

Abstract
CREB is a transcription factor implicated in the pathogenesis of multiple cancers. Targeting CREB is a promising strategy to develop potential cancer therapeutics. Previously, we identified 666-15 as a potent CREB inhibitor. Herein, we designed an ester prodrug of 666-15 through a long-range O,N-acyl transfer reaction for improved aqueous solubility. Unexpectedly, we discovered a small molecule 11 (653-47) that can potentiate the CREB inhibitory activity of 666-15 although 653-47 alone does not inhibit CREB.
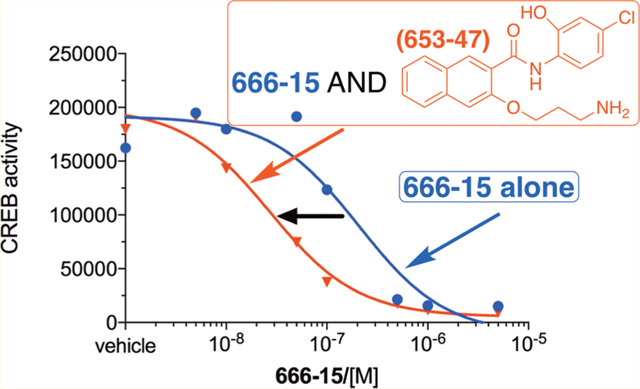
Graphical Abstract
INTRODUCTION
cAMP-response element binding protein (CREB) is a critical nuclear transcription factor, enabling cells to detect and respond to the extracellular microenvironment through transcriptional regulation.1 In an unstimulated cell, CREB constitutively binds to the cAMP-response elements (CRE) in the genome in a transcriptionally inactive state. Its transcription activity is activated upon phosphorylation mediated by a number of protein serine/threonine kinases, which are collectively called CREB kinases.1 Once phosphorylated, CREB can bind to the transcription coactivators, including CREB-binding protein (CBP) and its paralogue p300 through their KIX domain and kinase-inducible domain (KID) in CREB.2,3 The known CREB kinases include protein kinase A (PKA), protein kinase B (PKB/Akt), mitogen-activated protein kinases (MAPKs), and ribosomal S6 kinase (pp90RSK). The CREB’s transcription activity and phosphorylation status are tightly regulated in normal cells, allowing activation of CREB in a timely and pulsative fashion. The key signaling event to turn off CREB’s transcription activity is dephosphorylation. Three phosphatases, including protein phosphatase 1 (PP1),4 protein phosphatase 2 (PP2),5 and phosphatase and tensin homologue (PTEN),6 are known to dephosphorylate CREB and consequently inactivate CREB’s transcription activity.
The CREB kinases are often overactivated in cancer cells through either mutation or excessive growth signals. On the other hand, the phosphatases that can dephosphorylate CREB are known as potent tumor suppressors and often inactivated in the cancer cells through both genetic and nongenetic mechanisms.7 As a consequence of the positive and negative regulation of CREB, cancer tissues from different organs have been consistently shown to present higher expression levels of CREB and/or phosphorylated CREB than their normal counterparts.8 These cancers include acute myeloid leukemia (AML), breast cancer, prostate cancer, nonsmall cell lung cancer, glioblastoma (GBM), and kidney cancer.8–10 Very interestingly, the CREB signaling has also been shown to mediate melanoma cell resistance to BRAF(V600E) and MAPK inhibitors as well as the progression of castration-resistant prostate cancer.11,12 Therefore, targeting CREB has been pursued as an attractive strategy to develop novel cancer therapies that may ultimately overcome therapy resistance.8,13–15
Recently, we described the identification of 666-15 as a potent and selective inhibitor of CREB-mediated gene transcription (Figure 1).13,16 Among a panel of other transcription factors, it was found that 666-15 selectively inhibited CREB-mediated gene transcription.16 With 666-15 as a chemical tool, we showed that in vivo systemic inhibition of CREB was well-tolerated and produced efficacious anti-breast cancer activity.13,16 Others have also shown that 666-15 inhibited AML, GBM, and pancreatic cancer cell growth in different preclinical models.17–19 Furthermore, recent studies have shown that 666-15 could target the tumor microenvironment to present beneficial anticancer effect. For example, 666-15 was shown to inhibit pro-tumoric IL-6 expression from cancer-associated fibroblasts20 and reverse the immunosuppressive effect of myeloid-derived suppressor cells (MDSC),21 suggesting the potential of 666-15 to modulate tumor microenvironment and immunotherapy.
