

Abstract
Background:
Increased aurora A kinase (AAK) expression occurs in acute myeloid leukemia (AML); AAK inhibition is a promising therapeutic target in this disease. The aim of this phase II study was to assess activity of this combination in high-risk AML.
Methods:
This Phase II trial was conducted at Dana-Farber/Harvard Cancer Center in Boston, MA. We included untreated adult AML patients, ECOG 0–2, harboring an adverse-risk karyotype, secondary AML, therapy-related AML, and/or age ≥65 years. Patients received continuous infusion cytarabine 100mg/m2/day days 1–7 and idarubicin 12mg/m2/day days 1–3. Alisertib 30mg was administered orally twice daily days 8–15. Patients could receive up to 4 consolidation cycles with cytarabine and alisertib, and alisertib maintenance for 12 months. The primary endpoint was the rate of composite complete remission (CR+CRi). We used a Simon two-stage design, assuming a null CR+CRi of 45%. Analyses were per-protocol. The trial is registered (clinicaltrials.gov, NCT02560025) and has completed enrollment.
Findings:
We enrolled 39 eligible patients between December 31, 2015 and August 1, 2017; median follow up was 13.7mo (IQR 12.7–14.4). 19 patients (49%) had secondary AML and 3 had therapy-related AML. At mid-induction, 33 (85%) demonstrated marrow aplasia, 6 (15%) received re-induction. Mortality at 30 and 60 days was 8% and 13%, respectively. The composite remission rate was 64%: 20 (51%) CR and 5 (13%) CRi. There was 1 PR, 8 (21%) were refractory, and 5 (13%) died before response assessment, with no treatment-related deaths. The most common grade 3 or 4 adverse events included febrile neutropenia (n=14), neutropenia (n=12), thrombocytopenia (n=12), anemia (n=11), anorexia (n=9), and oral mucositis (n=4).
Interpretation:
Alisertib combined with induction chemotherapy demonstrated a rate of remission of 64% in this unfavorable leukemia subset. This study met criteria to move forward to a future randomized trial in previously untreated high-risk AML.
Keywords: Acute Myeloid Leukemia, Aurora A Kinase, Alisertib, Spindle Apparatus, Induction Chemotherapy
Introduction:
The majority of patients with acute myeloid leukemia (AML) will die of their disease. Rates of remission and long term survival are especially poor among those who are older, have therapy-related AML or secondary (post-myelodysplastic syndromes (MDS) or post-myeloproliferative neoplasm (MPN)) AML, as well as those with adverse cytogenetic features.1 Cytarabine- and anthracycline-based induction regimens have been the mainstay of initial AML therapy in all patients with AML for decades,2 but have been associated with poorer responses in these higher-risk populations. Recent efforts have focused on novel approaches to poor-risk subgroups, such as CPX-351, a liposomal anthracycline- and cytarabine-containing product, which, among patients aged 60–75 years with secondary AML, was associated with improved survival compared to conventional induction chemotherapy.3 Nevertheless, more effective and durable therapies are urgently needed for higher-risk disease.
Alisertib is a potent and selective oral inhibitor of aurora A kinase (AAK). AAK is essential for various processes associated with mitotic progression, including centrosome function, spindle assembly, and chromosome alignment.4, 5 AAK over-expression has been noted in a variety of hematologic malignancies, including AML,6, 7 in which overexpression is associated with unfavorable clinical features.8 In preclinical models, effective inhibition of AAK causes substantial mitotic defects, followed by cell death via deleterious aneuploidy; alisertib, in particular, potently reduces the viability of AML cells.9, 10 Using in vitro and murine models, alisertib has also been shown to increase the activity of the anti-leukemic chemotherapy cytarabine10 and diminish cell viability and survival via increased expression of pro-apoptotic proteins, p27 and BIM.10
Phase 1 studies of alisertib monotherapy in solid tumors11, 12 and myeloid malignancies13 revealed tolerability for most patients, with diarrhea, cytopenias, fatigue, and stomatitis representing the most common adverse events. A phase 2 study of alisertib monotherapy among those with advanced myeloid neoplasms included 35 AML patients, among whom 5 partial responses and one prolonged complete remission (CR) lasting several months were documented. Furthermore, 34% of these patients achieved transfusion-independence, and 49% experienced stable disease.13 We conducted a dose-escalation study of alisertib combined with conventional 7+3 induction chemotherapy for patients with previously untreated AML, excluding those with core-binding factor alterations or acute promyelocytic leukemia (APL). Alisertib was administered for 7 consecutive days, following the conclusion of cytarabine infusion on day 7. A dose of 30mg twice daily was deemed the recommended phase II dose (RP2D). A composite remission rate (CCR) of 86% (64% CR, 23% CRi) was observed, and all but one response-evaluable patient achieved CR or CRi,14 with a 12-month PFS of 42% and OS of 62%.
These results prompted us to conduct the current phase 2 study, to assess the activity of alisertib combined with 7+3 induction chemotherapy among those patients with higher-risk AML, defined as therapy-related AML, AML arising from an antecedent myeloid malignancy, those with an adverse-risk karyotype, and/or those aged ≥ 65 years.
Methods:
Study Design and Participants
Eligible patients had previously untreated AML by World Health Organization (WHO) criteria15, and were high-risk as defined by either the presence of an adverse-risk karyotype,16, 17 the presence of secondary AML arising from an antecedent myelodysplastic syndrome or myeloproliferative neoplasm, the presence of therapy-related AML, and/or age ≥65 years. One patient was allowed to enroll at age 64 but turned 65 years at time of induction. Patients age ≥65 years were included due to historically poor outcomes reported across ELN risk groups for these patients.18, 19 Patients were required to have an Eastern Cooperative Oncology Group performance status of ≤2, left ventricular ejection fraction ≥50%, and intact organ function including aminotransferases and alkaline phosphatase < 2.5 times the upper limit of normal (ULN) unless thought to be related to leukemic infiltration of liver (in which case < 5.0 ULN was allowed), direct bilirubin < 2.0mg/dL, and creatinine clearance ≥ 40 mL/min. Patients must have been off systemic chemotherapy or radiotherapy within 14 days prior to the study, except for hydroxyurea or 6-mercaptopurine. Cytogenetic and molecular testing was performed at diagnosis. Patients with APL or with core-binding factors alterations [t(8;21) or inv(16)/t(16;16)] were excluded. Patients could be enrolled prior to cytogenetic classification, but those without adverse-risk karyotype and who lacked other eligibility criteria were removed before day 8 and replaced (n=3). This study was approved by the local Institutional Review Board at the Dana-Farber/Harvard Cancer Center, and all patients provided written informed consent. The study was conducted in accordance with the Declaration of Helsinki.
Procedures
Induction chemotherapy (“7+3”) consisted of continuous infusion cytarabine at 100mg/m2/day on days 1 through 7 (D1–7) and idarubicin 12mg/m2/day (daunorubicin at 60mg/m2 was allowed if idarubicin was unavailable or at investigator discretion) administered on D1–3 by intravenous bolus. One day following conclusion of cytarabine infusion, alisertib was initiated orally at 30mg twice daily (BID) for 7 days (D8–14). A bone marrow biopsy was performed at mid-induction (between D13–16). If marrow cellularity was ≥20% and myeloblast involvement was >5%, “5+2” re-induction, with cytarabine at 100mg/m2/day on D1–5 and idarubicin 12mg/m2/day (or daunorubicin at 60mg/m2/day) intravenously on D1–2, was administered. If bone marrow cellularity was <20% but with >5% blasts, 5+2 re-induction was allowed per clinician discretion. Those receiving 5+2 re-induction did not receive additional alisertib. A bone marrow evaluation was performed at time of peripheral hematologic recovery (absolute neutrophil count [ANC] > 1000/μL and platelet count > 100,000/μL) to assess for response, or by day 40 (or 60 if 5+2 was administered) in the absence of optimal hematologic recovery. Responses were assessed by the treating investigator. Response criteria were categorized as complete remission (CR), complete remission with incomplete neutrophil or platelet recovery (CRi), or refractory disease.20, 21 All patients were followed for relapse and survival.
