

Abstract
A lentivirus-mediated doxycycline-inducible pTRIPZ shRNAmir plasmid targeting IGF1R transcript was transfected into two head and neck squamous cell carcinoma (HNSCC) cell lines to silence IGF1R expression and to assess the effect of its downregulation on cisplatin sensitivity in vitro. In Cal27-regIGF1R and SCC25-regIGF1R cell lines, IGF1R protein expression was reduced by more than 90% after 72 h of incubation with doxycycline. Both basal and IGF-stimulated pIGF1R, pAKT, and pERK were significantly reduced, without influence on total AKT and ERK expression. Downregulation of the IGF1R was associated with decreased proliferation and cell viability in both cell lines. Reduced IGF1R expression was also associated with increased sub-G0/G1-phase and G0/G1-phase populations and decreased S-phase and G2/M-phase populations. IGF1R downregulation enhanced sensitivity to cisplatin with decrease of cisplatin IC50 from 15 to 7.1 in Cal27-regIGF1R cells and from 11 to 6.3 in SCC25-regIGF1R cells. Cisplatin exhibited increased pro-apoptotic activity by annexin V staining and PARP cleavage in both cells lines when cultured in doxycycline. Thus, in two HNSCC cell lines in vitro, reduced IGF1R expression results in reduced growth rate and increased sensitivity to cisplatin. Thus, IGF1R downregulation and/or inhibition may serve as a useful adjunct to platinum-based cytotoxic chemotherapy.
Keywords: IGF1R, Oral cavity cancer, Squamous cell carcinoma
Introduction
Head and neck squamous cell carcinoma (HNSCC) include tumors that involve the mucosal surfaces of the oral cavity, pharynx, larynx, and nasal cavity [1]. Current therapeutic options for HNSCC include surgery, radiation therapy, chemotherapy, or a combination of treatments [2]. Chemotherapy plays an important role in treatment, especially of advanced or recurrent disease; cisplatin is commonly used, either alone or in combination with other chemotherapeutic drugs. The effectiveness of chemotherapy is often inadequate due to development of drug resistance. Thus, exploration of the molecular basis of HNSCC chemotherapeutic resistance is an important step toward improving survival of patients who suffer from this cancer [3, 4].
The insulin-like growth factor-1 receptor (IGF1R) is a hetero-tetrameric transmembrane glycoprotein. Once a ligand is bound, the IGF1R is autophosphorylated, activating its tyrosine kinase function. The IGF1R then engages adaptor molecules such as insulin receptor substrates (IRSs) and Shc, leading to activation of downstream protein kinases, including the phosphatidylinositol-3-kinase (PI3K)/AKT and mitogen activated protein kinase (MAPK)/ERK1/2 signaling pathways that regulate growth and survival of cancer cells [5].
Abnormal expression and/or activity of the IGF1R has been associated with tumor growth and poor prognosis in several human cancers, including the myeloma, lung, and prostate [6, 7]. Recent studies in HNSCC demonstrated that IGF1R expression was significantly higher in grade II and III tumors than in patients with grade I disease and that elevated IGF1R expression correlated with poorer survival [8]. In other cancer types, IGF1R activity may be more important and may be elevated without increased IGF1R expression level. Studies using siRNA, dominant negative mutants, and antisense molecules directed against the IGF1R mRNA have shown that decreased IGF1R expression is associated with decreased radio- and chemoresistance and with induction of apoptosis [9].
Hence, the IGF1R is regarded as a target for the development of novel anticancer therapy in selected tumor types, and multiple IGF1R antagonist agents are being tested clinically for their therapeutic potential [10, 11]. These include small molecule inhibitors of the IGF1R tyrosine kinase, anti-IGF1R antibodies, and anti-IGF antibodies [13]. These agents have entered phase II and III clinical trials for various human cancers [14, 15]. An important feature of the anti-IGF1R antibodies is their ability to downregulate IGF1R by promoting receptor internalization; their anti-tumor activity is existing in vitro and in vivo model systems, with favorable safety profile [16]. IGF1R tyrosine kinase inhibitors (that also inhibit the insulin receptor) and antibodies that neutralize both IGF-1 and IGF-2 (which can act through the insulin receptor) have the potential therapeutic advantage of targeting both IGF1R and insulin signaling pathways [17]. The available data from the clinical trials with agents targeting the IGF1R have been promising [18]; nevertheless, several clinical development programs have since been discontinued due to unsatisfactory results [12, 13].
We hypothesize that suppression of IGF1R expression in HNSCC will increase sensitivity to conventional chemotherapy. To test this hypothesis, we used an inducible shRNAmir expression system targeting the IGF1R in two oral cavity squamous cell carcinoma (HPV-negative) cell lines and assess whether downregulation of IGF1R expression increases sensitivity of these cell lines to cisplatin.
Materials and Methods
Tissue Culture and Reagents
Cal27, SCC25 and HEK293T cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and were grown in DMEM/F12 medium supplemented with 5% FBS and 1% penicillin/streptomycin. Cell cultures were maintained in a humidified atmosphere of 5% CO2 and 95% air at 37C. Cisplatin, doxycycline, puromycin, crystal violet, and 5-bromo-2′-deoxyuridine (BrdU) were purchased from Sigma-Aldrich (St. Louis, MO, USA), alamarBlue and trypan blue were obtained from Invitrogen (Carlsbad, CA, USA). Propidium Iodide (PI), FITC mouse anti-BrdU, and FITC Annexin V (BD Biosciences, San Jose, CA, USA), anti-AKT, anti-pAKT (Ser473), anti-IGF1R, anti-ERK1/2, and anti-pERK1/2, were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti-pIGFR and anti-β-actin were obtained from Millipore (Bedford, MA, USA).
pTRIPZ-shRNAmir Lentivirus Production and Cell Line Transduction
Inducible shRNA constructs (TRIPZ shRNAmir) targeting the IGF1R (catalog numbers RHS4696, RHS4697, and RHS4743), negative control shRNA (non-silencing-TRIPZ lentiviral inducible shRNAmir control), and RNAintro TRIPZ-inducible lentiviral shRNAmir transfection kit were obtained from Open Biosystems (Thermo Fisher Scientific, Rockford, IL, USA). IGF1R-TRIPZ lentiviral shRNAmir starter kit was used to produce the inducible shRNA according to the manufacturer’s instructions. One clone from Escherichia coli stocks containing TRIPZ shRNAmir plasmids was grown in 5 ml 2× LB low salt on a shaker at 37C for 18 h. pTRIPZ shRNA DNA was isolated using a Qiagen Maxprep Kit. DNA concentration was measured using a Nano Drop H spectrophotometer (Thermo Scientific). HEK293T cells (5.0 × 103) were seeded in a 10-cm plate for 24 h and then transfected with 37.5 μg trans-lentiviral TM packaging mix, 9 μg of pTRIPZ shRNA plasmids, and 187.5 ml of Arrest-In TM transfection reagent in serum and antibiotic-free medium. Medium was replaced with fresh serum and antibiotic-containing medium 4 h later. Medium containing viruses was harvested 48 and 72 h after transfection, filtered (0.45 mm falcon filters, Millex HV; Millipore), and concentrated by ultracentrifugation at 3 × 104 RPM at 4 C for 12 h. Cal27 and SCC25 cells were plated at 30% density in 24-well plate, and 24 h later, virus at multiplicity of infection (MOI) of 10 for IGF1R and 20 for negative control was added to the cells for 4 h and then replaced with fresh media and cultured for 48 h. Cells were trypsinized and reseeded in 6-well plate, and cells were selected for the puromycin resistance gene by adding 3 μg/ml puromycin for negative control and the IGF1R shRNA-transfected cells. Cells were maintained in puromycin for at least three successive passages to ensure full selection of the resistant cells. Cells were induced by adding 2 μg/ml doxycycline (Sigma) in the culture media for 3 days and examined by fluorescence microscopy to detect expression of the turboRFP shRNA reporter gene.
