

Abstract

Overdose of acetaminophen, a widely used antipyretic and analgesic drug, is one of the leading causes of drug-induced acute liver injury in the United States and worldwide. Phase-I metabolism of acetaminophen generates the toxic N-acetyl-p-benzoquinone imine (NAPQI) intermediate. Reactions of NAPQI with a wide range of biomolecules cause increased oxidative stress, endoplasmic reticulum (ER) stress, inflammation, and mitochondrial dysfunction, some of the cellular events contributing toward liver toxicity. Previously, we evaluated the potential of an FDA-approved, ER stress-modulating antihypertensive drug, Wytensin (trans-guanabenz, E-GA), as an antidote for acetaminophen hepatotoxicity. E-GA prevented elevation of the liver enzyme alanine aminotransferase (ALT), even when administered up to 6 h after acetaminophen overdose, and exhibited synergistic analgesic interactions. However, the commercially available guanabenz exists solely as a trans-isomer and suffers from sedative side effects resulting from the inhibition of central α2A-adrenergic receptors in locus coeruleus. Here, we studied the utility of the relatively unexplored cis-isomer of guanabenz as a treatment option for acetaminophen-induced liver toxicity. cis(Z)-Guanabenz acetate (Z-GA) lacks interaction with α2A-adrenoreceptors and is thus devoid of sedative, blood-pressure-lowering side effects of E-GA. Treatment of mice with Z-GA (10 mg/kg) before acetaminophen overdose and up to 6 h post APAP administration prevented liver injury and suppressed the elevation of serum ALT levels. Mechanistically, hepatoprotective effects of both isomers are similar and partly attributed to attenuation of the ER stress and oxidative stress in the liver. The results of this study suggest that Z-GA may be a safer, effective antidote for the clinical management of acute liver injury resulting from acetaminophen overdose. It also raises a tantalizing possibility of a prophylactic combination of the geometric isomer of the approved drug guanabenz with acetaminophen in a clinical setting.
Introduction
Drug-induced liver injury, a well-recognized problem in the clinical practice, is caused by acute or chronic reactions to drugs or natural medicines. It is one of the leading causes of acute liver failure in the US and Europe.1 Acetaminophen (APAP) is one of the most widely used antipyretic and analgesic over-the-counter medication and is generally considered safe for doses up to 4,000 mg every 24 h. Although safe at such doses, intentional or unintentional overdose of APAP is hepatotoxic and may lead to acute liver injury and subsequent death due to liver failure. In the United States alone, about 56,000 emergency room visits, 2,600 cases of hospitalizations, and 500 deaths are reported every year due to APAP-induced hepatotoxicity.2,3 Half of these are due to inadvertent administration of APAP contained in various formulations, including prescription combination products with opioids. The dangers of prescription APAP combinations caused the United States FDA to revise guidelines for the manufacturers of APAP-containing prescription drug products in 2011. The dose per tablet in such products is now limited to 325 mg, not exceeding 3,000 mg in 24 h time period, with a warning clearly stating the potential of APAP for severe liver injury.4 Treatment with an antidote is the most effective if administered within 8 h of APAP overdose, necessitating accurate diagnosis and availability of safe, effective antidotes.5
Metabolic activation of APAP has been extensively studied and well elucidated. There is clear evidence that supports the formation of a toxic metabolic intermediate, N-acetyl-p-benzoquinone imine (NAPQI), responsible for hepatotoxic consequences of APAP overdose.6,7 Under normal physiological conditions, the majority (90%) of APAP is subjected to phase II metabolic transformation, which results in conjugation of APAP by UDP-glucuronosyl transferase (UGT) and sulfotransferase (SULT) to the corresponding glucuronidated and sulfated metabolites.8 These metabolites along with a small amount of unmodified APAP (2%) are excreted in the urine. The remaining APAP (∼10%) is channeled through hepatic cytochrome CYP 2E1 to undergo phase-I oxidation, forming highly reactive NAPQI.9,10 Hepatic glutathione (GSH) is able to neutralize NAPQI by forming an irreversible, water-soluble conjugate that is excreted in the urine.11−14 However, APAP overdose resulting in excessive quantities of NAPQI overwhelms the liver content of GSH. This is additionally exacerbated by GSH depletion caused by increased intrahepatic oxidative stress. Hepatotoxic doses of APAP saturate glucuronidation and sulfation pathways, pushing the majority of APAP through phase I oxidation causing increased NAPQI generation.7 As a result, sufficient quantities of free NAPQI are available to covalently modify hepatic and mitochondrial proteins, leading to further oxidative stress and mitochondrial dysfunction resulting in the depletion of ATP stores.15−18 Effective treatment of this clinical scenario is possible by administration of the GSH precursor, N-acetylcysteine (NAC), either by intravenous (IV) or oral route. However, large oral doses of NAC employed are often poorly tolerated. IV NAC suffers from dose-limiting side effects such as anaphylaxis, nausea, vomiting, drowsiness, or hypotension.11,19,20 Thus, there is a dire need for the identification of a safer, effective antidote that could be useful in a clinical setting. Such an antidote could be combined with APAP in various formulations to prevent or reduce incidences of APAP overdose.
Development of effective antidotes for APAP-induced liver injury requires deep understanding of the intracellular and extracellular mechanisms involved in this complex toxicological process. Apart from the involvement of cellular and mitochondrial oxidative stress, APAP metabolism, autophagy, inflammation, and liver regeneration, the role of endoplasmic reticulum (ER) stress in APAP hepatotoxicity is increasingly being understood.21,22 Covalent binding of NAPQI to hepatic proteins triggers a cascade of stress responses, collectively termed the unfolded protein response (UPR), which constitutes the impaired protein folding process in the ER. The mammalian UPR system consists of three transmembrane ER sensors, i.e., protein kinase RNA-like endoplasmic reticulum kinase (PERK), the activating transcription factor 6 (ATF6); and inositol requiring kinase 1α (IRE1α).23,24 Activation of the PERK pathway leads to the phosphorylation of eukaryotic initiation factor 2α (eIF2α). In the short term, phosphorylation of eIF2α causes global reduction in protein synthesis, but the translation of specific genes like the activating transcription factor 4 (ATF4) is spared. Another effect of eIF2α phosphorylation is the increase in the expression of CCAAT-enhancer-binding protein homologous protein (CHOP), which is pro-apoptotic. Phosphorylated eIF2α (p-eIF2α) also enhances the expression of Growth Arrest and DNA Damage 34 (GADD34), a regulatory subunit of the phosphatase complex that dephosphorylates p-eIF2α, thereby downregulating PERK signaling (negative feedback). Thus, the net effect in the short term of eIF2α phosphorylation is stress adaptation. However, if stress is intense or chronic, increasingly high CHOP expression overwhelms adaptive processes and liver damage results.25,26 CHOP is a pro-damage protein in APAP and knockout of CHOP in mice caused partial resistance to hepatic injury caused by APAP.27 The IRE1α-XBP1 pathway was recently found important in the UPR response to APAP liver injury.28,29 Activation of the transcription factor ATF6 by sublethal doses of APAP was also demonstrated.30 Taken together, these studies put forth a rationale for the mitigation of ER stress as a potential therapeutic strategy for antidote development.