Figure 1.

Chemical structures of CREB inhibitors 666-15, 1, 2, and designed potential ester prodrug 3. The migrating acyl group is highlighted in red.
666-15 was derived from optimization of a less potent derivative 1,13 which was initially identified through our discovery of a rapid long-range O,N-acyl transfer reaction of the ester compound 2 (Figure 1).15 One advantage of compound 2 over compound 1 is its high aqueous solubility (>100 mg/mL in ddIH2O).15 In this report, we took advantage of the fact of high aqueous solubility of 2 and designed a corresponding ester compound 3 as a potential prodrug of 666-15 with improved aqueous solubility. Unexpectedly, compound 3 was found to undergo a different conversion reaction at physiologically relevant pH (7.40). Furthermore, we discovered a novel compound that can potentiate 666-15’s inhibitory effect against CREB-mediated gene transcription in living cells.
RESULTS AND DISCUSSION
The synthesis of compound 3 is presented in Scheme 1. Building blocks 4 and 5 were prepared as described before.13 The ester coupling reaction between phenol 4 and acid 5 was found to be problematic. Under the coupling conditions we used before (BOP, DIPEA, room temperature),13 a mixture of compositional isomers 6 and 7 involving acyl group swapping was obtained. These two isomers were only partially separable by conventional silica gel column chromatograph. Other coupling conditions (EDCI, DIPEA; DCC, DIPEA; MsCl, Et3N) were also attempted, but the results were found to be similar. The formation of the unanticipated alternative ester 7 was likely a result of generation of imide intermediate 8, although it was not detected from the reaction mixture. The intermediate 8 could be generated either before the formation of 6 or after 6 was formed. Once 8 was produced, it could generate either 6 or 7 through an ester bond formation (Scheme 1). After considerable experimentation involving the modifications of solvents, coupling reagents, and temperature, we found that lowering the reaction temperature from room temperature to 0 °C and shortening the reaction time to 1 h could significantly inhibit the formation of 7, although complete reaction conversion was not achieved under these conditions. The Boc groups in 6 were then deprotected under acidic conditions (HCl, DCM) to give 3 uneventfully (Supporting Information (SI), Figure S1). As expected, compound 3 was found to be highly soluble in ddIH2O (>100 mg/mL). This is in strong contrast to 666-15, which is much less soluble in ddIH2O (<0.5 mg/mL).
Scheme 1. Synthesis of Ester Compound 3.

With the desired compound 3 in hand, we first investigated the possibility of transformation of 3 into 666-15 in the complete cell culture media (Dulbecco’s Modified Eagle Medium with 10% fetal bovine serum (FBS)). When 3 was incubated with the complete cell culture media at 37 °C, it was rapidly converted into four new product peaks as assessed by HPLC (Figure 2B). In stark contrast to the structural congener 2,15 only a small amount of 666-15 was generated under the reaction condition. To investigate if simple ester hydrolysis was occurring to generate products 9 and 10 (Figure 2A), we compared the main peaks eluted at 10.5 and 11.2 min with standard samples of 9 and 10 by HPLC. However, these peaks did not correspond to either compound 9 or 10 (data not shown).
Figure 2.

Compound 3 was converted into 666-15, 11, and 12 in the complete cell culture media. (A) Reaction conversion of 3 in complete cell culture media to generate a mixture of 666-15, 11, and 12. (B) HPLC chromatograms of compound 3 incubated in complete cell culture media for 5 and 30 min.
To identify the products formed from incubating 3 in the complete cell culture media, we treated compound 3 in phosphate-buffered saline (PBS, pH = 7.40) (Scheme 2). An additional Boc protection step was added to facilitate purification of compounds containing free amino groups. After careful silica gel column purification and NMR analyses, we obtained an inseparable mixture of 13 and 14 in 18% total yield with a ratio of 3:1 as assessed by 1H NMR. Furthermore, two unexpected products, 15 (47%) and 12 (29%), were formed. The identity of 12 was further confirmed by an independent synthesis from 1613 through Boc deprotection and cyclization (Scheme 2).
Scheme 2. Reaction Conversion of 3 in PBS.

