

INTRODUCTION
β-thalassemia is one of the world’s most common inherited anemias, affecting millions of individuals worldwide.1,2 The disease is caused by more than 300 different mutations in the β-globin gene (HBB), all of which cause quantitative deficiencies of β-globin protein and adult-type hemoglobin (HbA, α2β2). Concomitantly, the accumulation of free α-globin forms toxic intracellular inclusions resulting in hemolysis and ineffective erythropoiesis IE. The clinical consequences are anemia, bone disease, extramedullary erythropoiesis, and iron overload. Medical therapy includes red blood cell (RBC) transfusions and iron chelation, both of which cause major toxicities.
Allogeneic hematopoietic stem cell transplantation (HSCT) is an effective cure for β-thalassemia, although not all patients have ideal donors and the procedure is associated with serious immune complications including, graft versus host disease, and graft rejection.3 These problems may be circumvented by experimental therapies in which autologous hematopoietic stem cells (HSCs) are isolated, genetically altered ex vivo and reintroduced into the patient after the administration of myelotoxic bone marrow conditioning to facilitate engraftment of modified cells. Current methods for genetic modification, referred to collectively here under the term “gene therapy”, include lentiviral transduction, genome editing and base editing, aim to either restore β-globin production during erythropoiesis or to reactivate the production of fetal γ-globin, which binds α-globin to form fetal hemoglobin (HbF, α2γ2). Here we provide a succinct review of ongoing preclinical and clinical studies, problems and future directions related to gene therapy for transfusion dependent β-thalassemia (TDT). Most aspects of the field have been summarized in recently published review articles and in other chapters in this volume. Due to word limitations, we refer to these reviews and other chapters rather than citing all original research publications.
Gene Manipulation Strategies for β-thalassemia
Figure 1 shows the overall strategy for current autologous HSC gene therapy. Gene manipulation strategies for treating TDT are shown in Figure 2; similar strategies are being used to treat sickle cell disease (SCD). The field is advancing rapidly as new and improved tools for autologous HSCT and HSC gene manipulation become available.4
Figure 1. Gene therapy for transfusion-dependent β-thalassemia.

The multi-step process includes: (I) Informed consent, including patient/family education and written consent; (II) mobilization, apheresis collection and enrichment of patient (autologous) hematopoietic stem cells (HSCs); (III) Ex vivo genetic manipulation of HSCs to restore erythroid expression of β-globin or induce fetal hemoglobin (HbF) expression; (IV) Administration of bone marrow conditioning followed by infusion of the modified HSCs.
Figure 2. Genetic manipulation of autologous HSCs for TDT gene therapy.
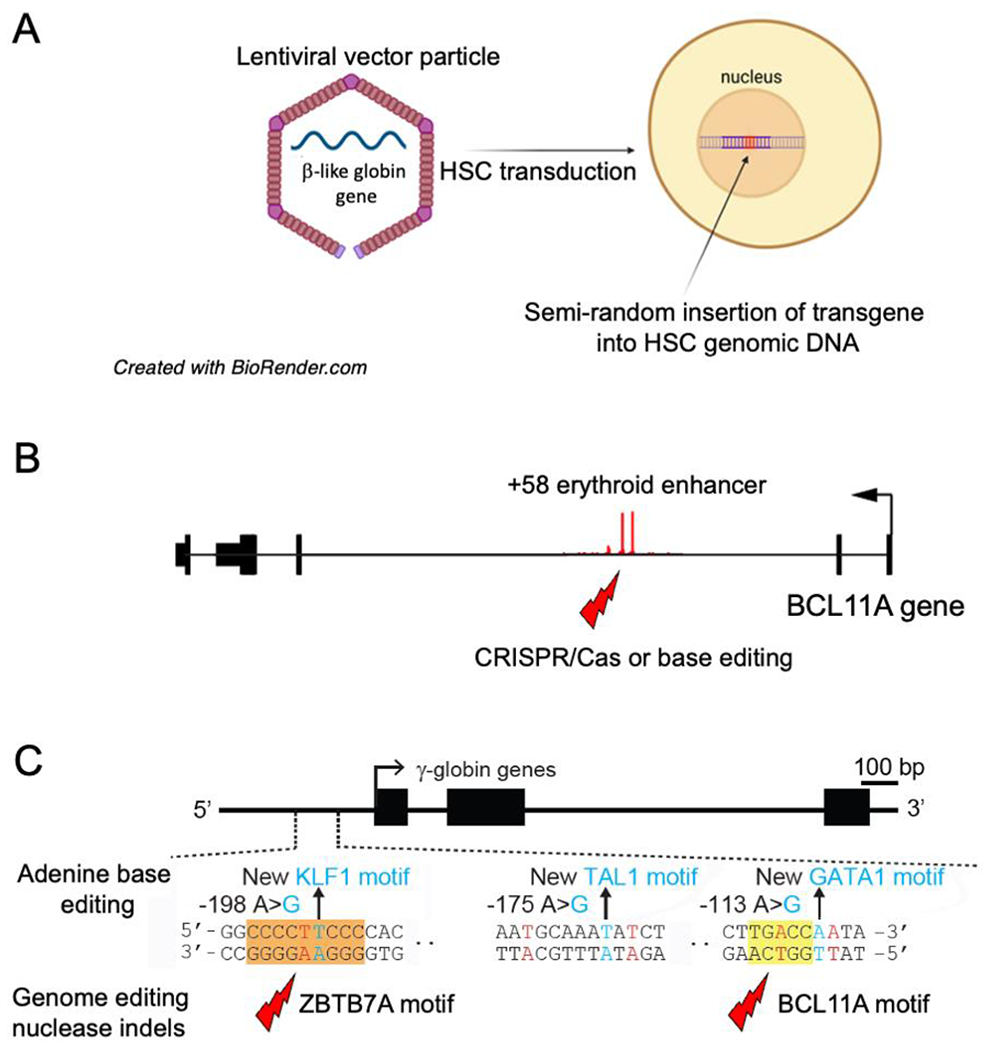
(A) Patient HSCs are transduced with lentiviral vector (LVV) particles encoding a β-like globin gene. (B) Induction of fetal hemoglobin (HbF, α2γ2) by disrupting the +58 erythroid enhancer in intron 2 of the BCL11A by genome editing nuclease mediated non-homologous end joining (NHEJ) or base editing. (C) Alteration of the γ-globin promoter. Bottom shows disruption of BCL11A or ZBTZ7A repressor binding motifs via genome editing nuclease-mediated NHEJ. Top shows installation of new binding motifs for one of several erythroid transcription factors with adenine base editors.