In our previous study, we examined the utility of an FDA-approved antihypertensive therapeutic, guanabenz acetate (GA, Wytensin), an established ER stress modulator as a treatment option for APAP overdose.31 Guanabenz was developed during the 1980s as a CNS-acting α2-adrenergic receptor (α2-AR) agonist to treat hypertension, with documented safety and pharmacokinetic information in humans.32−34 Guanabenz inhibits GADD34 and elevates the levels of p-eIF2α, independent of its α2-adrenoreceptor interaction, which is responsible for the amelioration of ER stress in various pathophysiological conditions. We showed that the treatment with GA protected against liver enzyme elevation and hepatocyte necrosis, even when administered up to 6 h after APAP overdose.31 Amelioration of ERS and oxidative stress was apparent in the liver tissue of mice treated with a combination of GA with APAP. The direct use of GA as an antidote for APAP hepatotoxicity is impractical because of its potent hypotensive and sedative effect arising from agonism at the α2A-adrenergic receptors expressed in locus coeruleus. Although GA dosage used in our study was within the clinically tolerated range, it raises questions about its potential to be administered along with APAP as a combination treatment wherein repeated intake could result in severe dose-limiting sedative effects of the antidote itself. An analogue of GA, devoid of sedative side effects would be an ideal candidate for APAP combination therapy. The clinically available guanabenz exists exclusively in thermodynamically stable trans-form. The geometric cis-isomer (Z-form) has been reported to form through photolytic degradation of E-GA (Figure 1).35 Of interest, the Z-isomer lacks the hypotensive activity of the parent E-isomer.36 We observed the retention of the anti-ER stress property by the Z-isomer in the tunicamycin-induced cellular model of ERS.37 Our pharmacokinetic analysis of Z-GA showed a distinct pharmacokinetic profile of this isomer and concluded that the cis-isomer is metabolically more stable against hepatic microsomal degradation and is significantly less protein bound in plasma compared to the trans-isomer.37 This study formed the basis of further investigation of Z-GA in efforts directed at repurposing of the clinically used guanabenz.
Figure 1.

Chemical structures of the geometric isomers of guanabenz.
In this study, we evaluated the efficacy of the cis-isomer of guanabenz (Z-GA) against acute liver injury caused by APAP poisoning. Delayed administration of the antidote has been attempted to mirror a clinical scenario. Mechanistic elucidation of the role of ER and oxidative stress in APAP-induced hepatotoxicity and effect of Z-GA treatment have been studied. This is the first report of the pharmacological efficacy of the relatively unexplored isomer of guanabenz. The results of this study suggest mitigation of ER stress as an effective strategy for the management of acute liver toxicity resulting from APAP overdose and support further investigation of Z-GA as a therapeutic agent for conditions wherein ER stress is indicated.
Materials and Methods
Materials and Their Sources
Acetaminophen (Spectrum Chemical Corp, New Brunswick, NJ, #AC100), E-guanabenz acetate (Far top limited, Nanjing, China, #427223S), Z-guanabenz was synthesized in our laboratory.37l-(−)-Norepinephrine-16-bitartrate (Millipore Sigma, St. Louis, MO, #489350), tunicamycin (Millipore Sigma, St. Louis, MO, #T7765), CCK-8 solution (Dojindo, Rockville, MD, #CK04), total–IRE1α (Novus Biologicals, Littleton, CO, #NB100-2324), phospho-IRE1α (Novus Biologicals, #NB100–2323), phospho and total eIF2α (Cell Signaling Technology, Danvers, MA, #9721, #9722), BiP (Cell Signaling Technology, #3177), ATF6 (Abcam, Cambridge, MA, #ab37149), CHOP (Cell Signaling Technology, #5554), α-tubulin (Abcam, #ab4074), anti-rabbit lgG (Cell Signaling Technology, #7074), triethanolamine (Sigma-Aldrich Corp, #t58300), 2-vinylpyridine (Alfa Aesar, Ward Hill, MA, #A14056), RIPA buffer (Cayman Chemical Co., Ann Arbor, MI, #10010263), GSH MES buffer (Cayman Chemical Co., #703010), cOmpleteProtease Inhibitor Cocktail (Roche, Penzberg, Germany, #4693124001), lipofectamine 2000 transfection reagent (Invitrogen, #11668027), Subcloning efficiencyTM DH5α-competent cells (Invitrogen, #18265017), ZymoPURE II plasmid midiprep Kit (Zymo Research, #D4200), SuperScript VILO cDNA synthesis kit (Invitrogen, #11754050), and PureLink RNA mini kit (Invitrogen, #12183018A) were used and both forward and reverse primers were synthesized by Integrated DNA Technologies (Coralville, IA).
Assessment of Cell Viability
HeLa Cells were seeded in 96 well plates at a density of 2000 cells/well and incubated at 37 °C in a humidified incubator with 5% CO2 for 24 h. The cells were then changed to a fresh medium containing 1 μg/mL tunicamycin in the presence of varying concentrations of E-GA and Z-GA. After incubation for 48 h, CCK-8 solution was added to each well to measure the cell viability. Cell viability is expressed as the mean percent of cell growth with respect to control with the standard error of the mean (n = 3).
Plasmid
The full-length of adrenergic receptor α 2A gene was amplified from cDNA by the polymerase chain reaction (forward primer, 5′-ATCGGCGGCCGCA ATGGGCTCCCTGCAGCCGGACGCGGGCAA -3′ (NotI site with underline); reverse primer, 5′-ATCGCTCGAG TCACACGATCCGCTTCCTGTCCCCCCGACAGA -3′ (XhoI site with underline)). The PCR product was then inserted into NotI and XhoI sites of the pCMV-3tag-6 vector to construct the full length of pCMV-3tag-6-ADRA2A. The sequence of the constructed plasmid was confirmed by Sanger DNA sequencing. The constructed plasmid was then transformed into a DH5α competent cell. After plasmid amplification, plasmid DNA was extracted using ZymoPURE II Plasmid Midiprep Kit (Zymo Research, #D4200) following the manufacturer’s protocol.
Transfection and Protein Preparation
pCMV-3tag-6-ADRA2A was transiently transfected into HEK 293T cells using the Lipofectamine 2000 reagent following the manufacturer’s protocol. After 48 h post transfection, the cells were washed with PBS (pH 7.4), harvested with a cell scraper, and then resuspended in lysis buffer (5 mM Tris–HCl, 5 mM EDTA, 5 mM EGTA, pH 7.4) containing the cOmplete protease inhibitor cocktail. After homogenization of cell suspension in 50 mM Tris–HCl (pH 8.0) with a mechanical homogenizer, membrane protein was extracted by ultracentrifugation (speed-100,000 rcf, for 30 min at room temperature). The obtained pellet was resuspended in reaction buffer containing 50 mM Tris–Hcl, 0.6 mM EDTA, and 5 mM MgCl2 (pH 7.4). Protein concentration was determined using a bicinchoninic acid assay kit.
Radioligand Binding Assay
The affinity of Z-GA and E-GA for the human α2A-adrenergic receptor was determined by the displacement assay by [3H]-yohimbine binding to membrane protein prepared from transiently transfected HEK 293T cells with the α2A-adrenergic receptor as described above following the manufacturer’s protocol.38,39 A total of 15 μg of membrane protein was used in each reaction, and [3H]-yohimbine (10 nM) in the presence or absence of compound (1 μM E-GA or Z-GA) was incubated at ambient temperature. Nonspecific binding was measured in the presence of norepinephrine (100 μM). After 30 min incubation, the sample was isolated by rapid filtration over GF/B glass fiber filter strips (Whatman, Inc.; Clifton, NJ) and the filter strips were washed with 2 mL of reaction buffer three times. The strips were dried at ambient temperature for 15 min and then transferred to 10 mL EcoLite scintillation cocktail glass vials. The vials were read with a PerkinElmer Tri-Carb 2900TR Liquid Scintillation analyzer.
