascular Ischemia, hemorrhage, vascular malformation Demyelinating Multiple sclerosis, neuromyelitis optica, acute disseminated encephalomyelitis Infectious Viral cerebellitis: VZV, EBV, influenza, EV71, parvovirus B19, RSV Bacterial: Listeria monocytogenes, Mycoplasma pneumoniae, Borrelia burgdorferi, abscess Fungal: Aspergillus, Histoplasmosis Parainfectious Bacterial: Mycoplasma pneumoniae Viral: measles, mumps, rubella, EBV, pertussis, rotavirus, coxsackie virus Toxic/metabolic Antiepileptic drugs, chemotherapy, benzodiazepines, alcohol intoxication, heavy metals, lithium, Wernicke encephalopathy |
| Subacute | Immune-mediated SLE, Sjögren syndrome, gluten ataxia, thyroid antibody mediated, GAD-65 antibody, Miller-Fisher syndrome Paraneoplastic Anti-Hu, Yo, Ri, mGluR1, Ma, CRMP, VGKC antibodies Neoplastic-posterior fossa tumors Children: medulloblastoma, astrocytomas, ependymoma, brainstem glioma Adults: metastasis, hemangioblastoma, lymphoma Infectious HIV, CJD, Whipple disease, JC virus/PML Toxic/metabolic Vitamin deficiencies (B1, B12, E), hypothyroidism, hyperparathyroidism |
| Chronic | Neurodegenerative Multiple system atrophy, progressive supranuclear palsy (less commonly) Idiopathic late onset cerebellar ataxia Hereditary Toxic/metabolic Heavy metals, solvents, phenytoin, alcohol Structural/Static Dandy Walker cyst, hydrocephalus, Joubert syndrome, cerebral palsy, Arnold-Chiari malformation, hypoxic ischemic encephalopathy |
Abbreviations: CJD, Creutzfeldt-Jakob disease; EBV, Epstein-Barr virus; EV71, enterovirus 71; HIV, human immunodeficiency virus; PML, progressive multifocal leukoencephalopathy; RSV, respiratory syncytial virus; VZV, varicella zoster virus.
Cerebellar ataxia can present episodically with normal examination or milder ataxia between episodes, outlined in Table 4. The episodic ataxias can be distinguished clinically based on precipitating factors, duration, and associated clinical phenomenon. Some episodic ataxias are inherited in an autosomal dominant fashion and are associated with mutations in genes encoding ion channels. Episodic ataxia types 1 (EA1) and 2 (EA2) and glucose transporter-1 (GLUT-1) deficiency are the most common types. EA1 attacks are triggered by exercise and startle; EA2 attacks are triggered by stress and alcohol; and GLUT-1 deficiency attacks are triggered by fasting or exercise and is associated with epilepsy.36 Mitochondrial disorders have notable neurologic features, including epilepsy, polyneuropathy, myopathy, and hearing or vision loss. Mitochondrial disorders may present with an episodic onset, then progress into a chronic form of ataxia. Finally, functional movement disorders should be included in the differential diagnosis of episodic ataxia.
Table 4.
Episodic causes of cerebellar ataxia
| Episodic Causes of Ataxia | Features |
|---|---|
| Genetic Episodic ataxia type 1 Episodic ataxia type 2 Episodic ataxia types 3–7 Glucose transporter-1 deficiency |
Lasts seconds-minutes + myokymia between attacks Lasts hours-days + nystagmus between attacks Rare Recurrent attacks ataxia ± vertigo +/− normal examination between attacks Paroxysmal exertional dyskinesias Ataxia with fasting, exercise, or illness Epilepsy |
| Inborn errors of metabolism Hartnup disease | Skin photosensitivity, pellagra-like rash |
| Mitochondrial disorders MELAS MERRF NARP |
Short stature Hearing and/or vision loss Polyneuropathy, myopathy +/− Epilepsy |
| Demyelinating Multiple sclerosis Neuromyelitis optica |
Holmes tremor, dysarthria, incoordination Cerebellar lesions Can progress to chronic ataxia Bulbar dysfunction, diplopia, vertigo, hiccups Brainstem, periaqueductal, & peri-third and fourth ventricular lesions |
| Functional movement disorder | Variability in phenomenology Distractable Entrainment |
Abbreviations: MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes; MERRF, myoclonus epilepsy with ragged red fibers; NARP, neuropathy, ataxia, and retinitis pigmentosa.
Age of Onset
Age of symptom onset can narrow the differential diagnosis. Common causes of cerebellar ataxia in children are toxic ingestion or infectious.37 Childhood chronic ataxia with a static disease course is consistent with perinatal insults. Childhood progressive ataxias should be evaluated for a genetic or metabolic basis. Children and young adults younger than 25 years should particularly be evaluated for autosomal recessive cerebellar ataxia if family history does not suggest an autosomal dominant inheritance. Adult-onset chronic progressive cerebellar ataxia should be investigated for genetic causes after acquired causes have been largely excluded. Generally, autosomal dominant cerebellar ataxias are more likely to have an age of onset in adulthood. In adults older than 50 years, degenerative causes, including MSA, idiopathic late-onset cerebellar ataxia, and fragile X-associated tremor and ataxia syndrome (FXTAS) should be considered. Regardless of age of onset, reversible causes, including immune-mediated or nutritional, should be investigated.
DIAGNOSTIC INVESTIGATION OF CEREBELLAR ATAXIA
Identifying clinical features of cerebellar ataxia warrants further investigation of the underlying diagnosis of cerebellar dysfunction. Imaging and serum laboratory workup are part of the initial evaluation. Particularly, the clinician should first look for reversible causes. A stepwise approach is outlined in Fig. 2.
Fig. 2.