Determination of Functional Titer and MOI
To establish a functional titer of lentiviral particles, HEK293 cells in 24-well plates were plated at a density of 5 × 104 cells/well and cultured overnight in humidified 37C incubator with 5% CO2. Transfection media (TM) containing 10 μg/ml polybrene in serum-free and antibiotic-free DMEM was freshly prepared. Culture medium was removed, and 225 μl TM was added to each well of the 24-well plate. Lentiviral particles were diluted to 5-fold serial dilutions in TM (5, 25, 125, 625-fold) using a flat-bottom 96-well plate. Twenty-five microliters of diluted lentiviral particles was added to each well. Following incubation for 24 h, doxycycline 2μg/ml was added to the wells to induce the expression of Turbo-RFP. Turbo-RFP expression in the cells was observed under a fluorescence microscope 72 h after transduction with the TRIPZ lentivirus. Turbo-RFP positive colonies were counted at × 10 magnification in three arbitrary fields using ImageJ software (National Institutes of Health, Bethesda, MD, USA). The functional titer was calculated as transducing units per milliliter (TU/ml) using the following formula: TU/ml = no. of TurboRFP positive colonies counted × dilution factor / volume of diluted lentiviral particles used. This functional titer was used to calculate the volume of lentiviral particles required to transfect Cal27 and SCC25 cells at the desired MOI. MOI was calculated from the formula MOI = (TU/ml) / no. of cells [19, 20].
Western Blot Analysis
Western immunoblots were obtained from total protein extracts as described previously [21, 22]. Briefly, treated cells in 6-cm dishes were washed with ice-cold phosphate-buffered saline (PBS) containing 2 mmol/l sodium orthovanadate. Cells were collected by scrapping in 1 ml cold PBS, centrifuged at 13,000 rpm for 5 min at 4 C and the supernatants were discarded. Cells pellets were re-suspended in 50 μl lysis buffer, vortexed, and incubated at 4 C for 30 min followed by centrifugation at 13,000 rpm for 15 min at 4 C. The supernatants were collected, and the concentrations of the supernatant were measured with a BCA protein assay kit (Pierce, Rockford, lL, USA). Proteins were resolved on a 12% SDS-PAGE by loading 25 μg protein and then transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, (Bedford, MA, USA). The membranes were incubated with blocking buffer (1% casein in PBS) (Bio-Rad, Hercules, CA, USA) for 60 min at room temperature (RT). The membranes were incubated with primary antibody overnight at 4 C. Proteins were visualized using an infrared-emitting conjugated secondary antibodies anti-mouse 680 Alexa Fluor (Molecular Probes, Eugene, OR, USA) or anti-rabbit IRDYE 800 (Rockland Immunochemicals, Gilbertsville, PA, USA) diluted 1:15,000 for 60 min. Band intensities were quantified using Odyssey imaging system (LI-COR Biosciences, Lincoln, NE, USA).
alamarBlue Cell Proliferation Assay
Cell suspensions treated with either vehicle or 2 μg/ml doxycycline were plated at 5 × 103 cells per well in 96-well plates and were incubated for 72 h. alamarBlue was added to each well according to the manufacturer’s protocol. Cells were incubated for 3 h at 37 C, and the fluorescence at 540 nm was recorded.
Trypan Blue Cell Viability Assay
Cells were plated at 5 × 104 cells per well in 12-well plates. Wells in triplicates were treated with either vehicle or 2 μg/ml doxycycline and incubated for 72 h. Medium was evacuated from each well were collected and wells were trypsinized; anchored and floating cells were combined and centrifuged at 1000 rpm for 3 min at 4 C. Cells were resuspended in 1 ml PBS. One hundred microliters of the suspension was stained with 100 μl trypan blue. Living and dead (blue) cells were counted using the TC20 Automated Cell Counter (Bio-Rad, Hercules, CA, USA).
Colony Formation Assay
Cells treated with vehicle or 2 μg/ml doxycycline were seeded at 250 cells per well in 6-well plates and incubated for 14 days; during this period, the medium was changed twice per week with fresh media ± 2 μg/ml doxycycline. Plates were washed with ice-cold PBS; the colonies were fixed with ice cold methanol for 15 min, stained with 2% crystal violet, and counted. Colonies consisting of ≥ 50 cells were scored.
Flow Cytometry Analysis of Cell Cycle
Cells were seeded in 6-cm dishes at 2.5 × 104/dish, treated with vehicle or 2 μg/ml doxycycline for 72 h to achieve a steady and maximal state of IGF1R knockdown. Cells were treated with cisplatin for 72 h, followed by incubation with BrdU at 1:1000 for 6 h. Cells were trypsinized, collected, washed twice in cold PBS solution, and then fixed in 95% ethanol. Cells pellets were re-suspended in 1 ml of 2 N HCl and 0.5% Triton X-100, incubated at (RT) for 30 min. Cells were pelleted and re-suspended in 1 ml of 0.1 M NaB4O7 (pH 8.5) and incubated for 30 min at RT. Cells were pelleted and re-suspended in 0.5 ml of anti-BrdU antibody dilute 1:50 in PBS containing 1% BSA and 0.2% Tween-20 and then incubated at RT for 60 min. Cell pellets were re-suspended in 10 ml of PBS/BSA/Tween containing FITC-conjugated rabbit anti-mouse secondary antibodies (1:25) and incubated at RT for 30 min in the dark. Cell suspensions were passed through a cell strainer to remove aggregates and collect single cells in proper flow cytometry tubes. The relative proportions of cells in the G0/G1, S, and G2/M phases were determined by FACS Calibur™ Flow Cytometry. Analysis was performed using Flow Jo v8.5 (Flowjo, LLC—Ashland, OR, USA).
Flow Cytometry Evaluation of Apoptosis
Cells were grown in 6 cm dishes to 50% confluence in 5% FBS ± 2 μg/ml doxycycline for 72 h. Cells were incubated with the desired concentration of cisplatin for another 72 h. The conditioned medium from each well was collected; cell monolayers were trypsinized, resuspended in the corresponding conditioned medium, centrifuged at 3000 rpm for 3 min at 4 C, washed twice with cold PBS, and resuspended in annexin V-FITC and PI according to the manufacturer’s recommendations (Millipore). Flow cytometry was conducted in the University of Virginia (UVA) Flow Cytometry Core Facility with marker combinations as follows: annexin V and PI negative; cells in early apoptosis are annexin V positive and PI negative; cells in late apoptosis are annexin V and PI positive; necrotic/dead cells are annexin V negative and PI positive [23]. Acquisition was performed on a FACS Calibur™ Flow Cytometry. Analysis was performed using Flow Jo v8.5 (Flowjo, LLC—Ashland, OR, USA).