Animals and In Vivo Treatment
Male Swiss Webster mice were purchased from the Envigo Company (Indianapolis, IN) at 6–7 weeks old. The mice were allowed at least 1-week acclimation prior to experimentation. All experimental protocols were as per the standard dictated by the Protocol Requirements at the University of Minnesota, Minneapolis, MN (IACUC). All mice were maintained and handled in accordance with the national ethics guidelines. Food was removed 16 h prior to experimentation and then mice were weighed prior to separation into treatment groups. APAP was prepared in warm saline and was injected intraperitoneally at a dose of 370 mg/kg (2.45 mmol/kg). E-GA and Z-GA were dissolved in saline and administered at a 10 mg/kg dose at the indicated time point (N = 10–15/group). Food was returned to the cages after the final injection, and animals were monitored every 30 min for the first 2 h. The mice were euthanized and blood and liver tissues were collected for biochemical and histopathological analysis 24 h after the APAP overdose.
Liver Enzyme Assessment
After coagulation of blood at room temperature for 30 min, the blood samples were centrifuged to obtain serum. The serum was stored at 4 °C before being submitted to the Veterinary Clinical Pathology Laboratory of the University of Minnesota for serum alanine aminotransferase (ALT) measurements.
Histology
Following 24 h APAP overdose, livers of mice were harvested and then fixed in 10% neutral buffered formalin for histopathological evaluation. After 24 h fixation, the mice liver tissues were submitted to the Comparative Pathology Shared Resource at the University of Minnesota for hematoxylin & eosin (H&E) staining to examine morphology.
Analysis of Lipid Peroxidation
TBARS assay was used to investigate liver lipid peroxidation through colorimetric measurements of malondialdehyde40 content. The protocol described in the kit was followed. Briefly, the mouse liver was homogenized in RIPA buffer with a protease inhibitor. After centrifugation, the supernatant was used for the measurement of the lipid peroxidation level in the mouse liver using the TBARS Assay Kit (Cayman Chemical Co., # 10009055).
Quantitation of GSH in the Mouse Liver
Measurement of mice liver GSH levels was performed using a Glutathione assay kit (Cayman Chemical Co., # 703002) according to the manufacturer’s instructions. Briefly, the mouse liver was homogenized in 50 mM MES buffer (pH = 6.0, containing 1 mM EDTA). After centrifugation, the supernatants were deproteinized with metaphosphoric acid, followed by neutralization with 4 M triethanolamine. The homogenate was then diluted with 50 mM MES buffer (pH = 6.0, containing 1 mM EDTA) prior to the assay. For GSH disulfide quantitation, samples were mixed with 1 M 2-vinylpyridine solution and incubated for 1 h at ambient temperature. Total GSH and GSSG levels were measured through the reaction of the GSH sulfhydryl group with 5,5′-dithiobis-2-nitrobenzoic acid (DTNB), producing a yellow-colored 5-thio-2-nitrobenzoic acid (TNB) which has absorbance at 412 nm.
Analysis of the Protein Carbonyl Content
To measure protein carbonyl levels in the mouse liver, we used a protein carbonyl colorimetric assay kit (Cayman Chemical Co., #10005020). Following the manufacturer’s protocol, the liver tissue was homogenized in 2 mL of 50 mM MES buffer (pH = 6.0, containing 1 mM EDTA). The homogenate was derivatized by DNPH, followed by the measurement of the amount of protein-hydrazone products at 370 nm using a SpectraMax M5 plate reader. The values then were normalized to protein concentration in these samples.
Western Blot
Mouse liver tissue was homogenized in fresh RIPA buffer containing a protease inhibitor cocktail tablet. The liver homogenate was denatured in reduced Laemmli sample buffer (62.5 mM Tris–HCl, pH 6.8, 2% SDS, 10% glycerol, 5% 2-mercaptoethanol, 0.002% bromophenol blue). The same amount of the sample was loaded onto the gel. After separation by sodium dodecyl sulfate poly(acrylamide) gel electrophoresis (SDS-PAGE), proteins were transferred from the SDS-PAGE gel to a poly(vinylidene difluoride) (PVDF) membrane. The membranes were incubated with the respective primary antibodies and then horseradish peroxidase-linked secondary antibodies (Cell Signaling). The membranes were developed with Clarity Western ECL substrate (Bio-rad), and the images were acquired with the ChemiDoc MP Imaging System (Bio-rad). Anti-α-tubulin antibody was used to confirm equal loading.
Behavioral Analysis
Antinociceptive response was assessed using the tail flick test as described previously.31,41 Briefly, mice were placed on top of the tail flick instrument (Columbus Instruments, Columbus, OH) and gently restrained in a soft cloth. The high-intensity light beam was focused on the area close to the dorsal surface of the tail (about 2 cm from the end of the tail). The time taken for the withdrawal of the tail away from the light beam was recorded after the heat source was activated. The light intensity of the instrument was adjusted so that the average baseline response latency of mice was 3–4 s. Mice were tested before (0 min) and 15, 30, 45, 60, 75, and 90 min after the compound administration. To minimize tissue injury, the light source was shut off after 10 s if the animal did not remove its tail away from the light beam. The antinociceptive response was expressed as percent maximal effect (%MPE), where %MPE = [(test – baseline)/(cut-off – baseline)] ×100.
Heart Rate Measurement
The heart rate of mice was measured using a MouseOx Plus Oximeter (Starr Life Sciences Corp). The collar sensor was placed around the necks of the animals to record the heart rate (beats per minute (bpm)). The heart rate of mice was calculated using MouseOx software and recorded at 1 Hz. The baseline was recorded, followed by injection of saline, E-GA (10 mg/kg, i.p.) or Z-GA (10 mg/kg, i.p.). The heart rate was recorded at 15, 30, 45, and 60 min after the injection.
Body Temperature Measurement
To determine the mouse body temperature, the mouse was restrained by grasping the skin at the nape of the neck. The mouse ventral side was placed approximately 6 inches away from the infrared TW2 thermometer (ThermoWorks, Alpine, UT), the mouse ventral side and the beam aimed below the base of the sternum. Mouse body temperature was sequentially measured before (time = 0 min) and 15, 30, 45, 60, 75, 90, and 120 min after drug administration.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism version 9.5.0 (GraphPad Software, San Diego, California). Data were expressed as means ± standard error of the mean (SEM). Statistical significance was analyzed by two-way analysis of variance (ANOVA) or one-way ANOVA, wherever appropriate. For all statistics, a p value < 0.05 was considered statistically significant.
Results
Geometric Isomer of Guanabenz Lacks Interaction with α2A-Adrenergic Receptor and Is Non-Sedative
To evaluate the binding of trans- and cis-isomers of guanabenz to α2A-adrenergic receptors, a radioligand displacement assay was used. [3H]-Yohimbine, a selective α2-adrenoreceptor antagonist, was used as a radiotracer to determine receptor binding in α2A-adrenoreceptor overexpressing cells. E-GA inhibited yohimbine-adrenoreceptor binding by 50%, while there was no effect of Z-GA treatment on yohimbine binding at the same concentration (Figure 2A). Since agonism of central α2A-receptors is one of the mechanisms behind analgesic and sedative behavior exhibited by guanabenz, we determined the analgesic potency of the two isomers. In agreement with the receptor binding properties, E-GA displayed analgesic efficacy in the tail flick nociception assay with 100% maximal possible effect achieved at 45 min post compound administration by the subcutaneous route. Z-GA was devoid of antinociceptive properties under similar experimental conditions (Figure 2B). Further, mice were co-administered with Z-GA and APAP to determine the effect of Z-GA on latter’s analgesic activity. APAP itself exhibited a marginal analgesic effect in this particular test, and its analgesic response was unaffected by combination with Z-GA (Supporting Information, Figure S1). Examination of sedative side effects was conducted by measuring the heart rate of the mice injected with the geometric isomers of guanabenz. A time-dependent reduction in the heart rate was apparent in E-GA (10 mg/kg, i.p.)-treated mice compared to saline-treated mice, while the mice treated with Z-GA at the same dose did not show significant depression of heart function (Figure 2C). A lower dose of E-GA (2 mg/kg, i.p.) also displayed similar reduction in the heart rate of mice (Supporting Information, Figure S2). Our previous study displayed distinct pharmacokinetic profiles of the two geometric isomers. Tissue distribution analysis of E- and Z-GA after oral administration offered similar concentrations of both isomers in tissues such as heart, spleen, and kidney (Supporting Information, Figure S3). Z-GA showed 1.69 fold higher Cmax in the liver, while E-GA showed the highest accumulation in the lungs (3-fold higher Cmax compared to Z-GA). This experiment also confirmed that less than 10% of the guanabenz isomer administered was converted to the other isomer (E-GA to Z-GA and vice versa; Supporting Information, Table 1) in all of the tissues analyzed, supporting the need to investigate the isomers individually for their pharmacological activity.