β-globin Replacement Therapy with Lentiviral Vectors
Lentiviral vectors (LVVs) mediate the semirandom integration of a therapeutic transgene and associated regulatory elements into the genomic DNA of dividing and nondividing cells, the latter being important for manipulating quiescent HSCs.5,6 Compared to γ-retroviral vectors that were used in early autologous HSC gene therapy protocols, LVVs have a reduced propensity to cause insertional activation of oncogenes and malignant transformation. Historically, the development of LVVs capable of driving sustained, high-level erythroid-specific expression of a β-like-globin transgene was a major challenge that was overcome by rational vector design and trial and error. Most current LVVs used to treat β-thalassemia and SCD include a β-like globin gene and promoter linked to a modified locus control region (LCR), a powerful enhancer in the β-like globin gene cluster. Another major challenge was to attain efficient transduction of HSCs with post-therapy vector copy numbers > 1, which has been achieved by improved process development, including the use of transduction enhancing reagents.7,8 In November 2022, 10 clinical trials using LVV gene therapy to treat TDT were registered on clinicaltrials.gov (Table 1).
Table 1.
Gene therapy and genome editing clinical trials for TDT.
| Strategy | Modality | Sponsoring Agent | Clinical trial ID Status | Estimated participants | Results/notes |
|---|---|---|---|---|---|
| β-Like globin gene replacement | Lentiviral Vector (LVV) | ||||
| TNS9.3.55 LVV/βA |
Memorial Sloan Kettering Cancer Center (MSKCC) |
NCT01639690 Phase I |
10 | 4 participants followed for 90 months had stable engraftment. Transfusion requirements were reduced by 35-57% in 2 individuals. HSC transduction and LVV copy number were low. | |
| BB305 LVV/βA-T87Q |
bluebird bio |
NCT01745120 Phase I, II |
22 | Reduced or eliminated RBC transfusions in 22 patients with TDT without LVV-related severe adverse events. | |
| BB305 LVV/βA-T87Q |
bluebird bio |
NCT02151526 Phase I, II |
7 | 4 patients followed for a approximately 4.5 years became transfusion independent with reductions in dyserythropoiesis and iron overload. | |
| OTL-300 GLOBE LVV/βA |
IRCCS San Raffaele & Orchard Therapeutics |
NCT02453477 Phase I/II |
10 | Modified cell product administered into bone marrow. All three adults treated exhibited reduced transfusion requirements. Three of 4 evaluable pediatric patients became transfusion-independent. | |
| BB305 LVV/βA-T87Q |
bluebird bio |
NCT02906202 Northstar-2 Phase III |
23 | 23 individuals treated, including children (4-34 years old). 91% became transfusion-independent with median follow-up of 29.5 months. | |
| BB305 LVV/βA-T87Q |
bluebird bio |
NCT03207009 Northstar-3 Phase III |
18 | No results reported | |
| LVV/Undisclosed | Shenzhen Geno-Immune Medical Institute China |
NCT03351829 Phase I/II |
20 | No results reported | |
| LVV/βA | Nanfang Hospital of Southern Medical University China |
NCT03276455 Phase I/II |
10 | No results reported | |
| LVV/βA-T87Q | BGI-research & Shenzhen Children’s Hospital China |
NCT04592458 Phase I |
10 | No results reported | |
| LVV/βA-T87Q | Shanghai BDgene Co., Ltd |
NCT05015920 Phase I |
10 | No results reported | |
| HbF Induction | Nuclease/Target | ||||
| Cas9 disruption of BCL11A erythroid enhancer via NHEJ | CRISPR Therapeutics; Vertex Pharmaceuticals |
NCT03655678 Phase I, II, III |
45 | 42 of 44 pts stopped RBC transfusions; 2 pts had 75% and 89% reductions in RBC transfusions | |
| BCL11A -Targeted zinc finger disruption of BCL11A erythroid enhancer via NHEJ (ST-400) | Sangamo Therapeutics and Sanofi |
NCT03432364 Phase I, II |
6 | 5 patients had transient elevation of that was not sustained. No long-term therapeutic benefit due to low HSC transduction efficiency. | |
| Cpf1 NHEJ mediated disruption of BCL11A binding site (EDIT-301) | Editas Medicine Inc. |
NCT05444894 Phase I, II |
6 | No results reported | |
| Cas9 disruption of BCL11A erythroid enhancer (ET01) | Edigene & Institute of Hematology & Blood Disease Hospital, Tianjin. China. |
NCT04390971 Phase I |
8 | No results reported | |
| γ-Globin reactivation using Glycosylate Base Editors (exact mechanism not specified) | Bioray Laboratories. Shanghai, China |
NCT05442346. Phase I/II Clinical Trial |
5 | No results reported | |
| Cas9 disruption of BCL11A erythroid enhancer via NHEJ | Bioray Laboratories Shanghai China |
NCT04211480 Phase I, II |
12 | 2 children with TDT achieved transfusion independence with normal hemoglobin levels after f>18 months follow-up | |
Current clinical trials for TDT on ClinicalTrials.gov as of November 2022.