Statistical Analysis
All statistical analyses were performed using Prism version 3.0 (GraphPad Software, Inc., La Jolla, CA, USA). Data were analyzed using two-way analysis of variance (ANOVA), followed by post-hoc Bonferroni to test significances between treatment groups. Data are presented as means ± standard error or the mean, and p < 0.05 was considered significant.
Results
Establishment of Doxycycline-Inducible IGF1R-shRNAmir Expressing Cells Lines
Lentiviral doxycycline-inducible pTRIPZ-shRNAmir constructs directed against IGF1R were used to transfect Cal27 and SCC25 HNSCC cells. The construct also contains a doxycycline-inducible TurboRFP as a reporter gene co-expressed with the shRNA sequence. A negative control pTRIPZ plasmid containing a non-silencing shRNAmir construct was also used. The transfected cells were selected with puromycin to generate Cal27-regIGF1R and SCC25-regIGF1R cells with the IGF1R-shRNAmir construct and Cal27-nonreg and SCC25-nonreg with the non-silencing shRNAmir construct. Cells were seeded in 6-well plates with vehicle or 2 μg/ml doxycycline for 3 days to induce shRNAmir and TurboRFP expression. Cells were then examined by florescence microscope for the expression of the reporter Turbo-RFP protein as marker of successful transfection and doxycycline inducibility (Fig. 1a). Immunoblot with anti-IGF1R showed that doxycycline-induced IGF1R-shRNAmir reduced the IGF1R protein level by 90% relative to the untreated cells (Fig. 1b, c). Untransfected parental cells and cells transfected with the non-silencing control did not show any changes in the level of the IGF1R in the absence or the presence of doxycycline (Fig.1d, e), indicating that doxycycline alone has no effect on IGF1R expression and that the downregulation of IGF1R occurs only when the IGF1R-shRNAmir is expressed.
Fig. 1.

Induction of shRNAmir-IGF1R with doxycycline reduces IGF1R expression. Indicated cell lines were grown in the absence or presence of 2 μg/ml doxycycline for 3 days. Cal27 and SCC25, parental cell lines. Cal27-regIGF1R and SCC25-regIGF1R, cell lines transfected with IGF1R-pTRIPZ-shRNAmir. Cal27-nonreg and SCC25-nonreg, cell lines transfected with non-silencing pTRIPZ-shRNAmir (negative control). a Fluorescence microscopy demonstrates expression of the reporter TurboRFP in the presence of doxycycline. b Representative immunoblot demonstrating regulation of IGF1R expression by doxycycline. c Quantification of densitometric analysis of three such immunoblots showing relative intensity of IGF1R band normalized to β-actin band. d, e Representative immunoblots demonstrating no impact of doxycycline or negative control plasmid on IGF1R expression. f, g Parental Cal27, Cal27-nonreg, SCC25, and SCC25-nonreg cells were grown in 96-well plates in the absence or presence of 2 μg/ml doxycycline as indicated. Proliferation was measured using the alamarBlue assay at 24, 48, and 72 h. Data are expressed as net fluorescence. Values represent means ± SEM of three independent experiments performed in triplicate and demonstrate no effect of non-silencing shRNAmir expression or dox treatment. h Cell viability by trypan blue assay at 72 h. Data are expressed as average live cell counts ± SEM for three independent experiments performed in triplicate. No effect of non-silencing shRNAmir expression or dox treatment. Asterisk indicates significant difference between −dox and +dox, p < 0.05
Parental cells and cells transfected with the non-silencing shRNAmir (Cal27-nonreg and SCC25-nonreg) were treated with vehicle or 2 μg/ml doxycycline and cell proliferation was determined at 24, 48, and 72 h using alamarBlue (Fig. 1f, g). Cell viability was evaluated at 72 h by trypan blue assay (Fig. 1h). Parental Cal27 and SCC25 cells or cells expressing the non-silencing shRNA do not show any significant difference in the cell proliferation or cell viability, indicating that transfection and doxycycline treatment, separately or combined, do not affect the proliferative and survival behavior of the parental cell lines.
Downregulation of IGF1R Decreases Phosphorylation of AKT and ERK
Cal27-regIGF1R and SCC25-regIGF1R cells in 6 cm dishes were incubated with 2 μg/ml doxycycline for 3 days to induce IGF1R-shRNAmir expression. Ten nanomolars of des[1–3]IGF-1 (desIGF1) was used to activate the IGF1R. des[1–3]IGF-1 is an N-terminally truncated form of IGF-1 that binds to the IGF1R with high affinity but has substantially reduced affinity for the IGFBPs. (Residues 1–3 and 49–51 of IGF-1 are crucial for interaction with the IGFBPs, and mutations in these domains reduce IGFBP affinity [24].) As some HNSCC cell lines are known to secrete high levels of IGFBPs, we routinely use desIGF1 to stimulate the IGF1R, thus eliminating IGFBP effects on IGF1R signaling. Cells were collected after 10 min of IGF1R activation and processed for immunoblot (Fig. 2). Stimulation with desIGF1 increased IGF1R, AKT, and ERK1/2 phosphorylation in cells not treated with doxycycline. In cells treated with doxycycline and thus expressing shRNAmir, the level of IGF1R was reduced with concomitant decrease in IGF-stimulated pIGF1R, pAKT, and pERK1/2 levels; levels of total AKT and ERK1/2 were unaffected by doxycycline.
Fig. 2.
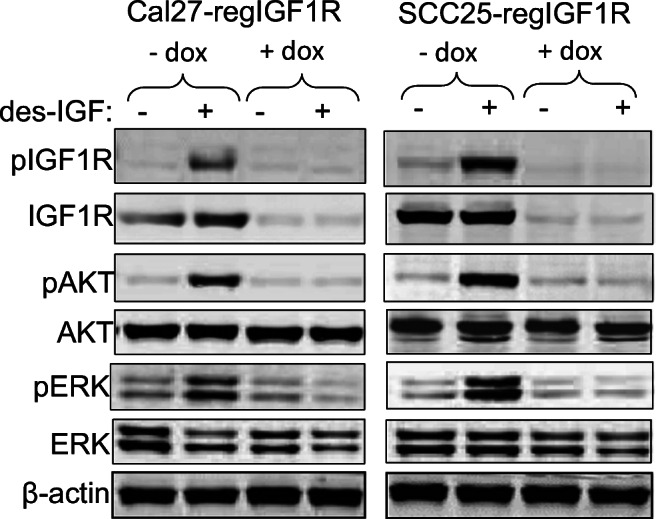
Suppression of IGF1R expression reduces IGF-induced AKT and ERK phosphorylation. Cal27-regIGF1R and SCC25-regIGF1R cells were incubated in the absence or presence of 2 μg/ml doxycycline for 3 days and then treated with vehicle or 10 nM des [1–3] IGF for 10 min. Immunoblots were performed on whole cell lysates with the indicated antibodies. The blots shown are representative of three independent replicate experiments
IGF1R Downregulation Exhibits Growth Inhibitory Effect on HNSCC Cells
Cal27 and SCC25 cells transfected with IGF1R-shRNAmir (Cal27-regIGF1R and SCC25-regIGF1R) or with non-silencing shRNAmir (Cal27-nonreg and SCC25-nonreg) were treated with vehicle or 2 μg/ml doxycycline, and cell proliferation was determined 72 h by alamarBlue. Expression of IGF1R-shRNAmir and resulting downregulation of the IGF1R results in a reduction in cell proliferation that is detected after 3 days of continuous exposure to doxycycline (p < 0.05) (Fig. 3a). This reduction in the cell proliferation was accompanied by a significant reduction in cell viability as evaluated by trypan blue assay (Fig. 3b). Cells expressing the non-silencing shRNAmir did not demonstrate any significant difference in the cell proliferation or cell viability, indicating that the growth effect is dependent on knockdown of the IGF1R.