Figure 2.

Cis-Isomer of guanabenz is devoid of hemodynamic modulatory activity of trans-guanabenz. (A) Radioligand displacement assay in the α2A-adrenergic receptor overexpressing cells showed a lack of interaction of the receptor with Z-GA, evident from the absence of radioligand displacement compared to control cells (***p < 0.001, when compared to the control group). (B) Tail flick analgesia assay confirmed analgesic potency of E-GA, while Z-GA was completely inactive. (C) cis-isomer of guanabenz showed no significant depression of heart rate, which was apparent in mice treated with trans-guanabenz. (D) Combination of low and high-dose Z-GA with APAP did not affect antipyretic activity of APAP.
Finally, we evaluated the effect of co-administration of Z-GA on the antipyretic effect of APAP. No significant change in the body temperature (Figure 2D) was observed in mice exposed to either APAP or a combination of APAP and Z-GA.
cis-Guanabenz Prevents Elevation of Liver Enzyme and Hepatotoxicity Resulting from APAP Overdose
An acute model of APAP hepatotoxicity used in our previous studies with the glutathione analogue42 and E-GA31 was employed in this study. Eight-week-old Swiss Webster mice that were fasted overnight were subjected to an overdose of APAP (370 mg/kg) to induce liver toxicity. Only male mice were used in our experiments because several reports have documented lower susceptibility of female mice in comparison to male mice to liver injury induced by acetaminophen overdose.43,44 This is attributed mainly to differences in the metabolic neutralization of the hepatotoxic intermediate and hormone levels. Specifically, it has been shown that acetaminophen causes severe liver injury in male mice compared to female mice. In fact, hepatotoxicity induced by APAP in female mice was substantially (69–77%) lower compared to male mice. To determine the effect of Z-GA on APAP toxicity, mice were pretreated with Z-GA 30 min prior to APAP administration. Elevation of serum ALT levels (4501.2 U/L, 184-fold over control mice, Figure 3A) was observed in saline-treated high-dose APAP-only mice, indicative of acute liver damage in these mice. Treatment with Z-GA (i.p. and oral) significantly reduced such increases in ALT levels induced by high-dose APAP (Figure 3A), thus displaying protection against APAP-induced liver injury. The ALT level in the oral Z-GA + APAP group (123.5 U/L) was significantly lower than that in the intraperitoneal Z-GA + APAP group (535.4 U/L, p < 0.05).
Figure 3.

Effect of administration of Z-GA by intraperitoneal and oral routes on serum ALT levels in APAP-overdosed mice. Serum samples were collected from mice 24 h post overdose of APAP (370 mg/kg) to determine ALT levels. At 24 h after APAP overdose, serum was collected from mice for serum ALT levels. (A) Serum ALT levels when Z-GA (10 mg/kg, i.p. or p.o.) was administered 30 min prior to injection of APAP (370 mg/kg). (B) Serum ALT levels in the group receiving delayed Z-GA treatment (10 mg/kg, i.p.), at 0.5, 1, 2, 4, and 8 h post administration of high-dose APAP. (C) Serum ALT levels in the group receiving Z-GA (20 mg/kg, i.p.) 2 and 4 h after treatment with a high dose of APAP (370 mg/kg, **, p < 0.01; ***, p < 0.001; ****, p < 0.0001; when compared to APAP-only group).
To evaluate the efficacy of Z-GA as a potential antidote under a simulated clinical scenario, treatment with Z-GA (10 mg/kg, i.p.) was initiated after APAP overdose at 1/2, 1, 2, 4, and 8 h post APAP administration. Significant reduction in the ALT level was observed in Z-GA treated mice; by 77, 72, 68, and 54%, respectively, for time points up to 4 h (Figure 3B). Although the effect on the ALT level by Z-GA did not reach statistical significance at 8 h time point, the trend in the ALT reduction was apparent (36% ALT level reduction). A higher dose of Z-GA (20 mg/kg, i.p.) was well tolerated in these mice. When administered at this higher dose (20 mg/kg) post APAP overdose, Z-GA partially protected the liver from APAP toxicity. This dose reduced the ALT level by 55 and 19% when administered 2 and 4 h after APAP overdose, respectively (Figure 3C). The protective effect disappeared when the same dose of intraperitoneal Z-GA (20 mg/kg) was administered 8 h post APAP overdose. No mortality occurred in the APAP + Z-GA combination group, while the combination of clinically used antidote NAC with APAP resulted in a survival rate of only 10–25% in our previous study.42
The histological analysis of liver tissues from APAP-only treated mice showed marked multifocal centrilobular or massive necrosis, correlating with the ALT elevations observed in these mice (Figure 4). Administration of Z-GA alone by intraperitoneal or oral route did not exhibit any adverse effects in the liver at doses tested and appeared similar to the saline-treated controls. The combination of Z-GA with APAP mitigated the extent and severity of necrotic lesions in the liver tissue. Minimal to no significant centrilobular hepatocyte degeneration and necrosis were apparent in this combination treatment group. The oral Z-GA-APAP combination group also showed minimal findings with several foci of neutrophil infiltration and the absence of hepatocyte necrosis. Both, liver enzyme and histological findings suggest protective effects of Z-GA treatment on APAP-induced liver damage (Figures 3 and 4).
Figure 4.

Histopathological examination of the liver tissues from mice subjected to high-dose APAP toxicity. H&E staining of liver sections around the centrilobular area: (A) saline; (B) Z-GA (i.p.); (C) Z-GA (p.o.); (D) APAP; (E) APAP + Z-GA (i.p.); and (F) APAP + Z-GA (p.o.). Scale bar: 150 μm.
Z-GA Mitigated APAP-Induced Oxidative Stress in Liver Tissue
One of the consequences of APAP-induced acute liver injury is increased hepatic oxidative stress. Several indices of oxidative stress were assessed to evaluate the protective effect of Z-GA (Figure 5). The high dose of APAP caused a significant reduction in the hepatic content of reduced glutathione (GSH, 36.1 μmol/g protein) when compared to saline-treated controls (70.6 μmol/g protein, Figure 5A). This depletion in GSH stores was replenished by pretreatment with Z-GA when administered by i.p. or oral routes. Consequently, the ratio of reduced to oxidized glutathione (GSH/GSSG, redox ratio), which is adversely affected by APAP overdose (APAP vs saline; 8 vs 3, respectively), was restored to saline control levels by Z-GA treatment (6.5 and 6 for i.p and oral Z-GA, respectively, Figure 5B). Other markers of oxidative stress such as lipid peroxidation and protein carbonyl content were elevated in the APAP-only treated group by 183 and 72%, respectively (Figure 5C,D), over saline-treated animals. Intraperitoneal Z-GA treatment attenuated the levels of lipid peroxides and protein carbonyls to those comparable to levels in the saline (control)-treated group. The effect of Z-GA treatment on GSH levels and TBARS was irrespective of the route of administration (intraperitoneal or oral route), while i.p. Z-GA was more effective in attenuating protein carbonyl levels than oral Z-GA. Administration of Z-GA alone did not affect these oxidative stress markers.