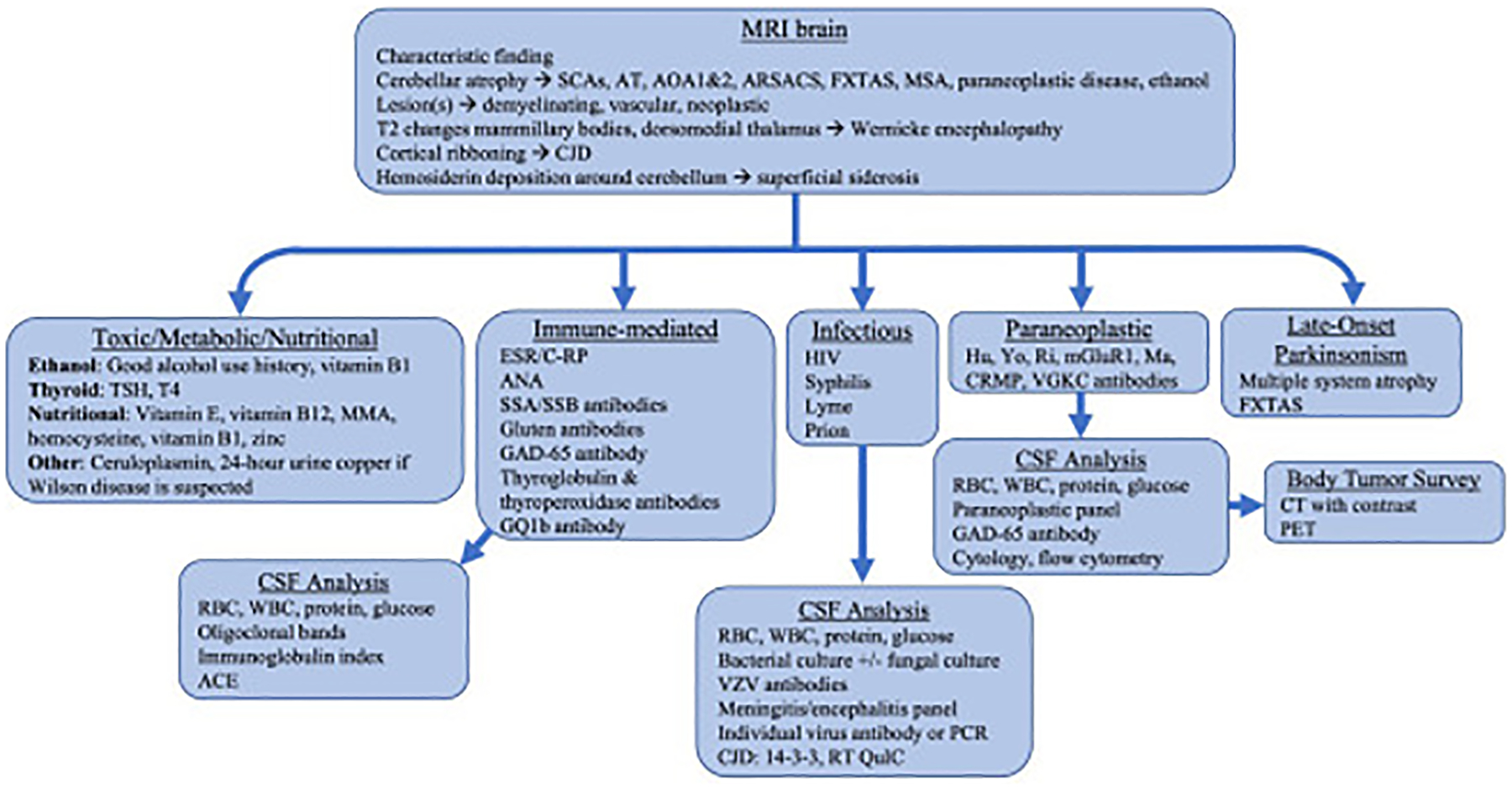
Investigatory workup for cerebellar ataxia. AOA, ataxia and oculomotor apraxia; ARSACS, autosomal recessive spastic ataxia of Charlevoix-Saguenay; AT, ataxia telangiectasia; CJD, Creutzfeldt-Jakob disease; CSF RT-QuIC, cerebrospinal fluid real-time quaking-induced conversion; TSH, thyroid stimulating hormone; FXTAS, fragile X tremor ataxia syndrome; MMA, methylmalonic acid; MSA, multiple system atrophy; SCA, spinocerebellar ataxia.
MRI of the brain is the best initial imaging modality to assess cerebellar atrophy and associated features. A contrasted study should be ordered when there is suspicion for an immune-mediated, neoplastic/paraneoplastic, or infectious processes. Several unique MRI features are associated with specific cerebellar diseases. Cerebellar atrophy is one of the most common imaging features of cerebellar ataxia (Fig. 3A). The pattern of cerebellar atrophy can also be helpful; notably, superior vermian atrophy is a hallmark finding in ARSACS (Fig. 3B). Pontine atrophy and “hot cross buns sign” can be seen in MSA (Fig. 3C) and in some cases of SCA2. Dopamine terminal degeneration in MSA can be evaluated by DaTSCAN (Fig. 3D). T2 FLAIR hyperintensities in the cerebellar peduncles are pathognomonic for FXTAS (Fig. 3E), and hyperintensities of the deep cerebellar nuclei can be seen in cerebrotendinous xanthomatosis (Fig. 3F). In patients with ataxia and palatal tremor, T2 hyperintensity in the inferior olives indicates hypertrophic degeneration (Fig. 3G).
Fig. 3.
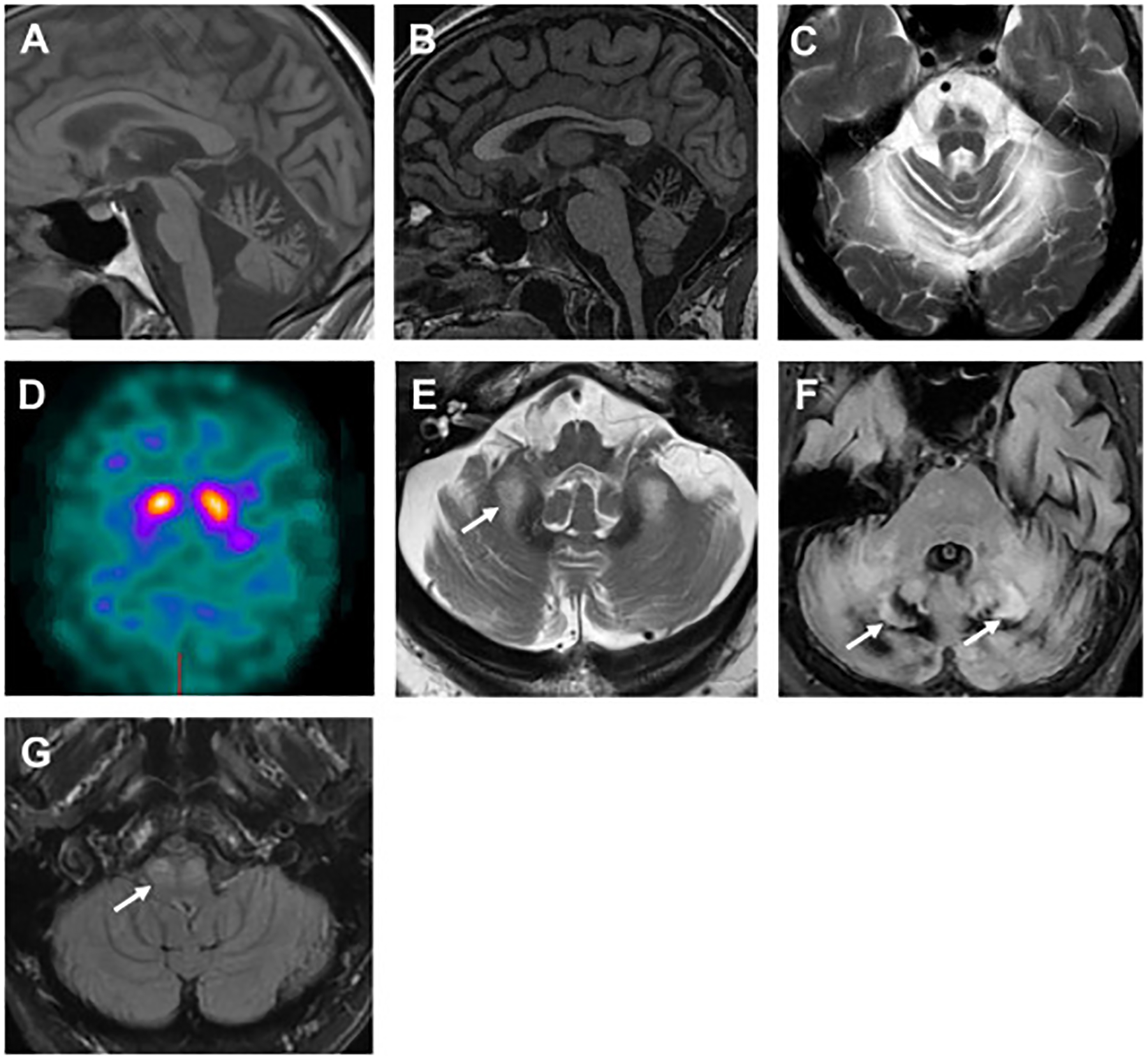
MRI findings that aid in the diagnosis of a specific ataxia syndrome. (A) Cerebellar atrophy in a patient with SCA2. (B) Superior cerebellar vermian atrophy in a patient with ARSACS. (C) “Hot cross buns” sign in a patient with MSA. (D) Asymmetric uptake DaTSCAN in a patient with MSA. (E) T2 hyperintensities of middle cerebellar peduncles in a patient with FXTAS (arrows). (F) T2 hyperintensities of dentate nuclei in a patient with cerebrotendinous xanthomatosis (arrows). (G) Inferior olive hypertrophy in a patient with progressive ataxia and palatal tremor (arrows). ARSACS, autosomal recessive spastic ataxia of Charlevoix-Saguenay; FXTAS, fragile X tremor ataxia; MSA, multiple system atrophy; SCA, spinocerebellar ataxia.
Serum laboratory investigation should be performed based on clinical suspicion (see Fig. 2). Reversible and acquired causes should be screened for, including vitamin deficiencies, such as vitamin B1, vitamin B12, and vitamin E; immune-mediated causes, including antinuclear antibody, Sjogren antibodies, gluten antibodies, anti-GAD-65 antibodies, and antithyroperoxidase and antithyroglobulin antibodies; and paraneoplastic causes. As the neuropsychological impairments of Wilson disease are treatable, clinicians should screen with serum ceruloplasmin and 24-hour urine copper in patients with ataxia and predominant dystonia.