Abbreviations: HSC, hematopoietic stem cell; TDT, transfusion dependent β-thalassemia; NHEJ, non-homologous end joining; HDR, homology directed repair; LVV: Lentiviral vector; CRISPR, Clustered regularly interspaced short palindromic repeats, HbF, fetal hemoglobin.
The most extensive study of LVV gene therapy for treating TDT utilizes the cellular drug product LentiGlobin (betibeglogene autotemcel), generated by transfecting patient CD34+ hematopoietic stem and progenitor cells (HSPCs) with an LVV encoding a modified β-globin gene (βA-T87Q) that inhibits polymerization of sickle hemoglobin5,9-13. Therefore, the same LVV and a similar manufacturing process are under investigation for treating sickle cell disease.14 Two companion phase I/II clinical trials sponsored by bluebird bio (ClinicalTrials.gov: NCT01745120, HBG-204 NORTHSTAR and NCT02151526, HBG-205) treated 22 patients (ages 12 to 35 years) with TDT genotypes including β0/β0 or IVS1-110, which produces very low levels of β-globin (n=9), βE/β0 (n=9) or other (n=4).13 Of the 9 individuals with β0/β0 or IVS1-110 genotypes, 3 became transfusion-independent and 6 experienced reductions (median 73%) in RBC transfusion requirements. Of the remaining 13 patients with less severe genotypes (mainly βE/β0), 12 became RBC transfusion independent. In general, the clinical efficacy correlated with vector copy number in peripheral blood mononuclear cells at 6 months, which reflects LVV transduction efficiency of HSCs. The adverse events were due to myeloablative busulfan conditioning and not attributed to the drug product.
More recently, a completed phase III study sponsored by bluebird bio evaluated Lentiglobin therapy in 23 individuals (4-34 years-old) with non-β0/β0 TDT (NCT02906202, HGB207, NORTHSTAR-2).9 Transfusion independence and normal or near-normal blood hemoglobin levels occurred in 91% of individuals with up to 4 years follow-up. The improved clinical efficacy compared to the previous phase I/II studies was largely due to manufacturing improvements that resulted in more efficient transduction of HSCs and higher post-therapy vector copy number. Another phase III study for TDT, Northstar-3 (HGB-212; NCT03207009), is ongoing.
The GLOBE phase I/II clinical trial (NCT02453477) utilized a LVV harboring wild-type βA-globin.15 Three adult participants experienced reduced transfusion requirements and 3 of 4 pediatric participants achieved transfusion independence (4-13 years old) over 1 to 28 months follow-up. The MSKCC phase I clinical trial (NCT01639690) examined the β-globin LVV TNS9.3.55 and reduced intensity busulfan conditioning in 4 individuals with TDT.16 After 6-8 years follow-up, RBC transfusion requirements were reduced by approximately 50% in 2 individuals. The modest therapeutic response was associated with low post-therapy vector copy number, which most likely resulted from inefficient transduction of patient HSCs rather than reduced intensity conditioning. Several LVV gene therapy trials for severe β-thalassemia have opened recently in China, where the disease is very common (Table 1).
In August 2022, the US Food and Drug Administration approved betibeglogene autotemcel (Zynteglo) for treating patients with TDT.17 This represents a major milestone and a culmination of more than a decade of intensive research. Updated information on clinical trials for Zynteglo and its regulatory approval process can be found on the FDA website [https://www.fda.gov/vaccines-blood-biologics/zynteglo].
Genome Editing for β-thalassemia
Genome editing nucleases
Genome editing strategies utilize targeted nucleases to introduce sequence-specific double stranded DNA breaks (DSBs) into the genome of live cells. The first genome editors to be developed were zinc finger nucleases, followed by transcription activator-like effector nucleases (TALENs); both require sophisticated protein engineering for DNA targeting.18,19 The field was revolutionized by the discovery and adaptation of bacterial-derived CRISPR/Cas nucleases, which are rapidly and easily programmed by a single guide RNA (sgRNA) that directs the nuclease to a specified DNA target site via nucleotide sequence complementarity.20,21 For current clinical applications, a ribonucleic acid-protein complex consisting of targeting sgRNA bound to a CRISPR/Cas nuclease are delivered into HSCs by electroporation.
Two major cellular mechanisms exist to repair DSBs: 1) error prone non-homologous end joining (NHEJ), which introduces small insertions or deletions (indels); and 2) homology directed repair (HDR), which employs a donor DNA template to introduce precise genomic alterations. Of the two cellular repair pathways, HSCs most frequently utilize NHEJ, with on-target efficiencies that can exceed 90%. Genome edited-mediated NHEJ is being used to induce HbF for treating TDT and SCD. This versatile approach can be applied for most severe β-thalassemia mutations. In principle, HDR can be used to treat β-thalassemia by correcting individual mutations or by introducing a β-globin transgene into an α-globin locus (HBA1),22 although this occurs at lower efficiency than NHEJ.
Genetic activation of HbF expression for β-thalassemia
Hereditary persistence of fetal hemoglobin (HPFH), a benign, naturally occurring genetic condition associated with high HbF levels throughout life, alleviates the symptoms of co-inherited SCD or β-thalassemia.23 Hence, considerable research effort has been dedicated to understanding and manipulating the perinatal γ-to-β globin switch that causes replacement of HbF with HbA.24 This switch is mediated by repressor proteins BCL11A and ZBTB7A, which bind distinct cognate motifs in the γ-globin promoter to inhibit gene transcription.25,26 Details of this process and the regulation of HbF are discussed in Chapter 5.