With the products identified from Scheme 2 and standard samples from the Boc-deprotected products, we were able to assign the peak eluted at 10.5 min being 12, 11.2 min being 11, and the shoulder peak close to 666-15 being 17 (Figure 2B and Scheme 3). On the basis of our previous results with compound 2,15 we initially hypothesized that the mechanism of formation of 666-15 from 3 would be through pathway A by a long-range O,N-acyl transfer reaction (Scheme 3). Although we could not completely rule out this possibility, a more likely scenario is through pathway B as shown in Scheme 3. In this mechanism, imide 18 was first formed from 3. Three possibilities exist for imide 18. Pathway a would generate 11 and 12, and this route was found to be the major pathway. Minor pathways b and c would generate 17 and 666-15, respectively.
Scheme 3. Proposed Mechanistic Pathways of Conversion of 3 into 666-15, 11, 12, and 17 in Physiologically Relevant Buffers.

Because generation of the potent CREB inhibitor 666-15 from 3 in the complete tissue culture media was not the major pathway (Figure 2B), we expected that compound 3 would not be a potent CREB inhibitor. To test this possibility, we employed a transcription luciferase reporter assay in HEK 293T cells.22 Briefly, the cells were transfected with a CREB Renilla luciferase reporter plasmid (CRE-RLuc). This plasmid expresses Renilla luciferase under the control of three tandem copies of CRE in the promoter region. After transfection, the cells were treated with different concentrations of compound 3 for 30 min when forskolin (Fsk) was added to stimulate CREB phosphorylation and its subsequent transcription activity. In this assay, 666-15 has an IC50 ∼ 80 nM (Table 1).13 Surprisingly, compound 3 was found to be quite potent, with an IC50 = 0.25 ± 0.11 μM (Table 1 and Figure 3A) despite the fact that only a small fraction of 3 was converted into 666-15 under these conditions (Figure 2B). This result suggested that other components in the reaction mixture might inhibit CREB-mediated gene transcription. Thus, the major compounds 11 and 12 were separately evaluated for their CREB inhibition potency. Compound 11 was found to be a very weak inhibitor with IC50 = 26.3 ± 13.6 μM, while compound 12 was inactive (Table 1 and Figure 3A). Consistent with its weak activity in inhibiting CREB’s transcription, compound 11 did not inhibit KIX–KID interaction as assessed in a biochemical split Renilla luciferase complementation assay (IC50 > 50 μM).22 The rather weak CREB inhibition activity associated with 11 and 12 suggested other possibilities to account for the potent CREB inhibition activity of 3. One of the possibilities was that the mixture might have a synergistic activity. To test this possibility, we evaluated the combination of 666-15 with 11 or 12. As shown in Figure 3B, combination of 666-15 with 12 did not produce further inhibition of CREB-mediated gene transcription. However, when 666-15 was combined with 11, it significantly potentiated activity of 666-15 in inhibiting CREB-mediated gene transcription. Importantly, the synergistic inhibition occurred at concentrations where 11 alone was not inhibiting CREB-mediated gene transcription (Figures 3A,B). The synergy between 666-15 and 11 was further demonstrated by the low combination indexes (CI) calculated using the Chou–Talalay method (SI, Figure S2 and Table S1), where CI > 1.0 indicates antagonism, CI = 1.0 indicates additive effect, and CI < 1.0 indicates synergism.23 These results indicated that the potent CREB inhibition activity of 3 is primarily a result of the combination of in situ generated 666-15 and 11.
Table 1.
CREB Inhibitory Activity of Different Compounds
| compd | CREB inhibition (IC50, μM)a |
|---|---|
| 666-15 | 0.081 ± 0.04 |
| 3 | 0.25 ± 0.11 |
| 11 | 26.3 ± 13.6 |
| 12 | >50 |
The IC50 refers to the concentration needed to inhibit 50% of the CREB-mediated gene transcription in HEK 293T cells using a transcription Renilla luciferase reporter assay. The data are presented as mean ± SD of at least two independent experiments, which were performed in triplicate.
Figure 3.

Inhibition of CREB-mediated gene transcription. HEK 293T cells were transfected with CRE-RLuc. Then the cells were treated with increasing concentrations of individual compounds (A) or drug combinations (B) for 30 min before the addition of Fsk (10 μM) for 5 h. The Renilla luciferase activity was normalized to the protein concentration in each well and expressed as relative luciferase unit (RLU)/μg protein. The errors are SEM from one representative experiment in triplicates.
