

SUMMARY
Antigen-specific T-cell receptor (TCR) sequences can have prognostic, predictive, and therapeutic value, but decoding the specificity of TCR recognition remains challenging. Unlike DNA strands that base pair, TCRs bind to their targets with different orientations and different lengths, which complicates comparisons. We present Scanning PArametrized by Normalized TCR Length (SPAN-TCR) to analyze antigen-specific TCR CDR3 sequences and identify patterns driving TCR-pMHC specificity. Using entropic analysis, SPAN-TCR identifies 2-mer motifs that decrease the diversity (entropy) of CDR3s. These motifs are the most common patterns that can predict CDR3 composition, and we identify ‘essential’ motifs that decrease entropy in the same CDR3 or chain containing the 2-mer, and ‘super-essential’ motifs that decrease entropy in both chains. Molecular dynamics analysis further suggests that these motifs may play important roles in binding. We then employ SPAN-TCR to resolve similarities in TCR repertoires against different antigens using public databases of TCR sequences. A record of this paper’s Transparent Peer Review process is included in the Supplementary Information.
eTOC Blurb
The specificity of T cell immunotherapy is encoded by T-cell Receptor (TCR) sequences, so decoding and ultimately engineering TCR specificity is a priority. Here we present SPAN-TCR to compare TCRs and perform entropic analysis to extract essential subsections of TCRs for binding.
INTRODUCTION
T cell receptors (TCRs), comprised of paired and chains, recognize peptide antigen targets presented on Major Histocompatibility Complexes (pMHCs)1,2. TCR-pMHC binding is dominated by the highly variable TCR domains called Complementarity Determining Region 3 (CDR3), which generate sequence diversity through the V-D-J recombination mechanism3–5. In addition to considerable V- and J-gene diversity, the insertion and deletion of nucleotides results in the remarkable heterogeneity of CDR3 length and peptide composition. Structural analyses6–8 have shown that binding orientations of different TCRs and pMHCs can be conserved (Fig. 1A)9,10,11,12. However, relationships between the TCR sequence and antigen binding have been much more difficult to establish.
Figure 1. A length-agnostic framework to characterize TCRs.

A. Crystal structures of different CMV pp65-specific TCRs show that TCR (blue) and (red) chains bind in similar conformations. B. A wide range of CDR3 lengths are reported for sets of TCRs that bind to a specific target (red, CMV pp65 epitope), TCRs reported in the VDJdb database (blue), and sequenced TCRs from a human donor (green). The relative frequency of amino acids across the entire CDR3 is shown on the Logo plot (right). C. CDR3 sequences that bind the same target share common amino acid features (highlighted in red). The chemical contexts of these features such as CDR3 length or amino acid hydrophobiciity are associated with specificity to antigen. D. TCRs of different lengths are difficult to compare. SPAN-TCR normalizes the length of TCR CDR3s, then searches for amino acids or k-mer subsequences at similar positions, which may be likely to interact with the same section of the p-MHC (highlighted in red). For each k-mer, we determine first if the k-mer appears frequently at a position. Next, we determine if sequences that have the k-mer at the position are diverse or repetitive using entropic analysis.
When next generation sequencing is coupled to strategies for isolating antigen-specific T cells, databases13–16 of antigen-specific TCR sequences can be built17–20 and mined for patterns in amino acid usage and CDR3 structure21–24. Such analyses reveal a high CDR3 diversity25 even for TCRs binding the same pMHC target (Fig. 1B, detailed metrics at https://vdjdb.cdr3.net/overview). While some TCRs are ‘public’ (shared between individuals25,26), most reported TCRs are unique, and CDR3 domains can vary in size. Tools that can capture common features shared between otherwise diverse TCRs are needed to identify how specificity is achieved (Fig. 1C)27. These tools often utilize protein sequence alignment (TCRdist),28,29 or incorporate features such as amino-acid chemical similarity,30–33 shared-motif identification (GLIPH),34,35 and machine learning techniques36. The common thread between these methods is a sequence-based framework where the single residue is the basic unit. However, structural analysis and molecular simulation demonstrates variability and “jitter” between amino acid residue positions in TCR/pMHC binding. Many sequence alignment tools employ a rigid representation of peptides in exact sequence, somewhat akin to queries for similar gene sequences. However, in protein-protein interactions, secondary structure can play important roles in bringing non-adjacent residues into proximity, and strategies that can computationally account for such possibilities may yield new insights when comparing and analyzing TCR amino acid sequences. Further, identifying, through computational analysis of large data sets, which regions of a CDR3 domain most strongly influence TCR:pMHC binding remains a challenge that is not addressed by current TCR analysis tools.
Here we present Scanning PArametrized by Normalized TCR Length (SPAN-TCR) as a tool for extracting structural and chemical insights from groups of antigen-specific TCR sequences in a length-agnostic fashion. SPAN-TCR is based upon two hypotheses. First, when multiple TCRs bind a single pMHC target, the structure of the protein complex will likely be similar across the diverse CDR3 lengths and sequences, especially as the CDR3s become more similar16,37. Second, a tool that successfully extracts such similarities can be used to describe entire sets of CDR3s by the frequency and location of these structural similarities. Thus, by analyzing an entire set of antigen-specific TCRs, we may find patterns of specific amino acids at specific positions along the CDR3 that serve an essential role in forming the TCR-pMHC complex. The two practical challenges of SPAN-TCR are: 1) to identify amino acid patterns of interest by their location and not their ordinal sequence (1st, 2nd, nth); and 2) to generate metrics to assess the importance of such amino acid patterns to antigen-specificity.
We use SPAN-TCR to first describe the relative positions of amino acids and amino acid k-mers in CDR3 chains (Fig. 1D, Sup. Fig. 1A). Although k-mers can be of variable length, we focus on 2-mers, since longer motifs are much less common. For example, unique 3-mers are found at ~1:20 the frequency of 2-mers. We postulate that if an amino acid 2-mer (YZ) is important for binding to a specific pMHC, then YZ in XYZX is likely performing a similar function to YZ in XXYZXX. We also compare k-mers identified by SPAN-TCR to similar motifs identified by GLIPH234,35. We then use SPAN-TCR to calculate informational entropy to identify high frequency 2-mers that we label as ‘essential’ or ‘super-essential’. An essential 2-mer is one that lowers the informational entropy (sequence diversity) within its own ( or ) CDR3 chain, while a super-essential 2-mer lowers the informational entropy within both CDR3 chains. We hypothesize that such 2-mers are important for TCR-pMHC binding, and we test this hypothesis by probing for 2-mer interfacial chemical interactions using molecular dynamics simulations. Finally, we extend these SPAN-TCR algorithms to yield comparisons between sets of TCRs known to bind to different antigens. SPAN-TCR is first validated through the analysis of public data bases of TCRs specific to viral antigens, followed by explorations of newly sequenced putative antigen-specific CDR3s against COVID-19.
RESULTS
Antigen-Specific TCR Structural Landscapes Using SPAN-TCR
For CDR3 domains that bind the same pMHC target, SPAN-TCR assumes that binding-essential amino acids are at similar relative positions. However, for CDR3s of different length, their counting positions from the C-terminus may differ. Thus, we first normalize CDR3s of TCR and chains (as defined by MiXCR38) by length. Positions 0 and 1 refer to the N- and C-termini of the CDR3, 0.5 refers to the center, etc. A logo plot can show amino acid usage across a set of CDR3s to illustrate this binning approach.39,40 In Fig 2A, 10-mer CDR3s align with a 10-bin logo plot. However, sequences of different length are not simply placed in such bins21,31. Thus, a 1% increment bin resolution better resolves how amino acid composition diverges between TCRs (Fig. 2A, Sup Fig. 1A).
Figure 2.

The landscape of amino acid usage within CMV pp65 antigen-specific TCRs. A. SPAN-TCR analyzes the amino acid composition of TCR sets using bins. By increasing bin number from 10 to 100, differences are finely resolved. SPAN-TCR further targets k consecutive amino acids (k-mers). The weighted influence of a k-mer is strongest at its center and decays as the next consecutive k-mer increases its influence. B. The SPAN-TCR formula used to generate a weighted display of k-mers for k=2. Low frequency (<3%) 2-mers are shown in gray and otherwise colored by hydrophobicity (linear scale, hydrophobicity index relative to glycine). Common 2-mers near the center of the chain, and the CDR3s containing those 2-mers are shown. C. Comparison of SPAN-TCR and GLIPH. GLIPH identifies enriched subsequences, which can be projected onto SPAN-TCR representations of TCR sets. SPAN-TCR k-mers for k=4 found in GLIPH groups, and number of GLIPH groups each 4-mer belongs to, are plotted.
A second refinement is to consider motifs comprised of consecutive amino acids. At each position (besides the beginning and end), two 2-mer CDR3s are found. To represent the change from one 2-mer to the next, a linear growth/decay weighting function is added to smooth the transition. Physically, this represents the flexibility of TCR chains used to achieve optimal binding conformations. We also sought to highlight stretches of chemical relevance by ordering the amino acids according to physico-chemical metrics31 such as hydrophobicity and molecular weight (Sup. Fig. 1B, C), or relative differences such as those calculated from PAM41 or BLOSUM42 algorithms. Finally, we choose a sequence-specific weight for each 2-mer, which represents the quality of the CDR3 contributing each 2-mer, either by a read percentage or confidence score.
The public data bases VDJdb13,16 (our primary reference), McPAS-TCR15 and TBAdb14 were used. We used SPAN-TCR to identify all 2-mers in CMV pp65-specific CDR3s in VDJdb (Fig. 2B for 2-mers with >3% frequency, linear decay weighting. Sup. Fig. 1D for k=3). VDJdb provides a confidence score from 0–3, with approximately 100x as many sequences reported with score=0 as score=3. Thus, we chose a sequence-specific weight of 5score such that the net contribution of score=0 and score=3 sequences was similar. This score can be adapted for different scenarios. The blue highlighted region shows the more conserved sequences at the CDR3 N-terminus, consisting primarily of hydrophobic 2-mers (CA, CI). The second region shows heavy use of hydrophilic chains such as TG/GN/NN at the center of the chain. The six most frequently observed 2-mers at the center of the CDR3 chain are listed to the right of Fig. 2B, along with representative CDR3s that reflect the different CDR3 lengths that contribute these 2-mers.