The CLIMB THAL-111 study (NCT03655678), sponsored by Vertex Pharmaceuticals and CRISPR Therapeutics, induces HbF by using CRISPR/Cas9 to disrupt an erythroid-specific enhancer in the BCL11A gene.27 In a recent meeting abstract, the authors reported that 44 individuals with TDT who received the gene modified cellular drug product CTX001 achieved either transfusion independence (n=42) or reduced transfusion needs (n=2) over 0.8-36.2 months follow-up.28 Shanghai Bioray Laboratories Inc. used a very similar approach to treat 2 pediatric TDT patients (7 and 8 years old) who achieved transfusion independence and normal hemoglobin levels at 18 months follow-up.29 The THALES trial (NCT03432364, ST-400) sponsored by Sangamo Therapeutics & Sanofi, used a zinc finger nuclease to disrupt the BCL11A erythroid enhancer in 5 individuals with TDT.30 Fetal hemoglobin levels rose transiently, but fell to near baseline after approximately one year with no long-term clinical improvement, reflecting the failure to achieve genetic modification of long-term bone marrow-repopulating HSCs.
Some HPFH variants disrupt BCL11A or ZBTB7A binding motifs in the γ-globin promoter, indicating that disruption of the same motifs by genome editing-mediated NHEJ could induce HbF therapeutically without eliminating expression of the erythroid repressor.31-36 This is particularly important for ZBTB7A, which acts through non-globin target genes to prevent apoptosis of erythroid precursors.37 The EDITHAL study sponsored by Editas Medicine (NCT05444894), utilizes the autologous HSPC drug product EDIT-301 generated by using Cas12a to disrupt the BCL11A binding motif in the γ-globin promoter.
Base editing
Base editors (BEs) consist of catalytically impaired Cas9 fused to a deaminase domain that generates A-to-G (Adenine Base Editors, ABEs) or C-to-T (Cytidine Base Editors, CBEs) at precise positions in the genome.38-40 Unlike Cas9 nuclease, BEs do not act through DSBs that can cause potentially deleterious chromosome-scale abnormalities or DNA damage responses. Base editors can be used to correct some common β-thalassemia mutations, although these approaches have not yet been adapted for clinical application.41
Base editors can induce HbF by generating point mutations in the γ-globin promoter that either disrupt BCL11A or ZBTB7A binding, or create new binding motifs for transcriptional activators, such as KLF1, TAL1, or GATA1.42-45 Preliminary studies indicate that creation of new binding motifs, which cannot be achieved by Cas9-mediated NHEJ, induces HbF more potently than disruption of repressor binding motifs or interfering with BCL11A expression by targeting its erythroid enhancer. A recently opened clinical trial sponsored by Beam Therapeutics seeks to induce HbF in individuals with SCD by using an adenine base editor to modify the γ-globin promoter at an undisclosed site (NCT05456880, BEACON study). If successful, this approach could be adapted for β-thalassemia.
Gene therapy genotoxicities
All gene therapy strategies cause genotoxicities with theoretical risks. Pre-HSCT myeloablative conditioning, usually with the alkylating agent busulfan, is common to most gene therapy approaches and predisposes to MDS/AML.46
Lentiviral vectors insert semi-randomly into the genome and can potentially activate oncogenes or inactivate tumor suppressor genes, although this occurs less frequently than with γ-retroviral vectors.47,48 No individuals with β-thalassemia have been reported to develop leukemia or myelodysplastic syndrome (MDS) after LVV gene therapy, although concerning events have occurred. One individual with β-thalassemia developed a dominant HSC clone caused by LVV insertional activation of HMGA2, a transcription factor that has been linked to tumorigenesis.49,50 This was detected at year 2 and resolved by year 8.12 In 44 subjects with SCD who underwent LVV β-globin replacement gene therapy, two developed acute myeloid leukemia (AML) after 3 and 5.5 years.14 These cases are not believed to be caused by LVV insertion.51,52 The etiology remains uncertain, but may be due to SCD-specific predisposing factors.53 Current research on LVV safety includes defining the integration profile of specific vectors in biologically relevant cell types, developing preclinical assays to detect oncogenicity and tracking hematopoietic clones in vivo post-gene therapy by analyzing LW integration sites.54
Genome editing nucleases cause several genotoxicities that carry theoretical risks.38-40,55-59 Unintended “off-target” DSBs, usually in genomic regions with partial homology to the sgRNA, can potentially interfere with normal gene function. Additionally, DSBs at on- or off-target sites can cause large chromosomal deletions, rearrangements, aneuploidy or chromothripsis with TP53 activation.60-64,65 Compared to Cas9 nuclease, base editors produce substantially lower rates of DSBs, which reduces certain genotoxicities.38-40 However, BEs cause unique genotoxicities, including bystander mutations that occur at nucleotides adjacent to the target site, and Cas9/sgRNA independent deamination of DNA and/or RNA.66-68
Current research is focused on developing sensitive assays to detect genome editing genotoxicities and designing modified versions of nucleases and BEs with enhanced specificity.55,56,59,68-76 Available methods can detect off-target genome editing activities or DSB-induced chromosomal rearrangements at a sensitivity of 0.1% genome-wide. Most genotoxic events are likely to be benign or result in cell death. However, rare unintended genetic events could predispose to malignancy by activating oncogenes or inactivating tumor suppressors. Such theoretical events are likely to be specific for the genome editor and sgRNA used and may be difficult to predict in advance of genome editing therapy. For this reason, long term follow-up for the development of clonal hematopoiesis, MDS or AML is required for all clinical gene therapy studies.
Perspectives and Future
Despite considerable advances in the treatment of severe β-thalassemia, life expectancy and quality of life remain lower than population norms.77 Less than 25% of patients have access to an HLA-matched donor for allogeneic HSCT, which was the only approved cure until very recently. The emerging prospect of gene therapy cures has generated considerable excitement and the FDA approval of betibeglogene autotemcel/Zynteglo LW gene therapy for TDT in August 2022 represents “the end of the beginning”. Genome editing/base editing approaches are not far behind, and the field is advancing at lightning speed.