To further test that compound 11 could potentiate 666-15’s CREB inhibitory activity, we evaluated the endogenous CREB target gene expression in HEK 293T cells. Nuclear receptor subfamily 4 group A member 2 (NR4A2) is a well-established CREB target gene in HEK 293T cells.24 We have shown that NR4A2 was robustly stimulated by Fsk, and its expression was suppressed by 666-15.13 Therefore, we investigated the expression level of NR4A2 in HEK293T cells with the combination of 666-15 and 11 using quantitative reverse transcription polymerase chain reaction (qRT-PCR). As reported previously,13 the expression level of NR4A2 was stimulated by Fsk (Figure 4). When the cells were pretreated with low concentration of 666-15 (50 nM), the expression level of NR4A2 was not significantly reduced. At higher concentration (100 nM), its transcript level was significantly inhibited. Consistent with the transcription reporter assay results shown in Figure 3B, 5 μM of compound 11 alone did not inhibit the expression of NR4A2. Instead, a minor increase of its expression was observed. However, when low concentration of 666-15 (50 nM) was combined with compound 11 (5 μM), significant inhibition of NR4A2 transcription was observed (Figure 4). Together with the transcription reporter assay results shown in Figure 3B, these data support that while compound 11, which was named as 653-47, was not active alone, it could significantly potentiate the CREB inhibitory activity of 666-15.
Figure 4.

Synergistic inhibition of CREB-mediated gene transcription in HEK 293T cells by 666-15 and 11. HEK 293T cells were treated with 666-15 and 11 for 1 h, when the cells were stimulated with Fsk (10 μM) for 45 min. Then the cells were subjected to qRT-PCR analysis after the total RNA was isolated using HPRT as a reference gene. The errors are SEM of two independent experiments performed in triplicates. * P < 0.05 by Student t-test.
We previously showed that 666-15 potently inhibited breast cancer cell growth.13 Our discovery of 11 as a potentiator to enhance 666-15’s potency in inhibiting CREB-mediated gene transcription suggested that it might also potentiate 666-15’s anticancer potential in breast cancer cells. To test this hypothesis, we treated MDA-MB-231 cells with different concentrations of 666-15 along with or without 11 for 72 h. Then the antiproliferative effect was evaluated using MTT assay as described before.13 As shown in SI, Figure S3, 666-15 was able to dose-dependently inhibit the cell growth. Compound 11 alone did not appreciably inhibit the cell growth at low concentrations (0.5 or 1.0 μM). However, when 11 was combined with 666-15, it further enhanced 666-15’s antiproliferative effect as indicated by the CI values. The CI values are all less than 1.0, indicating synergism between 666-15 and 11.
CONCLUSIONS
In this paper, we described our unexpected discovery of a potentiator compound 11 or 653-47 to synergistically inhibit CREB-mediated gene transcription with 666-15. We initially set out to improve the aqueous solubility of 666-15 by designing an ester prodrug 3, which was envisioned to undergo a long-range O,N-acyl transfer reaction. Our results showed that a modified O,N-acyl transfer process through an imide intermediate is a more likely mechanism of formation of 666-15 from the ester prodrug 3 (Scheme 3). Unexpectedly, we discovered compound 653-47 as an enhancer of 666-15’s activity in inhibiting CREB-mediated gene transcription and breast cancer cell growth even though 653-47 alone does not inhibit these activities. This discovery opens new avenues to further target CREB-mediated gene transcription with small molecules. Because CREB’s transcription activity is regulated at multiple levels, including phosphorylation, nuclear translocation, chromatin modulation, and DNA-binding, this discovery of 653-47 shall also provide a unique tool to further study CREB regulation.
EXPERIMENTAL SECTION
Chemistry: General.
A Glass Contour solvent purification system was used to purify all the anhydrous solvents to be used for reactions. Melting points were determined in capillary tubes using Mel-Temp and are uncorrected. All 1H and 13C NMR spectra were obtained in a Bruker Avance 400 MHz spectrometer using CDCl3 or DMSO-d6 as the solvent, and the chemical shifts of the residual CHCl3 (δ 7.24) or DMSO (δ 2.50) were taken as reference. Chemical shifts (δ) are reported in parts per million (ppm), and the signals are described as brs (broad singlet), d (doublet), dd (doublet of doublet), td (triplet of doublet), m (multiplet), q (quartet), s (singlet), and t (triplet). Coupling constants (J values) are given in Hz. Silica gel flash chromatography was performed using 230–400 mesh silica gel (EMD). All reactions were monitored using thin-layer chromatography (TLC) on silica gel plates (EMD). Yields were of purified compounds. All final compounds for biological evaluations were confirmed to be of >95% purity based on reverse phase HPLC (Waters, Milford, MA) analysis using an XBridge C18 column (4.6 mm × 150 mm) and detected at 254 nm. The mobile phases for HPLC are water and acetonitrile, both of which contained 0.1% TFA. The mass spectra were obtained from a Thermo Electron LTQ-Orbitrap Discovery high resolution mass spectrometer (Thermo Scientific) with electrospray operated either in positive or negative mode.