The positional flexibility of SPAN-TCR most resembles analysis by the GLIPH family of methods34,35, which also focuses on local sub-regions of CDR3 sequences. GLIPH2 is a method to extract previously unknown antigen-specific TCRs by identifying long, variable-length strings of residues shared by many TCRs. SPAN-TCR utilizes a complementary approach, whereby putative or known antigen-specific TCRs are analyzed to identify smaller, fixed strings of residues important for TCR binding. We compared the two methods by first using GLIPH2 to identify amino acid subsequences of note. We then applied SPAN-TCR using longer 4-mers to determine the locations and compositions of these subsequences (Fig. 2C, Sup. Table 1). GLIPH2 identified that motifs varying from 3–10 amino acids are usually found between the 0.25 and 0.75 relative positions. After the most frequent 4-mers (>0.5% abundance) were identified using SPAN-TCR between relative positions 0.25 and 0.75, the majority of these were a member of at least one and up to 10 GLIPH2 groups (Sup. Table 2). Given these similarities between SPAN-TCR and GLIPH2, the differences are that SPAN-TCR is applied to putative antigen-specific sequences, retains the spatial position of the motifs, and illustrates the TCR composition of a set using the Logo-esque plot (Fig. 2 A, B).
Quantifying Similarity Between TCR CDR3s Using SPAN-TCR
SPAN-TCR was used within an experimental workflow to compare CDR3s in a length-agnostic fashion by step-wise 1% bin increments (Sup. Fig. 2A). At each bin, 2-mers between 2 TCRs are compared, with the cumulative differences providing the distance metric between TCRs (Fig. 3A). The distance metric was computed using binary replacement and BLOSUM-derived chemical metrics42, and compared to Levenshtein distance (Fig. 3B, Sup. Table 3). A similar algorithm (TCRMatch) has been recently reported33, and so we carried out this analysis only to validate our core algorithm by analyzing VDJdb-reported CMV pp65 CDR3s specific to the epitope NLVPMVATV presented on HLA-A02, showing V/J gene usage of similarly clustered TCRs (Sup. Fig. 2 C–E).
Figure 3.
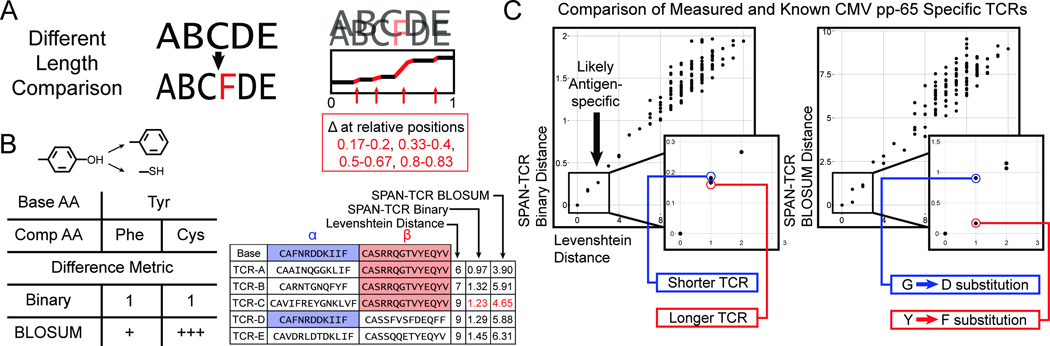
SPAN-TCR Distance Measurements. A. Using fine bins, SPAN-TCR compares CDR3 chains of variable lengths. B. Comparison of distance metrics. While a binary metric does not discriminate between a Tyrosine substituted with either Phenylalanine or Cysteine, the BLOSUM metric quantifies the chemical difference (ΔTyr-Phe < ΔTyr-Cys). C. The distance between a putative antigen-specific repertoire and the existing set of reported antigen-specific CDR3s is represented by Levenshtein distance (x-axis) and Binary and BLOSUM SPAN-TCR metrics (y-axis of separate plots) to resolve finer differences. CDR3s found in the lower left quadrant are most similar to the known CDR3s and thus more likely to share antigen specificity. SPAN-TCR reveals that among the nearest Levenshtein neighbors, the Y->F replacement produces the nearest neighbor.
To perform an in vitro validation, we isolated CMV pp65-specific (NLVPMVATV) T cells using MHC-peptide dextramer constructs, and sequenced the CDR3s. After calculating the difference between these CDR3s and CMV-specific, HLA-A02-specific VDJdb TCRs, we found an enrichment of CMV-specific TCRs (Fig. 3C, inset), which was not seen when comparing the experimental results to known TCRs specific to a different antigen or a negative control set of TCRs (Sup. Fig. 2B). Of the entire TCR repertoire, most sequences were not similar to any sequences found in VDJdb, which indicates that they were either nonspecifically captured, or they are patient-specific TCRs unreported in VDJdb. The SPAN-TCR analyses resolved certain fine features to reflect differences in amino acid biochemistry (Fig. 3C). As an example, for two highlighted putative pp65 TCRs, a VDJdb match was found at Levenshtein distance of 1. With the binary replacement metric, the longer TCR (red) was judged closer to its VDJdb match than the short TCR (blue), as a single substitution is less disruptive to a longer sequence. Using the BLOSUM metric, a CDR3 Y-F substitution (red) was distinguished from a G-D substitution (blue). SPAN-TCR determined that the Y-F substitution resulted in a more similar TCR.
Sequence Entropy and Essential Motifs
We coupled SPAN-TCR with information theory to identify 2-mers likely essential to antigen-specific binding. We hypothesized that there can exist “essential” 2-mers in a TCR that, once identified, can be used to predict the rest of the CDR3 sequence. By contrast, non-essential k-mers provide little information about the rest of the TCR. For a set of TCRs, we calculate the Shannon informational entropy of k-mers found at each 1% increment bin of the CDR3 (Fig. 4A). High entropy suggests a large diversity of 2-mers, as observed at the center of CDR3s. Low entropy signifies similar 2-mer usage, as found at the N- and C-termini.
Figure 4.
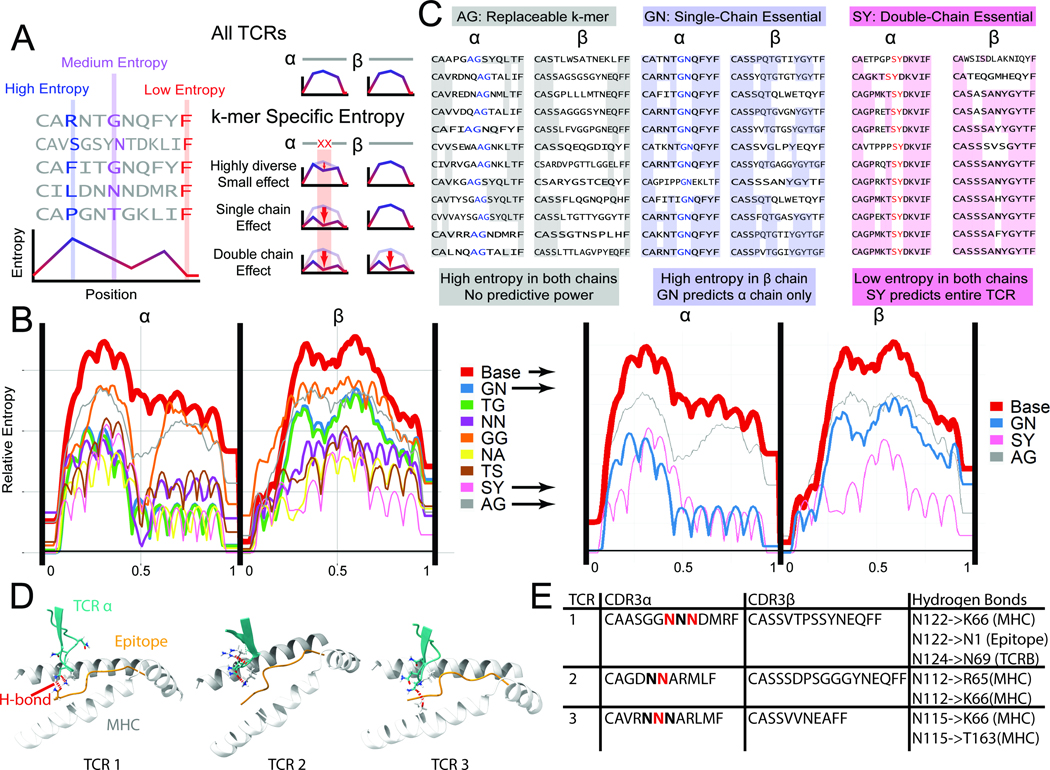
Entropy-based analysis of TCR repertoires. A. In a set of TCR sequences, the entropy at each position measures the diversity of amino acid (or 2-mer) usage. Low entropy at a position indicates repeated usage of 2-mers, possibly important for epitope binding and recognition. To determine if a 2-mer is important for binding, the subset of TCRs containing a 2-mer is isolated. Entropy calculated at every position can be unaffected in the subset relative to the whole set, reduced in the same chain, or reduced in both chains. B. The entropy for all CMV pp65-specific TCRs, as well as subsets of antigen-specific TCRs with common 2-mers near the center of the chain are plotted (N>10 TCRs per 2-mer). We observe 2-mers for which the set of CDR3s containing that 2-mer at a specific location has very low entropy (i.e. SY, NA, NN), denoting a specific sequence used for binding. The thickness of the lines is correlated to the number of TCRs containing the 2-mer at the center of the chain. C. 2-mers with minimal entropy reduction (AG), single-chain reduction (GN, essential), and double-chain reduction (SY, super-essential) are shown. See also Figs. S3–5. D. Simulated TCR-pMHC complexes are shown at the -chain CDR3/epitope/MHC interface. For the essential 2-mer NN, hydrogen bonds (red dashed lines) were found between the N residues and the MHC, the epitope, and/or the TCR chain. E. A tabulation of the specific H-bonding interactions observed in the three structures of panel D.
To determine if a 2-mer is essential, the entropy is calculated for the set of TCRs containing that 2-mer relative to the full set (Fig. 4A). A binding-essential 2-mer would be expected to lower the entropy (or sequence diversity) of antigen-specific CDR3s that possess that 2-mer at a similar relative location. We can further identify “super-essential” 2-mers that restrict the sequences of not only their own chains but also of their paired or chains. Entropy plots often have a characteristic humped structure, due to some lengths of CDR3s being more frequently reported in databases. For pp65 CDR3s, 11-mer chains are most frequent (Fig. 1B), and this leads to the 11 peaks and 10 dips observed in Fig. 4B (red line).