Cerebrospinal fluid (CSF) analysis is reserved for suspected immune-mediated, paraneoplastic, or infectious processes. For infectious processes, individual viral antibodies and a meningitis/encephalitis panel can identify specific organisms. In cases of suspected Creutzfeldt-Jacob disease, 14-3-3 and real-time quaking-induced conversion (RT-QuIC) should be tested. Elevated oligoclonal bands and immunoglobulin index suggest an immune-mediated process. CSF paraneoplastic and anti-GAD65 antibodies sometimes can be found when serum testing is negative.
HEREDITARY CAUSES OF CEREBELLAR ATAXIA
Genetic forms of cerebellar ataxia are categorized based on inheritance pattern: autosomal dominant, autosomal recessive, X-linked, and mitochondrial inheritance. A detailed family history can reveal an inheritance pattern that directs genetic testing. Notably, autosomal dominant cerebellar ataxia could still be found in 5% of cases without a family history.38 Determining the underlying genetic diagnosis is prudent for counseling on disease progression, family planning, and/or enrollment in clinical trials for disease-modifying therapies.
Autosomal Dominant Cerebellar Ataxia
Autosomal dominant cerebellar ataxias are labeled SCA nomenclature and numbered by order of discovery. To date, 48 types have been found. The average prevalence is 2.7 (1.5–4.0) per 100,000.39 Pathologic CAG repeat expansions are the most common genetic mutations found in the SCAs, and SCA1, SCA2, SCA3, SCA6, SCA7, and SCA17 belong to this category. Some SCA trinucleotide repeat expansions result in anticipation or increase in the repeat number with each generation, causing earlier presentation with subsequent generations. SCA3 (also known as Machado-Joseph disease) is the most prevalent and has ethnic predilection in Brazil, Portugal, and China. SCA2 is the second most common autosomal dominant cerebellar ataxia and has ethnic predilection in Spain, Cuba, and Italy.39,40 Each SCA has different rates of disease progression: SCA1 has the fastest decline followed by SCA3 and SCA2, with SCA6 having the slowest disease progression.41 Each CAG-repeat SCA has certain unique clinical feature as delineated in Table 5. For example, parkinsonism can be seen in SCA2, SCA3, and SCA1720,42; chorea can be found in SCA17. SCA2 patients commonly have slowed saccades, hyporeflexia, and tremor, whereas SCA3 patients can have muscle fasciculations, bulging eyes, and restless leg syndrome. SCA6 is a pure cerebellar syndrome associated with downbeating nystagmus.43 SCA7 causes vision loss due to pigmentary retinal macular degeneration.44 The second most common type of genetic mutations in SCAs are repeat expansions in noncoding regions, and this category includes SCA8, SCA10, and SCA12. The third category is SCAs with coding sequence alterations, including SCA13, SCA14, SCA15, and SCA35.45
Table 5.