Newer, more versatile technologies, such as advanced generation base editors, prime editing, and adapted transposases will enhance further our ability to manipulate the genome40,41. Other components of the gene therapy process (Figure 1) are being refined to improve safety and efficacy. This includes new drugs to enhance the collection of autologous HSCs for gene correction,78,79 and antibody-based approaches for bone marrow conditioning that are less toxic than current myeloablative protocols.80,81 The development of “in vivo gene therapies” that can be administered parentally via modified viral vectors or lipid nanoparticles promises to simplify administration and increase access to curative therapy in low- and middle- income countries where β-thalassemia is prevalent.82,83 Most likely, the entire HSC gene therapy process (Figure 1) will continue to be refined and improved over time as new technologies are adapted into clinical use. The best approaches are not yet known. Ideal HSC gene therapies for TDT and other blood disorders will demonstrate long-term safety and efficacy over at least 5 years and be relatively straightforward to manufacture and administer so as to be accessible for all patients.
While the science and technology of gene therapy for β-thalassemia and other blood disorders have made exciting strides in the past few years, there have been recent setbacks in delivering these new medicines to patients.84 This year, bluebird bio closed its operations in Europe due to difficulties in negotiating with payors for pricing of gene therapies for β-thalassemia and adrenoleukodystrophy, a rare neurodegenerative disorder. Orchard Therapeutics will discontinue gene therapy for three rare immunodeficiencies. Reasons for these disinvestments include the high costs of developing, manufacturing and administering autologous cell-based therapies, regulatory and reimbursement complexities that vary across different countries77, and commercialization by companies whose survival may depend on short-term profits. Solving these problems for the benefit of all patients will require close collaboration between many stakeholders. Ultimately, society will profit from one-treatment cures for chronic diseases such as TDT by eliminating the cost of therapy over the entire lifespan and improving lives.
Summary
After many years of painstaking research, the potential of gene therapy to cure severe β-thalassemia is now becoming evident through recent clinical trials. Therapeutic protocols will continue to evolve and improve as new innovations are incorporated over time. Establishing the most safe and effective approaches will require long-term comparative studies. Ideally, pharmaceutical companies, governments, payors, and health care reimbursement systems will collaborate and adapt to optimize the delivery of these new personalized medicines.
Synopsis.
After many years of intensive research, emerging data from clinical trials indicate that gene therapy for transfusion-dependent β-thalassemia is now possible. Strategies for therapeutic manipulation of patient hematopoietic stem cells include lentiviral transduction of a functional β-globin gene and genome editing to activate fetal hemoglobin production in patient red blood cells. Gene therapy for β-thalassemia and other blood disorders will invariably improve as experience accumulates over time. The best overall approaches are not known and likely not yet established. Gene therapy comes at a high cost, and collaboration between multiple stakeholders is required to ensure that this new medicine is administered equitably.
Key points.
β-thalassemia is a common, frequently devastating hemoglobinopathy caused by HBB gene mutations that reduce or eliminate β-globin synthesis.
Allogenic hematopoietic stem cell transplantation can cure transfusion-dependent β-thalassemia (TDT) but has several major problems, including lack of HLA-matched donors for most patients and immune complications.
Genetic manipulation of patient hematopoietic stem cells (HSCs) to restore β-globin gene expression or induce fetal hemoglobin expression can cure β-thalassemia while circumventing some problems associated with allogeneic HSCT.
Methods for therapeutic manipulation of HSCs include lentiviral vectors, genome editing nucleases, and base editors.
Technical complexity and high costs associated with gene therapies threaten to restrict their commercialization and availability.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Taher AT, Musallam KM, Cappellini MD. β-Thalassemias. N Engl J Med. February 25 2021;384(8):727–743. doi: 10.1056/NEJMra2021838 [DOI] [PubMed] [Google Scholar]
- 2.Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. Jun 03 2010;115(22):4331–6. doi: 10.1182/blood-2010-01-251348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strocchio L, Locatelli F. Hematopoietic Stem Cell Transplantation in Thalassemia. Hematol Oncol Clin North Am. April 2018;32(2):317–328. doi: 10.1016/j.hoc.2017.11.011 [DOI] [PubMed] [Google Scholar]
- 4.Leonard A, Tisdale JF, Bonner M. Gene Therapy for Hemoglobinopathies: Beta-Thalassemia, Sickle Cell Disease. Hematol Oncol Clin North Am. Aug 2022;36(4):769–795. doi: 10.1016/j.hoc.2022.03.008 [DOI] [PubMed] [Google Scholar]
- 5.Magrin E, Miccio A, Cavazzana M. Lentiviral and genome-editing strategies for the treatment of β-hemoglobinopathies. Blood. October 10 2019;134(15):1203–1213. doi: 10.1182/blood.2019000949 [DOI] [PubMed] [Google Scholar]