Those achieving CR or CRi could come off study treatment to pursue hematopoietic cell transplantation (HCT). Responding patients were eligible for up to four cycles of consolidation therapy, at their treating clinicians’ discretion. Consolidation therapy consisted of cytarabine intravenously dosed at 3g/m2 every twelve hours on days 1, 3, and 5 in patients aged <60, or 2g/m2 daily on D1–5 in those aged ≥60. Starting on day 6 of each consolidation cycle, alisertib was administered at 30mg BID for 7 days (D6–12). After completing consolidation, patients were eligible for alisertib maintenance, which was administered at 30mg BID, on D1–7 of 21-day cycles, and continued until 12 months after initiation of induction in the absence of relapse (Supplemental Figure 1). Toxicities were graded and categorized according to common criteria (CTCAE version 4.0) during induction for all patients, and during consolidation and maintenance.
Studies to assess target (AAK) inhibition were performed on marrow or peripheral blood blasts obtained at diagnosis. Patient samples taken at time of diagnosis were first cultured ex vivo. Cryopreserved primary bone marrow samples, collected at baseline, were thawed and cultured for 36–48h on confluent MMC (2μg/ml) treated mouse M2–10B4 stromal cells in RPMI medium supplemented with 20% fetal bovine serum, 1% penicillin/streptomycin, 50μM β-mercaptoethanol, and human cytokines including stem cell factor (100ng/ml), interleukin −3 (10ng/ml), IL-6 (20ng/ml), thrombopoietin (10ng/ml), and FLT3 ligand ( 10ng/ml) (Peprotech). Once cells were proliferative, each sample was transferred to two new wells of MMC treated M2–10B4 stroma and left untreated, or treated with 100nM alisertib for 18 hrs.14,10, 22 Patient leukemic cells were then spun at 1,000rpm for 3 minutes onto poly-lysine coated coverslips and fixed in ice-cold methanol. Coverslips were blocked in tris buffered saline /bovine serum albumin , stained for tubulin (dmlα: Sigma) and DNA (0.2 μg/mL 4′,6-diamidino-2-phenylindole), and mounted on coverslips using ProLong Antifade mounting medium (Molecular Probes). A Nikon Ti-E fluorescence microscope equipped with a 60x objective and a Zyla sCMOS camera was used to identify and capture images of mitotic cells in each sample. The identification and characterization of the mitotic spindle structure was performed on a minimum of 20 mitotic figures per condition.
Sequencing of 95 genes recurrently mutated in myeloid malignancies was performed on diagnostic samples as well as at the end of induction using a clinically validated amplicon-based targeted sequencing platform (TruSeq Custom Amplicon, Illumina).23 Variants detected at ≥5% variant allele fraction (VAF) were classified as pathogenic using gene-specific criteria, as previously described.25
Outcomes
The primary endpoint of the study is the rate of complete remission (CR) or complete remission with incomplete count recovery (CRi) in higher risk patients receiving alisertib in combination with 7+3 induction chemotherapy. Secondary endpoints include 1-year overall survival (OS), relapse free survival (RFS), remission duration, and toxicity for patients receiving alisertib in combination with induction. Prespecified correlative studies included gene panel sequencing on leukemia samples to assess associations with treatment response and progression, as well as ex vivo studies to assess aurora kinase inhibition on patient-derived leukemic blasts.
Statistical Analysis
We hypothesized that alisertib with chemotherapy would have a CR+CRi rate of 0.70, compared to a historical remission rate of 0.45. A total of 39 evaluable patients were planned, assuming the null CR+CRi rate (CCR) of 0.45 with 7+3 chemotherapy alone and a rate of 0.70 for alisertib with 7+3 induction. This would have a 90% power to detect a difference at the 0.05 significance level using a one-sided single proportion test. In order to not expose patients to an inactive regimen, a Simon two-stage design was used. The first stage would accrue 13 patients and, if more than 6 CR+CRi’s were observed, would then accrue 26 additional patients. If more than 22 CR+CRi’s were observed out of 39, then alisertib combined with 7+3 induction would be considered active. Under this design, the probability of stopping after the first stage was 0.64. The rate of CR+CRi at the end of induction was estimated, as well as a bias-corrected maximum likelihood CR+CRi estimate and 2-stage 95% confidence interval.24
One-year overall survival (OS) and relapse free survival (RFS) after treatment were estimated using the Kaplan-Meier method, and presented as a 90% confidence interval using Greenwood’s formula for standard errors of the estimate. OS was defined as the time from study entry to death from any cause, and RFS was defined as the time from achieving a CR until disease recurrence or death. Remission duration was estimated using the method of Kaplan-Meier.
All toxicities encountered during the study were evaluated according to the NCI criteria (CTCAE version 4.0); grade 1–2 occurring in at least 10% of the patients, and all grade 3 or higher toxicities that were possibly, probably, or definitely attributed to alisertib by the treating investigator were presented. Exploratory outcomes were prespecified (protocol page 61–62). Pharmacodynamic and correlative studies, as well as patient and disease characteristics, including the number of patients completing each stage of therapy (induction, consolidation, and maintenance) were presented descriptively. To investigate associations with CR+CRi, we used a Student t-test for quantitatively measured correlatives and Fisher exact test for categorized correlatives. No adjustments were made for multiplicity of testing. For associations of post-induction VAF23 with RFS, we dichotomized patients at pre-specified VAF levels of 10%, 5% and 1% (exploratory analyses) and obtained p-values from the log rank test. Statistical analyses were performed using R version 3.4.3 (www.R-project.org). The trial is registered with ClinicalTrials.gov, NCT02560025.
Role of the Funding Source:
The funder of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. The corresponding author had full access to all of the data and the final responsibility to submit for publication.
Results:
A total of 39 eligible patients enrolled between December 31, 2015 and January 8, 2017 (Figure 1) and are included in this analysis. Three patients were excluded after starting screening; two did not meet inclusion criteria and one was lost to follow up. The median age was 67 (IQR 63–71; range 33–83); 25 (64%) were male, and 38 (97%) were white (Table 1). Patients were eligible if they had high risk AML. 19 patients (49%) had secondary AML (16 with antecedent MDS, 2 with antecedent chronic myelomonocytic leukemia (CMML), 1 with antecedent MPN), and 3 (8%) had therapy related AML; 6 patients met eligibility based on secondary AML alone. Thirteen (33%) exhibited adverse-risk karyotype, two of which had this as the sole inclusion.16, 17 27 patients were 65 or older, 12 of whom had age as their sole risk factor. Molecular profiling of recurrently mutated somatic genes was enriched for mutations associated with secondary AML,25 including DNMT3A (12, 31%), ASXL1 (12, 31%), TET2 (7, 18%), RUNX1 (8, 21%), and in pre-RNA splicing (SRSF2 7, 18%, SF3B1 3, 8%, ZRSR2 1, 3%) (Table 1). FLT3 mutations were seen in 7 patients (18%), NPM1 in 7 (18%), IDH1 in 5 (13%), IDH2 in 5 (13%), CEBPA in 3 (8%), and TP53 mutations in 4 (10%) patients. Of the 17 patients with a normal karyotype, 8 (47%) had ELN high risk AML due to their molecular profile.
Figure 1.