Fig. 3.

IGF1R downregulation reduces HNSCC cell growth and viability. Cal27-regIGF1R, SCC25-regIGF1R, and their negative controls Cal27-nonreg and SCC25-non-reg respectively were grown in 96-well plates in the absence or presence of 2 μg/ml doxycycline as indicated. a Proliferation was measured using the alamarBlue assay at 72 h. Data are expressed as net fluorescence after subtracting background. Values represent means ± SEM of three independent experiments performed in triplicate. b Cell viability by trypan blue assay at 72 h. Data are expressed as average live cell counts ± SEM for three independent experiments performed in triplicate. Asterisk indicates significant difference between −dox and +dox, p < 0.05
IGF1R Downregulation Alters Cell Cycle Distribution in HNSCC Cells
The effect of IGF1R downregulation on the cell cycle distribution was determined 72 h after doxycycline treatment using flow cytometric analyses of Cal27-regIGF1R and SCC25-regIGF1R cells labeled with BrdU/FITC. Compared to cells without doxycycline treatment, doxycycline-treated Cal27-regIGF1R and SCC25-regIGF1R cells showed the following: (1) slight increase in the fraction of cells in Sub-G0/G1 phase (4.1% and 5.1%, respectively with dox vs. 3.2% and 2.6%, respectively without dox); (2) an increase in the fraction of cells in the G0/G1 phase (49% and 54%, respectively with dox vs. 38% and 39%, respectively without dox, p < 0.05); (3) a decrease in the proportion of cells in the S phase (26% and 30%, respectively with dox vs. 36% and 35%, respectively without dox, p < 0.05); and (4) a decrease in the G2/M phase (14.7% and 15%, respectively with dox vs. 22% and 20%, respectively without dox). Control cells without doxycycline treatment had a significantly lower G0/G1 cell population and higher S population (Table 1).
Table 1.
Comparison of cell cycle distribution in OSCC cells (mean ± SD) (*P < 0.05)
| Cell type | Cell cycle phases % M ± SEM | |||
|---|---|---|---|---|
| Sub-G1 | G0/G1 | S | G2/M | |
| Cal27-regIGF1R, −dox | 3.2 ± 0.1 | 38.7 ± 0.9 | 36.3 ± 2.3 | 21.7 ± 1.2 |
| Cal27-regIGF1R, +dox | 4.1 ± 0.6 | 49.0 ± 2.1* | 26.3 ± 0.9* | 14.7 ± 1.5 |
| SCC25-regIGF1R, −dox | 2.6 ± 0.9 | 39.7 ± 1.2 | 35.3 ± 1.9 | 19.7 ± 0.9 |
| SCC25-regIGF1R, +dox | 5.1 ± 0.6 | 54.3 ± 1.5* | 30.0 ± 3.5* | 15.0 ± 0.6 |
IGF1R Downregulation Enhances Growth Inhibitory Effect of Cisplatin on HNSCC Cells
Cal27-regIGF1R and SCC25-regIGF1R (or Cal27-nonreg and SCC25-nonreg) cells were treated with increasing concentrations (2.5, 5, 10, 15, and 25 μM) of cisplatin in the absence and presence of 2 μg/ml doxycycline. Cell proliferation was determined after 72 h using alamarBlue (Fig. 4a). Treatment with cisplatin alone caused a dose-dependent reduction in cell number at 72 h. The cisplatin IC50 was 15 and 11 μM for Cal27-regIGF1R and SCC25-regIGF1R, respectively. After reduction in IGF1R expression, the cisplatin IC50 decreased to 7.1 and 6.3 μM, respectively. There was a significant difference between the growth inhibition caused by cisplatin in IGF1R-depleted cells (+dox) compared to cisplatin in control cells (−dox, p < 0.05). Table 2 summarizes the growth inhibitory effect at different concentrations of cisplatin in the presence or absence of doxycycline. The enhanced sensitivity to cisplatin with IGF1R reduction was accompanied by a significant reduction in the cell viability as determined by trypan blue assay (Fig. 4b). This effect was absent in cells transfected with non-silencing shRNAmir. The effect of chronic exposure (14 days) of Cal27-regIGF1R cells to 2.5, 5, and 10 μM cisplatin in the presence or absence of 2 μg/ml doxycycline on clonogenic survival was evaluated using a colony formation assay. Doxycycline alone caused a non-significant reduction in the number of colonies formed. Cisplatin at 2.5, 5, and 10 μM resulted in 17%, 33%, and 92% reductions respectively in colony formation compared to untreated cells (p < 0.05). In the presence of doxycycline resulting in reduced IGF1R expression, reductions of 33%, 57%, and 97% were observed (p < 0.05). Control cell expressing non-silencing shRNAmir showed no difference in the number of colonies formed with or without dox (Fig. 4c).
Fig. 4.

Impact of IGF1R knockdown on sensitivity to cisplatin. Cal27-regIGF1R and SCC25-regIGF1R cells were grown for 3 days in the absence or presence of 2 μg/ml doxycycline to alter IGF1R expression. They were then treated with cisplatin for 72 h. a Proliferation was measured using the alamarBlue assay. Data are expressed as average net fluorescence as a percentage of cells not treated with cisplatin (each cell line and dox treatment set to 100%). b Cell viability was measured using the trypan blue assay. Data are expressed as average live cell counts ± SEM for three independent experiments performed in triplicate. Asterisk indicates p < 0.05 compared to control. c Colony formation assay was performed on Cal27-regIGF1R cells and Cal27-nonreg cells in the absence or presence of 2 μg/ml doxycycline. Cells were treated with cisplatin dose as indicated. Image shows representative stained colonies. Graph quantifies three independent experiments showing mean colony number ± SEM. Asterisk indicates significant difference between −dox and +dox at given cisplatin concentration, p < 0.05
Table 2.