- 6.Naldini L Genetic engineering of hematopoiesis: current stage of clinical translation and future perspectives. EMBO Mol Med. March 2019;11(3)doi: 10.15252/emmm.201809958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delville M, Soheili T, Bellier F, et al. A Nontoxic Transduction Enhancer Enables Highly Efficient Lentiviral Transduction of Primary Murine T Cells and Hematopoietic Stem Cells. Mol Ther Methods Clin Dev. Sep 21 2018;10:341–347. doi: 10.1016/j.omtm.2018.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hauber I, Beschorner N, Schrödel S, et al. Improving Lentiviral Transduction of CD34. Hum Gene Ther Methods. Apr 2018;29(2):104–113. doi: 10.1089/hgtb.2017.085 [DOI] [PubMed] [Google Scholar]
- 9.Locatelli F, Thompson AA, Kwiatkowski JL, et al. Betibeglogene Autotemcel Gene Therapy for Non-beta(0)/beta(0) Genotype beta-Thalassemia. N Engl J Med. Feb 3 2022;386(5):415–427. doi: 10.1056/NEJMoa2113206 [DOI] [PubMed] [Google Scholar]
- 10.Magrin E, Semeraro M, Hebert N, et al. Long-term outcomes of lentiviral gene therapy for the beta-hemoglobinopathies: the HGB-205 trial. Nat Med. Jan 2022;28(1):81–88. doi: 10.1038/s41591-021-01650-w [DOI] [PubMed] [Google Scholar]
- 11.Negre O, Eggimann AV, Beuzard Y, et al. Gene Therapy of the beta-Hemoglobinopathies by Lentiviral Transfer of the beta(A(T87Q))-Globin Gene. Hum Gene Ther. Feb 2016;27(2):148–65. doi: 10.1089/hum.2016.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pawliuk R, Westerman KA, Fabry ME, et al. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science. Dec 14 2001;294(5550):2368–71. doi: 10.1126/science.1065806 [DOI] [PubMed] [Google Scholar]
- 13.Thompson AA, Walters MC, Kwiatkowski J, et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N Engl J Med. April 19 2018;378(16):1479–1493. doi: 10.1056/NEJMoa1705342 [DOI] [PubMed] [Google Scholar]
- 14.Kanter J, Walters MC, Krishnamurti L, et al. Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease. N Engl J Med. February 17 2022;386(7):617–628. doi: 10.1056/NEJMoa2117175 [DOI] [PubMed] [Google Scholar]
- 15.Marktel S, Scaramuzza S, Cicalese MP, et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent ß-thalassemia. Nat Med. February 2019;25(2):234–241. doi: 10.1038/s41591-018-0301-6 [DOI] [PubMed] [Google Scholar]
- 16.Boulad F, Maggio A, Wang X, et al. Lentiviral globin gene therapy with reduced-intensity conditioning in adults with β-thalassemia: a phase 1 trial. Nat Med. Jan 2022;28(1):63–70. doi: 10.1038/s41591-021-01554-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rubin R New Gene Therapy for β-Thalassemia. JAMA. Sep 20 2022;328(11):1030. doi: 10.1001/jama.2022.14709 [DOI] [PubMed] [Google Scholar]
- 18.Ochiai H, Yamamoto T. Construction and Evaluation of Zinc Finger Nucleases. Methods Mol Biol. 2017;1630:1–24. doi: 10.1007/978-1-4939-7128-2_1 [DOI] [PubMed] [Google Scholar]
- 19.Hensel G, Kumlehn J. Genome Engineering Using TALENs. Methods Mol Biol. 2019;1900:195–215. doi: 10.1007/978-1-4939-8944-7_13 [DOI] [PubMed] [Google Scholar]
- 20.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. Nov 28 2014;346(6213):1258096. doi: 10.1126/science.1258096 [DOI] [PubMed] [Google Scholar]
- 21.Doudna JA. The promise and challenge of therapeutic genome editing. Nature. Feb 2020;578(7794):229–236. doi: 10.1038/s41586-020-1978-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cromer MK, Camarena J, Martin RM, et al. Gene replacement of α-globin with β-globin restores hemoglobin balance in β-thalassemia-derived hematopoietic stem and progenitor cells. Nat Med. Apr 2021;27(4):677–687. doi: 10.1038/s41591-021-01284-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forget BG. Molecular basis of hereditary persistence of fetal hemoglobin. Ann N Y Acad Sci. Jun 30 1998;850:38–44. doi: 10.1111/j.1749-6632.1998.tb10460.x [DOI] [PubMed] [Google Scholar]
- 24.Vinjamur DS, Bauer DE, Orkin SH. Recent progress in understanding and manipulating haemoglobin switching for the haemoglobinopathies. Br J Haematol. Mar 2018;180(5):630–643. doi: 10.1111/bjh.15038 [DOI] [PubMed] [Google Scholar]
- 25.Martyn GE, Wienert B, Yang L, et al. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat Genet. April 2018;50(4):498–503. doi: 10.1038/s41588-018-0085-0 [DOI] [PubMed] [Google Scholar]
- 26.Masuda T, Wang X, Maeda M, et al. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science. Jan 15 2016;351(6270):285–9. doi: 10.1126/science.aad3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frangoul H, Altshuler D, Cappellini MD, et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N Engl J Med. January 21 2021;384(3):252–260. doi: 10.1056/NEJMoa2031054 [DOI] [PubMed] [Google Scholar]
- 28.F L, F H, C S, et al. Efficacy and safety of a single dose of CTX for transfusion-dependent beta-thalassemia and severe sickle cell disease. EHA Library. Locatelli F. June/12/22; 366210; LB2367; 2022. [Google Scholar]
- 29.Fu B, Liao J, Chen S, et al. CRISPR-Cas9-mediated gene editing of the BCL11A enhancer for pediatric beta(0)/beta(0) transfusion-dependent beta-thalassemia. Nat Med. Aug 2022;28(8):1573–1580. doi: 10.1038/s41591-022-01906-z [DOI] [PubMed] [Google Scholar]
- 30.Walters MC , Smith AR , Schiller GC , et al. Updated Results of a Phase 1/2 Clinical Study of Zinc Finger Nuclease-Mediated Editing of BCL11A in Autologous Hematopoietic Stem Cells for Transfusion-Dependent Beta Thalassemia. Blood (2021) 138 (Suppl 1): 3974. [Google Scholar]