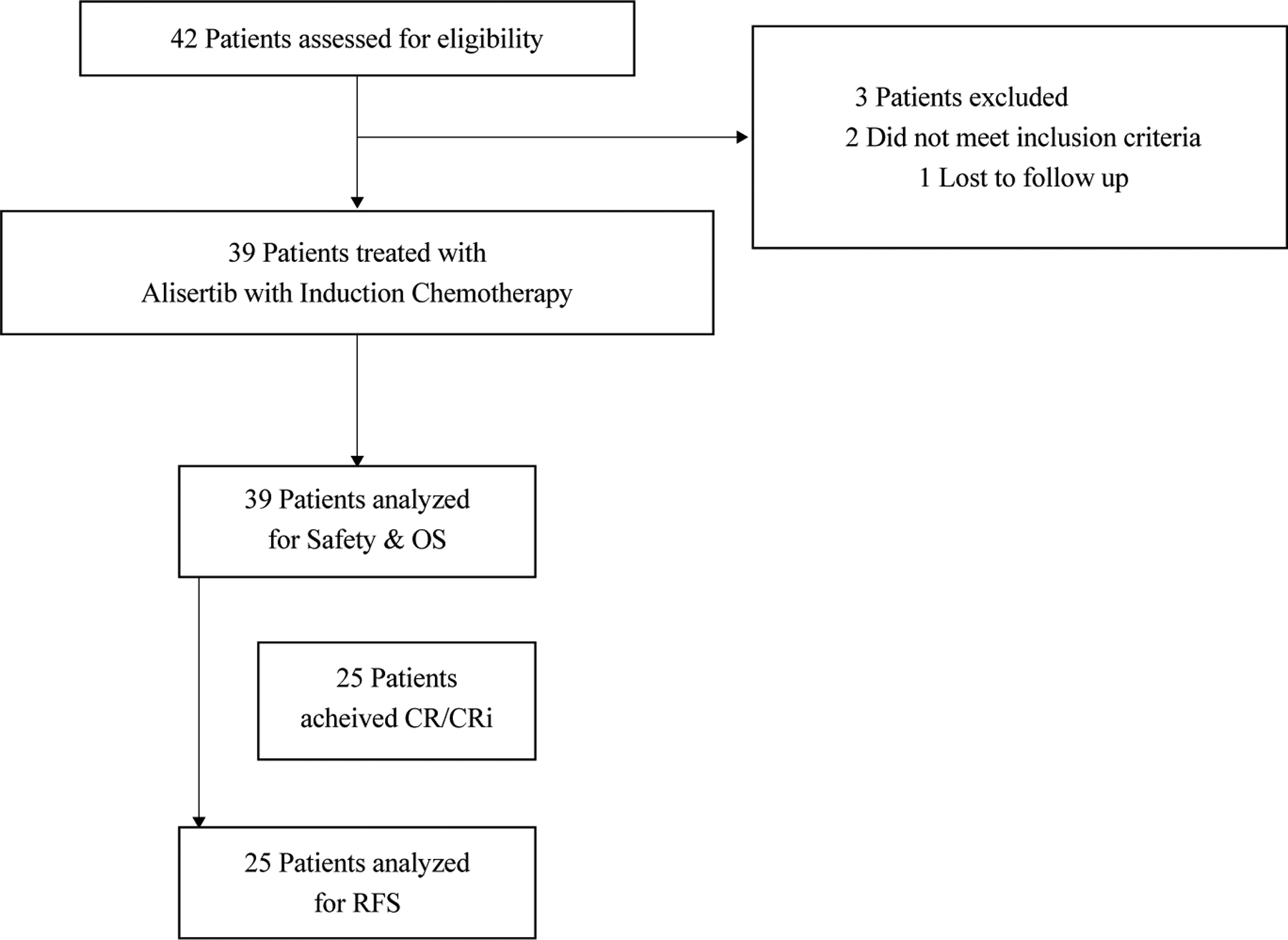
CONSORT diagram.
Table 1:
Patient Characteristics
| N (%) | |
|---|---|
| Number Enrolled | 39 |
| Median age (IQR, range) | 67 (8, 33–83) |
| Male | 25 (64%) |
| Caucasian | 38 (97%) |
| Median WBC (x10 3 ) (IQR, range) | 3.6 (7.15, 0.4–59.7) |
| Secondary AML | 19 (49%) |
| Prior MDS | 16 (41%) |
| Prior CMML | 2 (5%) |
| Prior MPN | 1 (3%) |
| Therapy-related AML | 3 (8%) |
| Cytogenetic Risk | |
| High | 13 (33%) |
| Intermediate | 26 (67%) |
| Normal Karyotype | 17 (44%) |
| ELN 2017 Risk Category | |
| Favorable* | 5 (13%) |
| Intermediate | 9 (23%) |
| Adverse | 25 (64%) |
| Mutation Profile | |
| ASXL1 | 12 (31%) |
| DNMT3A | 12 (31%) |
| RUNX1 | 8 (21%) |
| TET2 | 7 (18%) |
| NPM1 | 7 (18%) |
| SRSF2 | 7 (18%) |
| NRAS | 6 (15%) |
| FLT3 ITD | 5 (13%) |
| FLT3 TKD | 3 (8%) |
| IDH1 | 5 (13%) |
| IDH2 | 5 (13%) |
| STAG2 | 5 (13%) |
| TP53 | 4 (10%) |
| PTPN11 | 4 (10%) |
| BCOR | 4 (10%) |
| EZH2 | 4 (10%) |
| SF3B1 | 3 (8%) |
| BCORL1 | 3 (8%) |
| CEBPA | 3 (8%) |
| CBL | 3 (8%) |
| ZRSR2 | 1 (3%) |
| RIT1 | 1 (3%) |
| KRAS | 1 (3%) |
| ETV6 | 1 (3%) |
| Other | 7 (18%) |
All favorable risk patients were 65 or older at the time of induction, and none had core binding factor-altered AML.
Of the 39 treated patients, 33 (85%) demonstrated marrow aplasia at mid-induction, while the other six (15%) required re-induction with 5+2. Five patients (13%) died prior to response assessment, all before day 32; deaths were due to infectious or bleeding complications, and this rate was similar to that predicted in patients >60 years of age using the validated TRM score.26 The toxicity profile was consistent with that seen during the phase I study and consistent with that of 7+3 used in this setting (Supplementary Table 1); the most frequent attributable toxicities (possibly, probably, or definitely related to alisertib) included grade 4 neutropenia (28%), grade 3 anemia (28%), grade 4 thrombocytopenia (26%), and febrile neutropenia (36%). Grade 3 anorexia was reported in 23% of patients while grade 3 mucositis was seen in 10% (Table 2). A total of 2 patients required dose modification during their time on study, and 4 discontinued due to toxicity. Reported drug-related SAEs included G3 febrile neutropenia n=4, G3 increased bilirubin n=2, G3 diarrhea n=2, G3 ALT and AST elevation n=1, G2 rash n=1, and G2 abdominal pain n=1. There were zero treatment-related deaths, and six deaths overall during the study period (sepsis n=3, respiratory failure n=1, GI bleed n=1, hemorrhage n=1). The median time to ANC recovery to 500/mm3 in responding patients was 28 days (range 22–93 days), and to 1000/mm3 was 30 days (range 22–94). The median time to platelet recovery of 50,000/mm3 was 31 days (range 22–57) and to 100,000/mm3 was 33 days (range 22–51). Mortality at 30-days was 8% (95%CI 0–16%) and at 60-days was 13% (95%CI 2–23%). The composite CCR was 64% (66% per bias-corrected maximum likelihood estimation, 2-stage 95% CI 48–79%) with 20 patients (51%) achieving CR and 5 (13%) achieving CRi. One patient achieved a partial remission (3%). A total of 8 patients (21%) had progressive/refractory disease at the end of the 7+3 induction period; 6 of those patients had mid-induction marrow aplasia while 2 received 5+2 re-induction.
Table 2.