Figure 5.

Effect of administration of the cis-isomer of guanabenz through intraperitoneal and oral routes in APAP-overdosed mice on the levels of liver oxidative stress markers. All liver tissues for the oxidative stress assay were collected at 24 h post overdose of APAP (370 mg/kg). Z-GA (10 mg/kg, i.p. or p.o.) was administered 30 min prior to the APAP injection. (A) GSH levels (μmol GSH per g of protein). (B) Liver GSH/GSSG ratio. (C) Liver TBARS levels (μmol/g of protein). (D) Liver protein carbonyl (nmol/mg of protein). (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; when compared to the APAP-only group).
The protective effect of Z-GA against oxidative stress was also observed when administered after APAP overdose (Supporting Information, Figure S4). Reduced cellular GSH levels and redox potential (GSH/GSSG) were restored when Z-GA was administered up to 4 h after APAP. Similarly, the liver lipid peroxidation level was significantly lower than the APAP-only treated group when Z-GA treatment was delayed for up to 4 h. At 8 h post APAP overdose, however, the restoration of redox status by Z-GA was ineffective, mimicking the ALT elevation observed in the APAP + Z-GA group at this time point (Figure 3B).
Z-GA Alleviates ER Stress Resulting from High-Dose APAP Challenge
Apart from elevated oxidative stress, induction of ER stress after APAP overdose is observed in the acute liver injury mouse model of APAP hepatotoxicity by others and us previously.31,45 At the tested dose, APAP strongly upregulated the protein expression of ER-chaperone Bip/GPR78 (13 fold), with the effect on the PERK pathway (Figure 6A,B). A marginal increase in the protein levels of eIF2α was detected in the liver tissue, however, Z-GA treatment did not influence the eIF2α protein expression (Figure 6A). Increased phosphorylation of eIF2α indicates the inhibition of protein translation, however, administration of APAP reduced eIF2α phosphorylation; potentially contributing toward ER stress propagation (Figure 6A,C). Guanabenz is a known inhibitor of dephosphorylase GADD34. Treatment with Z-GA resulted in the accumulation of the phosphorylated eIF2α protein, thus relieving the ER stress by attenuation of new protein synthesis. Increased expression of p-eIF2α has been noted in other studies investigating APAP-induced ER stress,46−48 which could stem from differences in exposure time and the animal model used. However, the inhibition of eIF2α dephosphorylation caused by Z-GA is hepatoprotective, suggesting an important role for the PERK pathway in APAP toxicity. The effect of Z-GA treatment was also reflected on the UPR target CHOP. Increased expression of CHOP (2.4 fold over saline controls) after APAP overdose was reversed completely in mice treated with Z-GA to levels comparable to saline-treated controls (Figure 7A,C).
Figure 6.

cis-Isomer of guanabenz treatment significantly inhibits APAP-induced ER stress markers. All liver tissues for western blot were collected at 24 h post overdose of APAP (370 mg/kg). Z-GA (10 mg/kg; i.p.) was administered 30 min prior to a 370 mg/kg APAP injection. (A) Representative immunoblots of ER stress-related proteins (BiP, p-eIF2α, and eIF2α) in liver homogenates from mice treated with saline, APAP (370 mg/kg), or the combination Z-GA (10 mg/kg; i.p.) was administered 30 min prior to APAP (370 mg/kg) overdose. (B, C) Quantification of protein expression is shown for each blot after normalization to the reference protein, α-tubulin (*p < 0.05; **p < 0.01; ****p < 0.0001, when compared to the APAP-only group). Adapted with permission from ref (31). Copyright 2019 American Chemical Society.
Figure 7.
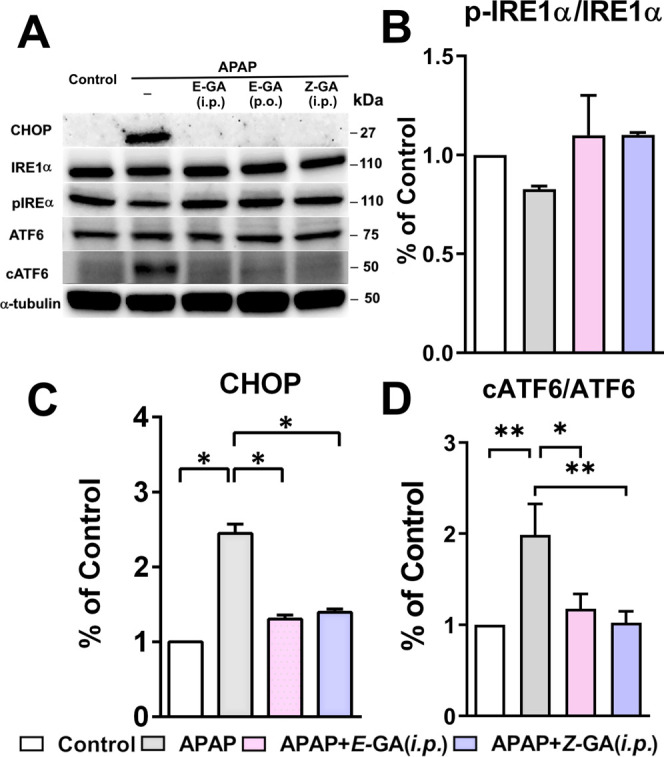
cis-Isomer of guanabenz treatment significantly inhibits the APAP-induced ER stress marker. All liver tissues for western blot were collected at 24 h post overdose of APAP (370 mg/kg). Z-GA (10 mg/kg; i.p.) was administered 30 min prior to a 370 mg/kg APAP injection. (A) Representative immunoblots of ER stress-related proteins (CHOP, IRE1α, pIRE1α, ATF6, and cleaved ATF6) in liver homogenates from mice treated with saline, APAP (370 mg/kg), or the combination Z-GA (10 mg/kg; i.p.) was administered 30 min prior to APAP (370 mg/kg) overdose. (B–D) The quantification of protein expression is shown for each blot after normalization of the reference protein, α-tubulin (*p < 0.05; **p < 0.01; when compared to APAP-only group). Adapted with permission from ref (31). Copyright 2019 American Chemical Society.
In addition to the effect on the PERK pathway, high-dose APAP activated other two ER transmemebrane sensors, IRE1α and ATF6. Elevation in the levels of both of these proteins caused by APAP was prevented in the Z-GA treated group. Activation of ATF6 by its cleavage was also observed in the APAP-overdose group while the combination of Z-GA and APAP did not induce ATF6 cleavage in the liver tissue (Figure 7A,D). Additionally, the phosphorylation of IRE1α, which leads to the regulation of genes involved in restoring protein folding or degradation of misfolded proteins, was marginally suppressed by high-dose APAP. Relative changes in the levels of phospho-IRE1α between APAP-only group and mice treated with a combination of APAP and Z-GA did not reach statistical significance (Figure 7A,B).