We used SPAN-TCR to scan paired and chain TCRs and measure the binned 2-mer entropy of TCR sets (Fig. 4B, Sup. Table 4), and extracted the most common 2-mers at the center of the chain (>10 appearances in VDJdb, bin =0.5). We plotted the bin entropy for TCRs containing that central 2-mer (colored lines). Note that in all plots, the entropy dips in the middle because of the forced presence of the specific 2-mer. However, the overall contour of these entropy plots provides a metric for the essential nature of the 2-mer.
For some 2-mers (GG, AG), the entropy is minimally reduced (Fig. 4B, grey and orange lines), and so these 2-mers are replaceable (Fig 4C, left). For GN and TG, the entropy of the chain only is reduced, suggesting an essential role only for the chain (Fig 4C, middle). The NN, NA, TS, and SY 2-mers are super-essential, lowering both and -chain entropy (Fig 4C, right). Note that “essential” does not reference the TCR:pMHC binding affinity or the frequency of the 2-mer usage.
We used molecular dynamics simulations to explore the role of k-mers in TCR-pMHC interfaces. Crystal structures of the pMHC-bound TCR complex are rare and we were unable to find structures that contained the same k-mers. Thus, we used the AlphaFold molecular dynamics simulation algorithm to simulate the folding of TCR-pMHC complexes. First, using three existing structures with no shared 2-mers, we observed nearly identical structure between the simulated folded molecules and the measured crystal structures with an average RMS error of ~ 1 Angstrom (Sup. Fig. 3A). After determining that the algorithm reproduced TCR-pMHC complexes, we explored how the super-essential 2-mer NN may interact with the CMV pp65 epitope43. We collected 10 reported paired CDR3s that utilized NN 2-mers near the center of the chain. Using the simulated folded complexes, we observed repeated interaction motifs, including hydrogen bonds between the 2-mer and the epitope, MHC, and/or TCR chain (Fig. 4D, E, Sup. Fig. 3B). These calculations suggest that a super-essential 2-mer may participate in bonding interactions but that the details of those interactions are context-dependent.
We extended this entropic analysis to identify essential 2-mers for paired and pp65-specific CDR3s (Sup. Fig. 3C), and essential 2-mers at the center of the chain of other major VDJdb epitopes (Sup. Fig. 3D), showing some epitope-dependent similarities (Sup. Fig. 4A). Hydrophilic 2-mers appear more essential than neutral 2-mers (G, A-containing 2-mers). 2-mers with charged amino acids (N, K, Q, R) are most frequently associated with large entropy reductions. Clustering k-mers by BLOSUM metrics provides more chemical insight into TCR composition (Sup. Fig. 4B). These general findings were generally consistent across all three databases (VDJdb, McPAS-TCR, TBAdb) (Sup. Fig. 5) with differences emerging due to distinct CDR3s contained in one data set but not the others.
SARS-CoV-2 TCR Analysis
We analyzed >150,000 recently published putative SARS-CoV-2 epitope-specific TCR chain sequences using SPAN-TCR, arranged by specificity to 269 peptides or peptide groups44,45. Considering only sets with >500 reported TCRs (n = 61, Sup. Table 5), we grouped TCRs by their antigen specificity and performed SPAN-TCR entropy analysis to identify essential 2-mers. For each antigen, we selected 2-mers at each 1% increment of CDR3 position with >3% frequency and plotted the reduction in entropy to identify the most essential 2-mers (see the YLNTLTLAV example of Fig. 5A). The most essential 2-mers were adjacent to the center of the CDR3, such as “GE” in the YLNTLTLAV epitope group (Fig. 5B).
Figure 5.
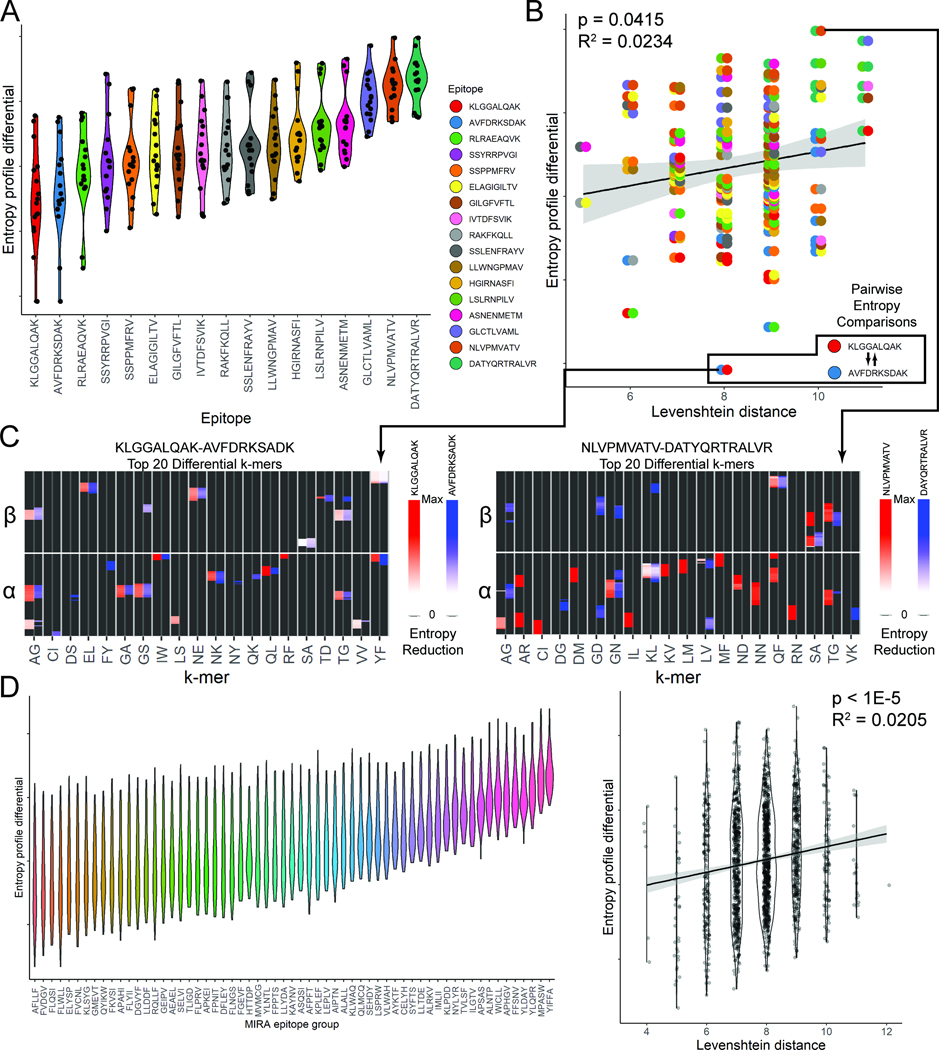
SPAN-TCR Entropy Analysis of Putative SARS-CoV-2 TCRs. A. Contributions to entropy for 2-mers in YLNTLTLAV TCRs at each relative position of the chain identify 2-mers essential to binding. B. A large entropy reduction is associated with 2-mer GE. C. The frequency and average entropy reduction of 2-mers are plotted (error bars = standard deviation) for all SARS-CoV-2 epitopes contained in MIRA. YE is notable as a 2-mer with high frequency and a strong entropy reducing influence. D. The frequency and entropy reduction of the YE 2-mer in different epitopes is plotted. Two epitope groups which contain YE at similar frequency but different entropy reduction are circled. The inset shows that YE only restricts the V gene usage for epitope group [SEHDY], while J gene usage and entropy are similar. E. Within CDR3s specific to a single epitope group, YE is the most essential. F. Plot showing that for CDR3s containing YE between positions 0.7–0.8, the CDR3 composition is restricted.
We identified 2-mers (>3% frequency) that produced the largest average entropy reductions across all MIRA epitopes. As a case study, we explored the 2-mer YE in Fig 5C because it has both high frequency and a large entropic effect. Next, we determined the epitopes where specific 2-mers are most essential. After plotting the frequency and entropy reduction of YE 2-mers in 1% bins across all epitopes (Fig. 5D), a clear outlier was observed, identified as the MIRA group [SEHDY], which is specific to epitopes SEHDYQIGGYTEKW, YQIGGYTEK, and YQIGGYTEKW. Only in [SEHDY] is YE both frequent and essential. In fact, among high frequency 2-mers in [SEHDY]-specific TCRs, YE has the greatest impact on entropy (Fig. 5E). Finally, by plotting the composition of all TCRs containing YE in the position range from 0.7–0.8 (Fig. 5F), we identified the frequent appearance of GXG motifs at the center as GX 2-mers followed by XG 2-mers, with more diverse yet mostly hydrophobic k-mers in the 0.5–0.6 position range (GL, GV, GI, indicated by the blue sections).
We contrasted VJ gene usage among YE TCRs in [SEHDY] with another epitope group (AFPFTIYSL, GYINVFAFPF, INVFAFPFTI, MGYINVFAF, NVFAFPFTI, NVFAFPFTIY, YINVFAFPF) [AFPFT], which is also circled in Fig. 5D and appears at similar frequency. The entropy of J genes is similar between the two epitopes (Fig. 5D inset), but the entropy of V gene is 0.64 in [SEHDY] and 3.29 in [AFPFT]. Thus, YE is far more essential to binding in [SEHDY] than [AFPFT].
Epitope Sequence Correlation to TCR Sequence Entropy
We sought to establish relationships between different epitopes and their associated T cell clonotypes using SPAN-TCR entropy analysis. We defined the entropy reduction profile for a given epitope by first calculating the reduction in entropy induced by each 2-mer at each position. We then calculated the Euclidean distance between profiles for each of 17 major epitopes with >200 paired chains in VDJdb. These entropy reduction profiles reveal which epitope-specific TCR groups utilize essential 2-mers at similar positions in their and chains (Fig. 6A). Here, each dot on the violin plot represents a cross-epitope comparison. A violin plot with lower values, exhibited by KLGG… and AVFD…, suggests that similar essential 2-mers are shared at the same position between the TCR sets specific to these epitopes and those specific to the 16 other epitopes. The profiles of certain epitopes (NLVP… and DATY…) were consistently more different than others, likely reflecting an increased usage of rare k-mers.
Figure 6.