Notable autosomal dominant cerebellar ataxia syndromes
| Genetic Disorder | Mutation | Other Movement Disorder Features | Neurologic Features |
|---|---|---|---|
| SCA1 | CAG expansion ATXN1 |
Early bulbar symptoms Dysarthria, dysphagia Hypermetric saccades |
|
| SCA2 | CAG expansion ATXN2 |
Parkinsonism Truncal titubation Postural & rest tremors |
Slowed saccades Ophthalmoplegia Hyporeflexia Motor neuron disease |
| SCA3 (Machado-Joseph disease) | CAG expansion ATXN3 |
Parkinsonism Dystonia Restless leg syndrome |
Fasciculations Bulging eyes Ophthalmoplegia |
| SCA6 | CAG expansion CACN1A |
Pure cerebellar syndrome | Downbeating nystagmus |
| SCA7 | CAG expansion ATXN7 |
Pigmentary retinal macular degeneration Slowed saccades Spasticity |
|
| SCA17 (Huntington disease-like) | CAG expansion TBP |
Parkinsonism Chorea |
Early dysarthria Early dementia Psychiatric disorder |
Abbreviation: SCA, spinocerebellar ataxia.
Autosomal Recessive Cerebellar Ataxia
The prevalence of autosomal recessive cerebellar ataxias is 3.3 (1.8–4.9) per 100,000.39 The most commonly inherited recessive ataxia is Friedreich ataxia followed by AOA and ataxia telangiectasia. Friedreich ataxia and cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS) are due to repeat expansions, whereas the most of the remaining autosomal recessive cerebellar ataxias are due to sequence alterations.
Peripheral neuropathy is common in the autosomal recessive cerebellar ataxias and can be a defining feature for categorization into 3 types: (1) associated sensory neuropathy, which includes Friedreich ataxia and POLG ataxia; (2) associated sensorimotor neuropathy, which includes ataxia telangiectasia, cerebrotendinous xanthomatosis, AOA1, AOA2, and ARSACS; and (3) no neuropathy, which includes autosomal recessive cerebellar ataxia type 1 and 2 and NPC.
Friedreich ataxia patients often have skeletal abnormalities of pes cavus (Fig. 4) and scoliosis. Other systemic issues in Friedreich ataxia are cardiomyopathy and glucose intolerance. Ocular apraxia can be seen in AOA1, AOA2, and ataxia telangiectasia. AOAs are slowly progressive diseases, and ocular apraxia may progress to external ophthalmoplegia. The hallmark of NPC is vertical supranuclear ophthalmoplegia.46 CANVAS is characterized by cerebellar, vestibular, and sensory neuronopathy degeneration. Oculocephalic reflex produces saccadic eye movements.47 Selective clinical characteristics in autosomal recessive cerebellar ataxia are listed in Table 6.
Fig. 4.
General examination finding of pes cavus seen in Friedreich ataxia. AD, autosomal dominant; AOA, ataxia with oculomotor apraxia; AR, autosomal recessive; ARSACS, autosomal recessive spastic ataxia of Charlevoix-Saguenay; AT, ataxia telangiectasia; CANVAS, cerebellar ataxia with neuropathy and vestibular areflexia syndrome; CT, cerebrotendinous xanthomatosis; DRPLA, dentatorubral-pallidoluysian atrophy; FA, Friedreich ataxia; FXTAS, fragile X tremor ataxia; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes; MERRF, myoclonus epilepsy with ragged red fibers; NARP, neuropathy, ataxia, and retinitis pigmentosa; NPC, Niemann-Pick type C; SANDO, sensory ataxic neuropathy, dysarthria, and ophthalmoparesis; SCA, spinocerebellar ataxia.
Table 6.
Notable autosomal recessive cerebellar ataxia features
| Genetic Disorder | Mutation | CNS Manifestations | Extra-CNS Manifestations | Further Diagnostics |
|---|---|---|---|---|
| Friedreich ataxia | GAA repeat FXN | Sensory axonal neuropathy Proprioceptive loss, areflexia |
Diabetes mellitus Hypertrophic cardiomyopathy Pes cavus, scoliosis |
|
| Ataxia telangiectasia | ATM | Dystonia, choreoathetosis Oculomotor apraxia |
Recurrent infections (pulmonary) Risk for cancer (lymphoma) Telangiectasias |
Elevated CEA and α-fetoprotein |
| AOA type 1 AOA type 2 |
APTX
SETX |
Mental disability (type 1) Oculomotor apraxia Sensorimotor neuropathy Choreoathetosis |
Low albumin Elevated α-fetoprotein |
|
| Niemann-Pick type C | NPC1 | Supranuclear ophthalmoplegia Dystonia Dementia, epilepsy |
Hepatosplenomegaly | Elevated oxysterols |
| CANVAS | RFC1 repeat | Impaired visually enhanced VOR Sensory neuronopathy |
Dorsal vermis and lateral hemispheric atrophy | |
| POLG ataxia/SANDO | POLG | Sensory axonal polyneuropathy Epilepsy Chorea, dystonia, myoclonus |
Chronic external ophthalmoplegia Bilateral ptosis | |
| Autosomal recessive cerebellar ataxia type 1 | SYNE1 | Motor neuron disease | Cardiomyopathy Arthrogryposis, pes cavus | |
| Abetalipoproteinemia | MTTP | Retinitis pigmentosa Dystonia, chorea | Steatorrhea, failure to thrive Vitamin E malabsorption |
Acanthocytosis Low TG & cholesterol |
| ARSACS | SACS | Sensorimotor neuropathy Loss of vibration sense Spasticity |
Distal amyotrophy of feet | Superior cerebellar vermian atrophy, linear T2 pontine hypointensities |
| Cerebrotendinous xanthomatosis | CYP27A1 | Cataracts Epilepsy, cognitive impairment |
Diarrhea Tendinous xanthomas |
Elevated cholestanol Elevated bile alcohols |
Abbreviations: AOA, ataxia with oculomotor apraxia; ARSACS, autosomal recessive spastic ataxia of Charlevoix-Saguenay; CANVAS, cerebellar ataxia with neuropathy and vestibular areflexia syndrome; CEA carcinoembryonic antigen; SANDO, sensory ataxic neuropathy, dysarthria, and ophthalmoparesis; TG, triglycerides; VOR, vestibulo-ocular reflex.
X-Linked Cerebellar Ataxia
The most notable X-linked inherited ataxia is FXTAS, which is due to a premutation range CGG repeat expansion (55–200 repeats) in the FMR1 gene. FXTAS usually presents as action tremor in men in their 60s. As women have 2 X chromosomes, they are less likely to be affected. Nevertheless, clinicians should investigate family history of premature ovarian insufficiency. FXTAS may present with parkinsonism and ataxia in 20% of cases.48 Neuropathy and cognitive impairment are also common features.48 MRI T2 hyperintensities in the middle cerebellar peduncles is a significant clue (see Fig. 3E).
Ataxias in Mitochondrial Disorders
Mitochondrial disorders are maternally inherited. Cerebellar ataxia, neuropathy, myopathy, myoclonus, and epilepsy are common neurologic signs. Patients may also have short stature, hearing or vision loss.