2-(3-(2-Aminoethoxy)-2-naphthamido)-5-chlorophenyl 3-(3-Aminopropoxy)-2-naphthoate Dihydrochloride (3).
An HCl solution in Et2O (2 M, 3 mL) was added to a stirred solution of 6 (75 mg, 0.095 mmol) in DCM (3 mL) at room temperature. The resulting mixture was stirred at room temperature for 4 h. The solvent was removed and the residue was treated with acetone and the precipitate was collected by filtration to give product 3 (8 mg, 13%) as a white solid; mp 216–217 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.18 (s, 1 H), 8.75 (s, 1 H), 8.24 (brs, 3 H), 8.09 (s, 1 H), 8.01–7.78 (m, 8 H), 7.66–7.60 (m, 3 H), 7.58–7.45 (m, 4 H), 7.40 (t, J = 7.6 Hz, 1 H), 7.35 (t, J = 7.7 Hz, 1 H), 4.28 (t, J = 5.4 Hz, 2 H), 4.23 (t, J = 5.4 Hz, 2 H), 3.21 (q, J = 5.8 Hz, 2 H), 3.01 (q, J = 5.8 Hz, 2 H), 2.03 (quintet, J = 6.0 Hz, 2 H). HRESIMS calcd for C33H31ClN3O5 [M + H]+ 584.1947, found 584.1943.
2-(3-(2-((tert-Butoxycarbonyl)amino)ethoxy)-2-naphthamido)-5-chlorophenyl 3-(3-((tert-Butoxycarbonyl)amino)propoxy)-2-naphthoate (6).
To a stirred solution of 515 (173 mg, 0.5 mmol) in dichloromethane (10 mL) was added BOP (221 mg, 0.5 mmol) and DIPEA (90 μL, 0.5 mmol). The reaction mixture was stirred at room temperature for 5 min. Then the reaction mixture was cooled down to 0 °C. Compound 413 (229 mg, 0.5 mmol) and another portion of DIPEA (120 μL, 0.65 mmol) were added sequentially. The resulting mixture was stirred for 1 h at 0 °C. The solvent was removed under reduced pressure, and the residue was purified by flash column chromatography, eluting with dichloromethane–ethyl acetate (20:1) to give the crude product 6, which was further purified by the Biotage purification system, eluting with hexanes–ethyl acetate (20:1 to 4:1) to give pure product 6 (75 mg, 19%) and some less pure product (170 mg, 43%). 1H NMR (400 MHz, CDCl3) δ 9.81 (s, 1 H), 8.74 (s, 1 H), 8.58 (s, 1 H), 8.41 (d, J = 8.8 Hz, 1 H), 7.87 (d, J = 8.2 Hz, 1 H), 7.81 (d, J = 8.2 Hz, 1 H), 7.76 (d, J = 8.2 Hz, 1 H), 7.67 (d, J = 8.2 Hz, 1 H), 7.59 (t, J = 7.6 Hz, 1 H), 7.51 (t, J = 7.5 Hz, 1 H), 7.44–7.37 (m, 3 H), 7.34 (dd, J = 8.8, 2.3 Hz, 1 H), 7.24 (s, 1 H), 7.16 (s, 1 H), 5.40 (brs, 1 H), 4.73 (brs, 1 H), 4.12 (t, J = 7.1 Hz, 2 H), 3.91 (t, J = 5.4 Hz, 2 H), 3.33–3.07 (m, 4 H), 1.89 (brs, 2 H), 1.35 (s, 9 H), 1.33 (s, 9 H). 13C NMR (101 MHz, CDCl3) δ 163.67, 163.47, 156.28, 155.88, 155.05, 153.27, 142.16, 136.73, 135.79, 134.20, 129.69, 129.62, 129.58, 129.10, 128.98, 128.68, 128.35, 127.33, 126.70, 126.60, 126.38, 125.13, 125.03, 124.84, 122.73, 108.42, 107.92, 79.62, 78.88, 68.33, 67.55, 39.37, 38.30, 29.05, 28.36, 28.27.
3,4-Dihydronaphtho[2,3-f ][1,4]oxazepin-5(2H)-one (12).