- 31.Métais JY, Doerfler PA, Mayuranathan T, et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. November 12 2019;3(21):3379–3392. doi: 10.1182/bloodadvances.2019000820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weber L, Frati G, Felix T, et al. Editing a γ-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci Adv. Feb 2020;6(7)doi: 10.1126/sciadv.aay9392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Humbert O, Radtke S, Samuelson C, et al. Therapeutically relevant engraftment of a CRISPR-Cas9-edited HSC-enriched population with HbF reactivation in nonhuman primates. Sci Transl Med. July 31 2019;11(503)doi: 10.1126/scitranslmed.aaw3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Traxler EA, Yao Y, Wang YD, et al. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med. September 2016;22(9):987–90. doi: 10.1038/nm.4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ye L, Wang J, Tan Y, et al. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: An approach for treating sickle cell disease and β-thalassemia. Proc Natl Acad Sci U S A. September 20 2016;113(38):10661–5. doi: 10.1073/pnas.1612075113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lux CT, Pattabhi S, Berger M, et al. TALEN-Mediated Gene Editing of. Mol Ther Methods Clin Dev. Mar 15 2019;12:175–183. doi: 10.1016/j.omtm.2018.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maeda T, Ito K, Merghoub T, et al. LRF is an essential downstream target of GATA1 in erythroid development and regulates BIM-dependent apoptosis. Dev Cell. Oct 2009;17(4):527–40. doi: 10.1016/j.devcel.2009.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gaudelli NM, Komor AC, Rees HA, et al. Programmable base editing of A• T to G• C in genomic DNA without DNA cleavage. Nature. 2017;551(7681):464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533(7603):420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. Jul 2020;38(7):824–844. doi: 10.1038/s41587-020-0561-9 [DOI] [PubMed] [Google Scholar]
- 41.Antoniou P, Miccio A, Brusson M. Base and Prime Editing Technologies for Blood Disorders. Front Genome Ed. 2021;3:618406. doi: 10.3389/fgeed.2021.618406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L, Li L, Ma Y, et al. Reactivation of γ-globin expression through Cas9 or base editor to treat β-hemoglobinopathies. Cell research. 2020;30(3):276–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li C, Georgakopoulou A, Mishra A, et al. In vivo HSPC gene therapy with base editors allows for efficient reactivation of fetal γ-globin in β-YAC mice. Blood advances. 2021;5(4):1122–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ravi NS, Wienert B, Wyman SK, et al. Identification of novel HPFH-like mutations by CRISPR base editing that elevate the expression of fetal hemoglobin. Elife. February 11 2022;11 doi: 10.7554/eLife.65421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Antoniou P, Hardouin G, Martinucci P, et al. Base-editing-mediated dissection of a gamma-globin cis-regulatory element for the therapeutic reactivation of fetal hemoglobin expression. Nat Commun. Nov 4 2022;13(1):6618. doi: 10.1038/s41467-022-34493-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhatia S Therapy-related myelodysplasia and acute myeloid leukemia. Semin Oncol. Dec 2013;40(6):666–75. doi: 10.1053/j.seminoncol.2013.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cesana D, Ranzani M, Volpin M, et al. Uncovering and dissecting the genotoxicity of self-inactivating lentiviral vectors in vivo. Mol Ther. Apr 2014;22(4):774–85. doi: 10.1038/mt.2014.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baum C, Kustikova O, Modlich U, Li Z, Fehse B. Mutagenesis and oncogenesis by chromosomal insertion of gene transfer vectors. Hum Gene Ther. Mar 2006;17(3):253–63. doi: 10.1089/hum.2006.17.253 [DOI] [PubMed] [Google Scholar]
- 49.Cavazzana-Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature. Sep 16 2010;467(7313):318–22. doi: 10.1038/nature09328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bonner MA, Morales-Hernandez A, Zhou S, et al. 3' UTR-truncated HMGA2 overexpression induces non-malignant in vivo expansion of hematopoietic stem cells in non-human primates. Mol Ther Methods Clin Dev. Jun 11 2021;21:693–701. doi: 10.1016/j.omtm.2021.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hsieh MM, Bonner M, Pierciey FJ, et al. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv. May 12 2020;4(9):2058–2063. doi: 10.1182/bloodadvances.2019001330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goyal S, Tisdale J, Schmidt M, et al. Acute Myeloid Leukemia Case after Gene Therapy for Sickle Cell Disease. N Engl J Med. January 13 2022;386(2):138–147. doi: 10.1056/NEJMoa2109167 [DOI] [PubMed] [Google Scholar]
- 53.Jones RJ, DeBaun MR. Leukemia after gene therapy for sickle cell disease: insertional mutagenesis, busulfan, both, or neither. Blood. September 16 2021;138(11):942–947. doi: 10.1182/blood.2021011488 [DOI] [PubMed] [Google Scholar]
- 54.Lidonnici MR, Paleari Y, Tiboni F, et al. Multiple Integrated Non-clinical Studies Predict the Safety of Lentivirus-Mediated Gene Therapy for β-Thalassemia. Mol Ther Methods Clin Dev. Dec 14 2018;11:9–28. doi: 10.1016/j.omtm.2018.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim D, Lim K, Kim ST, et al. Genome-wide target specificities of CRISPR RNA-guided programmable deaminases. Nat Biotechnol. May 2017;35(5):475–480. doi: 10.1038/nbt.3852 [DOI] [PubMed] [Google Scholar]
- 56.Kim D, Luk K, Wolfe SA, Kim JS. Evaluating and Enhancing Target Specificity of Gene-Editing Nucleases and Deaminases. Annu Rev Biochem. June 20 2019;88:191–220. doi: 10.1146/annurev-biochem-013118-111730 [DOI] [PubMed] [Google Scholar]