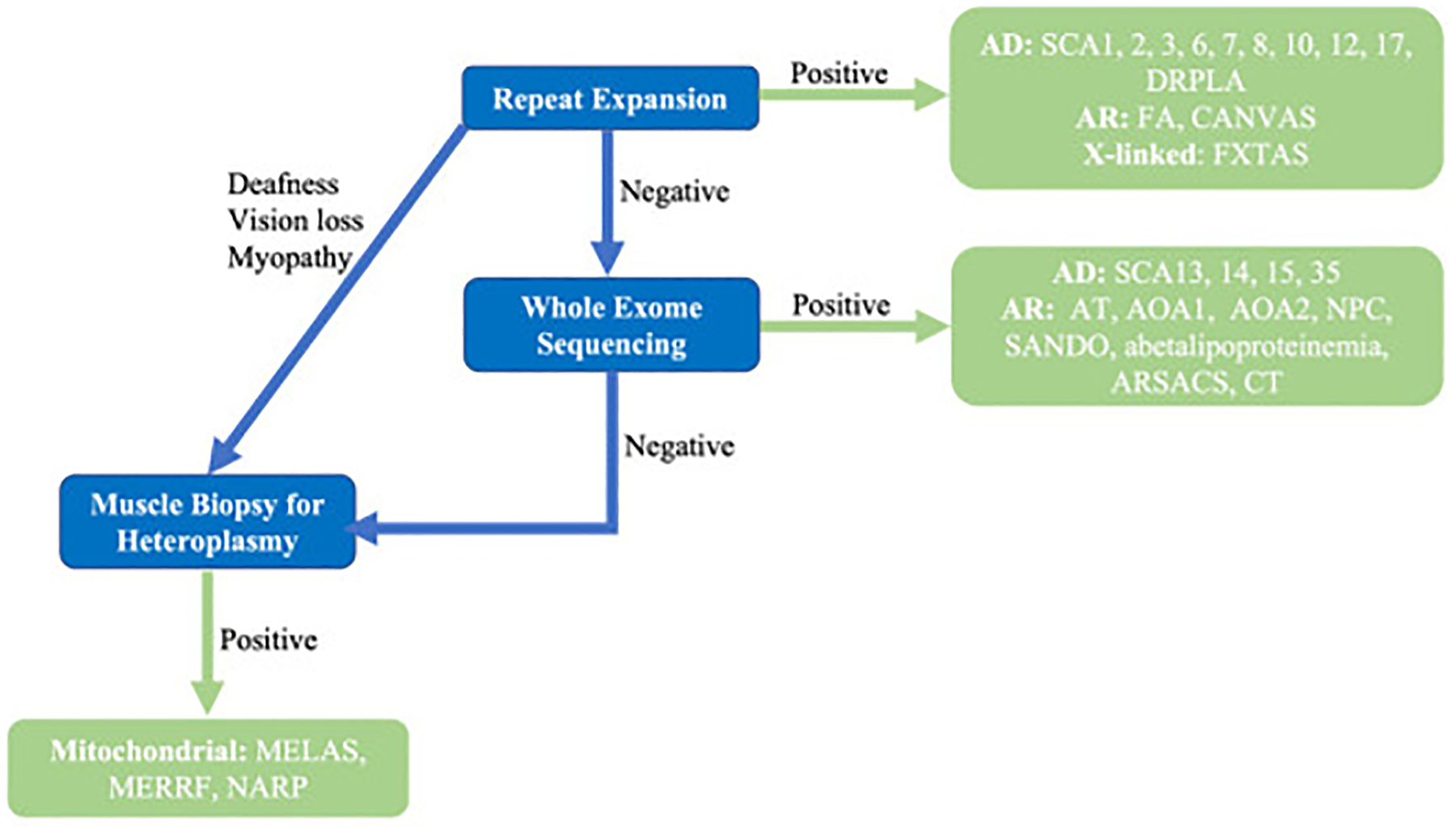
Genetic Testing Algorithm
A brief genetic test algorithm is provided in Fig. 5. The predominant autosomal dominant, autosomal recessive, and X-linked cerebellar ataxias are repeat expansions and thus are not commonly detected by whole exome sequencing. Thus, the first step of genetic evaluation for cerebellar ataxias is repeat expansion panels, which should include both coding and noncoding repeat expansions. The second step is to pursue either genetic sequence determination of individual ataxia genes or whole exome sequencing to detect sequence alterations and point mutations. If the clinical presentation and inheritance is consistent with mitochondrial disease, muscle biopsy should be performed to detect mitochondrial abnormality.
Fig. 5.
Algorithm for suspected inherited cerebellar ataxia syndrome.
THERAPEUTIC MANAGEMENT OF CEREBELLAR ATAXIA
Management of ataxia includes a two-pronged approach of medication and physical, occupational, and speech therapies. Physical therapy has been shown to improve balance and mobility in both hereditary and degenerative cerebellar ataxia.49,50 Patients should be encouraged to perform home exercises, which can improve balance and gait.51 These evidence-based exercise programs for cerebellar ataxia are demonstrated online and accessible to patients (https://www.ataxia.org/11-exercises-forataxia-patients/). In addition, high-intensity exercise improves mobility and balance in cerebellar ataxia52 and should also be recommended to patients as tolerated.
Pharmacologic therapies for cerebellar ataxia include both symptomatic and potentially disease-modifying therapies. Evidence often comes from clinical trials with small numbers of patients. Selective pharmacologic therapies for cerebellar ataxias are listed in Table 7. Immune-mediated cerebellar ataxias and paraneoplastic disorders can be treated with intravenous methylprednisolone or immunoglobulins; plasmapheresis is required in some cases. Certain autosomal recessive ataxias have targeted therapies, including miglustat for NPC, vitamin E for abetalipoproteinemia, and chenodeoxycholic acid for cerebrotendinous xanthomatosis. Ketogenic diet for GLUT-1 deficiency and gluten-free for gluten-associated ataxias are recommended.
Table7.