Compound 16 (103 mg, 0.3 mmol) was treated with TFA (1 mL) for 1 h at room temperature. TFA was removed under reduced pressure, and the residue was redissolved in MeOH (4 mL) and DIPEA (1 mL). The mixture was heated under reflux for overnight. The solvent was removed under reduced pressure, and the residue was purified by flash column chromatography, eluting with dichloromethane–methanol (20:1) to give product 12 (25 mg, 39%) as a yellow solid; mp 128–129 °C. 1H NMR (400 MHz, CDCl3) δ 8.39 (s, 1 H), 7.91 (d, J = 8.2 Hz, 1 H), 7.78 (d, J = 8.3 Hz, 1 H), 7.56–7.51 (m, 1 H), 7.50 (s, 1 H), 7.45 (m, 1 H), 7.07 (s, 1 H), 4.39 (t, J = 5.4 Hz, 2 H), 3.48 (q, J = 5.6 Hz, 2 H). 13C NMR (101 MHz, CDCl3) δ 171.14, 151.27, 136.06, 132.05, 130.11, 128.93, 128.09, 127.50, 126.81, 125.55, 118.36, 73.79, 40.50. HRESIMS calcd for C13H12NO2 [M + H]+ 214.0863, found 214.0858.
Biology.
General.
HEK293T cells were from ATCC and MDA-MB-231 cells were from the Development Therapeutics Program at the National Cancer Institute. The cells were routinely cultured in DMEM (Life Technologies) with 10% fetal bovine serum (FBS, Hyclone) in a humidified incubator with 5% CO2 at 37 °C. The cells were confirmed to be mycoplasma negative and authenticated through STR profiling. pCRE-RLuc was reported before.22 All the compounds for biological testing were dissolved in DMSO as stock solutions except compound 3, which was dissolved in DMF because compound 3 was found to be unstable in DMSO for long-term storage. Compounds 11 and 17 were reported previously.13
Inhibition of CREB-Mediated Gene Transcription.
HEK293T cells in a well of 6-well plate were transfected with a plasmid (1 μg) encoding Renilla luciferase under the control of three copies of CRE (pCRE-RLuc) using Lipofectamine2000 (Life Technologies) for 3 h. Then the cells were replated into a 96-well plate, and the cells were allowed to attach to the bottom of the plate for overnight. The cells were treated with different concentrations of the compounds for 30 min, when Fsk (10 μM) was added to the cells. The cells were further incubated for 5 h at 37 °C. The media were removed and the cells were lysed in 30 μL of 1× Renilla luciferase lysis buffer (Promega) and the Renilla luciferase activity was measured using the Renilla luciferase assay system (Promega). The protein concentration in each well was determined using the Protein Assay Dye Reagent Concentrate (Biorad). The luciferase activity in each well was normalized to the protein concentration and expressed as relative luciferase unit (RLU)/μg protein. The IC50 was calculated in Prism 5.0 using the nonlinear regression analysis.
Conversion of Ester 3 in DMEM.
Compound 3 (200 μM) was incubated with complete tissue culture media (DMEM with 10% FBS) at 37 °C for various time periods. An aliquot of 10 μL was taken at a given time point and mixed with acetonitrile (90 μL). The mixture was centrifuged at 14000× rpm for 5 min at room temperature in a tabletop centrifuge to precipitate proteins. The supernatant was collected and analyzed by HPLC eluting with a linear of water and acetonitrile, both of which contained 0.1% TFA.
Inhibition of Endogenous CREB Target Gene Expression.
HEK 293T cells in 6-well plates were treated with different concentrations of 666-15 along with or without 11 for 1 h. Then the cells were stimulated with Fsk (10 μM) for 45 min. The total RNA was isolated using NucleoSpin RNA isolation kit (Takara Bio). Following RNA isolation, the first strand cDNA was synthesized using a PrimeScript first strand cDNA synthesis kit (Takara Bio) with random hexamers as the primers. Quantitative PCR reactions were performed with TB Green Advantage qPCR Premix (Takara Bio). The relative mRNA level was determined with ΔΔCt method using HPRT as a reference gene. Primers used for qRT-PCR are available upon request.
Cell Growth Inhibition Assay.