Grade 2 or higher toxicities attributable (Possibly, Probably, Definitely) to Alisertib (n=39).
| Toxicity Description | All Cause Toxicities | Attributed to Alisertib | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Grade 1–2 | Grade 3 | Grade 4 | Grade 5 | Grade 1–2 | Grade 3 | Grade 4 | ||||||||
| n | % | n | % | n | % | n | % | n | % | n | % | n | % | |
| Abdominal distension | 4 | 10 | ||||||||||||
| Abdominal pain | 7 | 18 | 1 | 3 | ||||||||||
| Acute kidney injury | 4 | 10 | 1 | 3 | 2 | 5 | ||||||||
| Adult respiratory distress syndrome | 1 | 3 | ||||||||||||
| Alanine aminotransferase increased | 11 | 28 | 4 | 10 | 5 | 13 | 4 | 10 | ||||||
| Alkaline phosphatase increased | 15 | 38 | 11 | 28 | ||||||||||
| Anemia | 7 | 18 | 19 | 49 | 4 | 10 | 11 | 28 | ||||||
| Anorexia | 23 | 59 | 11 | 28 | 17 | 44 | 9 | 23 | ||||||
| Anxiety | 5 | 13 | ||||||||||||
| Aspartate aminotransferase increased | 10 | 26 | 3 | 8 | 5 | 13 | 2 | 5 | ||||||
| Atrial fibrillation | 1 | 3 | 2 | 5 | ||||||||||
| Blood bilirubin increased | 13 | 33 | 4 | 10 | 2 | 5 | 9 | 23 | 3 | 8 | 1 | 3 | ||
| Bronchopulmonary hemorrhage | 1 | 3 | ||||||||||||
| Cardiac arrest | 1 | 3 | 2 | 5 | ||||||||||
| Catheter related infection | 1 | 3 | ||||||||||||
| Chills | 7 | 18 | ||||||||||||
| Colitis | 4 | 10 | 2 | 5 | 1 | 3 | ||||||||
| Confusion | 4 | 10 | 2 | 5 | ||||||||||
| Constipation | 7 | 18 | ||||||||||||
| Cough | 8 | 21 | ||||||||||||
| Creatinine increased | 1 | 3 | ||||||||||||
| Delirium | 2 | 5 | ||||||||||||
| Diarrhea | 28 | 72 | 4 | 10 | 15 | 38 | 2 | 5 | ||||||
| Dizziness | 6 | 15 | ||||||||||||
| Duodenal obstruction | 1 | 3 | ||||||||||||
| Dysgeusia | 4 | 10 | 4 | 10 | ||||||||||
| Dysphagia | 1 | 3 | 1 | 3 | ||||||||||
| Dyspnea | 9 | 23 | 2 | 5 | ||||||||||
| Edema limbs | 7 | 18 | ||||||||||||
| Enterocolitis | 3 | 8 | ||||||||||||
| Epistaxis | 5 | 13 | ||||||||||||
| Esophagitis | 5 | 13 | 2 | 5 | ||||||||||
| Fatigue | 22 | 56 | 1 | 3 | 14 | 36 | ||||||||
| Febrile neutropenia | 27 | 69 | 1 | 3 | 16 | 41 | ||||||||
| Fever | 10 | 26 | 1 | 3 | ||||||||||
| Gastroesophageal reflux disease | 4 | 10 | ||||||||||||
| Gastrointestinal disorders - Other | 6 | 15 | ||||||||||||
| Headache | 8 | 21 | ||||||||||||
| Hypernatremia | 4 | 10 | ||||||||||||
| Hypoxia | 8 | 21 | 1 | 3 | ||||||||||
| Hematuria | 1 | 3 | ||||||||||||
| Hepatic failure | 1 | 3 | ||||||||||||
| Hypertension | 1 | 3 | ||||||||||||
| Hypocalcemia | 1 | 3 | ||||||||||||
| Hyponatremia | 1 | 3 | ||||||||||||
| Hypophosphatemia | 1 | 3 | 2 | 5 | 1 | 3 | ||||||||
| Hypotension | 3 | 8 | 2 | 5 | ||||||||||
| Infections and infestations - Other | 1 | 3 | ||||||||||||
| Insomnia | 9 | 23 | ||||||||||||
| Laryngeal mucositis | 1 | 3 | 1 | 3 | ||||||||||
| Lung infection | 9 | 23 | ||||||||||||
| Lipase increased | 1 | 3 | ||||||||||||
| Multi-organ failure | 1 | 3 | ||||||||||||
| Mucositis oral | 14 | 36 | 5 | 13 | 11 | 28 | 4 | 10 | ||||||
| Nausea | 21 | 54 | 3 | 8 | 15 | 38 | 3 | 8 | ||||||
| Neutrophil count decreased | 3 | 8 | 19 | 49 | 1 | 3 | 11 | 28 | ||||||
| Pain | 1 | 3 | ||||||||||||
| Pericardial effusion | 1 | 3 | ||||||||||||
| Peripheral sensory neuropathy | 1 | 3 | ||||||||||||
| Platelet count decreased | 6 | 15 | 8 | 21 | 20 | 51 | 3 | 8 | 10 | 26 | ||||
| Pleural effusion | 6 | 15 | ||||||||||||
| Pneumonitis | 1 | 3 | ||||||||||||
| Pulmonary edema | 5 | 13 | ||||||||||||
| Pulmonary fibrosis | 1 | 3 | ||||||||||||
| Rash maculo-papular | 7 | 18 | 8 | 21 | ||||||||||
| Respiratory failure | 3 | 8 | 1 | 3 | ||||||||||
| Respiratory, thoracic and mediastinal disorders | 1 | 3 | ||||||||||||
| Sepsis | 2 | 5 | 3 | 8 | 2 | 5 | 1 | 3 | ||||||
| Sinus bradycardia | 1 | 3 | ||||||||||||
| Sinus tachycardia | 2 | 5 | ||||||||||||
| Skin infection | 1 | 3 | ||||||||||||
| Skin/subcutaneous tissue disorders; Other | 8 | 21 | 1 | 3 | ||||||||||
| Small intestine ulcer | 1 | 3 | ||||||||||||
| Sore throat | 4 | 10 | ||||||||||||
| Somnolence | 1 | 3 | ||||||||||||
| Supraventricular tachycardia | 1 | 3 | ||||||||||||
| Syncope | 2 | 5 | ||||||||||||
| Thromboembolic event | 1 | 3 | ||||||||||||
| Typhlitis | 1 | 3 | ||||||||||||
| Vascular access complication | 1 | 3 | ||||||||||||
| Vasovagal reaction | 1 | 3 | ||||||||||||
| Ventricular arrhythmia | 1 | 3 | ||||||||||||
| Ventricular tachycardia | 1 | 3 | ||||||||||||
| Vomiting | 7 | 18 | ||||||||||||
| White blood cell decreased | 1 | 3 | 2 | 5 | ||||||||||
Following induction chemotherapy, 10 patients (26%) received at least 1 cycle of consolidation therapy (1 cycle n=6, 2 cycles n=2, 3 cycles n=2). A total of 16 patients (41%) proceeded to allogeneic HCT, 7 of which received prior consolidation chemotherapy; all HCT patients had achieved prior CR/CRi (n=15) or PR (n=1). One patient received maintenance alisertib on study. A total of five patients have relapsed to date, 3 of whom were post-HCT, while 18 patients were alive at last follow-up. The median RFS of patients achieving remission was not reached (Figure 2A; 95%CI 7.1mo-N/A), and at 9 months the RFS was 56% (95%CI 37%−76%). With a median follow-up of 14 months, the median overall survival was 12.3 months (95% CI 8.1-N/A), with a one-year survival of 50% (95%CI 34%−66%) (Figure 2B).
Figure 2.