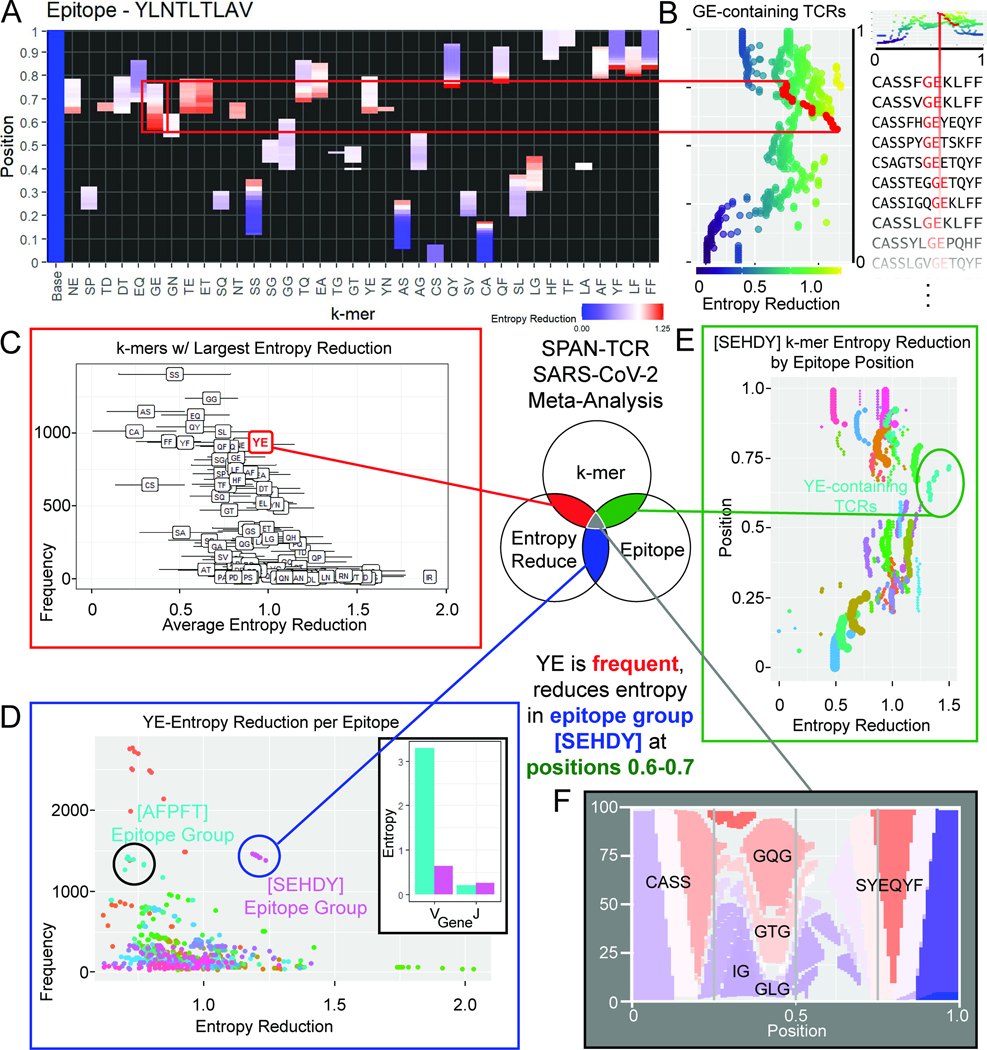
Epitope sequence and TCR entropy correlation. A. The TCR entropy reduction profile similarities between major epitopes (epitopes with >200 paired chains, n=17) in VDJdb are plotted. The entropy reduction profile similarity falls within a range for each epitope(136 comparisons). B. Entropy profile differences are plotted against the Levenshtein distance between epitope sequences. There is a significant correlation between Levenshtein distance between epitopes and TCR entropy profile differences. A triad of 3 epitopes, AVFDRKSDAK, KLGGALQAK, and RLRAEAQVK, has the smallest difference by entropy profile (linear regression). C. Entropy profiles for the most differential k-mers are shown for epitopes KLGGALQAK and AVFDRKSDAK together, and KLGGALQAK and DATYQRTRALVR (arbitrary units, linear scale). D. Entropy reduction analysis was performed using MIRA TCR chains, showing the same trends (n=61, 1830 comparisons, linear regression). See also Fig. S6.
Plotting the entropy profile difference against the Levenshtein distance between epitopes, we observed a weak but significant correlation (Fig. 6B, Sup. Fig. 6A). This relationship was maintained despite TCR sharing between epitopes, which was not correlated with the Levenshtein distance between epitopes (Sup. Fig. 6B, C). The entropy reduction profiles of two epitopes with small entropic differences (KLGG… vs AVFD…) are plotted side-by-side (Fig 6C, left, top 20 differential k-mers). Here, we find similar essential k-mers. This contrasts with two epitopes with a large difference (Fig. 6C, KLGG… vs DATY…). Three epitopes (KLGG…, AVFD…, and RLRA…) form a triad of the smallest entropy profile differences, perhaps due to common TCRs reported in the database or cross-reactivity of similar TCR sequences.
A similar correlation of epitope-epitope Levenshtein distance and entropy profile difference was observed (Fig. 6D, Sup. Fig. 6D–F) using the entropy reduction profiles of chains from MIRA epitope groups (Sup. Fig. 7). However, in MIRA, there was very little sharing of TCRs between epitope groups (Sup. Fig. 6D, E). The correlation of epitope Levenshtein distance and entropy profile difference was also observed at the individual epitope group level (Sup. Fig. 6F). Relationships between two sets of TCR sequences can be challenging to extract. Here we distill, from a set of disparate TCR sequences, their most influential features, the essential k-mers. This approach reveals relationships across epitopes and their cognate TCRs.
DISCUSSION
SPAN-TCR is a suite of tools for the analysis of large databases of diverse TCR CDR3 sequences. It is based on the observation that the TCR-pMHC interaction is somewhat constrained by the TCR-pMHC binding conformation, even for TCRs of different length. The primary goal of SPAN-TCR is to identify subsequences of TCRs that have outsized influence on TCR-pMHC interfaces. Our strategy is complementary to other TCR analysis such as GLIPH35, TCRdist31, TCRMatch33, or ALICE32, which each compare TCR sequences to each other. The use case for SPAN-TCR that we propose is to study sets of putative antigen-specific sequences, to generate visual representations of TCR composition, and to use entropic analysis to stratify k-mer motifs by their contributions to antigen-specificity. The challenge of understanding how different TCRs bind the same pMHCs is simplified by SPAN-TCR, which provides comparative TCR:pMHC binding insights from analysis of large TCR databases using multiple strategies. These include a normalization step to compare variable length CDR3s, the incorporation of smoothing functions to simulate peptide flexibility, and the incorporation of specific amino acid physico-chemical metrics. While these steps do not eliminate the variability of TCR-pMHC binding orientation from the model, essential k-mers discovered through SPAN-TCR appear to reflect groups of TCRs where the binding orientation is most similar.
TCR-pMHC binding can involve solvent-removing hydrophobic effect, hydrogen bonds, or salt bridges46, all of which may require more than one amino acid to initiate, thus motivating our use of k-mers. A distinguishing feature of this work is the use of informational entropy to distinguish between the distinct concepts of enriched k-mers and essential (or super-essential) k-mers. K-mers that are not enriched cannot be essential, but k-mers can be enriched but not essential. The characteristic of a k-mer being essential likely has implications. For example, super-essential k-mers may generate low affinity TCRs in one chain and require a specific paired chain, whereas essential k-mers define a single high affinity chain giving more flexibility to the paired chain sequence. We believe that further designs for TCRs may be centered around these essential k-mers as anchor points or sources of intermolecular bonds. Further experiments are needed to verify this hypothesis, and other types of entropy measurements may reveal further insights47. Interestingly, while hydrophilic 2-mer motifs are relatively low in frequency compared to hydrophobic motifs48–50, they can be deemed “essential” by SPAN-TCR51,52. In fact, large scale MD simulations suggest that these hydrophilic essential 2-mers participate in interfacial hydrogen bonding.
By mining databases of TCRs specific to SARS-CoV-2 viral antigens, we identified that essential k-mers are most likely to be found adjacent to the center of the chain (positions 0.35–0.45, 0.55–0.65). Entropic analyses of TCRs specific to diverse epitopes identified similarities in the identity and locations of essential k-mers between specific but very different epitopes. Such relationships were found in both VDJDB and MIRA databases, which contain epitopes from a variety of pathogens and SARS-CoV-2 epitope specific CDR3s, respectively. While VDJDB contains many shared TCRs between antigens, MIRA does not (Sup. Fig. 6C, E), suggesting that this finding is robust and independent of the degree of TCR sequence sharing between datasets. The viral antigens explored here represent well-studied examples of epitope-specific TCRs. As novel data is generated on emerging epitopes and immune-oncology-related neoepitopes, tools like SPAN-TCR should provide insights and permit comparisons across data sets53,54.
SPAN-TCR’s niche lies between CDR3 sequence alignment strategies55, which can oversimplify a complex binding interface that includes CDR3-HLA interactions and non-CDR3 interactions56,57, and full molecular simulations9,58,59, which remain expensive60. Unlike certain machine learning tools61–66, we do not try to predict TCR specificity but instead identify k-mers that are likely important for TCR specificity. One common ground between SPAN-TCR and machine learning is our reliance on reference data such as VDJdb, and standardization of quality control in these data will be an important topic for the field at large.
Another biological limitation that affects the entire field of TCR sequence analysis is HLA specificity. Epitopes do not present universally across HLA alleles and measuring the extent of shared TCR specificity across HLAs is a complementary field of research67,68 that is being propelled by TCR-affinity capture and TCR sequencing strategies18,69,70. The collective advances of these experimental and computational toolsets are beginning to reveal new patterns and insights into the vast diversity of adaptive immunity.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, James Heath (jheath@isbscience.org).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Single-cell TCR-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. TCR sequence data was obtained from VDJdb, McPAS-TCR, TBAdb, and MIRA databases. Links are provided in the key resources table.