The growth inhibitory effect of cisplatin in IGF1R downregulated Cal27-regIGF1R and SCC25-regIGF1R cells (*P < 0.05)
| Cisplatin/μM | Cal27-regIGF1R cell growth inhibition (%) | SCC25-regIGF1R, cell growth inhibition (%) | ||||
|---|---|---|---|---|---|---|
| −dox | +dox | p | −dox | +dox | p | |
| 2.5 | 6.5 | 21.5 | < 0.05 | 9 | 15 | < 0.05 |
| 5 | 20 | 42 | < 0.05 | 20 | 41 | < 0.05 |
| 10 | 31.5 | 62.5 | < 0.05 | 45 | 72.5 | < 0.05 |
| 15 | 50 | 85 | < 0.05 | 72.5 | 89.7 | < 0.05 |
| 25 | 90 | 95 | < 0.05 | 90 | 95 | < 0.05 |
IGF1R Knockdown Enhances the Apoptotic Effect of Cisplatin on HNSCC Cell Lines
The effect of IGF1R downregulation on the induction of apoptosis by cisplatin treatment of Cal27-regIGF1R and SCC25-regIGF1R cells was examined by flow cytometry. Cells were incubated with 5, 10, and 25 μM cisplatin either alone or in combination with 2 μg/ml doxycycline for 72 h. Cells were processed for flow cytometry using annexin V-FITC and PI as described in “Materials and Methods.” Figure 5a demonstrates the cell populations in early (lower right quadrant) and late (upper right quadrant) apoptosis at 10 μM cisplatin dose. The apoptotic rates of Cal27-regIGF1R and SCC25-regIGF1R at various concentrations of cisplatin are summarized in Table 3 and Fig. 5b. The apoptotic rates (early apoptotic and late apoptotic) of Cal27-regIGF1R and SCC25-regIGF1R 72 h after doxycycline induced IGF1R downregulation increased from 3 and 5 to 3.5 and 7% (P > 00.5) respectively. The percentage of apoptotic cells treated with 5, 10, and 25 μM cisplatin alone increased to 8% and 19%, and 40%, respectively in Cal27-regIGF1R and to 12%, 25%, and 33% in SCC25-regIGF1R (p < 0.05). With IGF1R downregulation, this apoptotic rate respectively increased to 14%, 40%, and 52% in Cal27-regIGF1R and to 25%, 50%, and 56% in SCC25-regIGF1R (p < 0.05).
Fig. 5.

IGF1R downregulation enhances cisplatin-induced apoptosis. Cal27-regIGF1R and SCC25-regIGF1R cells were plated in the absence (−dox) or presence (+dox) of 2 μg/ml doxycycline for 3 days followed by treatment with cisplatin for 72 h. a Cells were collected, stained with annexin V-FITC and PI, and subjected to flow cytometry. Representative dot blots are shown for treatment with 10 μM cisplatin (Cisp10), with percentage population in early and late apoptosis indicated in the lower right and upper right corners, respectively. b Quantification of three independent experiments using 5 μM (Cisp5), 10 μM (Cisp10), and 25 μM (Cisp25) cisplatin. Data are shown as mean ± SEM. Asterisk indicates significant difference between −dox and +dox at given cisplatin concentration, p < 0.05. c Adherent and non-adherent cells were collected, and whole cell lysates were subjected to immunoblot with the indicated antibodies. Fold change in the PARP cleavage was determine by densometric analysis of the band intensity
Table 3.
Effect of cisplatin on the apoptotic rate in IGF1R downregulated Cal27-regIGF1R and SCC25-regIGF1R cells
| Cal27-regIGF1R | SCC25-regIGF1R | |||||
|---|---|---|---|---|---|---|
| Cell treatment | Apoptosis M ± SEM | p value | Apoptosis M ± SEM | p value | ||
| Early | Late | Early | Late | |||
| −dox | 1.2 ± 0.3 | 3.7 ± 0.4 | > 0.05 | 1.2 ± 0.2 | 1.8 ± 0.1 | > 0.05 |
| +dox | 1.1 ± 0.3 | 3.6 ± 0.5 | 2.6 ± 0.3 | 2.9 ± 0.3 | ||
| Cisp5 | 2.7 ± 0.3 | 5.5 ± 0.4 | < 0.05 | 3.7 ± 0.3 | 8.4 ± 0.8 | < 0.05 |
| Cisp5 + dox | 5.8 ± 0.7 | 8.2 ± 0.8 | 9.7 ± 0.7 | 15.6 ± 1.1 | ||
| Cisp10 | 7.1 ± 0.5 | 11.5 ± 1.7 | < 0.05 | 11.7 ± 0.9 | 21.0 ± 1.8 | < 0.05 |
| Cisp10 + dox | 15.0 ± 0.9 | 25.1 ± 0.8 | 23.4 ± 1.4 | 27.1 ± 0.9 | ||
| Cisp 25 | 16.0 ± 0.9 | 25.5 ± 1.6 | < 0.05 | 23.9 ± 1.3 | 27.2 ± 1.4 | < 0.05 |
| Cisp25 + dox | 19.5 ± 1.3 | 27.9 ± 0.8 | 26.5 ± 0.6 | 30.6 ± 2.0 | ||
Apoptosis was also assessed using immunoblot for PARP cleavage as shown in Fig. 5c. Cal27-regIGF1R and SCC25-regIGF1R cells either untreated or pretreated with doxycycline for 3 days were incubated with 2.5, 5, 10, or 25 μM cisplatin for 72 h followed by immunoblot analysis of whole cell lysates. Cisplatin caused a dose-dependent induction in PARP cleavage. Combined treatment with cisplatin and doxycycline caused a cisplatin dose-dependent significant increase in PARP cleavage with 3–4-fold increases in the PARP cleavage (determined by densometric analysis of the band intensity) was noticed at 5, 10, and 25 μM. Cisplatin also induced pIGF1R, pAKT, and pERK in dose dependent. IGF1R downregulation suppressed the cisplatin induced IGF1R, AKT, and ERK activation which shows that induction of apoptosis via PARP cleavage was associated with decrease in the survival signals via decrease phosphorylation of IGF1R/AKT/ERK pathway.
Discussion
The effects of IGF1R downregulation on the sensitivity of HNSCC to the chemotherapeutic agent cisplatin have been tested in vitro. Two HNSCC cell lines that express high levels of IGF1R were used. A post-transcriptional gene silencing mechanism was used to target the IGF1R via a doxycycline-inducible short hairpin RNA construct (TRIPZ shRNAmir). The resultant cell lines, Cal27-regIGF1R and SCC25-regIGF1R, demonstrate reduction in IGF1R with exposure to doxycycline. IGF1R protein expression was reduced by more than 90% within 72 h. The decrease in IGF1R level is associated with abrogation of IGF-stimulated growth and survival signals, including the AKT and ERK pathways. These signaling changes were mirrored by the suppressed proliferation, decreased cell viability, and reduced clonogenicity after continuous IGF1R downregulation.
Exploration of cell cycle changes associated with IGF1R downregulation showed increases in the sub-G0/G1 and G0/G1 fractions. There was also a decrease in the proportion of cells in the S and G2/M phases. These changes in cell cycle distribution are consistent with the inability of the IGF1R downregulated cells to promote G1 to S transition resulting in G0/G1 cell cycle delay and a subsequent reduction in the S phase and G2/M population. This is consistent with the finding of inhibited cell proliferation.
Cisplatin is the mainstay of many chemotherapeutic regimens used in a wide variety of solid tumors, including HNSCC. The suppressing effect on tumor growth is limited due to severe toxicity particularly nephrotoxicity and hepatotoxicity [25]. Thus, enhanced of the chemotherapeutic effects of cisplatin at the tumor would be expected to have clinical relevance. The effect of IGF1R downregulation on chemosensitivity to cisplatin was tested in Cal27-regIGF1R and SCC25-regIGF1R cell lines. Cisplatin inhibited proliferation, viability, and colony formation in a dose-dependent fashion. Downregulation of the IGF1R enhanced the growth inhibitory effect of cisplatin with significant 50% reduction in IC50 in both cell lines. At all dose tested, IGF1R downregulation increased chemosensitivity to cisplatin. Flowcytometric examination of different cell populations treated with cisplatin demonstrated that cisplatin significantly increases the rate of apoptosis. IGF1R downregulation in the presence of doxycycline significantly augmented the pro-apoptotic effect of cisplatin; this was assessed by flow cytometry for annexin V staining and immunoblot for PARP cleavage.