The cells were plated in 96-well plates at 1000 cells/well and allowed to attach to the bottom of the plate for overnight. Then the cells were treated with different concentrations of compounds for 72 h at 37 °C. At the end of treatment, the viable cells were determined by incubating cells with MTT reagent (0.5 mg/mL) in the complete tissue culture media for 3 h. Then the media were removed, and the formed purple formazan was dissolved in DMSO for quantification by absorbance at 570 nm using a SpectraMax i3 plate reader (Molecular Devices). The percent of growth is defined as 100 × (Atreated – Ainitial)/(Acontrol – Ainitial), where Atreated represents absorbance in wells treated with a compound, Ainitial represents the absorbance at time 0, and Acontrol denotes media-treated cells.
Statistics.
The statistics was computed using student t-test in Microsoft Excel, and P < 0.05 was considered significant. The CI values were calculated using Chou–Talalay method implemented in CompuSyn program.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH R01GM122820 and R21CA220061. Oregon Health & Science University School of Medicine, and Office of the Technology Transfer and Business Development also partially supported this research. We thank Ian Munhenzva (Portland State University) for determining the high-resolution mass spectra.
ABBREVIATIONS USED
- AML
acute myeloid leukemia
- CRE
cAMP-response element
- CREB
cAMP-response element binding protein
- CBP
CREB-binding protein
- Fsk
forskolin
- GBM
glioblastoma
- HPLC
high-performance liquid chromatography
- MAPK
mitogen-activated protein kinase
- NR4A2
nuclear receptor subfamily 4 group A member 2
- PBS
phosphate buffered saline
- PKA
protein kinase A
- PKB
protein kinase B
- RSK
ribosomal S6 kinase
- PP1
protein phosphatase 1
- PP2A
protein phosphatase 2A
- PTEN
phosphatase and tensin homologue
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
- SD
standard deviation
- SEM
standard error of the mean
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01207.
HPLC traces, CI values, and cell growth inhibition data (PDF)
Molecular formula strings and their associated data (CSV)
The authors declare no competing financial interest.
REFERENCES
- (1).Mayr B; Montminy M. Transcriptional Regulation by the Phosphorylation-Dependent Factor Creb. Nat. Rev. Mol. Cell Biol 2001, 2, 599–609. [DOI] [PubMed] [Google Scholar]
- (2).Cardinaux JR; Notis JC; Zhang Q; Vo N; Craig JC; Fass DM; Brennan RG; Goodman RH Recruitment of Creb Binding Protein Is Sufficient for Creb-Mediated Gene Activation. Mol. Cell. Biol 2000, 20, 1546–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Radhakrishnan I; Perez-Alvarado GC; Parker D; Dyson HJ; Montminy MR; Wright PE Solution Structure of the Kix Domain of Cbp Bound to the Transactivation Domain of Creb: A Model for Activator:Coactivator Interactions. Cell 1997, 91, 741–752. [DOI] [PubMed] [Google Scholar]
- (4).Hagiwara M; Alberts A; Brindle P; Meinkoth J; Feramisco J; Deng T; Karin M; Shenolikar S; Montminy M. Transcriptional Attenuation Following Camp Induction Requires Pp-1-Mediated Dephosphorylation of Creb. Cell 1992, 70, 105–113. [DOI] [PubMed] [Google Scholar]
- (5).Wadzinski BE; Wheat WH; Jaspers S; Peruski LF; Lickteig RL; Johnson GL; Klemm DJ Nuclear-Protein Phosphatase-2a Dephosphorylates Protein Kinase a Phosphorylated Creb and Regulates Creb Transcriptional Stimulation. Mol. Cell. Biol 1993, 13, 2822–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Gu T; Zhang Z; Wang J; Guo J; Shen WH; Yin Y. Creb Is a Novel Nuclear Target of Pten Phosphatase. Cancer Res. 2011, 71, 2821–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Meeusen B; Janssens V. Tumor Suppressive Protein Phosphatases in Human Cancer: Emerging Targets for Therapeutic Intervention and Tumor Stratification. Int. J. Biochem. Cell Biol 2018, 96, 98–134. [DOI] [PubMed] [Google Scholar]
- (8).Xiao X; Li BX; Mitton B; Ikeda A; Sakamoto KM Targeting Creb for Cancer Therapy: Friend or Foe. Curr. Cancer Drug Targets 2010, 10, 384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Rodon L; Gonzalez-Junca A; Inda MDM; Sala-Hojman A; Martinez-Saez E; Seoane J. Active Creb1 Promotes a Malignant Tgf-Beta2 Autocrine Loop in Glioblastoma. Cancer Discovery 2014, 4, 1230–1241. [DOI] [PubMed] [Google Scholar]