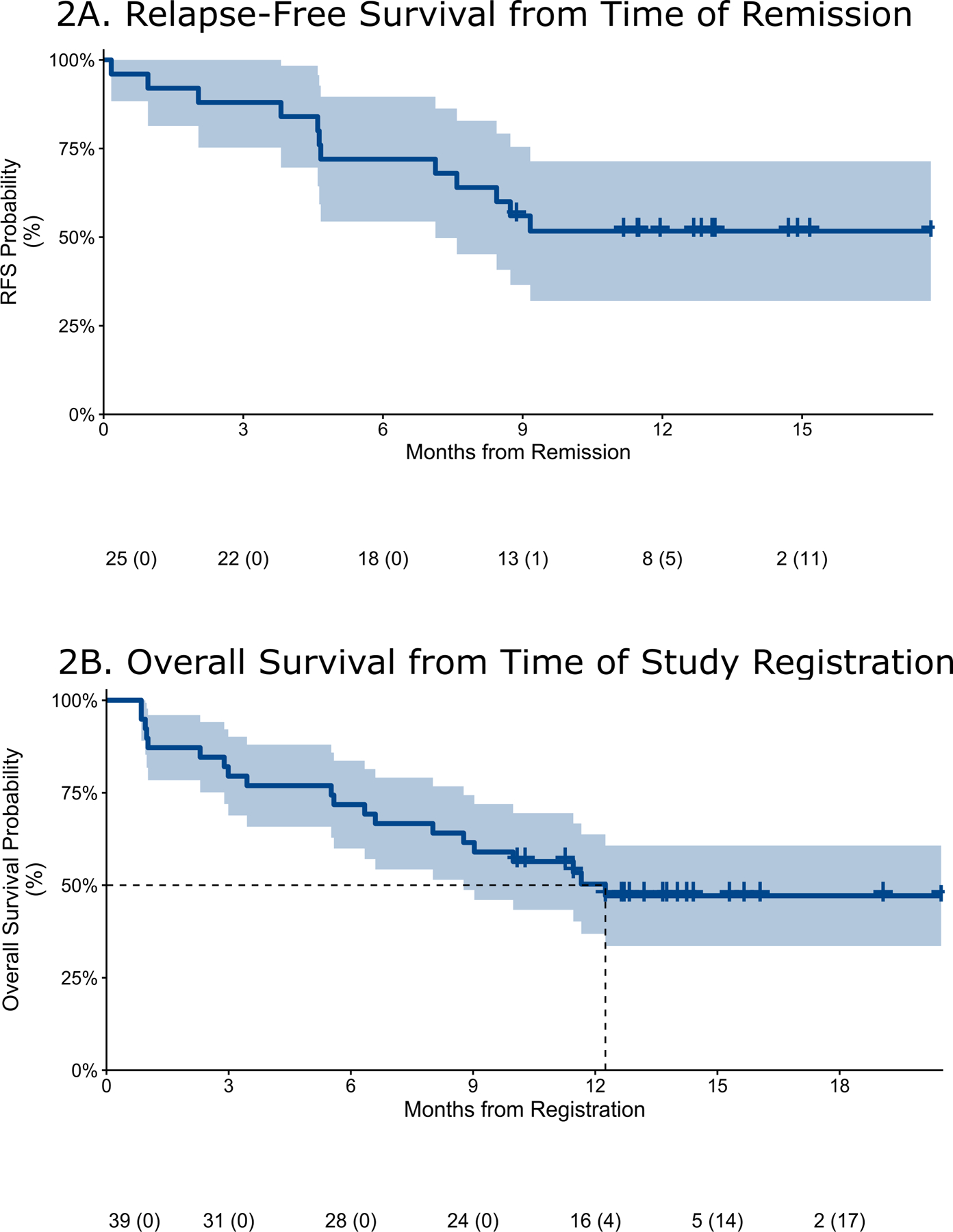
Relapse-free (A) and Overall (B) survival of the entire cohort (Kaplan-Meier estimate and 90% confidence intervals). The median RFS was not reached (90%CI 7.6mo-N/A), while at 9 months was 56% (90%CI 40%−72%). The median OS was 12.3 months (90% CI 8.8-NA); at one year, OS was 50.3% (90%CI 36.9%−63.7%).
We assessed remission rates according to AML subgroups of interest. There was no significant variation in the CCR according to whether patients were aged 65 or older (67%, n=18/27), had an antecedent MDS or MPN (58%, n=11/19), had therapy-related AML (67%, n=2/3), or had adverse-risk cytogenetics (77%, n=10/13). The CCR for TP53-mutated patients was 75% (3 of 4). We explored responses according to AML mutation profiles (Figure 3); because of the heterogeneity of mutation profiles, we were underpowered to detect associations between mutations and response; however, univariate analysis identified the presence of DNMT3A mutations as predictive of response to therapy compared to those without DNMT3A mutations (p=0.01). Since alisertib disrupts spindle formation during mitosis and the cohesin complex is involved in the separation of sister chromatids during mitosis, we asked whether cohesin mutations were predictive of response. Interestingly, of the 5 patients with STAG2 mutations, 3 achieved CR and 1 CRi, while the other patient died during treatment.
Figure 3.
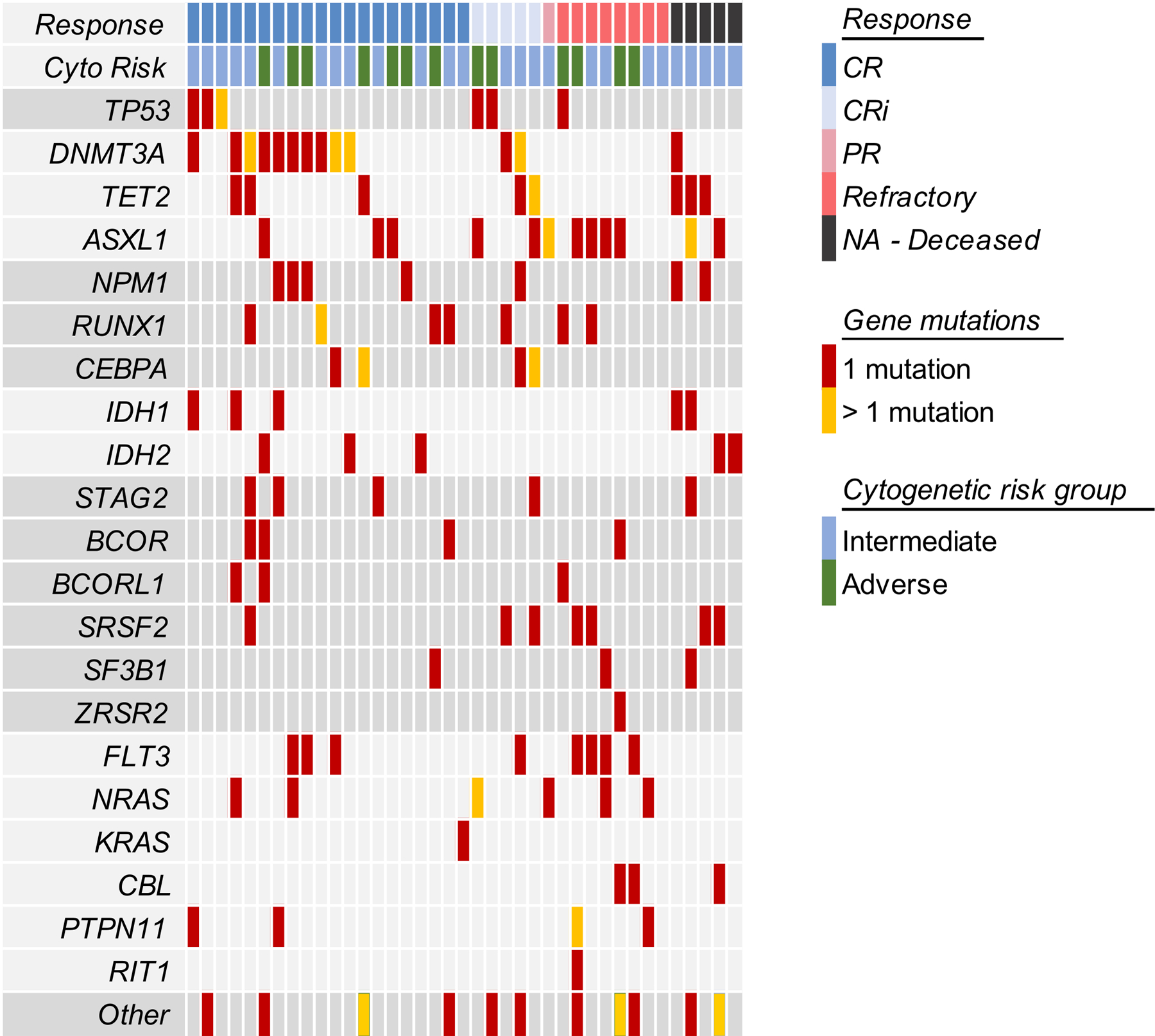
Response to Induction according to Patient Profile. Mutations in DNMT3A were associated with achievement of CR/CRi on univariate analysis (p = 0.0132).