All original code has been deposited at Github and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Healthy HLA-A02 PBMCs | StemCell Technologies | Cat#70025 |
| Chemicals, peptides, and recombinant proteins | ||
| Biotinylated HLA-A2 MHC-I Monomer | Fred Hutchinson Cancer Research Center Immune Monitoring Shared Resource | N/A |
| NLVPMVATV peptide | IBA-Lifesciences | N/A |
| PE-conjugated streptavidin | Thermo Fisher | Cat#SA10041 |
| APC-conjugated streptavidin | Thermo Fisher | Cat#SA1005 |
| 500 kDa biotin-dextran | Nanocs | Cat#DX500-BN-1 |
| Critical commercial assays | ||
| Single Cell Immune Profiling Kit | 10X | Cat#1000542 |
| Deposited data | ||
| VDJDB | Shugay et al.13 | https://vdjdb.cdr3.net/ |
| TBAdb | Zhang et al.14 | https://db.cngb.org/pird/home/ |
| McPAS-TCR | Tickotsky et al.15 | http://friedmanlab.weizmann.ac.il/McPAS-TCR/ |
| MIRA COVID sequences | Nolan et al.44 | https://doi.org/10.21203/rs.3.rs-51964/v1 |
| Raw and Analyzed Data | This paper | SRA: PRJNA918821 |
| Software and algorithms | ||
| R v4.1.1 | CRAN | https://cran.r-project.org/ |
| RStudio v1.4.1717 | RStudio | https://www.rstudio.com |
| uwot R package | James Melville | https://github.com/jlmelville/uwot |
| data.table R package | Matt Dowle | https://cran.r-project.org/web/packages/data.table/index.html |
| dplyr R package | Hadley Wickham | https://cran.r-project.org/web/packages/dplyr/index.html |
| ggplot2 R package | Thomas Lin Pedersen | https://cran.r-project.org/web/packages/ggplot2/index.html |
| pheatmap R package | Raivo Kolde | https://cran.r-project.org/web/packages/pheatmap/index.html |
| ggnewscale R package | Elio Campitelli | https://cran.r-project.org/web/packages/ggnewscale/index.html |
| GLIPH | Glanville et al.34 | http://50.255.35.37:8080/ |
| AlphaFold v2.0 | Deepmind | https://github.com/deepmind/alphafold |
| SPAN-TCR | This paper | https://doi.org/10.5281/zenodo.7637683 |
| Other | ||
| UV Crosslinker | Boekel | Cat#234100 |
Method Details
Publicly available data from four TCR databases, VDJdb, McPAS-TCR, TBAdb, and the MIRA database44 were used. VDJdb chains were all MHC Class I specific. SPAN-TCR analysis can be performed for paired chains only, or all and chains. Each and chain was first normalized by its length. Within each chain, for each relative position from 0 to 1, by increments of 0.01 or 0.1, the k-mers that overlap with that position were recorded. The contribution of each k-mer at each relative position is summarized by the function:
Where frequency is either the number of reads for the TCR, or a function based on the quality of the TCR within the database. Here we applied exponential frequency functions (VDJdb: 5score, McPAS-TCR: 2id.method, TBAdb: 3grade). This function is malleable, the exponential terms here were chosen to allow high score values and low score values to collectively contribute similar weights, i.e. there were approximately 100x as many score=0 terms in VDJdb as score=3 terms. The choice of function depends on the nature of the input data, with the overall goal to emphasize the contribution from high quality CDR3s and limit noisy, low confidence data. The symbol denotes the k-mers that intersect with the position being investigated, and weight is a function based on the location of each k-mer and the relative position being analyzed. Here, weight is based on the formula:
Where position is the relative position within the sphere of influence of each k-mer. This is a linear decay function from the center point 0.5. For paired chain analysis, and chains were individually analyzed and then concatenated.
Individual k-mers were analyzed as the sum of the properties of its component amino acids. Each k-mer was assigned either a universal value for comparison purposes such as hydrophobicity, or a relative distance matrix was constructed to compare k-mers. An example distance matrix derived from BLOSUM62 is presented in Sup. Table 3. The entropy is a standard Shannon entropy calculation:
Where is the contribution for a specific k-mer at the position: . This calculation is performed on the contributing k-mers at each relative position. Other entropy or diversity metrics may be substituted.
SPAN-TCR Parameters
The SPAN-TCR representation of sequence diversity reveals common amino acid k-mers utilized for binding a pMHC target at relative positions of the CDR3. It can be interpreted as a logo plot. A large block of color indicates k-mers that are used most frequently, and the color indicates a property of the k-mer such as hydrophobicity. Our computational model is designed to capture the critical chemical interactions that happen at the TCR-pMHC interface, and to account for the variable lengths of TCR CDR3 regions that are experimentally found, while capturing the elements of CDR3s that are shared between different TCRs recognizing the same antigens. To this end, there are two critical parameters.
The first parameter considers how consecutive amino acid residues coordinate their chemical characteristics to comprise a binding motif. Do we consider each residue separately (k=1), or do we consider pairs or longer strings of residues (k=2, 3, etc.)? The second is the position of k-mers at the binding interface, which uses the concept of binning and weighting. Increasing binning increases the spatial resolution of the interface to resolve CDR3s of unequal lengths, and weighting accounts for flexibility within the TCR peptide chain, whereby the local influence of a k-mer at the interface can be adjusted.
To illustrate this model, one can consider a simple example: amino acid 1-mers for a set of two 10-amino acid CDR3s in a 10-bin comparison (k=1, 10 bins). Since all CDR3 regions are the same length, binning is trivial, with each residue comprising one bin. This results in one-to-one comparisons between each CDR3 residue. Graphically, bars represent the percentage of CDR3s with an amino acid at each position (Fig. 2A). Common amino acids are taller regions on the SPAN-TCR plot, and the width of a region indicates the relative length of the CDR occupied by the amino acid (10% for k=1, 10 amino-acid CDR3s). This is equivalent to a logo plot.
Next, consider a set of 2 CDR3s of unequal length, again binned into a 10-bin comparison. For a CDR3 shorter than 10 amino acids, the 1st amino acid is found in bin 1 and bin 2, muddling the position of residue #2 (Fig. 2A). Parsing this comparison into 100 bins does a better job of locating the relative positions of each residue (Sup. Fig. 1A for CMV pp65-specific TCRs). Thus, increasing binning resolves the differences between TCRs and shows the structural similarities where sequence-based comparisons do not.
SPAN-TCR Options
Each dataset, whether obtained using the 10X Immune Profiling platform or one of the 3 databases, was preprocessed. Paired chains containing multiple or chains were considered as multiple paired chains (i.e. chain 1 and chain 1 and 2 = chain 1 and chain 1; chain 1 and chain 2). Separate weight functions were used for each data format. For VDJdb, a vdjdb.score metric was supplied ranging from 0 to 3. The scoring is described here: https://github.com/antigenomics/vdjdb-db/blob/master/README.md, and the weight function applied was equal to 5^vdjdb.score. The McPAS database uses a similar metric ranging from 1 to 3, with subcategories within each score. PIRD uses a cumulative grading metric described here: https://db.cngb.org/pird/tbadb/. The BLOSUM-derived distance matrix is created by setting all BLOSUM62 values greater than 3 to equal 431, and subtracting these values from 4 to create a distance matrix where 0 denotes an amino acid match. The matrix for k-mers is calculated by the sum of pairwise distances at each position of k-mers, for each combination of k-mers. For entropy contribution analysis, only epitopes that contributed over 250 paired entries were considered. For unpaired TCRs, cutoffs of 250 entries for VDJdb and 100 for MCPAS and PIRD were used.
GLIPH2 Analysis
GLIPH2 was performed using the executable version and the default parameters (see Data and Code Availability). The default reference files for both CD4 and CD8 T cells were used, available from the same website. VDJdb pp65 CMV epitope GLIPH2 analysis was performed using all pp65-specific TCRB sequences reported in VDJdb. SARS-CoV-2 GLIPH2 analysis was performed using input data from 10X TCR sequencing. To compare GLIPH2 results with SPAN-TCR, all SPAN-TCR 4-mers that were found in any GLIPH2 group were recorded. As GLIPH2 does not analyze the beginning and end of the CDR3 chain, only SPAN-TCR k-mers in the middle half of CDR3s were considered.
Dextramer Pulldown and Sequencing
UV-mediated peptide exchange was used to generate HLA-A2 MHC-I monomers presenting peptides of interest (pMHC). 50 μl of 2 μM biotinylated HLA-A2 MHC-I monomers with UV-cleavable peptides (Fred Hutchinson Cancer Research Center Immune Monitoring Shared Resource) was combined with 2.5 μl of 1mM CMV pp65 peptide NLVPMVATV (IBA-Lifesciences) in a single well of a 96-well v-bottom polypropylene plate (Corning) and exposed to UV (360 nm) for 1 hour in a UV Crosslinker (Boekel). UV-exchanged pMHC were used immediately for dextramer formation and cell labeling.
Dextramers were made by MHC-I multimerization and dextran doping71. pMHC monomers were combined with PE- or APC-conjugated streptavidin (Thermo Fisher Scientific) at a 3:1 molar ratio in the dark at room temperature for 10 minutes. Streptavidin/pMHC complexes were combined with biotin-dextran 500 kDa (Nanocs) at a 20:1 molar ratio in the dark at room temperature. The dextramer reagent was then clarified for 2 minutes at 15,000 g.
PBMCs from HLA-A2 healthy donors (StemCell Technologies) were cultured in R10 media (RPMI 1640 supplemented with 10% heat-inactivated FBS, glutamax, penicillin and streptomycin) stimulated with 1000 U/ml recombinant IL-2 (PeptroTech) and 1 μg/ml peptide for 14 days. IL-2 and peptide were replenished every 3–4 days and the culture volume was expanded 2x on day 10.
5 × 107 cells from the expansion culture were washed with 1X PBS and resuspended in 100 μl of 50 nM Dasatinib (Cayman Chemical) for 30 minutes. Clarified PE- and APC-conjugated dextramer reagents were diluted 2.5X with 1X PBS and 100 μl of each was added directly to the Dasatinib treated cells for 1 hour. Cells were washed 2X with 1X PBS and incubated with a cocktail of anti-PE, anti-APC, anti-CD3-APC eFlour 780 (Clone SK7; Invitrogen), and anti-CD8-FITC (clone RPA T8; BD Biosciences) as per manufacturer’s instructions for 30 minutes. Cells were washed and resuspended in 1X PBS at 1 × 107 cells/ml for immediate FACS sorting. CD3+ CD8+ dual PE and APC+ cells were sorted into R10 media using a MA900 Cell Sorter (Sony). Fluorochrome compensation was performed using UltraComp eBeads Compensation Beads (Thermo Fisher Scientific). Cells were pelleted, counted, and used directly for Single Cell Immune Profiling (10X Genomics).
Distance Function
The distance function between two TCRs was calculated by scanning along the relative position of the CDR3s. At each position range, i.e. the nth 1% of the CDR3, the k-mers of each chain that influence that position are recorded for the initial chain and the comparison chain. For each k-mer of the initial chain, the distance to any k-mer of the comparison chain is recorded as the accumulated distance by the formula:
Where diff is a function describing the distance between two k-mers. Two difference functions were demonstrated here, one using a simple replacement matrix:
And one using the BLOSUM62 derived matrix in Sup. Table 3.