- (10).Zhuang H; Meng X; Li Y; Wang X; Huang S; Liu K; Hehir M; Fang R; Jiang L; Zhou JX; Wang P; Ren Y. Cyclic Amp Responsive Element-Binding Protein Promotes Renal Cell Carcinoma Proliferation Probably Via the Expression of Spindle and Kinetochore-Associated Protein 2. Oncotarget 2016, 7, 16325–16337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Johannessen CM; Johnson LA; Piccioni F; Townes A; Frederick DT; Donahue MK; Narayan R; Flaherty KT; Wargo JA; Root DE; Garraway LA A Melanocyte Lineage Program Confers Resistance to Map Kinase Pathway Inhibition. Nature 2013, 504, 138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhang Y; Zheng D; Zhou T; Song H; Hulsurkar M; Su N; Liu Y; Wang Z; Shao L; Ittmann M; Gleave M; Han H; Xu F; Liao W; Wang H; Li W. Androgen Deprivation Promotes Neuroendocrine Differentiation and Angiogenesis through Creb-Ezh2-Tsp1 Pathway in Prostate Cancers. Nat. Commun 2018, 9, 4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Xie F; Li BX; Kassenbrock A; Xue C; Wang X; Qian DZ; Sears RC; Xiao X. Identification of a Potent Inhibitor of Creb-Mediated Gene Transcription with Efficacious in Vivo Anticancer Activity. J. Med. Chem 2015, 58, 5075–5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Xie F; Li BX; Xiao X. Design, Synthesis and Biological Evaluation of Regioisomers of 666-15 as Inhibitors of Creb-Mediated Gene Transcription. Bioorg. Med. Chem. Lett 2017, 27, 994–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Li BX; Xie F; Fan Q; Barnhart KM; Moore CE; Rheingold AL; Xiao X. Novel Type of Prodrug Activation through a Long-Range O,N-Acyl Transfer: A Case of Water-Soluble Creb Inhibitor. ACS Med. Chem. Lett 2014, 5, 1104–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Li BX; Gardner R; Xue C; Qian DZ; Xie F; Thomas G; Kazmierczak SC; Habecker BA; Xiao X. Systemic Inhibition of Creb Is Well-Tolerated in Vivo. Sci. Rep 2016, 6, 34513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kang X; Lu Z; Cui C; Deng M; Fan Y; Dong B; Han X; Xie F; Tyner JW; Coligan JE; Collins RH; Xiao X; You MJ; Zhang CC The Itim-Containing Receptor Lair1 Is Essential for Acute Myeloid Leukaemia Development. Nat. Cell Biol 2015, 17, 665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mantamadiotis T. Towards Targeting Pi3k-Dependent Regulation of Gene Expression in Brain Cancer. Cancers 2017, 9, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Srinivasan S; Totiger T; Shi C; Castellanos J; Lamichhane P; Dosch AR; Messaggio F; Kashikar N; Honnenahally K; Ban Y; Merchant NB; VanSaun M; Nagathihalli NS Tobacco Carcinogen-Induced Production of Gm-Csf Activates Creb to Promote Pancreatic Cancer. Cancer Res. 2018, 78, 6146–6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wiley SZ; Sriram K; Liang W; Chang SE; French R; McCann T; Sicklick J; Nishihara H; Lowy AM; Insel PA Gpr68, a Proton-Sensing Gpcr, Mediates Interaction of Cancer-Associated Fibroblasts and Cancer Cells. FASEB J. 2018, 32, 1170–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Rodriguez-Ubreva J; Catala-Moll F; Obermajer N; Alvarez-Errico D; Ramirez RN; Company C; Vento-Tormo R; Moreno-Bueno G; Edwards RP; Mortazavi A; Kalinski P; Ballestar E. Prostaglandin E2 Leads to the Acquisition of Dnmt3a-Dependent Tolerogenic Functions in Human Myeloid-Derived Suppressor Cells. Cell Rep. 2017, 21, 154–167. [DOI] [PubMed] [Google Scholar]
- (22).Li BX; Xiao X. Discovery of a Small-Molecule Inhibitor of the Kix-Kid Interaction. ChemBioChem 2009, 10, 2721–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chou TC Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol. Rev 2006, 58, 621–681. [DOI] [PubMed] [Google Scholar]
- (24).Conkright MD; Guzman E; Flechner L; Su AI; Hogenesch JB; Montminy M. Genome-Wide Analysis of Creb Target Genes Reveals a Core Promoter Requirement for Camp Responsiveness. Mol. Cell 2003, 11, 1101–1108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.