Mutation burden was followed serially using a clinical, next generation sequencing (NGS) assay during treatment to assess mutational persistence and impact on survival. As expected, decreases in mutational VAF tracked with clinical response (Appendix page 7). Mutations in DNMT3A, TET2, and ASXL1 (“DTA mutations”) that are associated with clonal hematopoiesis frequently persisted at a high VAF after induction, even among patients with morphological remission (Appendix page 8), while NPM1 mutations often declined below the level of detection of clinical assays. We assessed survival outcomes according to different thresholds of mutational persistence (Figure 4). Using a VAF detection level of 1% and excluding DTA mutations, there was an association between RFS at 12 months and lack of detection for somatic variants at time of remission (none detected at a VAF of 1%, p=0.03).
Figure 4:
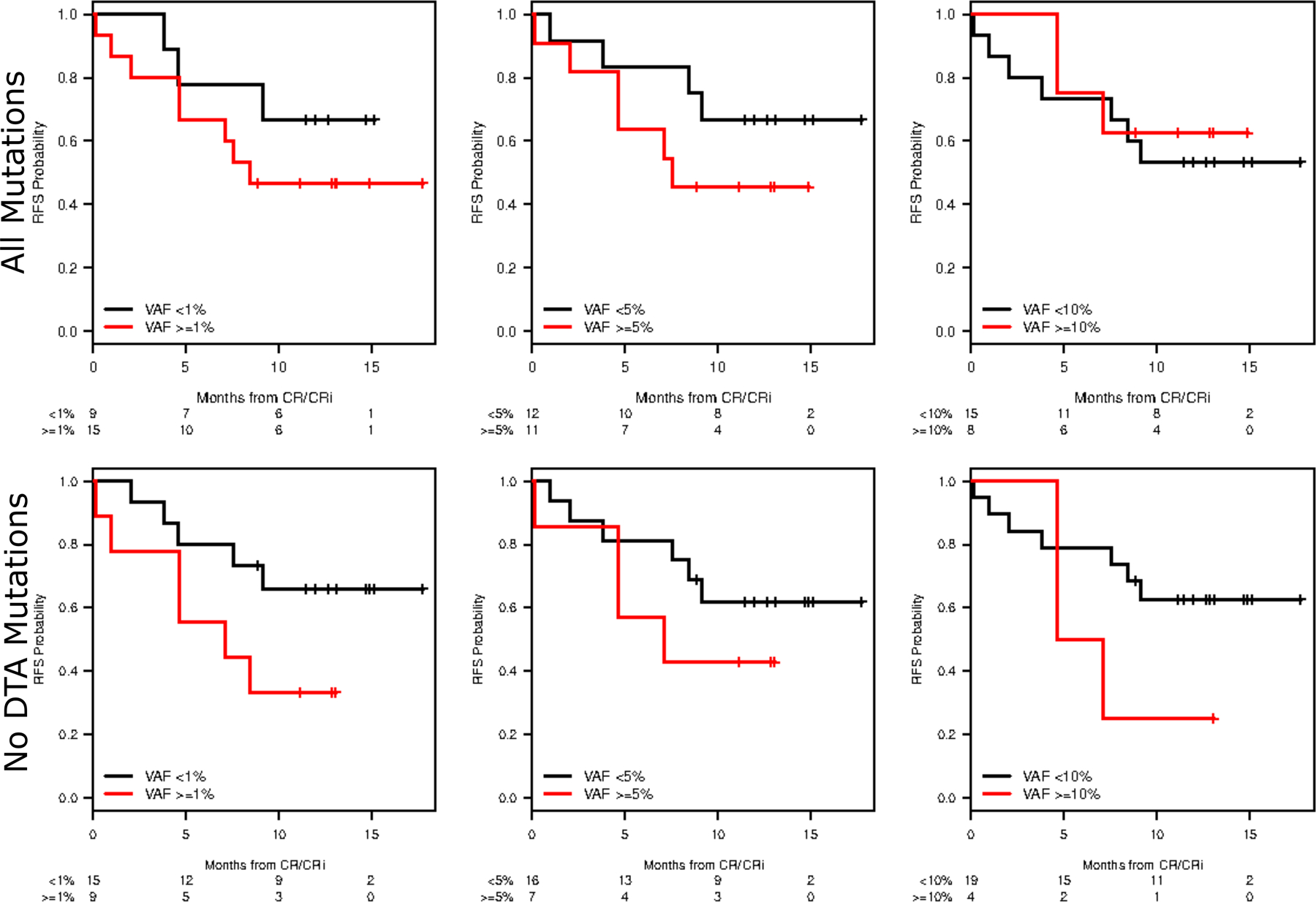
Relapse-Free Survival by Mutation Clearance. RFS was assessed using different MRD thresholds (VAF <1%, VAF < 5%, and VAF <10%) as well as with or without DTA mutations.
Patient samples at diagnosis were exposed to 100nM alisertib and assessed for spindle formation. Mitotic cells in all samples displayed a similar proportion of bipolar spindles in the absence of alisertib treatment (Appendix page 9), indicating that each sample was similarly competent to complete mitotic cell divisions. Inhibition of AAK is expected to corrupt mitotic spindle formation and promote mitotic arrest. To assess the in vitro response to alisertib, the frequency of normal bipolar spindle formation in the ex vivo alisertib-treated fraction was compared to that of the alisertib-untreated fraction from the same patient sample. Consistent with the expected mechanism of action for AAK inhibition, cultured samples from CR/CRi patients exhibited a 3 to 4-fold mitotic enrichment following AAK inhibition (Appendix page 10). Eleven of 15 of these samples showed no evidence of bipolar mitotic spindles or anaphase figures that would have been indicative of continued proliferation. Overall, on average, there was an ~87% (SD=29%) reduction in bipolar spindles following alisertib treatment in CR/CRi patients. In contrast, all of the available samples from patients with refractory disease (n=4) appeared resistant to alisertib-induced mitotic corruption, such that 4 of 4 samples remained competent to form bipolar spindles and complete mitosis, with a statistically significant difference in the average response ratio of ~0.5 (SD=0.2) (Appendix page 11; student t-test: p=0.04). Together, these data demonstrate correlation between ex vivo cellular response to alisertib and patient clinical response (Fisher exact test p= 0.02), indicating that assessment of mitotic spindle structure following AAK inhibition may be informative in identifying patients likely to benefit from alisertib.
Discussion:
The addition of alisertib, a specific and potent AAK inhibitor, to traditional 7+3 induction chemotherapy yielded a promising remission rate of 64% (CR+CRi) among patients with traditionally high-risk AML, including those with adverse-risk cytogenetics, secondary AML, and therapy-related AML. This translated to a sizeable proportion of treated patients being bridged to SCT and encouraging overall survival. Our data also suggests that duration of response may be associated with lower levels of detection of somatic mutations at the time of remission using a VAF threshold of 1 or 5%, consistent with most clinically available NGS assays; moreover, these responses appear to correlate ex vivo with alisertib-mediated disruption of the mitotic spindle.