Discussion
Clinical management of acetaminophen hepatotoxicity involves intravenous or oral administration of the antidote, N-acetylcysteine, depending on the severity of the symptoms and time-lapse from the administration of APAP. Anaphylactic reactions and unpleasantness of NAC formulations have demanded a search for safer, molecularly targeted antidotes for the prevention of acute liver failure due to APAP overdose.20,49 Contribution of ER stress toward hepatic injury caused by high-dose APAP has been reported by us and others.31,45 We were successful in preventing hepatic necrosis in a mouse model of APAP hepatotoxicity by interfering with ER stress using an FDA-approved α2-adrenergic agonist, guanabenz (GA).31 Approved clinically as an antihypertensive, GA displays a good safety profile, however, it also causes sedative side effects that are characteristic of this therapeutic class. Thus, further exploitation of guanabenz as a potential APAP antidote needs abrogation of the cardiovascular effects of the compound, while conserving its beneficial ERS modulatory effects.
The geometric isomer of the clinically used trans-guanabenz, viz. E-GA, was previously isolated and characterized as a photolytic degradation product during stability experiments.35Z-GA was also detected as a minor metabolite of E-GA in humans.50 The cis-isomer was found to be practically devoid of antihypertensive activity of the parent trans-isomer, however, further systematic pharmacological investigations of this compound were not conducted. We recently demonstrated the ERS-reducing ability of Z-GA in a cellular model of tunicamycin-induced ERS.37 Interconversion of E- and Z-isomers in the solution occurs upon exposure to UV and intense visible light, however, we observed minimal interconversion of these isomers in mouse plasma. The extent of conversion of E-GA → Z-GA was found to be 0.8%, while it was 2% for Z-GA → E-GA based on the area under the curve calculations of the converted isomer to the corresponding parent isomer. In vitro stability analysis in liver microsomes showed higher stability and low protein binding of Z-GA compared to E-GA, translating into a greater volume of distribution of Z-GA in the in vivo pharmacokinetic study.37 Additionally, both isomers displayed similar tissue distribution patterns (Supporting Information, Figure S3). Higher accumulation of Z-GA was found in the liver and kidneys compared to that of E-GA. Furthermore, interconversion of the two isomers in the analyzed tissues was less than 10% of the injected isomer. This prompted us to further explore the utility of Z-GA in pharmacological applications wherein α2-adrenergic receptor modulation is not needed.
We first confirmed the lack of analgesic activity of Z-GA in a tail flick nociceptive assay in contrast with E-GA (Figure 2). The activity corresponded with the binding affinity of these isomers for the α2A-adrenergic receptor. The cis-isomer was unable to displace the radiolabeled form of the α2-adrenorecptor antagonist, yohimbine, while a significant displacement of yohimbine was observed by E-GA. The lack of interaction with the adrenoreceptor was also apparent in the cardiovascular effects of Z-GA when compared to E-GA. The latter caused a significant heart rate depression even at a 2 mg/kg dose, which was not the case for Z-GA at 10 mg/kg. To be useful as a combination therapeutic with APAP, we also examined the effect of Z-GA on antipyretic activity of APAP. Similar to the parent trans-isomer, there was no effect of co-administration of Z-GA and APAP on the body temperature.
Our next experiments involved efficacy evaluation of Z-GA in an acute hepatotoxicity mouse model of APAP overdose (Figure 3). Elevation of the liver enzyme ALT was measured as a surrogate of liver injury caused by APAP. Intraperitoneal administration of Z-GA prevented the increase in APAP-induced ALT levels in these mice, even when the compound was administered up to 6 h post APAP overdose. Oral Z-GA was equally effective under similar experimental conditions. Histopathological examination of the liver tissues displayed a lack of centrilobular necrosis in the Z-GA + APAP group compared to the APAP-only group, confirming the hepatoprotective effect of Z-GA (Figure 4). A higher dose of Z-GA (20 mg/kg, i.p.,), however, was not effective at preventing APAP-induced ALT rise. This could be due to a biphasic dose–response curve of Z-GA, similar to that reported for E-GA against ER and oxidative stress,51 leading to partial protection in combination with APAP at a higher dose.
Mechanistic evaluation of Z-GA in comparison with E-GA offered no distinction between the activities of these two isomers, indicative of the absence of α2-adrenoreceptor involvement in hepatoprotection offered by these isomers. Both isomers mitigated the increased oxidative stress in liver tissue caused by high-dose APAP (Figure 5). Reduced intrahepatic GSH content was rectified by both isomers of GA, while the levels of protein and lipid oxidation products were significantly reduced, thus reducing damage caused by high-dose APAP. The contribution of ERS modulatory potential of Z-GA toward APAP hepatoprotection was investigated next (Figures 6 and 7). Activation of multipronged UPR was confirmed by the release of binding immunoglobulin protein/78 kDa glucose-regulated protein (BiP/GRP78) from the three transmembrane sensors of UPR in the APAP-only group. The levels of BiP were drastically reduced by the treatment of both isomers of GA. Though the goal of UPR is to restore protein homeostasis for cell survival, chronic unmitigated ERS after APAP overdose leads to the activation of the apoptotic cascade. Thus, APAP-mediated activation of PERK upregulated an apoptotic transcription factor, CHOP, which reversed phosphorylation of eIF2α by activating phosphatase GADD34. As an inhibitor of GADD34, treatment with GA raised the levels of p-eIF2α, sustaining translational inhibition and relieving ER stress. It is interesting to note that the GADD34 inhibitory activity was retained by both the isomers of GA. The effect of APAP alone or/and APAP + GA treatment were modest on the IRE1 arm of the UPR. However, a significant effect of the antidote was noted on the ATF6 arm of the UPR, which promotes protein folding and activates ER-associated degradation pathways. ER stress resulting from APAP overdose resulted in the cleavage of ATF6, thereby increasing the levels of cATF6. Both GA isomers prevented such proteolysis of ATF6, potentially contributing toward the mitigation of ER stress consequences.
Collectively, we have shown that antidote activity of GA is independent of its α2-adrenoreceptor modulation and stems from the mitigation of APAP overdose-induced oxidative and ER stress. Geometric isomerization of guanabenz to its cis-isomer abolished the interaction with the α2-adrenergic receptor but retained the ER stress and oxidative stress-modulatory activities. Mechanistic elucidation of the interactions of individual GA isomers with phosphatase GADD34 and ATF4, the primary transcriptional activator of GADD34, are currently underway in our laboratory. The results of this study pave the way for further exploitation of Z-GA in diseases wherein such pathologies play an important role. The study also provides a rational basis for utilizing geometric isomerization as a strategy to dissect the analgesic and ER stress-modulatory effects of guanabenz. In conclusion, Z-GA appears to be a promising antidote for the treatment of APAP poisoning in a clinical setting. A combination of APAP with Z-GA in the formulation itself could potentially reduce the occurrence of APAP overdose.
Acknowledgments
The authors would like to thank Dr. Gerry O’Sullivan, Director of Comparative Pathology Shared Resource, Masonic Cancer Center, the University of Minnesota, for histopathological analysis of the liver samples.
Glossary
Abbreviations
- APAP
acetaminophen
- GA
guanabenz
- ER
endoplasmic reticulum
- NAPQI
N-acetyl-p-benzoquinone imine
- ALT
alanine aminotransferase
- UGT
UDP-glucuronosyl transferase
- SULT
sulfotransferase
- GSH
glutathione
- ERS
endoplasmic reticulum stress
- UPR
unfolded protein response
- α2-AR
α2-adrenergic receptor
- PERK
protein kinase RNA-like ER kinase
- ATF6
activating transcription factor 6
- BiP/GRP78
binding immunoglobulin protein/78 kDa glucose-regulated protein
- eIF2α
eukaryotic initiation factor 2α
- XPB1
X-box binding protein 1
- IRE1α
inositol requiring enzyme 1α
- CYP
cytochrome P450
- H&E
hematoxylin & eosin
- GSSG
glutathione disulfide
- NAC
N-acetylcysteine
- CHOP
C/EBP homologous protein
- GADD34
growth arrest and DNA damage inducible 34
- BPM
beat per minutes
- MDA
malondialdehyde
- PK
pharmacokinetics
- UV
ultraviolet
- CNS
central nervous system
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.chemrestox.3c00047.