The results of this study are consistent with a large body of evidence which suggests that IGF1R signaling plays a vital role in chemotherapy resistance in a wide variety of tumors [26–28]. IGF1R inhibition sensitizes breast cancer cells to cisplatin [29]. Knockdown of IGF1R enhances chemosensitivity to cisplatin in human lung adenocarcinoma A549 cells [30]. Inhibition of IGF1R signaling using RNAi enhances sensitivity to adriamycin chemotherapy liver cancer cell lines [31, 32] and the effect of the 5-fluorouracil (5-FU) in SW480 colon cancer cells [33]. In pancreatic cancer xenografts, the chemosensitivity to gemcitabine was enhanced by concomitant treatment with an anti-IGF1R antibody [34]. Furthermore, inhibition of IGF1R with an antisense molecule increases sensitivity of bladder cancer cell [35] or prostate cancer cells to cisplatin, mitoxantrone, and paclitaxel [36]. This is the first demonstration of this phenomenon in HNSCC, a tumor type where cisplatin resistance portends very poor clinical outcomes.
In addition to apoptosis, additional mechanisms could be relevant to the enhancement of cisplatin sensitivity with IGF1R down regulation. Prior reports have indicated that cytotoxic drugs can induce IGF1R activation leading to drug resistance via activation of pro-survival/anti-apoptotic pathways [37]. In the present study, HNSCC cell lines treated with cisplatin demonstrated a dose-dependent increase in IGF1R, AKT, and ERK1/2 phosphorylation; this activation of pro-survival signaling was reduced by IGF1R downregulation. Thus, IGF1R downregulation or targeted IGF1R inhibition has the potential to block this compensatory pathway in HNSCC treated with cisplatin. Furthermore, there is evidence that a functional IGF1R is required for repair of DNA double strand breaks caused by chemotherapeutic drugs [38]. IGF1R downregulation or targeted IGF1R inhibition has the potential to interfere with DNA damage repair resulting in enhanced drug sensitivity and increased cell death.
Clinical studies have shown that upregulation of IGF1R expression during treatment is a poor prognostic factor, and such patients may benefit from targeted IGF1R inhibition [11, 39]. Though phase I/II studies in many cancer including breast, lung, prostate and pancreatic carcinoma with agents that target IGF1R have demonstrated a wide range of responses ranging from progressive disease to complete response [40–42], most of phase III clinical trials failed to show a clear benefit from targeting IGF1R in combination with conventional treatment [43]. However, IGF1R expression was not a selection factor in these clinical trials, and it is likely that IGF1R expression and/or activity are important predictors of response to IGF1R antagonists [44, 45]. Thus, understanding the cellular effects of IGF1R downregulation or inhibition and the related impact on sensitivity to chemotherapeutic and other targeted therapeutic agents is necessary for identification of reliable predictive biomarkers of response to combination therapy [5, 22, 46].
It should be noted that prior studies of HNSCC have reported that outcomes are particularly poor for patients with HPV-negative tumors. Dale et al. noted the association of high IGF1R expression with higher T-stage and HPV-negativity, with a tendency toward shorter OS and indicated the potential value of adding targeted anti-IGF1R therapy to selected HPV-negative tumors with high IGF1R expression [47]. Both Cal27 and SCC25 cell lines are HPV-negative, which may represent the HNSCC tumors most likely to respond to reduced IGF1R signaling. Further study should evaluate the comparative impact of IGF1R inhibition/antagonism in HPV-related vs. HPV-unrelated disease.
In conclusion, the results of this in vitro study provide the evidence that the IGF1R axis is an important mediator of cisplatin sensitivity in HNSCC through regulation of both cell cycle progression and apoptosis. It has been demonstrated that a doxycycline-inducible shRNAmir effectively suppresses IGF1R expression in Cal27 and SCC25 HNSCC cell lines, leading to enhanced cisplatin toxicity in vitro. These results encourage further exploration of IGF1R downregulation or inhibition as a potential method for chemosensitization in head and neck cancer treatment.
Author Contributions
Ashraf Khalil conceived and performed experiments and wrote the manuscript.
Mark Jameson secured funding and reagents, provided supervision and expertise, and reviewed and edited manuscript.
Funding Information
The study was supported by NIH grant K08-DE019477 and American Head and Neck Society pilot grant to M.J. Jameson.
Compliance with Ethical Standards
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Contributor Information
Ashraf Khalil, Email: ashkalil2010@gmail.com.
Mark J. Jameson, Email: mark.jameson@virginia.edu
References
- 1.Bower R, Green VL, Kuvshinova E, Kuvshinov D, Karsai L, Crank ST, Stafford ND, Greenman J. Maintenance of head and neck tumor on-chip: gateway to personalized treatment? Future Sci OA. 2017;3(2):FSO174. doi: 10.4155/fsoa-2016-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahn PH, Machtay M, Anne PR, Cognetti D, Keane WM, Wuthrick E, Dicker AP, Axelrod RS (2016) Phase I trial using induction cisplatin, docetaxel, 5-FU and erlotinib followed by cisplatin, bevacizumab and erlotinib with concurrent radiotherapy for advanced head and neck cancer. Am J Clin Oncol [DOI] [PubMed]
- 3.Yang J, Ju Z, Dong S. Cisplatin and paclitaxel co-delivered by folate-decorated lipid carriers for the treatment of head and neck cancer. Drug Deliv. 2016;24(1):792–799. doi: 10.1080/10717544.2016.1236849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang HM, Lin CY, Hsieh CH, Hsu CL, Fan KH, Chang JT, Huang SF, Kang CJ, Liao CT, Ng SH, Yen TC. Induction chemotherapy with dose-modified docetaxel, cisplatin, and 5-fluorouracil in Asian patients with borderline resectable or unresectable head and neck cancer. J Formos Med Assoc. 2017;116(3):185–192. doi: 10.1016/j.jfma.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 5.Denduluri SK, Idowu O, Wang Z, Liao Z, Yan Z, Mohammed MK, Ye J, Wei Q, Wang J, Zhao L, Luu HH. Insulin-like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis. 2015;2(1):13–25. doi: 10.1016/j.gendis.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Limesand KH, Chibly AM, Fribley A. Impact of targeting insulin-like growth factor signaling in head and neck cancers. Growth Horm IGF Res. 2013;23(5):135–140. doi: 10.1016/j.ghir.