In spite of many efforts to improve outcomes over the last decades, outcomes for AML patients aged over 65 years remain especially poor.27 Similarly, patients whose disease harbors adverse-risk cytogenetics, including complex abnormalities or alterations of chromosomes 3, 5, and 7, have poor outcomes16, 17 and low rates of response to traditional cytotoxic induction.17, 28, 29 These alterations are enriched in AML arising from antecedent myeloid malignancy or as a result of prior treatment. Failure to achieve remission (CR/CRi) also impacts bridging to curative HCT in otherwise eligible patients. A recent randomized phase 3 study specifically in 60–75 year-old patients with secondary AML compared traditional 7+3 induction chemotherapy with CPX-351, a liposomal nano-scale particle that releases a fixed-molar ratio of daunorubicin and cytarabine. Although a survival advantage was found for CPX-351, the majority in both arms did not achieve remission, with the CCR being 48% for those receiving CPX-351 and 33% for those receiving conventional 7+3 induction. In our current study, a total of 18 patients would have been eligible for entry on the trial of CPX-351, and of these patients the ORR was 11/18 (61%) with 9 CR and 2 CRi responses.
We previously reported that alisertib could be safely added to 7+3 induction in a newly diagnosed AML population, and identified an activity signal in less favorable AML populations.14 In the current study, we report similar responses to this combination in traditionally higher-risk patients. The time to count recovery of neutrophils and platelets were slightly longer than may be expected with conventional induction therapy in patients with de novo AML, although this may reflect enrichment of patients with antecedent MDS. The 30- and 60-day mortality rates were 8% and 13%, respectively, similar to other studies of induction chemotherapy alone.26 Additionally, deaths occurring during induction were uniformly related to severe cytopenias characteristic of induction chemotherapy, three caused by infection or sepsis and two by hemorrhage, and all occurring at day 32 or earlier, suggesting that prolonged cytopenias were less likely to be contributive. Because a high proportion of patients proceeded to transplant (41%), we are underpowered to assess the impact of alisertib during consolidation and maintenance.
The rates of composite remission appeared similar across multiple high-risk groups, including those with adverse-risk cytogenetics, those aged 65 and over, those with antecedent myeloid malignancy, and those harboring TP53 mutations. Patients over 65 could be included even if they had ELN favorable risk – in this case NPM1 mutated disease; only 5 patients met this criteria and since no response variation was seen across risk subgroups this is unlikely to have impacted our reported findings. This is consistent with data suggesting older patients with favourable ELN risk still have poor outcomes with survival less than 2 years.19 Nonetheless, we recognize the limitations of our findings given 44% of patients had a normal karyotype (though 8 of 17 had high ELN risk by molecular testing). We explored depth of remission in responding patients using a clinical NGS panel of myeloid genes. Excluding DTA (DNMT3A, TET2, ASXL1) mutations, known to be associated with clonal hematopoiesis,30 15 of 25 patients (62.5%) with CR/CRi did not have detectable pathogenic mutations using our clinical assay at the time of response. Although this clinical NGS platform has limited depth of coverage, it is an assay that is widely available and hence readily translatable to clinical care. We recognize that these subgroup analyses are underpowered in the current study and largely exploratory, but the uniformity of responses is encouraging and will inform future study. Our data also suggests that those achieving response were more likely to have disrupted mitotic spindle formation compared to patients with refractory disease, perhaps through on-target activity of alisertib in responders, and supports future exploration of spindle formation as a predictive biomarker.
There were limitations associated with this phase 2 study. Being a single-arm trial, data regarding activity cannot be fully attributed to alisertib in the absence of a monotherapy comparator; however, this study was sufficiently promising to pursue a randomized trial that is planned for launch in the near future. In addition, given the study size, subgroup analysis interpretations are underpowered and will also be explored further in future trials. Finally, this was a single-institution study, conducted at the hospitals of one cancer center, and as such, there are inherent limitations for generalizability of data, given the demographic populations and referral patterns seen at these large academic institutions.
In summary, the combination of the AAK inhibitor alisertib with conventional 7+3 chemotherapy has encouraging activity among patients with traditionally high-risk AML. The combination led to a rate of composite remission that is generally higher than historical experience with induction therapy alone in these populations, and it appeared that the majority of responding patients cleared characteristic pathogenic mutations at remission. A global, randomized, and placebo-controlled phase 3 study is planned to assess the efficacy of alisertib when combined with induction chemotherapy as a new approach for patients with high-risk AML.
Supplementary Material
Research in Context:
Evidence before this study
The initial treatment of acute myeloid leukemia (AML) with poor-risk features remains challenging. Several therapies have recently been approved for this setting. CPX-351, a liposomal formulation of a fixed ratio of daunorubicin and cytarabine, showed superior efficacy in patients age 60–75 with secondary AML or AML with myelodysplastic syndrome (MDS)-related features, compared to standard 7+3 induction. Other approaches have added agents to lower-intensity chemotherapies; the combination of venetoclax with azacitidine, decitabine, or low dose cytarabine resulted in a response rate of approximately 60% among predominantly older-aged patients. It is not known whether the benefits of these new therapies extend to younger patients or in other disease settings. Moreover, long-term survival remains low among all patients with AML with poor-risk features. We previously conducted a Phase 1 study of a novel inhibitor of aurora A kinase (AAK), alisertib, combined with standard induction chemotherapy, and found the combination to be tolerable, and associated with a suggestion of clinical activity. This served the basis for the current study. A formal search was not performed prior to this study.
Added value of this study
This current study is a phase II trial evaluating the addition of alisertib to standard 7+3 induction chemotherapy in patients with adverse-risk disease. The combination led to a promising rate of complete remission and of overall survival compared to other recently-reported data, including that seen with agents such as CPX-351. This suggests that further exploration of the combination of alisertib with induction chemotherapy in high-risk AML is merited.
Implications of all the available evidence
High-risk AML, including those who are older, those with secondary AML and therapy-related disease, as well as those with adverse-risk karyotypic alterations, is associated with poor rates of response and survival outcomes. Even with the development of newer agents, responses are often suboptimal and of limited duration. The findings from this phase II study suggest that AAK may be a therapeutic target for high-risk AML and form the basis for larger, confirmatory randomized studies.
Acknowledgements:
This work has been presented in part at the American Society of Hematology 2018 Annual Meeting, San Diego, CA. Biostatistical analysis was supported by NIH grant 5P30 CA 006516. This study was funded by Millennium Pharmaceuticals, Inc., Cambridge, MA, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Declaration of Interests:
Dr. Fathi reports grants and personal fees from Takeda, during the conduct of the study; grants and personal fees from Celgene, grants and personal fees from Seattle Genetics, personal fees from Jazz, personal fees from Medimmune, personal fees from Clear Creek, grants from Exelexis, grants and personal fees from Agios, personal fees from Boston Biomedical, personal fees from PTC Therapeutics, personal fees from NewLink Genetics, personal fees from Daiichi Sankyo, personal fees from Novartis, personal fees from Astellas, outside the submitted work. Dr. Brunner reports advisory board participation from Celgene, Jazz Pharmaceuticals, Forty Seven, research funding to his institution from Celgene, Novartis, Takeda, AstraZeneca, outside the submitted work. Dr. DeAngelo reports personal fees from Amgen, Autolus, Celgene, Forty-Seven, Incyte, Imunogen, Jazz, Pfizer, Shire, Takeda, personal fees and other from Novartis and Blueprint, other from Abbvie and Glycomimetics, outside the submitted work. Dr. Avigan reports grants from Celgene, grants from Pharmacyclics, other from Celgene, other from Juno, other from Partners TX, other from Karyopharm, other from BMS, other from Janssen, other from Parexel, other from Takeda, outside the submitted work. Dr. Steensma reports personal fees from Daiichi Sankyo, personal fees from Onconova, personal fees from Pfizer, personal fees from Amgen, other from Celgene, other from H3 Biosciences, other from Syros, other from Arrowhead, personal fees from Stemline, personal fees from Summer Road, outside the submitted work. Dr. Neuberg reports grants from NCI 5P30 CA006516, during the conduct of the study; grants from Pharmacyclics, other from Madrigal Pharmaceuticals, other from Celgene, outside the submitted work. Dr. Winer reports advisory boards for Jazz Pharmaceuticals and Pfizer Inc. Dr. Hobbs reports other from Incyte, other from Bayer, other from Merck, other from Agios, other from Jazz pharmaceuticals, outside the submitted work. Dr. Rosenblatt reports grants from Celegene, grants from BMS, other from Amgen, other from Merck, other from BMS, other from Partner TX, other from Parexel, other from Imaging Endpoint, other from Dava Oncology, outside the submitted work. Dr. Amrein reports grants from Millennium, during the conduct of the study; grants from Millennium, grants from Takeda, grants from Amgen, grants from Astex, grants from AstraZeneca, personal fees from AstraZeneca, outside the submitted work. Dr. Lindsley reports personal fees from Takeda, grants from Jazz Pharmaceuticals, grants from MedImmune, outside the submitted work. Dr. Stone reports personal fees and other from Abbive, personal fees from Actinium, personal fees and other from Agios, personal fees from Amgen, personal fees from Argenx, personal fees and other from Arog, personal fees from Astellas, personal fees from AstraZeneca, personal fees from Biolinerx, personal fees from Celgene, personal fees from Cornerstone, personal fees from Daiichi-Sankyo, personal fees from FujiFilm, personal fees from Jazz, personal fees from Macrogenics, personal fees and other from Novartis, personal fees from Ono/Theradex, personal fees from Orsenix, personal fees from Otsuka/Astex, personal fees from Pfizer, personal fees from Roche, personal fees from Stemline, personal fees from Takeda, personal fees from Trovagene, outside the submitted work. Dr. Chen reports personal fees from Takeda, during the conduct of the study; personal fees from Magenta, personal fees from Kiadis, personal fees from Incyte, personal fees from Abbvie, outside the submitted work. The other authors report no relevant conflicts of interest.