Tail flick analgesia of APAP and Z-GA combination; effect of APAP + Z-GA on the heart rate; tissue distribution analysis of Z-GA; effect of delayed Z-GA administration after APAP overdose on liver oxidative stress (PDF)
Author Present Address
† Drug Metabolism and Pharmacokinetics, Eisai Inc., Cambridge, Massachusetts 02140, United States
Author Contributions
The first draft of the manuscript was written by W.X. and S.S.M. The final draft was edited through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the research endowment funds from the Center for Drug Design (CDD) and the NIH Grant R01AG062469.
The authors declare no competing financial interest.
Supplementary Material
References
- Hoofnagle J. H.; Björnsson E. S. Drug-Induced Liver Injury — Types and Phenotypes. N. Engl. J. Med. 2019, 381, 264–273. 10.1056/NEJMra1816149. [DOI] [PubMed] [Google Scholar]
- Lee W. M. Acetaminophen and the U.S. Acute Liver Failure Study Group: lowering the risks of hepatic failure. Hepatology 2004, 40, 6–9. 10.1002/hep.20293. [DOI] [PubMed] [Google Scholar]
- Lee W. M. Acetaminophen toxicity: changing perceptions on a social/medical issue. Hepatology 2007, 46, 966–970. 10.1002/hep.21926. [DOI] [PubMed] [Google Scholar]
- US FDA . FDA Drug Safety Communication: Prescription Acetaminophen Products to be Limited to 325 mg Per Dosage Unit; Boxed Warning Will Highlight Potential for Severe Liver Failure, 2011. https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-prescription-acetaminophen-products-be-limited-325-mg-dosage-unit.
- Ogilvie J. D.; Rieder M. J.; Lim R. Acetaminophen overdose in children. CMAJ 2012, 184, 1492–1496. 10.1503/cmaj.111338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson S. D. Molecular mechanisms of the hepatotoxicity caused by acetaminophen. Semin. Liver Dis. 1990, 10, 267–278. 10.1055/s-2008-1040482. [DOI] [PubMed] [Google Scholar]
- Mazaleuskaya L. L.; Sangkuhl K.; Thorn C. F.; FitzGerald G. A.; Altman R. B.; Klein T. E. PharmGKB summary: pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenet. Genomics 2015, 25, 416–426. 10.1097/fpc.0000000000000150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson A. M. Acetaminophen hepatotoxicity. Clin. Liver Dis. 2007, 11, 525–548. 10.1016/j.cld.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Raucy J. L.; Lasker J. M.; Lieber C. S.; Black M. Acetaminophen activation by human liver cytochromes P450IIE1 and P450IA2. Arch. Biochem. Biophys. 1989, 271, 270–283. 10.1016/0003-9861(89)90278-6. [DOI] [PubMed] [Google Scholar]
- Mitchell J. R.; Jollow D. J.; Potter W. Z.; Davis D. C.; Gillette J. R.; Brodie B. B. Acetaminophen-induced hepatic necrosis. I. Role of drug metabolism. J. Pharmacol. Exp. Ther. 1973, 187, 185–194. [PubMed] [Google Scholar]
- Bunchorntavakul C.; Reddy K. R. Acetaminophen-related hepatotoxicity. Clin. Liver Dis. 2013, 17, 587–607. 10.1016/j.cld.2013.07.005. [DOI] [PubMed] [Google Scholar]
- Zaher H.; Buters J. T.; Ward J. M.; Bruno M. K.; Lucas A. M.; Stern S. T.; Cohen S. D.; Gonzalez F. J. Protection against acetaminophen toxicity in CYP1A2 and CYP2E1 double-null mice. Toxicol. Appl. Pharmacol. 1998, 152, 193–199. 10.1006/taap.1998.8501. [DOI] [PubMed] [Google Scholar]
- Patten C. J.; Thomas P. E.; Guy R. L.; Lee M.; Gonzalez F. J.; Guengerich F. P.; Yang C. S. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem. Res. Toxicol. 1993, 6, 511–518. 10.1021/tx00034a019. [DOI] [PubMed] [Google Scholar]
- Jollow D. J.; Mitchell J. R.; Potter W. Z.; Davis D. C.; Gillette J. R.; Brodie B. B. Acetaminophen-induced hepatic necrosis. II. Role of covalent binding in vivo. J. Pharmacol. Exp. Ther. 1973, 187, 195–202. [PubMed] [Google Scholar]
- Hinson J. A.; Roberts D. W.; James L. P. Mechanisms of acetaminophen-induced liver necrosis. Handb. Exp. Pharmacol. 2010, 196, 369–405. 10.1007/978-3-642-00663-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kon K.; Kim J. S.; Jaeschke H.; Lemasters J. J. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology 2004, 40, 1170–1179. 10.1002/hep.20437. [DOI] [PubMed] [Google Scholar]
- Kon K.; Ikejima K.; Okumura K.; Aoyama T.; Arai K.; Takei Y.; Lemasters J. J.; Sato N. Role of apoptosis in acetaminophen hepatotoxicity. J. Gastroenterol. Hepatol. 2007, 22, S49–S52. 10.1111/j.1440-1746.2007.04962.x. [DOI] [PubMed] [Google Scholar]
- Jaeschke H.; McGill M. R.; Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab. Rev. 2012, 44, 88–106. 10.3109/03602532.2011.602688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman D. N.; Woodhouse K. W.; Rawlins M. D. Adverse reactions to N-acetylcysteine. Lancet 1984, 3, 393–398. 10.1016/s0140-6736(84)90516-6. [DOI] [PubMed] [Google Scholar]
- Park B. K.; Dear J. W.; Antoine D. J. Paracetamol (acetaminophen) poisoning. BMJ Clin. Evidence 2015, 2015, 2101. [PMC free article] [PubMed] [Google Scholar]
- Du K.; Ramachandran A.; Jaeschke H. Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox Biol. 2016, 10, 148–156. 10.1016/j.redox.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M.; Huo Y.; Yin S.; Hu H. Mechanisms of acetaminophen-induced liver injury and its implications for therapeutic interventions. Redox Biol. 2018, 17, 274–283. 10.1016/j.redox.2018.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P.; Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Ron D.; Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Pakos-Zebrucka K.; Koryga I.; Mnich K.; Ljujic M.; Samali A.; Gorman A. M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wek R. C. Role of eIF2α Kinases in Translational Control and Adaptation to Cellular Stress. Cold Spring Harbor Perspect. Biol. 2018, 10, a032870 10.1101/cshperspect.a032870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzi D.; Barda L.; Scaiewicz V.; Mills M.; Mueller T.; Gonzalez-Rodriguez A.; Valverde A. M.; Iwawaki T.; Nahmias Y.; Xavier R.; et al. CHOP is a critical regulator of acetaminophen-induced hepatotoxicity. J. Hepatol. 2013, 59, 495–503. 10.1016/j.jhep.2013.04.024. [DOI] [PubMed] [Google Scholar]