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taunk NK, Goyal S, Moran MS, Yang Q, Parikh R, Haffty BG. Prognostic significance of IGF-1R expression in patients treated with breast-conserving surgery and radiation therapy. Radiother Oncol. 2010;96(2):204–208. doi: 10.1016/j.radonc.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 8.Lara PC, Bordon E, Rey A, Moreno M, Lloret M, Henriquez-Hernandez LA. IGF-1R expression predicts clinical outcome in patients with locally advanced oral squamous cell carcinoma. Oral Oncol. 2011;47(7):615–619. doi: 10.1016/j.oraloncology.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 9.Zhao S, Qiu Z, He J, Li L, Li W. Insulin-like growth factor receptor 1 (IGF1R) expression and survival in non-small cell lung cancer patients: a meta-analysis. Int J Clin Exp Pathol. 2014;7(10):6694–6704. [PMC free article] [PubMed] [Google Scholar]
- 10.Ryan PD, Goss PE. The emerging role of the insulin-like growth factor pathway as a therapeutic target in cancer. Oncologist. 2008;13(1):16–24. doi: 10.1634/theoncologist.2007-0199. [DOI] [PubMed] [Google Scholar]
- 11.Simpson A, Petnga W, Macaulay VM, Weyer-Czernilofsky U, Bogenrieder T (2017) Insulin-like growth factor (IGF) pathway targeting in cancer: role of the IGF axis and opportunities for future combination studies. Target Oncol [DOI] [PMC free article] [PubMed]
- 12.Chen HX, Sharon E. IGF-1R as an anti-cancer target—trials and tribulations. Chin J Cancer. 2013;32(5):242–252. doi: 10.5732/cjc.012.10263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qu X, Wu Z, Dong W, Zhang T, Wang L, Pang Z, Ma W, Du J. Update of IGF-1 receptor inhibitor (ganitumab, dalotuzumab, cixutumumab, teprotumumab and figitumumab) effects on cancer therapy. Oncotarget. 2017;8(17):29501–29518. doi: 10.18632/oncotarget.15704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beltran PJ, Mitchell P, Chung YA, Cajulis E, Lu J, Belmontes B, Ho J, Tsai MM, Zhu M, Vonderfecht S, Baserga R, Kendall R, Radinsky R, Calzone FJ. AMG 479, a fully human anti-insulin-like growth factor receptor type I monoclonal antibody, inhibits the growth and survival of pancreatic carcinoma cells. Mol Cancer Ther. 2009;8(5):1095–1105. doi: 10.1158/1535-7163.MCT-08-1171. [DOI] [PubMed] [Google Scholar]
- 15.Gualberto A. Figitumumab (CP-751,871) for cancer therapy. Expert Opin Biol Ther. 2010;10(4):575–585. doi: 10.1517/14712591003689980. [DOI] [PubMed] [Google Scholar]
- 16.King ER, Wong KK. Insulin-like growth factor: current concepts and new developments in cancer therapy. Recent Pat Anticancer Drug Discov. 2012;7(1):14–30. doi: 10.2174/157489212798357930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao J, Chesebrough JW, Cartlidge SA, Ricketts SA, Incognito L, Veldman-Jones M, Blakey DC, Tabrizi M, Jallal B, Trail PA, Coats S, Bosslet K, Chang YS. Dual IGF-I/II-neutralizing antibody MEDI-573 potently inhibits IGF signaling and tumor growth. Cancer Res. 2011;71(3):1029–1040. doi: 10.1158/0008-5472.CAN-10-2274. [DOI] [PubMed] [Google Scholar]
- 18.Arcaro A. Targeting the insulin-like growth factor-1 receptor in human cancer. Front Pharmacol. 2013;4:30. doi: 10.3389/fphar.2013.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sloutskin A, Yee MB, Kinchington PR, Goldstein RS. Varicella-zoster virus and herpes simplex virus 1 can infect and replicate in the same neurons whether co- or superinfected. J Virol. 2014;88(9):5079–5086. doi: 10.1128/JVI.00252-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aznan AN, Abdul Karim N, Wan Ngah WZ, Jubri Z. Critical factors for lentivirus-mediated PRDX4 gene transfer in the HepG2 cell line. Oncol Lett. 2018;16(1):73–82. doi: 10.3892/ol.2018.8650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jameson MJ, Beckler AD, Taniguchi LE, Allak A, Vanwagner LB, Lee NG, Thomsen WC, Hubbard MA, Thomas CY. Activation of the insulin-like growth factor-1 receptor induces resistance to epidermal growth factor receptor antagonism in head and neck squamous carcinoma cells. Mol Cancer Ther. 2011;10(11):2124–2134. doi: 10.1158/1535-7163.MCT-11-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khalil A, Jameson MJ (2017) The EGFR inhibitor gefitinib enhanced the response of human oral squamous cell carcinoma to cisplatin in vitro. Drugs R D [DOI] [PMC free article] [PubMed]
- 23.Khalil AA, Jameson MJ. Sodium Orthovanadate Inhibits Proliferation and Triggers Apoptosis in Oral Squamous Cell Carcinoma in vitro. Biochemistry (Mosc) 2017;82(2):149–155. doi: 10.1134/S0006297917020067. [DOI] [PubMed] [Google Scholar]
- 24.Horney MJ, Evangelista CA, Rosenzweig SA. Synthesis and characterization of insulin-like growth factor (IGF)-1 photoprobes selective for the IGF-binding proteins (IGFBPS). Photoaffinity labeling of the IGF-binding domain on IGFBP-2. J Biol Chem. 2001;276(4):2880–2889. doi: 10.1074/jbc.M007526200. [DOI] [PubMed] [Google Scholar]
- 25.Quintanilha JCF, Visacri MB, Sousa VM, Bastos LB, Vaz CO, Guarnieri JPO, Amaral LS, Malaguti C, Lima CSP, Vercesi AE, Moriel P (2017) Cisplatin-induced human peripheral blood mononuclear cells’ oxidative stress and nephrotoxicity in head and neck cancer patients: the influence of hydrogen peroxide. Mol Cell Biochem [DOI] [PubMed]
- 26.Wang T, Ge G, Ding Y, Zhou X, Huang Z, Zhu W, Shu Y, Liu P. MiR-503 regulates cisplatin resistance of human gastric cancer cell lines by targeting IGF1R and BCL2. Chin Med J (Engl) 2014;127(12):2357–2362. [PubMed] [Google Scholar]
- 27.Yang M, Shan X, Zhou X, Qiu T, Zhu W, Ding Y, Shu Y, Liu P. miR-1271 regulates cisplatin resistance of human gastric cancer cell lines by targeting IGF1R, IRS1, mTOR, and BCL2. Anticancer Agents Med Chem. 2014;14(6):884–891. doi: 10.2174/1871520614666140528161318. [DOI] [PubMed] [Google Scholar]
- 28.Zhuang M, Shi Q, Zhang X, Ding Y, Shan L, Shan X, Qian J, Zhou X, Huang Z, Zhu W, Ding Y, Cheng W, Liu P, Shu Y. Involvement of miR-143 in cisplatin resistance of gastric cancer cells via targeting IGF1R and BCL2. Tumour Biol. 2015;36(4):2737–2745. doi: 10.1007/s13277-014-2898-5. [DOI] [PubMed] [Google Scholar]