Footnotes
Primary Scientific Category: Myeloid Neoplasia
Secondary Scientific Category: Clinical Trials and Observations
Data sharing:
Individual participant data are not available. For original data please contact Amir Fathi (afathi@partners.org).
References:
- 1.Dohner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med 2015; 373(12): 1136–52. [DOI] [PubMed] [Google Scholar]
- 2.Yates J, Glidewell O, Wiernik P, et al. Cytosine arabinoside with daunorubicin or adriamycin for therapy of acute myelocytic leukemia: a CALGB study. Blood 1982; 60(2): 454–62. [PubMed] [Google Scholar]
- 3.Lancet JE, Uy GL, Cortes JE, et al. CPX-351 (cytarabine and daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients With Newly Diagnosed Secondary Acute Myeloid Leukemia. J Clin Oncol 2018; 36(26): 2684–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fu J, Bian M, Jiang Q, Zhang C. Roles of Aurora kinases in mitosis and tumorigenesis. Molecular cancer research : MCR 2007; 5(1): 1–10. [DOI] [PubMed] [Google Scholar]
- 5.Marumoto T, Honda S, Hara T, et al. Aurora-A kinase maintains the fidelity of early and late mitotic events in HeLa cells. J Biol Chem 2003; 278(51): 51786–95. [DOI] [PubMed] [Google Scholar]
- 6.Goldenson B, Crispino JD. The aurora kinases in cell cycle and leukemia. Oncogene 2015; 34(5): 537–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ye D, Garcia-Manero G, Kantarjian HM, et al. Analysis of Aurora kinase A expression in CD34(+) blast cells isolated from patients with myelodysplastic syndromes and acute myeloid leukemia. Journal of hematopathology 2009; 2(1): 2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lucena-Araujo AR, de Oliveira FM, Leite-Cueva SD, dos Santos GA, Falcao RP, Rego EM. High expression of AURKA and AURKB is associated with unfavorable cytogenetic abnormalities and high white blood cell count in patients with acute myeloid leukemia. Leukemia research 2011; 35(2): 260–4. [DOI] [PubMed] [Google Scholar]
- 9.Kelly KR, Ecsedy J, Mahalingam D, et al. Targeting aurora kinases in cancer treatment. Current drug targets 2011; 12(14): 2067–78. [DOI] [PubMed] [Google Scholar]
- 10.Kelly KR, Nawrocki ST, Espitia CM, et al. Targeting Aurora A kinase activity with the investigational agent alisertib increases the efficacy of cytarabine through a FOXO-dependent mechanism. Int J Cancer 2012; 131(11): 2693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cervantes A, Elez E, Roda D, et al. Phase I pharmacokinetic/pharmacodynamic study of MLN8237, an investigational, oral, selective aurora a kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res 2012; 18(17): 4764–74. [DOI] [PubMed] [Google Scholar]
- 12.Dees EC, Cohen RB, von Mehren M, et al. Phase I study of aurora A kinase inhibitor MLN8237 in advanced solid tumors: safety, pharmacokinetics, pharmacodynamics, and bioavailability of two oral formulations. Clin Cancer Res 2012; 18(17): 4775–84. [DOI] [PubMed] [Google Scholar]
- 13.Goldberg SL, Fenaux P, Craig MD, et al. An exploratory phase 2 study of investigational Aurora A kinase inhibitor alisertib (MLN8237) in acute myelogenous leukemia and myelodysplastic syndromes. Leukemia research reports 2014; 3(2): 58–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fathi AT, Wander SA, Blonquist TM, et al. Phase I study of the aurora A kinase inhibitor alisertib with induction chemotherapy in patients with acute myeloid leukemia. Haematologica 2017; 102(4): 719–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016; 127(20): 2391–405. [DOI] [PubMed] [Google Scholar]
- 16.Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017; 129(4): 424–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grimwade D, Hills RK, Moorman AV, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010; 116(3): 354–65. [DOI] [PubMed] [Google Scholar]
- 18.Lowenberg B, Ossenkoppele GJ, van Putten W, et al. High-dose daunorubicin in older patients with acute myeloid leukemia. N Engl J Med 2009; 361(13): 1235–48. [DOI] [PubMed] [Google Scholar]
- 19.Mrozek K, Marcucci G, Nicolet D, et al. Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J Clin Oncol 2012; 30(36): 4515–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheson BD, Bennett JM, Kopecky KJ, et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol 2003; 21(24): 4642–9. [DOI] [PubMed] [Google Scholar]
- 21.Cheson BD, Greenberg PL, Bennett JM, et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood 2006; 108(2): 419–25. [DOI] [PubMed] [Google Scholar]
- 22.Navarro-Serer B, Childers EP, Hermance NM, Mercadante D, Manning AL. Aurora A inhibition limits centrosome clustering and promotes mitotic catastrophe in cells with supernumerary centrosomes. Oncotarget 2019; 10(17): 1649–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kluk MJ, Lindsley RC, Aster JC, et al. Validation and Implementation of a Custom Next-Generation Sequencing Clinical Assay for Hematologic Malignancies. The Journal of molecular diagnostics : JMD 2016; 18(4): 507–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Atkinson EN, Brown BW. Confidence limits for probability of response in multistage phase II clinical trials. Biometrics 1985; 41(3): 741–4. [PubMed] [Google Scholar]
- 25.Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015; 125(9): 1367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walter RB, Othus M, Borthakur G, et al. Prediction of early death after induction therapy for newly diagnosed acute myeloid leukemia with pretreatment risk scores: a novel paradigm for treatment assignment. J Clin Oncol 2011; 29(33): 4417–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klepin HD, Rao AV, Pardee TS. Acute myeloid leukemia and myelodysplastic syndromes in older adults. J Clin Oncol 2014; 32(24): 2541–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Byrd JC, Mrozek K, Dodge RK, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461). Blood 2002; 100(13): 4325–36. [DOI] [PubMed] [Google Scholar]
- 29.Grimwade D, Walker H, Oliver F, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood 1998; 92(7): 2322–33. [PubMed] [Google Scholar]
- 30.Jongen-Lavrencic M, Grob T, Hanekamp D, et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N Engl J Med 2018; 378(13): 1189–99. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Individual participant data are not available. For original data please contact Amir Fathi (afathi@partners.org).