Treatments of cerebellar ataxia
| Therapeutic Interventions for Cerebellar Ataxia | ||
|---|---|---|
| Targeted disease-modifying therapy for acquired disorders | Wernicke encephalopathy & chronic alcohol use Immune-mediated Paraneoplastic Gluten-associated ataxia |
Thiamine, 1500 mg, IV daily × 2 d then 250 mg daily × 5 d Thiamine, 100 mg, PO daily Methylprednisolone, 1000 mg, IV × 3–5 d ± IVIG Methylprednisolone, 1000 mg, IV × 5 d + IVIG/plasmapheresis Gluten-free diet |
| Targeted disease-modifying therapy for inherited disorders | Niemann-Pick type C Abetalipoproteinemia GLUT-1 deficiency Cerebrotendinous xanthomatosis |
Miglustat 200 mg TID Vitamin E, 150 mg/kg/d Ketogenic diet, avoid fasting Chenodeoxycholic acid, 250 mg, TID |
| Symptomatic therapy | Cerebellar ataxia SCA3 Episodic ataxia type 1 Episodic ataxia type 2 |
Riluzole, 50 mg, BID Varenicline, 1 mg, BID Carbamazepine Acetazolamide, 250–1000 mg/d, 4-aminopyridine, 15 mg/d |
Abbreviations: BID, twice daily; d, day(s); GLUT, glucose transporter; IV, intravenous; IVIG, intravenous immunoglobulins; kg, kilograms; mg, milligrams; TID, three times per day.
Symptomatic therapies are also available for treating cerebellar ataxia. Riluzole improves gait and speech in a variety of ataxia diseases.53,54 Valproic acid55 and varenicline56 may improve ataxia symptoms in SCA3 patients; however, they both have notable side effects that should be discussed when weighing benefit and risk. Dietary supplementation with coenzyme Q10 (CoQ10) is associated with better outcomes in the natural history study of SCAs; however, randomized clinical trials are lacking.57 Patients with MSA have reduced cerebellar CoQ10 levels58,59; thus further studies are needed to determine supplementation benefits in this population. Carbamazepine decreases attacks in EA1, and both acetazolamide and 4-aminopyridine can decrease attacks in EA2.60 As SCA6 and EA2 are both caused by mutations in CACN1A gene, acetazolamide may mitigate episodic symptoms in SCA6.61,62 Other therapeutic options, such as amantadine or idebenone, have been reviewed by the American Academy of Neurology and deemed not enough evidence to determine benefit.63
Parkinsonism seen in MSA, SCA2, SCA3, SCA8, and SCA17 may be levodopa-responsive, so clinicians can trial levodopa starting 100 mg three times a day to assess the responses. Compared with patients with Parkinson disease, the response tends to be less robust and transient. Higher doses of levodopa up to 1,000–1,200 mg daily may be required to see effects, permitting side effects of sedation, orthostasis, and nausea. Vertigo may respond to benzodiazepines. Nystagmus may improve with symptomatic therapy for memantine, gabapentin,64 or 4-aminopyridine.65 Depression should be approached with cognitive behavioral therapy and pharmacologic therapy when indicated.
Active research is ongoing for gene therapy and antisense oligonucleotide targeting of specific genetic forms of cerebellar ataxias.66 Deep brain stimulation has recently been explored to treat cerebellar ataxias as well.67,68 Transcranial direct current stimulations and repetitive transcranial magnetic stimulation can have sustained benefits in patients with cerebellar ataxia.69–71
KEY POINTS.
Cerebellar ataxia is characterized by movement incoordination and variability.
The cerebellum plays an important role in scaling precise movements, mood, and cognition.
The causes of cerebellar ataxia are vast, and once cerebellar ataxia is established based on examination, workup for reversible and genetic causes should be pursued.
Important diagnostic clues for cerebellar ataxia that guide a diagnostic workup include age of onset, chronicity, and other associated movement disorders.
Some genetic and acquired causes of cerebellar ataxia have targeted disease-modifying therapies.
CLINICS CARE POINTS.
Once cerebellar ataxia is established based on examination, the etiology should be investigated.
Each patient should be evaluated for treatable or reversible causes of cerebellar ataxia.
More investigation is needed for therapeutic medications to treat ataxia symptoms of non-reversible causes of cerebellar ataxia.
FINANCIAL DISCLOSURES/CONFLICTS OF INTEREST
Dr S-H. Kuo has received funding from the National Institutes of Health: NINDS #R01 NS118179 (principal investigator), NINDS #R01 NS104423 (principal investigator), NINDS #R03 NS114871 (principal investigator), the Louis V. Gerstner Jr. Scholar Award, Parkinson’s Foundation, National Ataxia Foundation, and International Essential Tremor Foundation. Dr S-H. Kuo served as the Scientific Advisor for Praxis Precision Medicines and Sage Therapeutics. Dr T.A. Zesiewicz has received personal compensation for serving on the advisory boards of Boston Scientific, Reata Pharmaceuticals, and Steminent Biotherapeutics. Dr T.A. Zesiewicz has received personal compensation as senior editor for Neurodegenerative Disease Management and as a consultant for Steminent Biotherapeutics. Dr T.A. Zesiewicz has received royalty payments as co-inventor of varenicline for treating imbalance and nonataxic imbalance. Dr T.A. Zesiwicz has received research grant support as principle investigator for studies from AbbVie, Biogen, Biohaven Pharmaceuticals, Boston Scientific, Buk-wang Pharmaceuticals Co, Ltd, Cala Health, Inc, Cavion, Friedreich’s Ataxia Research Alliance; Houston Methodist Research Institute, National Institutes of Health, REtrotope Inc; and Takeda Development Center Americas, Inc.
Footnotes
Video content accompanies this article at http://www.neurologic.theclinics.com.
SUPPLEMENTARY DATA
Supplementary data related to this article can be found online at https://doi.org/10.1016/j.ncl.2022.05.002.