Entropy Calculations
The entropy at each relative position was calculated for groups of TCRs as well as subsets of TCRs that contained contributions from specific k-mers at specific relative positions (i.e.). Heatmaps were colored by their relative contributions to the average entropy across the entire CDR3 analyzed (paired or single) compared to the subset with the highest entropy (typically the full group of antigen-specific TCRs) or the complete set of CDR3s, with full data table examples for paired and unpaired TCRs in Sup. Tables 6 and 7. If the subset of TCRs containing a k-mer at a specific location was below a threshold that k-mer was removed from the analysis as the entropy would necessarily be low. To obtain entropy profiles, all k-mers in 1% bins that appeared above a threshold (3% of sequences) were found, and entropy reduction was calculated for subsets of CDR3s containing such k-mers. A matrix was constructed containing the entropy reduction per k-mer per 1% bin, vectorized, and compared to entropy profiles of other epitopes by Euclidean distance.
Entropy Metric Examples
To illustrate the entropy metric in TCR sequences, we consider the extremes among CMV pp65-specific TCRs. The simplest essential k-mer is a complete antigen-specific paired and chain. The set of TCRs that contains this k-mer is the complete TCR itself, so each SPAN-TCR bin has 1 or 2 k-mers only and minimal bin entropy.
In contrast, the initial C residue observed at the beginning of CDR3 and chains does not provide information about the rest of the TCR. Almost all CDR3s contain an initial C, so entropy in the first bin of these and chains is 0 for k=1 since all these chains begin with C. Entropy remains low for k=2 given the limited number of starting k=2 k-mers. The bin entropy rises as we continue down the TCR. The entropy at the center of the (bin =0.5) and (bin =0.5) chains is high with many different k-mers found, before the entropy falls in bins at the end of and (bin /=1) chains until the common terminal F is observed (Sup. Fig. 3A, k=1).
Entropy Considerations
We determined that for each k-mer, if the subset of TCRs that contains the k-mer at a specific location reduces the entropy of the subset substantially, this signifies that the k-mer is essential to binding. Another line of reasoning may conclude that for a k-mer such that the subset of TCRs with the k-mer at a specific location has high entropy, this k-mer may be essential for binding due to the “popularity” of the k-mer. That is, since many diverse TCRs have the k-mer at the position, the k-mer may be needed for binding for each of these TCRs.
We believe this second interpretation to be inaccurate, first due to conflation with general amino acid prevalence, as k-mers that do not reduce entropy often contain the most common amino acids, G, L, A, V, S. Thus, these k-mers are more likely to be observed frequently enough by random chance to pass the selection thresholds for our analysis but are otherwise similar to any other k-mer, which is supported by the minimal change in entropy for the subset. Second, while these k-mers that do not reduce entropy may appear in many diverse TCRs and assist in TCR binding to a pMHC, they do not seem to contribute to the specificity of the TCR to a specific epitope. This is illustrated by Fig. 5B, where the k-mers that do not reduce entropy are often observed widely across many peptide epitopes. These k-mers typically have minimal side chain functionality, and their function may be to add flexibility as posited in the discussion, or serve as spacers between more functionally active amino acids during binding with the pMHC to prevent overly tight binding6–8. Thus, we believe that while a k-mer that generates a high-entropy subset may either appear through chance or serve an important, albeit functionally quiet role, these k-mers do not appear to be responsible for the specific binding of TCR-pMHC complexes, in contrast to k-mers that are found in many TCRs which result in subsets of TCRs that have similar composition and low entropy.
Molecular Dynamics Simulations
Structures were generated using the multimer preset of Alphafold2. All settings were run at the default using structures found in the RCSB-PDB, and the max template date was set to June 20th 2022. Sequences for the variable, joining and constant regions of the TCR were found from IMGT. Sequences for the HLA-A2 region were found from https://www.rcsb.org/structure/4U6Y in the RCSB-PDB. To generate the alignment error data between known and unknown structures, ChimeraX’s Matchmaker tool was used, using the “Best Aligning Pairs of Chains Between Reference and Match Structure” setting, using the Needleman-Wunsch pairwise sequencing algorithm, and the BLOSUM-62 similarity matrix. Reference structures were found from the RCSB-PDB and used for comparison.
SARS-CoV-2 Analysis
MIRA TCR sequences were obtained and analyzed from the Immunoseq database. Each read from immunoseq was assigned a score of 1 for SPAN-TCR analysis. For each k-mer, if more than 3% of CDR3s contained that k-mer in a 1% increment, the k-mer was included for entropy analysis.
Entropy Reduction Profile
The entropy reduction profile comparison between TCR sets was obtained by calculating the entropy reduction among all n possible k-mers at each 1% increment. The n x 100 (200 for paired TCRs) matrix was converted into a single vector for each epitope TCR set, and the Euclidean distance between vectors was calculated. For Levenshtein distance between MIRA epitope groups, the minimum distance between the epitopes present in any group was used as the Levenshtein distance between epitope groups. Entropy reduction is a unit-less metric.
Software and Packages
Analysis was performed in R 4.1.1 and RStudio 1.4.1717 using packages dplyr, data.table, and uwot. Images were generated using packages ggplot2, pheatmap, and ggnewscale. GLIPH is available from http://50.255.35.37:8080/, SPANTCR code is available at https://github.com/alexandermxu/SPANTCR.
Quantification and Statistical Analysis
The correlation between the Levenshtein distance between epitopes and the distances between entropy reduction profiles of TCRs specific to epitopes and p-values were calculated using a linear regression model (lm function in R). As reported in Figure 6, n represents the number of groups of TCRs specific to a single epitope, and the total number of comparisons is calculated as nC2. Comparisons between epitopes use the entropy reduction profile, which is the Euclidean distance between two high-dimensional vectors. Thus, comparisons are relatively independent, i.e. knowing the distance between entropy reduction profiles between A and B, and B and C, provides little information on the distance between A and C. For some epitope-specific groups that share TCRs, we explore possible correlations in Supplementary Figure 6.
Supplementary Material
Supplementary Table 1. GLIPH2 analysis of CMV pp65 antigen-specific TCRs in VDJdb, related to Figure 2.
Supplementary Table 4. CMV pp65-specific TCRs in VDJdb enriched for high frequency k-mers at the center of the chain, related to Figure 4.
Supplementary Table 5. The number of TCRs associated with each MIRA epitope. Only epitopes with >500 sequences were analyzed, related to Figure 5.
Supplementary Table 6. Entropy contributions from paired chains in VDJdb, related to Figure 6. Epitopes had to have over 250 paired chains. Each k-mer at each position was only considered if over 3 percent of the total TCRs at that position contained a contribution from that k-mer. The average entropy over the paired chain was obtained for each subset of TCRs containing a k-mer.
Supplementary Table 7. Entropy contributions from unpaired chains in VDJdb, related to Figure 6. Epitopes had to have over 250 unpaired chains. Each k-mer at each position was only considered if over 2 percent of the total TCRs at that position contained a contribution from that k-mer. The average entropy over the paired chain was obtained for each subset of TCRs containing a k-mer. The and chains were analyzed separately.
Highlights.
T-cell receptors have subsections (CDR3s) of varying length used to bind antigen.
SPAN-TCR is a tool to compare amino acid composition of CDR3s of all lengths.
Entropic analysis reveals short amino acid motifs most essential to binding.
Binding motifs display patterns between / TCR chains and across epitopes.
ACKNOWLEDGEMENTS
We are grateful to all participants in this study and to the medical teams at Swedish Medical Center for their support. We thank the Northwest Genomic Center and Fred Hutchinson Cancer Research Center for help with sequencing services, and the ISB-Swedish COVID-19 Biobanking Unit. We thank the Fred Hutch Genomics Shared Resource (supported by the NIH/NCI Cancer Center Support Grant P30 CA015704) and the WA State Andy Hill Care Fund. We thank Amazon Web Services for their support through cloud computing credits provided by the AWS Diagnostic Development Initiative (DDI). We acknowledge funding support from the Parker Institute for Cancer Immunotherapy (J.R.H., M.M.D.), Merck, the Biomedical Advanced Research and Development Authority (HHSO10201600031C to J.R.H. and M.M.D.), the Department of Defense Prostate Cancer Research Program (W81XWH-20-1-0119 to D.C.D. and J.K.L.), and the NIH (P50 CA097186 Developmental Research Program to J.K.L., 1 R01 CA264090-01 to J.R.H., and CTSI UL1TR001881 to A.M.X).