- 57.Kim DY, Moon SB, Ko J-H, Kim Y-S, Kim D. Unbiased investigation of specificities of prime editing systems in human cells. Nucleic Acids Research. 2020-October-09 2020;48(18):10576–10589. doi: 10.1093/nar/gkaa764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsai SQ, Joung JK. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat Rev Genet. May 2016;17(5):300–12. doi: 10.1038/nrg.2016.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheng Y, Tsai SQ. Illuminating the genome-wide activity of genome editors for safe and effective therapeutics. Genome Biol. Dec 22 2018;19(1):226. doi: 10.1186/s13059-018-1610-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leibowitz ML, Papathanasiou S, Doerfler PA, et al. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat Genet. June 2021;53(6):895–905. doi: 10.1038/s41588-021-00838-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Enache OM, Rendo V, Abdusamad M, et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat Genet. July 2020;52(7):662–668. doi: 10.1038/s41588-020-0623-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med. July 2018;24(7):927–930. doi: 10.1038/s41591-018-0049-z [DOI] [PubMed] [Google Scholar]
- 63.Ihry RJ, Worringer KA, Salick MR, et al. p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nature medicine. 2018;24(7):939–946. [DOI] [PubMed] [Google Scholar]
- 64.Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nature biotechnology. 2018;36(8):765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Blattner G, Cavazza A, Thrasher AJ, Turchiano G. Gene Editing and Genotoxicity: Targeting the Off-Targets. Front Genome Ed. 2020;2:613252. doi: 10.3389/fgeed.2020.613252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grünewald J, Zhou R, Garcia SP, et al. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature. 2019-May-01 2019;569(7756):433–437. doi: 10.1038/s41586-019-1161-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou C, Sun Y, Yan R, et al. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature. July 2019;571(7764):275–278. doi: 10.1038/s41586-019-1314-0 [DOI] [PubMed] [Google Scholar]
- 68.Doman JL, Raguram A, Newby GA, Liu DR. Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat Biotechnol. May 2020;38(5):620–628. doi: 10.1038/s41587-020-0414-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jeong YK, Song B, Bae S. Current Status and Challenges of DNA Base Editing Tools. Mol Ther. September 02 2020;28(9):1938–1952. doi: 10.1016/j.ymthe.2020.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim D, Kim DE, Lee G, Cho SI, Kim JS. Genome-wide target specificity of CRISPR RNA-guided adenine base editors. Nat Biotechnol. April 2019;37(4):430–435. doi: 10.1038/s41587-019-0050-1 [DOI] [PubMed] [Google Scholar]
- 71.Yu Y, Leete TC, Born DA, et al. Cytosine base editors with minimized unguided DNA and RNA off-target events and high on-target activity. Nature Communications. 2020-December-01 2020;11(1)doi: 10.1038/s41467-020-15887-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zuo E, Sun Y, Yuan T, et al. A rationally engineered cytosine base editor retains high on-target activity while reducing both DNA and RNA off-target effects. Nat Methods. June 2020;17(6):600–604. doi: 10.1038/s41592-020-0832-x [DOI] [PubMed] [Google Scholar]
- 73.Liang M, Sui T, Liu Z, et al. AcrIIA5 Suppresses Base Editors and Reduces Their Off-Target Effects. Cells. 2020-July-27 2020;9(8):1786. doi: 10.3390/cells9081786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Coelho MA, De Braekeleer E, Firth M, et al. CRISPR GUARD protects off-target sites from Cas9 nuclease activity using short guide RNAs. Nat Commun. Aug 17 2020;11(1):4132. doi: 10.1038/s41467-020-17952-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Naeem M, Majeed S, Hoque MZ, Ahmad I. Latest Developed Strategies to Minimize the Off-Target Effects in CRISPR-Cas-Mediated Genome Editing. Cells. Jul 2 2020;9(7):1608. doi: 10.3390/cells9071608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Richter MF, Zhao KT, Eton E, et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat Biotechnol. July 2020;38(7):883–891. doi: 10.1038/s41587-020-0453-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Beaudoin FL, Richardson M, Synnott PG, et al. Betibeglogene Autotemcel for Beta Thalassemia: Effectiveness and Value. https://icer.org/beta-thalassemia-2022/#timeline: Institute for Clinical and Economic Review (ICER); 2022. [Google Scholar]
- 78.Fukuda S, Bian H, King AG, Pelus LM. The chemokine GROβ mobilizes early hematopoietic stem cells characterized by enhanced homing and engraftment. Blood. 2007-08-01 2007;110(3):860–869. doi: 10.1182/blood-2006-06-031401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hoggatt J, Singh P, Tate TA, et al. Rapid Mobilization Reveals a Highly Engraftable Hematopoietic Stem Cell. Cell. 2018-January-01 2018;172(1-2):191–204.e10. doi: 10.1016/j.cell.2017.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Czechowicz A, Palchaudhuri R, Scheck A, et al. Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nat Commun. Feb 06 2019;10(1):617. doi: 10.1038/s41467-018-08201-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Palchaudhuri R, Saez B, Hoggatt J, et al. Non-genotoxic conditioning for hematopoietic stem cell transplantation using a hematopoietic-cell-specific internalizing immunotoxin. Nat Biotechnol. Jul 2016;34(7):738–45. doi: 10.1038/nbt.3584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cullis PR, Hope MJ. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol Ther. Jul 05 2017;25(7):1467–1475. doi: 10.1016/j.ymthe.2017.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Raguram A, Banskota S, Liu DR. Therapeutic in vivo delivery of gene editing agents. Cell. Jul 21 2022;185(15):2806–2827. doi: 10.1016/j.cell.2022.03.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Aiuti A, Pasinelli F, Naldini L. Ensuring a future for gene therapy for rare diseases. Nat Med. Oct 2022;28(10):1985–1988. doi: 10.1038/s41591-022-01934-9 [DOI] [PubMed] [Google Scholar]