- 29.O’Flanagan CH, O’Shea S, Lyons A, Fogarty FM, McCabe N, Kennedy RD, O’Connor R. IGF-1R inhibition sensitizes breast cancer cells to ATM-related kinase (ATR) inhibitor and cisplatin. Oncotarget. 2016;7(35):56826–56,841. doi: 10.18632/oncotarget.10862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dong A, Kong M, Ma Z, Qian J, Cheng H, Xu X. Knockdown of insulin-like growth factor 1 receptor enhances chemosensitivity to cisplatin in human lung adenocarcinoma A549 cells. Acta Biochim Biophys Sin (Shanghai) 2008;40(6):497–504. doi: 10.1111/j.1745-7270.2008.00429.x. [DOI] [PubMed] [Google Scholar]
- 31.Niu J, Li XN, Qian H, Han Z. siRNA mediated the type 1 insulin-like growth factor receptor and epidermal growth factor receptor silencing induces chemosensitization of liver cancer cells. J Cancer Res Clin Oncol. 2008;134(4):503–513. doi: 10.1007/s00432-007-0314-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Niu J, Xu Z, Li XN, Han Z. siRNA-mediated type 1 insulin-like growth factor receptor silencing induces chemosensitization of a human liver cancer cell line with mutant P53. Cell Biol Int. 2007;31(2):156–164. doi: 10.1016/j.cellbi.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 33.Yavari K, Taghikhani M, Maragheh MG, Mesbah-Namin SA, Babaei MH, Arfaee AJ, Madani H, Mirzaei HR. SiRNA-mediated IGF-1R inhibition sensitizes human colon cancer SW480 cells to radiation. Acta Oncol. 2010;49(1):70–75. doi: 10.3109/02841860903334429. [DOI] [PubMed] [Google Scholar]
- 34.Maloney EK, McLaughlin JL, Dagdigian NE, Garrett LM, Connors KM, Zhou XM, Blattler WA, Chittenden T, Singh R. An anti-insulin-like growth factor I receptor antibody that is a potent inhibitor of cancer cell proliferation. Cancer Res. 2003;63(16):5073–5083. [PubMed] [Google Scholar]
- 35.Sun HZ, Wu SF, Tu ZH. Blockage of IGF-1R signaling sensitizes urinary bladder cancer cells to mitomycin-mediated cytotoxicity. Cell Res. 2001;11(2):107–115. doi: 10.1038/sj.cr.7290075. [DOI] [PubMed] [Google Scholar]
- 36.Hellawell GO, Ferguson DJ, Brewster SF, Macaulay VM. Chemosensitization of human prostate cancer using antisense agents targeting the type 1 insulin-like growth factor receptor. BJU Int. 2003;91(3):271–277. doi: 10.1046/j.1464-410X.2003.04061.x. [DOI] [PubMed] [Google Scholar]
- 37.Eckstein N, Servan K, Hildebrandt B, Politz A, von Jonquieres G, Wolf-Kummeth S, Napierski I, Hamacher A, Kassack MU, Budczies J, Beier M, Dietel M, Royer-Pokora B, Denkert C, Royer HD. Hyperactivation of the insulin-like growth factor receptor I signaling pathway is an essential event for cisplatin resistance of ovarian cancer cells. Cancer Res. 2009;69(7):2996–3003. doi: 10.1158/0008-5472.CAN-08-3153. [DOI] [PubMed] [Google Scholar]
- 38.Roos WP, Nikolova T, Quiros S, Naumann SC, Kiedron O, Zdzienicka MZ, Kaina B. Brca2/Xrcc2 dependent HR, but not NHEJ, is required for protection against O(6)-methylguanine triggered apoptosis, DSBs and chromosomal aberrations by a process leading to SCEs. DNA Repair (Amst) 2009;8(1):72–86. doi: 10.1016/j.dnarep.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 39.Heskamp S, Boerman OC, Molkenboer-Kuenen JD, Wauters CA, Strobbe LJ, Mandigers CM, Bult P, Oyen WJ, van der Graaf WT, van Laarhoven HW. Upregulation of IGF-1R expression during neoadjuvant therapy predicts poor outcome in breast cancer patients. PLoS One. 2015;10(2):e0117745. doi: 10.1371/journal.pone.0117745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olmos D, Postel-Vinay S, Molife LR, Okuno SH, Schuetze SM, Paccagnella ML, Batzel GN, Yin D, Pritchard-Jones K, Judson I, Worden FP, Gualberto A, Scurr M, de Bono JS, Haluska P. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: a phase 1 expansion cohort study. Lancet Oncol. 2010;11(2):129–135. doi: 10.1016/S1470-2045(09)70354-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pappo AS, Patel SR, Crowley J, Reinke DK, Kuenkele KP, Chawla SP, Toner GC, Maki RG, Meyers PA, Chugh R, Ganjoo KN, Schuetze SM, Juergens H, Leahy MG, Geoerger B, Benjamin RS, Helman LJ, Baker LH. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a phase II Sarcoma Alliance for Research through Collaboration study. J Clin Oncol. 2010;29(34):4541–4547. doi: 10.1200/JCO.2010.34.0000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tolcher AW, Sarantopoulos J, Patnaik A, Papadopoulos K, Lin CC, Rodon J, Murphy B, Roth B, McCaffery I, Gorski KS, Kaiser B, Zhu M, Deng H, Friberg G, Puzanov I. Phase I, pharmacokinetic, and pharmacodynamic study of AMG 479, a fully human monoclonal antibody to insulin-like growth factor receptor 1. J Clin Oncol. 2009;27(34):5800–5807. doi: 10.1200/JCO.2009.23.6745. [DOI] [PubMed] [Google Scholar]
- 43.Ramalingam SS, Spigel DR, Chen D, Steins MB, Engelman JA, Schneider CP, Novello S, Eberhardt WE, Crino L, Habben K, Liu L, Janne PA, Brownstein CM, Reck M. Randomized phase II study of erlotinib in combination with placebo or R1507, a monoclonal antibody to insulin-like growth factor-1 receptor, for advanced-stage non-small-cell lung cancer. J Clin Oncol. 2011;29(34):4574–4580. doi: 10.1200/JCO.2011.36.6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sclafani F, Kim TY, Cunningham D, Kim TW, Tabernero J, Schmoll HJ, Roh JK, Kim SY, Park YS, Guren TK, Hawkes E, Clarke SJ, Ferry D, Frodin JE, Ayers M, Nebozhyn M, Peckitt C, Loboda A, Mauro DJ, Watkins DJ. A randomized phase II/III study of dalotuzumab in combination with cetuximab and irinotecan in chemorefractory, KRAS wild-type, metastatic colorectal cancer. J Natl Cancer Inst. 2015;107(12):djv258. doi: 10.1093/jnci/djv258. [DOI] [PubMed] [Google Scholar]
- 45.Sclafani F, Kim TY, Cunningham D, Kim TW, Tabernero J, Schmoll HJ, Roh JK, Kim SY, Park YS, Guren TK, Hawkes E, Clarke SJ, Ferry D, Frodin JE, Ayers M, Nebozhyn M, Peckitt C, Loboda A, Watkins DJ. Dalotuzumab in chemorefractory KRAS exon 2 mutant colorectal cancer: Results from a randomised phase II/III trial. Int J Cancer. 2017;140(2):431–439. doi: 10.1002/ijc.30453. [DOI] [PubMed] [Google Scholar]
- 46.Bowers LW, Rossi EL, O’Flanagan CH, deGraffenried LA, Hursting SD. The role of the insulin/IGF system in cancer: lessons learned from clinical trials and the energy balance-cancer link. Front Endocrinol (Lausanne) 2015;6:77. doi: 10.3389/fendo.2015.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dale OT, Aleksic T, Shah KA, Han C, Mehanna H, Rapozo DC, Sheard JD, Goodyear P, Upile NS, Robinson M, Jones TM, Winter S, Macaulay VM. IGF-1R expression is associated with HPV-negative status and adverse survival in head and neck squamous cell cancer. Carcinogenesis. 2015;36(6):648–655. doi: 10.1093/carcin/bgv053. [DOI] [PubMed] [Google Scholar]