Footnotes
DECLARATION OF INTERESTS
J.R.H is a founder and board member of Isoplexis and PACT Pharma. M.M.D. is a member of the Scientific Advisory Board of PACT Pharma.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davis MM, and Bjorkman PJ (1988). T-cell antigen receptor genes and T-cell recognition. Nature 334, 395–402. 10.1038/334395a0. [DOI] [PubMed] [Google Scholar]
- 2.Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, Cantrell JR, Wheeler DK, Gabbard JL, Hix D, Sette A, and Peters B. (2015). The immune epitope database (IEDB) 3.0. Nucleic Acids Res 43, D405–412. 10.1093/nar/gku938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borg NA, Ely LK, Beddoe T, Macdonald WA, Reid HH, Clements CS, Purcell AW, Kjer-Nielsen L, Miles JJ, Burrows SR, et al. (2005). The CDR3 regions of an immunodominant T cell receptor dictate the ‘energetic landscape’ of peptide-MHC recognition. Nat Immunol 6, 171–180. 10.1038/ni1155. [DOI] [PubMed] [Google Scholar]
- 4.Blevins SJ, Pierce BG, Singh NK, Riley TP, Wang Y, Spear TT, Nishimura MI, Weng Z, and Baker BM (2016). How structural adaptability exists alongside HLA-A2 bias in the human alphabeta TCR repertoire. Proc Natl Acad Sci U S A 113, E1276–1285. 10.1073/pnas.1522069113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song I, Gil A, Mishra R, Ghersi D, Selin LK, and Stern LJ (2017). Broad TCR repertoire and diverse structural solutions for recognition of an immunodominant CD8(+) T cell epitope. Nat Struct Mol Biol 24, 395–406. 10.1038/nsmb.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garcia KC, and Adams EJ (2005). How the T cell receptor sees antigen--a structural view. Cell 122, 333–336. 10.1016/j.cell.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 7.Rudolph MG, Luz JG, and Wilson IA (2002). Structural and thermodynamic correlates of T cell signaling. Annu Rev Biophys Biomol Struct 31, 121–149. 10.1146/annurev.biophys.31.082901.134423. [DOI] [PubMed] [Google Scholar]
- 8.Rudolph MG, Stanfield RL, and Wilson IA (2006). How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol 24, 419–466. 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- 9.Leem J, de Oliveira SHP, Krawczyk K, and Deane CM (2018). STCRDab: the structural T-cell receptor database. Nucleic Acids Res 46, D406–D412. 10.1093/nar/gkx971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gowthaman R, and Pierce BG (2018). TCRmodel: high resolution modeling of T cell receptors from sequence. Nucleic Acids Res 46, W396–W401. 10.1093/nar/gky432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adams JJ, Narayanan S, Liu B, Birnbaum ME, Kruse AC, Bowerman NA, Chen W, Levin AM, Connolly JM, Zhu C, et al. (2011). T cell receptor signaling is limited by docking geometry to peptide-major histocompatibility complex. Immunity 35, 681–693. 10.1016/j.immuni.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gras S, Chadderton J, Del Campo CM, Farenc C, Wiede F, Josephs TM, Sng XYX, Mirams M, Watson KA, Tiganis T, et al. (2016). Reversed T Cell Receptor Docking on a Major Histocompatibility Class I Complex Limits Involvement in the Immune Response. Immunity 45, 749–760. 10.1016/j.immuni.2016.09.007. [DOI] [PubMed] [Google Scholar]
- 13.Shugay M, Bagaev DV, Zvyagin IV, Vroomans RM, Crawford JC, Dolton G, Komech EA, Sycheva AL, Koneva AE, Egorov ES, et al. (2018). VDJdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res 46, D419–D427. 10.1093/nar/gkx760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang W, Wang L, Liu K, Wei X, Yang K, Du W, Wang S, Guo N, Ma C, Luo L, et al. (2020). PIRD: Pan Immune Repertoire Database. Bioinformatics 36, 897–903. 10.1093/bioinformatics/btz614. [DOI] [PubMed] [Google Scholar]
- 15.Tickotsky N, Sagiv T, Prilusky J, Shifrut E, and Friedman N. (2017). McPAS-TCR: a manually curated catalogue of pathology-associated T cell receptor sequences. Bioinformatics 33, 2924–2929. 10.1093/bioinformatics/btx286. [DOI] [PubMed] [Google Scholar]
- 16.Bagaev DV., Vroomans RMA., Sami J., Stervbo U., Rius C., Dolton G., Greenshields-Watson A., Attaf M., Egorov ES., Zvyagin IV., et al. (2020). VDJdb in 2019: database extension, new analysis infrastructure and a T-cell receptor motif compendium. Nucleic Acids Res 48, D1057–D1062. 10.1093/nar/gkz874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Friedensohn S, Khan TA, and Reddy ST (2017). Advanced Methodologies in High-Throughput Sequencing of Immune Repertoires. Trends Biotechnol 35, 203–214. 10.1016/j.tibtech.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 18.Howie B, Sherwood AM, Berkebile AD, Berka J, Emerson RO, Williamson DW, Kirsch I, Vignali M, Rieder MJ, Carlson CS, and Robins HS (2015). High-throughput pairing of T cell receptor alpha and beta sequences. Sci Transl Med 7, 301ra131. 10.1126/scitranslmed.aac5624. [DOI] [PubMed] [Google Scholar]
- 19.Hou X, Wang M, Lu C, Xie Q, Cui G, Chen J, Du Y, Dai Y, and Diao H. (2016). Analysis of the Repertoire Features of TCR Beta Chain CDR3 in Human by High-Throughput Sequencing. Cell Physiol Biochem 39, 651–667. 10.1159/000445656. [DOI] [PubMed] [Google Scholar]
- 20.Calis JJ, and Rosenberg BR (2014). Characterizing immune repertoires by high throughput sequencing: strategies and applications. Trends Immunol 35, 581–590. 10.1016/j.it.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bradley P, and Thomas PG (2019). Using T Cell Receptor Repertoires to Understand the Principles of Adaptive Immune Recognition. Annu Rev Immunol 37, 547–570. 10.1146/annurev-immunol-042718-041757. [DOI] [PubMed] [Google Scholar]
- 22.Duez M, Giraud M, Herbert R, Rocher T, Salson M, and Thonier F. (2016). Vidjil: A Web Platform for Analysis of High-Throughput Repertoire Sequencing. PLoS One 11, e0166126. 10.1371/journal.pone.0166126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosati E, Dowds CM, Liaskou E, Henriksen EKK, Karlsen TH, and Franke A. (2017). Overview of methodologies for T-cell receptor repertoire analysis. BMC Biotechnol 17, 61. 10.1186/s12896-017-0379-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruggiero E, Nicolay JP, Fronza R, Arens A, Paruzynski A, Nowrouzi A, Urenden G, Lulay C, Schneider S, Goerdt S, et al. (2015). High-resolution analysis of the human T-cell receptor repertoire. Nat Commun 6, 8081. 10.1038/ncomms9081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyama T, Kawase T, Kitaura K, Chishaki R, Shibata M, Oshima K, Hamana H, Kishi H, Muraguchi A, Kuzushima K, et al. (2017). Highly functional T-cell receptor repertoires are abundant in stem memory T cells and highly shared among individuals. Sci Rep 7, 3663. 10.1038/s41598-017-03855-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pogorelyy MV, Fedorova AD, McLaren JE, Ladell K, Bagaev DV, Eliseev AV, Mikelov AI, Koneva AE, Zvyagin IV, Price DA, et al. (2018). Exploring the pre-immune landscape of antigen-specific T cells. Genome Med 10, 68. 10.1186/s13073-018-0577-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miho E, Yermanos A, Weber CR, Berger CT, Reddy ST, and Greiff V. (2018). Computational Strategies for Dissecting the High-Dimensional Complexity of Adaptive Immune Repertoires. Front Immunol 9, 224. 10.3389/fimmu.2018.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yokota R, Kaminaga Y, and Kobayashi TJ (2017). Quantification of Inter-Sample Differences in T-Cell Receptor Repertoires Using Sequence-Based Information. Front Immunol 8, 1500. 10.3389/fimmu.2017.01500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laydon DJ, Bangham CR, and Asquith B. (2015). Estimating T-cell repertoire diversity: limitations of classical estimators and a new approach. Philos Trans R Soc Lond B Biol Sci 370, 20140291. 10.1098/rstb.2014.0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim Y, Sidney J, Pinilla C, Sette A, and Peters B. (2009). Derivation of an amino acid similarity matrix for peptide: MHC binding and its application as a Bayesian prior. BMC Bioinformatics 10, 394. 10.1186/1471-2105-10-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dash P., Fiore-Gartland AJ., Hertz T., Wang GC., Sharma S., Souquette A., Crawford JC., Clemens EB., Nguyen THO., Kedzierska K., et al. (2017). Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 547, 89–93. 10.1038/nature22383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pogorelyy MV, Minervina AA, Shugay M, Chudakov DM, Lebedev YB, Mora T, and Walczak AM (2019). Detecting T cell receptors involved in immune responses from single repertoire snapshots. PLoS Biol 17, e3000314. 10.1371/journal.pbio.3000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chronister WD, Crinklaw A, Mahajan S, Vita R, Kosaloglu-Yalcin Z, Yan Z, Greenbaum JA, Jessen LE, Nielsen M, Christley S, et al. (2021). TCRMatch: Predicting T-Cell Receptor Specificity Based on Sequence Similarity to Previously Characterized Receptors. Front Immunol 12, 640725. 10.3389/fimmu.2021.640725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glanville J, Huang H, Nau A, Hatton O, Wagar LE, Rubelt F, Ji X, Han A, Krams SM, Pettus C, et al. (2017). Identifying specificity groups in the T cell receptor repertoire. Nature 547, 94–98. 10.1038/nature22976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang H, Wang C, Rubelt F, Scriba TJ, and Davis MM (2020). Analyzing the Mycobacterium tuberculosis immune response by T-cell receptor clustering with GLIPH2 and genome-wide antigen screening. Nat Biotechnol 38, 1194–1202. 10.1038/s41587-020-0505-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang W, Hawkins PG, He J, Gupta NT, Liu J, Choonoo G, Jeong SW, Chen CR, Dhanik A, Dillon M, et al. (2021). A framework for highly multiplexed dextramer mapping and prediction of T cell receptor sequences to antigen specificity. Sci Adv 7, eabf5835. 10.1126/sciadv.abf5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gorski J, Yassai M, Zhu X, Kissela B, Kissella B, Keever C, and Flomenberg N. (1994). Circulating T cell repertoire complexity in normal individuals and bone marrow recipients analyzed by CDR3 size spectratyping. Correlation with immune status. J Immunol 152, 5109–5119. [PubMed] [Google Scholar]
- 38.Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, and Chudakov DM (2015). MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods 12, 380–381. 10.1038/nmeth.3364. [DOI] [PubMed] [Google Scholar]
- 39.Minervina AA, Komech EA, Titov A, Bensouda Koraichi M, Rosati E, Mamedov IZ, Franke A, Efimov GA, Chudakov DM, Mora T, et al. (2021). Longitudinal high-throughput TCR repertoire profiling reveals the dynamics of T-cell memory formation after mild COVID-19 infection. Elife 10, e63502. 10.7554/eLife.63502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen G, Yang X, Ko A, Sun X, Gao M, Zhang Y, Shi A, Mariuzza RA, and Weng NP (2017). Sequence and Structural Analyses Reveal Distinct and Highly Diverse Human CD8(+) TCR Repertoires to Immunodominant Viral Antigens. Cell Rep 19, 569–583. 10.1016/j.celrep.2017.03.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dayhoff MO (1972). Atlas of protein sequence and structure (National Biomedical Research Foundation.). [Google Scholar]