REFERENCES
- 1.Koziol LF, Budding D, Andreasen N, et al. Consensus paper: the cerebellum’s role in movement and cognition. Cerebellum 2014;13(1):151–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guell X, Gabrieli JDE, Schmahmann JD. Embodied cognition and the cerebellum: Perspectives from the Dysmetria of Thought and the Universal Cerebellar Transform theories. Cortex 2018;100:140–8. [DOI] [PubMed] [Google Scholar]
- 3.Schmahmann JD. The cerebellum and cognition. Neurosci Lett 2019;688:62–75. [DOI] [PubMed] [Google Scholar]
- 4.Roostaei T, Nazeri A, Sahraian MA, et al. The human cerebellum: a review of physiologic neuroanatomy. Neurol Clin 2014;32(4):859–69. [DOI] [PubMed] [Google Scholar]
- 5.Manto M, Bower JM, Conforto AB, et al. Consensus paper: roles of the cerebellum in motor control–the diversity of ideas on cerebellar involvement in movement. Cerebellum 2012;11(2):457–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmahmann JD, Pandya DN. The cerebrocerebellar system. Int Rev Neurobiol 1997;41:31–60. [DOI] [PubMed] [Google Scholar]
- 7.Schmahmann JD, Pandya DN. Projections to the basis pontis from the superior temporal sulcus and superior temporal region in the rhesus monkey. J Comp Neurol 1991;308(2):224–48. [DOI] [PubMed] [Google Scholar]
- 8.Schmahmann JD, Pandya DN. Anatomic organization of the basilar pontine projections from prefrontal cortices in rhesus monkey. J Neurosci 1997;17(1):438–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dum RP, Strick PL. An unfolded map of the cerebellar dentate nucleus and its projections to the cerebral cortex. J Neurophysiol 2003;89(1):634–9. [DOI] [PubMed] [Google Scholar]
- 10.Schmahmann JD, Sherman JC. The cerebellar cognitive affective syndrome. Brain 1998;121(Pt 4):561–79. [DOI] [PubMed] [Google Scholar]
- 11.Amokrane N, Viswanathan A, Freedman S, et al. Impulsivity in Cerebellar Ataxias: Testing the Cerebellar Reward Hypothesis in Humans. Mov Disord 2020;35(8): 1491–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amokrane N, Lin CR, Desai NA, et al. The Impact of Compulsivity and Impulsivity in Cerebellar Ataxia: A Case Series. Tremor Other Hyperkinet Mov (N Y) 2020; 10:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lo RY, Figueroa KP, Pulst SM, et al. Depression and clinical progression in spinocerebellar ataxias. Parkinsonism Relat Disord 2016;22:87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benrud-Larson LM, Sandroni P, Schrag A, et al. Depressive symptoms and life satisfaction in patients with multiple system atrophy. Mov Disord 2005;20(8): 951–7. [DOI] [PubMed] [Google Scholar]
- 15.Luo L, Wang J, Lo RY, et al. The Initial Symptom and Motor Progression in Spinocerebellar Ataxias. Cerebellum 2017;16(3):615–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Termsarasab P, Thammongkolchai T, Rucker JC, et al. The diagnostic value of saccades in movement disorder patients: a practical guide and review. J Clin Mov Disord 2015;2:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rascol O, Sabatini U, Simonetta-Moreau M, et al. Square wave jerks in parkinsonian syndromes. J Neurol Neurosurg Psychiatry 1991;54(7):599–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leigh J, Zee DS. The neurology of eye movements. Oxford University Press; 2015. [Google Scholar]
- 19.Shaikh AG, Ramat S, Optican LM, et al. Saccadic burst cell membrane dysfunction is responsible for saccadic oscillations. J Neuroophthalmol 2008;28(4): 329–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim M, Ahn JH, Mun JK, et al. Extracerebellar Signs and Symptoms in 117 Korean Patients with Early-Stage Spinocerebellar Ataxia. J Clin Neurol 2021; 17(2):242–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stephen CD, Schmahmann JD. Eye Movement Abnormalities Are Ubiquitous in the Spinocerebellar Ataxias. Cerebellum 2019;18(6):1130–6. [DOI] [PubMed] [Google Scholar]
- 22.Love RJ, Webb WG. The Neuromotor Control of Speech. Neurology for the Speech-Language Pathologist. 2nd edition. Butterworth-Heinemann; 1992:81–111: Chapter 6. [Google Scholar]
- 23.Gan SR, Wang J, Figueroa KP, et al. Postural Tremor and Ataxia Progression in Spinocerebellar Ataxias. Tremor Other Hyperkinet Mov (N Y) 2017;7:492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lai RY, Tomishon D, Figueroa KP, et al. Tremor in the Degenerative Cerebellum: Towards the Understanding of Brain Circuitry for Tremor. Cerebellum 2019; 18(3):519–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoche F, Guell X, Vangel MG, et al. The cerebellar cognitive affective/Schmahmann syndrome scale. Brain 2018;141(1):248–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nie S, Chen G, Cao X, et al. Cerebrotendinous xanthomatosis: a comprehensive review of pathogenesis, clinical manifestations, diagnosis, and management. Orphanet J Rare Dis 2014;9:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmitz-Hübsch T, du Montcel ST, Baliko L, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 2006;66(11): 1717–20. [DOI] [PubMed] [Google Scholar]
- 28.Ashizawa T, Figueroa KP, Perlman SL, et al. Clinical characteristics of patients with spinocerebellar ataxias 1, 2, 3 and 6 in the US; a prospective observational study. Orphanet J Rare Dis 2013;8:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jacobi H, Bauer P, Giunti P, et al. The natural history of spinocerebellar ataxia type 1, 2, 3, and 6: a 2-year follow-up study. Neurology 2011;77(11):1035–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen ML, Lin CC, Rosenthal LS, et al. Rating scales and biomarkers for CAG-repeat spinocerebellar ataxias: Implications for therapy development. J Neurol Sci 2021;424:117417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmahmann JD, Gardner R, MacMore J, et al. Development of a brief ataxia rating scale (BARS) based on a modified form of the ICARS. Mov Disord 2009; 24(12):1820–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sawaishi Y, Takada G. Acute cerebellitis. Cerebellum 2002;1(3):223–8. [DOI] [PubMed] [Google Scholar]
- 33.Lancella L, Esposito S, Galli ML, et al. Acute cerebellitis in children: an eleven year retrospective multicentric study in Italy. Ital J Pediatr 2017;43(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nussinovitch M, Prais D, Volovitz B, et al. Post-infectious acute cerebellar ataxia in children. Clin Pediatr (Phila) 2003;42(7):581–4. [DOI] [PubMed] [Google Scholar]
- 35.Bozzola E, Bozzola M, Tozzi AE, et al. Acute cerebellitis in varicella: a ten year case series and systematic review of the literature. Ital J Pediatr 2014;40:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kipfer S, Strupp M. The Clinical Spectrum of Autosomal-Dominant Episodic Ataxias. Mov Disord Clin Pract 2014;1(4):285–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pavone P, Praticò AD, Pavone V, et al. Ataxia in children: early recognition and clinical evaluation. Ital J Pediatr 2017;43(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moseley ML, Benzow KA, Schut LJ, et al. Incidence of dominant spinocerebellar and Friedreich triplet repeats among 361 ataxia families. Neurology 1998;51(6): 1666–71. [DOI] [PubMed] [Google Scholar]