- Ye H.; Chen C.; Wu H.; Zheng K.; Martín-Adrados B.; Caparros E.; Francés R.; Nelson L. J.; Gómez del Moral M.; Asensio I.; et al. Genetic and pharmacological inhibition of XBP1 protects against APAP hepatotoxicity through the activation of autophagy. Cell Death Dis. 2022, 13, 143 10.1038/s41419-022-04580-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur K. Y.; So J. S.; Ruda V.; Frank-Kamenetsky M.; Fitzgerald K.; Koteliansky V.; Iwawaki T.; Glimcher L. H.; Lee A. H. IRE1alpha activation protects mice against acetaminophen-induced hepatotoxicity. J. Exp. Med. 2012, 209, 307–318. 10.1084/jem.20111298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K.; Sato T.; Matsui T.; Sato M.; Okada T.; Yoshida H.; Harada A.; Mori K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376. 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Xie W.; Xie J.; Vince R.; More S. S. Guanabenz Attenuates Acetaminophen-Induced Liver Toxicity and Synergizes Analgesia in Mice. Chem. Res. Toxicol. 2020, 33, 162–171. 10.1021/acs.chemrestox.9b00162. [DOI] [PubMed] [Google Scholar]
- Lasseter K. C.; Shapse D.; Pascucci V. L.; Chiang S. T. Pharmacokinetics of guanabenz in patients with impaired liver function. J. Cardiovasc. Pharmacol. 1984, 6, S766–770. 10.1097/00005344-198400065-00008. [DOI] [PubMed] [Google Scholar]
- Vieira F. G.; Ping Q.; Moreno A. J.; Kidd J. D.; Thompson K.; Jiang B.; Lincecum J. M.; Wang M. Z.; De Zutter G. S.; Tassinari V. R.; et al. Guanabenz Treatment Accelerates Disease in a Mutant SOD1 Mouse Model of ALS. PLoS One 2015, 10, e0135570 10.1371/journal.pone.0135570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino S.; Iwasaki Y.; Matsumoto S.; Satoh T.; Ozawa A.; Yamada E.; Kakizaki S.; Trejo J. A. O.; Uchiyama Y.; Yamada M.; Mori M. Administration of small-molecule guanabenz acetate attenuates fatty liver and hyperglycemia associated with obesity. Sci. Rep. 2020, 10, 13671 10.1038/s41598-020-70689-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer C. M.; Deangelis N. J. Guanabenz Degradation Products and Stability Assay. J. Pharm. Sci. 1979, 68, 1010–1012. 10.1002/jps.2600680824. [DOI] [PubMed] [Google Scholar]
- National Center for Biotechnology Information. PubChem Annotation Record for GUANABENZ ACETATE Source: Hazardous Substances Data Bank (HSDB), 2023. https://pubchem.ncbi.nlm.nih.gov.
- Xie J.; Jiang R.; Xie W.; Cao B.; More S. S. LC-MS/MS determination of guanabenz E/Z isomers and its application to in vitro and in vivo DMPK profiling studies. J. Pharm. Biomed. Anal. 2021, 205, 114331 10.1016/j.jpba.2021.114331. [DOI] [PubMed] [Google Scholar]
- Wu G.; Zhao G.; He Y. Distinct pathways for the trafficking of angiotensin II and adrenergic receptors from the endoplasmic reticulum to the cell surface: Rab1-independent transport of a G protein-coupled receptor. J. Biol. Chem. 2003, 278, 47062–47069. 10.1074/jbc.M305707200. [DOI] [PubMed] [Google Scholar]
- Duvernay M. T.; Zhou F.; Wu G. A conserved motif for the transport of G protein-coupled receptors from the endoplasmic reticulum to the cell surface. J. Biol. Chem. 2004, 279, 30741–30750. 10.1074/jbc.M313881200. [DOI] [PubMed] [Google Scholar]
- Patil V.; Noronha V.; Joshi A.; Chougule A.; Kannan S.; Bhattacharjee A.; Goud S.; More S.; Chandrasekharan A.; Menon N.; et al. Phase III Non-inferiority Study Evaluating Efficacy and Safety of Low Dose Gemcitabine Compared to Standard Dose Gemcitabine With Platinum in Advanced Squamous Lung Cancer. EClinicalMedicine 2019, 9, 19–25. 10.1016/j.eclinm.2019.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannon A. W.; Malmberg A. B. Models of nociception: hot-plate, tail-flick, and formalin tests in rodents. Curr. Protoc. Neurosci. 2007, 41, 8–9. 10.1002/0471142301.ns0809s41. [DOI] [PubMed] [Google Scholar]
- More S. S.; Nugent J.; Vartak A. P.; Nye S. M.; Vince R. Hepatoprotective Effect of psi-Glutathione in a Murine Model of Acetaminophen-Induced Liver Toxicity. Chem. Res. Toxicol. 2017, 30, 777–784. 10.1021/acs.chemrestox.6b00291. [DOI] [PubMed] [Google Scholar]
- Du K.; Williams C. D.; McGill M. R.; Jaeschke H. Lower susceptibility of female mice to acetaminophen hepatotoxicity: Role of mitochondrial glutathione, oxidant stress and c-jun N-terminal kinase. Toxicol. Appl. Pharmacol. 2014, 281, 58–66. 10.1016/j.taap.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai G.; He L.; Chou N.; Wan Y. J. Acetaminophen metabolism does not contribute to gender difference in its hepatotoxicity in mouse. Toxicol. Sci. 2006, 92, 33–41. 10.1093/toxsci/kfj192. [DOI] [PubMed] [Google Scholar]
- Uzi D.; Barda L.; Mills M.; Merquiol E.; Scaiewicz V.; Tirosh B.; Shibolet O. Endoplasmic reticulum (ER) stress and the unfolded protein response (UPR) are major regulators of acetaminophen (APAP)-induced hepatotoxicity: 45. Hepatology 2011, 54, 382A. [Google Scholar]
- Carraro V.; Combaret L.; Coudy-Gandilhon C.; Parry L.; Averous J.; Maurin A. C.; Jousse C.; Voyard G.; Fafournoux P.; Papet I.; Bruhat A. Activation of the eIF2α-ATF4 Pathway by Chronic Paracetamol Treatment Is Prevented by Dietary Supplementation with Cysteine. Int. J. Mol. Sci. 2022, 23, 7196 10.3390/ijms23137196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinec G. M.; Thein P.; Parsa A.; Yorgason J.; Luxford W.; Urrutia R.; Kalinec F. Acetaminophen and NAPQI are toxic to auditory cells via oxidative and endoplasmic reticulum stress-dependent pathways. Hear. Res. 2014, 313, 26–37. 10.1016/j.heares.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y.; He W.; Li X.; Huang J.; Wang J. Rosiglitazone Protects against Acetaminophen-Induced Acute Liver Injury by Inhibiting Multiple Endoplasmic Reticulum Stress Pathways. BioMed Res. Int. 2022, 2022, 6098592 10.1155/2022/6098592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr F.; Dawson A.; Whyte I. M.; Buckley N.; Murray L.; Graudins A.; Chan B.; Trudinger B. The Australasian Clinical Toxicology Investigators Collaboration randomized trial of different loading infusion rates of N-acetylcysteine. Ann. Emerg. Med. 2005, 45, 402–408. 10.1016/j.annemergmed.2004.08.040. [DOI] [PubMed] [Google Scholar]
- McEvoy G. K.American Society of Health-System, P. In AHFS Drug information 2002; American Society of Health-System Pharmacists, 2002. [Google Scholar]
- Neuber C.; Uebeler J.; Schulze T.; Sotoud H.; El-Armouche A.; Eschenhagen T. Guanabenz interferes with ER stress and exerts protective effects in cardiac myocytes. PLoS One 2014, 9, e98893 10.1371/journal.pone.0098893. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.