- 42.Henikoff S, and Henikoff JG (1992). Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A 89, 10915–10919. 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nolan S, Vignali M, Klinger M, Dines JN, Kaplan IM, Svejnoha E, Craft T, Boland K, Pesesky M, Gittelman RM, et al. (2020). A large-scale database of T-cell receptor beta (TCRbeta) sequences and binding associations from natural and synthetic exposure to SARS-CoV-2. Res Sq. 10.21203/rs.3.rs-51964/v1. [DOI] [Google Scholar]
- 45.Snyder TM., Gittelman RM., Klinger M., May DH., Osborne EJ., Taniguchi R., Zahid HJ., Kaplan IM., Dines JN., Noakes MT., et al. (2020). Magnitude and Dynamics of the T-Cell Response to SARS-CoV-2 Infection at Both Individual and Population Levels. medRxiv, 2020.2007.2031.20165647. 10.1101/2020.07.31.20165647. [DOI] [Google Scholar]
- 46.Singh NK, Riley TP, Baker SCB, Borrman T, Weng Z, and Baker BM (2017). Emerging Concepts in TCR Specificity: Rationalizing and (Maybe) Predicting Outcomes. J Immunol 199, 2203–2213. 10.4049/jimmunol.1700744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leinster T, and Cobbold CA (2012). Measuring diversity: the importance of species similarity. Ecology 93, 477–489. 10.1890/10-2402.1. [DOI] [PubMed] [Google Scholar]
- 48.Chowell D, Krishna S, Becker PD, Cocita C, Shu J, Tan X, Greenberg PD, Klavinskis LS, Blattman JN, and Anderson KS (2015). TCR contact residue hydrophobicity is a hallmark of immunogenic CD8+ T cell epitopes. Proc Natl Acad Sci U S A 112, E1754–1762. 10.1073/pnas.1500973112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Calis JJ, Maybeno M, Greenbaum JA, Weiskopf D, De Silva AD, Sette A, Kesmir C, and Peters B. (2013). Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput Biol 9, e1003266. 10.1371/journal.pcbi.1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wells DK, van Buuren MM, Dang KK, Hubbard-Lucey VM, Sheehan KCF, Campbell KM, Lamb A, Ward JP, Sidney J, Blazquez AB, et al. (2020). Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell 183, 818–834 e813. 10.1016/j.cell.2020.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yin L, Crawford F, Marrack P, Kappler JW, and Dai S. (2012). T-cell receptor (TCR) interaction with peptides that mimic nickel offers insight into nickel contact allergy. Proc Natl Acad Sci U S A 109, 18517–18522. 10.1073/pnas.1215928109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cole DK, Yuan F, Rizkallah PJ, Miles JJ, Gostick E, Price DA, Gao GF, Jakobsen BK, and Sewell AK (2009). Germ line-governed recognition of a cancer epitope by an immunodominant human T-cell receptor. J Biol Chem 284, 27281–27289. 10.1074/jbc.M109.022509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carter JA, Preall JB, Grigaityte K, Goldfless SJ, Jeffery E, Briggs AW, Vigneault F, and Atwal GS (2019). Single T Cell Sequencing Demonstrates the Functional Role of alphabeta TCR Pairing in Cell Lineage and Antigen Specificity. Front Immunol 10, 1516. 10.3389/fimmu.2019.01516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schumacher TN, and Schreiber RD (2015). Neoantigens in cancer immunotherapy. Science 348, 69–74. 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 55.Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990). Basic local alignment search tool. Journal of molecular biology 215, 403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 56.Chiu TP, Rao S, Mann RS, Honig B, and Rohs R. (2017). Genome-wide prediction of minor-groove electrostatic potential enables biophysical modeling of protein-DNA binding. Nucleic Acids Res 45, 12565–12576. 10.1093/nar/gkx915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Knapp B, van der Merwe PA, Dushek O, and Deane CM (2019). MHC binding affects the dynamics of different T-cell receptors in different ways. PLOS Computational Biology 15, e1007338. 10.1371/journal.pcbi.1007338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lanzarotti E, Marcatili P, and Nielsen M. (2019). T-Cell Receptor Cognate Target Prediction Based on Paired alpha and beta Chain Sequence and Structural CDR Loop Similarities. Front Immunol 10, 2080. 10.3389/fimmu.2019.02080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Milighetti M, Shawe-Taylor J, and Chain B. (2021). Predicting T Cell Receptor Antigen Specificity From Structural Features Derived From Homology Models of Receptor-Peptide-Major Histocompatibility Complexes. Front Physiol 12, 730908. 10.3389/fphys.2021.730908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bryant P, Pozzati G, and Elofsson A. (2022). Improved prediction of protein-protein interactions using AlphaFold2. Nat Commun 13, 1265. 10.1038/s41467-022-28865-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shoukat MS., Foers AD., Woodmansey S., Evans SC., Fowler A., and Soilleux EJ. (2021). Use of machine learning to identify a T cell response to SARS-CoV-2. Cell reports. Medicine 2, 100192. 10.1016/j.xcrm.2021.100192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sidhom JW, and Baras AS (2021). Deep learning identifies antigenic determinants of severe SARS-CoV-2 infection within T-cell repertoires. Sci Rep 11, 14275. 10.1038/s41598-021-93608-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fischer DS, Wu Y, Schubert B, and Theis FJ (2020). Predicting antigen specificity of single T cells based on TCR CDR3 regions. Molecular systems biology 16, e9416. 10.15252/msb.20199416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weber A, Born J, and Rodriguez Martinez M. (2021). TITAN: T-cell receptor specificity prediction with bimodal attention networks. Bioinformatics 37, i237–i244. 10.1093/bioinformatics/btab294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lu T, Zhang Z, Zhu J, Wang Y, Jiang P, Xiao X, Bernatchez C, Heymach JV, Gibbons DL, Wang J, et al. (2021). Deep learning-based prediction of the T cell receptor-antigen binding specificity. Nat Mach Intell 3, 864–875. 10.1038/s42256-021-00383-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moris P, De Pauw J, Postovskaya A, Gielis S, De Neuter N, Bittremieux W, Ogunjimi B, Laukens K, and Meysman P. (2021). Current challenges for unseen-epitope TCR interaction prediction and a new perspective derived from image classification. Brief Bioinform 22. 10.1093/bib/bbaa318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Logunova NN, Kriukova VV, Shelyakin PV, Egorov ES, Pereverzeva A, Bozhanova NG, Shugay M, Shcherbinin DS, Pogorelyy MV, Merzlyak EM, et al. (2020). MHC-II alleles shape the CDR3 repertoires of conventional and regulatory naive CD4(+) T cells. Proc Natl Acad Sci U S A 117, 13659–13669. 10.1073/pnas.2003170117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Odak I, Raha S, Schultze-Florey C, Tavil S, Ravens S, Ganser A, Forster R, Prinz I, and Koenecke C. (2019). Focusing of the regulatory T-cell repertoire after allogeneic stem cell transplantation indicates protection from graft-versus-host disease. Haematologica 104, e577–e580. 10.3324/haematol.2019.218206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ng AHC, Peng S, Xu AM, Noh WJ, Guo K, Bethune MT, Chour W, Choi J, Yang S, Baltimore D, and Heath JR (2019). MATE-Seq: microfluidic antigen-TCR engagement sequencing. Lab Chip 19, 3011–3021. 10.1039/c9lc00538b. [DOI] [PubMed] [Google Scholar]
- 70.Bentzen AK, Marquard AM, Lyngaa R, Saini SK, Ramskov S, Donia M, Such L, Furness AJ, McGranahan N, Rosenthal R, et al. (2016). Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat Biotechnol 34, 1037–1045. 10.1038/nbt.3662. [DOI] [PubMed] [Google Scholar]
- 71.Bethune MT, Comin-Anduix B, Hwang Fu YH, Ribas A, and Baltimore D. (2017). Preparation of peptide-MHC and T-cell receptor dextramers by biotinylated dextran doping. Biotechniques 62, 123–130. 10.2144/000114525. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. GLIPH2 analysis of CMV pp65 antigen-specific TCRs in VDJdb, related to Figure 2.
Supplementary Table 4. CMV pp65-specific TCRs in VDJdb enriched for high frequency k-mers at the center of the chain, related to Figure 4.
Supplementary Table 5. The number of TCRs associated with each MIRA epitope. Only epitopes with >500 sequences were analyzed, related to Figure 5.
Supplementary Table 6. Entropy contributions from paired chains in VDJdb, related to Figure 6. Epitopes had to have over 250 paired chains. Each k-mer at each position was only considered if over 3 percent of the total TCRs at that position contained a contribution from that k-mer. The average entropy over the paired chain was obtained for each subset of TCRs containing a k-mer.
Supplementary Table 7. Entropy contributions from unpaired chains in VDJdb, related to Figure 6. Epitopes had to have over 250 unpaired chains. Each k-mer at each position was only considered if over 2 percent of the total TCRs at that position contained a contribution from that k-mer. The average entropy over the paired chain was obtained for each subset of TCRs containing a k-mer. The and chains were analyzed separately.
Data Availability Statement
Single-cell TCR-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. TCR sequence data was obtained from VDJdb, McPAS-TCR, TBAdb, and MIRA databases. Links are provided in the key resources table.
All original code has been deposited at Github and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Healthy HLA-A02 PBMCs | StemCell Technologies | Cat#70025 |
| Chemicals, peptides, and recombinant proteins | ||
| Biotinylated HLA-A2 MHC-I Monomer | Fred Hutchinson Cancer Research Center Immune Monitoring Shared Resource | N/A |
| NLVPMVATV peptide | IBA-Lifesciences | N/A |
| PE-conjugated streptavidin | Thermo Fisher | Cat#SA10041 |
| APC-conjugated streptavidin | Thermo Fisher | Cat#SA1005 |
| 500 kDa biotin-dextran | Nanocs | Cat#DX500-BN-1 |
| Critical commercial assays | ||
| Single Cell Immune Profiling Kit | 10X | Cat#1000542 |
| Deposited data | ||
| VDJDB | Shugay et al.13 | https://vdjdb.cdr3.net/ |
| TBAdb | Zhang et al.14 | https://db.cngb.org/pird/home/ |
| McPAS-TCR | Tickotsky et al.15 | http://friedmanlab.weizmann.ac.il/McPAS-TCR/ |
| MIRA COVID sequences | Nolan et al.44 | https://doi.org/10.21203/rs.3.rs-51964/v1 |
| Raw and Analyzed Data | This paper | SRA: PRJNA918821 |
| Software and algorithms | ||
| R v4.1.1 | CRAN | https://cran.r-project.org/ |
| RStudio v1.4.1717 | RStudio | https://www.rstudio.com |
| uwot R package | James Melville | https://github.com/jlmelville/uwot |
| data.table R package | Matt Dowle | https://cran.r-project.org/web/packages/data.table/index.html |
| dplyr R package | Hadley Wickham | https://cran.r-project.org/web/packages/dplyr/index.html |
| ggplot2 R package | Thomas Lin Pedersen | https://cran.r-project.org/web/packages/ggplot2/index.html |
| pheatmap R package | Raivo Kolde | https://cran.r-project.org/web/packages/pheatmap/index.html |
| ggnewscale R package | Elio Campitelli | https://cran.r-project.org/web/packages/ggnewscale/index.html |
| GLIPH | Glanville et al.34 | http://50.255.35.37:8080/ |
| AlphaFold v2.0 | Deepmind | https://github.com/deepmind/alphafold |
| SPAN-TCR | This paper | https://doi.org/10.5281/zenodo.7637683 |
| Other | ||
| UV Crosslinker | Boekel | Cat#234100 |