- 39.Ruano L, Melo C, Silva MC, et al. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 2014;42(3):174–83. [DOI] [PubMed] [Google Scholar]
- 40.Schöls L, Bauer P, Schmidt T, et al. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol 2004;3(5):291–304. [DOI] [PubMed] [Google Scholar]
- 41.Jacobi H, du Montcel ST, Bauer P, et al. Long-term disease progression in spinocerebellar ataxia types 1, 2, 3, and 6: a longitudinal cohort study. Lancet Neurol 2015;14(11):1101–8. [DOI] [PubMed] [Google Scholar]
- 42.Park H, Kim HJ, Jeon BS. Parkinsonism in spinocerebellar ataxia. Biomed Res Int 2015;2015:125273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yabe I, Sasaki H, Takeichi N, et al. Positional vertigo and macroscopic downbeat positioning nystagmus in spinocerebellar ataxia type 6 (SCA6). J Neurol 2003; 250(4):440–3. [DOI] [PubMed] [Google Scholar]
- 44.Michalik A, Martin JJ, Van Broeckhoven C. Spinocerebellar ataxia type 7 associated with pigmentary retinal dystrophy. Eur J Hum Genet 2004;12(1):2–15. [DOI] [PubMed] [Google Scholar]
- 45.Ashizawa T,Öz G, Paulson HL. Spinocerebellar ataxias: prospects and challenges for therapy development. Nat Rev Neurol 2018;14(10):590–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gupta DK, Blanco-Palmero VA, Chung WK, et al. Abnormal Vertical Eye Movements as a Clue for Diagnosis of Niemann-Pick Type C. Tremor Other Hyperkinet Mov (N Y) 2018;8:560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Szmulewicz DJ, Roberts L, McLean CA, et al. Proposed diagnostic criteria for cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS). Neurol Clin Pract 2016;6(1):61–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leehey MA. Fragile X-associated tremor/ataxia syndrome: clinical phenotype, diagnosis, and treatment. J Investig Med 2009;57(8):830–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ilg W, Synofzik M, Brötz D, et al. Intensive coordinative training improves motor performance in degenerative cerebellar disease. Neurology 2009;73(22): 1823–30. [DOI] [PubMed] [Google Scholar]
- 50.Miyai I, Ito M, Hattori N, et al. Cerebellar ataxia rehabilitation trial in degenerative cerebellar diseases. Neurorehabil Neural Repair 2012;26(5):515–22. [DOI] [PubMed] [Google Scholar]
- 51.Keller JL, Bastian AJ. A home balance exercise program improves walking in people with cerebellar ataxia. Neurorehabil Neural Repair 2014;28(8):770–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Synofzik M, Ilg W. Motor training in degenerative spinocerebellar disease: ataxia-specific improvements by intensive physiotherapy and exergames. Biomed Res Int 2014;2014:583507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ristori G, Romano S, Visconti A, et al. Riluzole in cerebellar ataxia: a randomized, double-blind, placebo-controlled pilot trial. Neurology 2010;74(10):839–45. [DOI] [PubMed] [Google Scholar]
- 54.Romano S, Coarelli G, Marcotulli C, et al. Riluzole in patients with hereditary cerebellar ataxia: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2015;14(10):985–91. [DOI] [PubMed] [Google Scholar]
- 55.Lei LF, Yang GP, Wang JL, et al. Safety and efficacy of valproic acid treatment in SCA3/MJD patients. Parkinsonism Relat Disord 2016;26:55–61. [DOI] [PubMed] [Google Scholar]
- 56.Zesiewicz TA, Greenstein PE, Sullivan KL, et al. A randomized trial of varenicline (Chantix) for the treatment of spinocerebellar ataxia type 3. Neurology 2012; 78(8):545–50. [DOI] [PubMed] [Google Scholar]
- 57.Lo RY, Figueroa KP, Pulst SM, et al. Coenzyme Q10 and spinocerebellar ataxias. Mov Disord 2015;30(2):214–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kuo SH, Quinzii CM. Coenzyme Q10 as a Peripheral Biomarker for Multiple System Atrophy. JAMA Neurol 2016;73(8):917–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakamoto FK, Okamoto S, Mitsui J, et al. The pathogenesis linked to coenzyme Q10 insufficiency in iPSC-derived neurons from patients with multiple-system atrophy. Sci Rep 2018;8(1):14215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jen JC, Graves TD, Hess EJ, et al. Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain 2007;130(Pt 10):2484–93. [DOI] [PubMed] [Google Scholar]
- 61.Yabe I, Sasaki H, Yamashita I, et al. Clinical trial of acetazolamide in SCA6, with assessment using the Ataxia Rating Scale and body stabilometry. Acta Neurol Scand 2001;104(1):44–7. [DOI] [PubMed] [Google Scholar]
- 62.Jen JC, Yue Q, Karrim J, et al. Spinocerebellar ataxia type 6 with positional vertigo and acetazolamide responsive episodic ataxia. J Neurol Neurosurg Psychiatry 1998;65(4):565–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zesiewicz TA, Wilmot G, Kuo SH, et al. Comprehensive systematic review summary: Treatment of cerebellar motor dysfunction and ataxia: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology 2018;90(10):464–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thurtell MJ, Joshi AC, Leone AC, et al. Crossover trial of gabapentin and memantine as treatment for acquired nystagmus. Ann Neurol 2010;67(5):676–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tsunemi T, Ishikawa K, Tsukui K, et al. The effect of 3,4-diaminopyridine on the patients with hereditary pure cerebellar ataxia. J Neurol Sci 2010;292(1–2):81–4. [DOI] [PubMed] [Google Scholar]
- 66.Toonen LJ, Schmidt I, Luijsterburg MS, et al. Antisense oligonucleotide-mediated exon skipping as a strategy to reduce proteolytic cleavage of ataxin-3. Sci Rep 2016;6:35200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cury RG, França C, Duarte KP, et al. Safety and Outcomes of Dentate Nucleus Deep Brain Stimulation for Cerebellar Ataxia. Cerebellum 2021. 10.1007/s12311-021-01326-8. [DOI] [PubMed] [Google Scholar]
- 68.Teixeira MJ, Cury RG, Galhardoni R, et al. Deep brain stimulation of the dentate nucleus improves cerebellar ataxia after cerebellar stroke. Neurology 2015; 85(23):2075–6. [DOI] [PubMed] [Google Scholar]
- 69.Chen TX, Yang CY, Willson G, et al. The Efficacy and Safety of Transcranial Direct Current Stimulation for Cerebellar Ataxia: a Systematic Review and Meta-Analysis. Cerebellum 2021;20(1):124–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maas RPPW, Toni I, Doorduin J, et al. Cerebellar transcranial direct current stimulation in spinocerebellar ataxia type 3 (SCA3-tDCS): rationale and protocol of a randomized, double-blind, sham-controlled study. BMC Neurol 2019;19(1):149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Manor B, Greenstein PE, Davila-Perez P, et al. Repetitive Transcranial Magnetic Stimulation in Spinocerebellar Ataxia: A Pilot Randomized Controlled Trial. Front Neurol 2019;10:73. [DOI] [PMC free article] [PubMed] [Google Scholar]