

Abstract
Despite advances in surgical technique and adjuvant treatment, endometrial cancer has recently seen an increase in incidence and mortality in the USA. The majority of endometrial cancers can be cured by surgery alone or in combination with adjuvant chemo- or radiotherapy; however, a subset of patients experience recurrence for reasons that remain unclear. Recurrence is associated with chemoresistance to carboplatin and paclitaxel and consequentially, high mortality. Understanding the pathways involved in endometrial cancer chemoresistance is paramount for the identification of biomarkers and novel molecular targets for this disease. Here, we generated the first matched pairs of carboplatin-sensitive/carboplatin-resistant and paclitaxel-sensitive/paclitaxel-resistant endometrial cancer cells and subjected them to bulk RNA sequencing analysis. We found that 45 genes are commonly upregulated in carboplatin- and paclitaxel-resistant cells as compared to controls. Of these, the leukemia inhibitory factor, (LIF), the protein tyrosine phosphatase type IVA, member 3 (PTP4A3), and the transforming growth factor beta 1 (TGFB1) showed a highly significant correlation between expression level and endometrial cancer overall survival (OS) and can stratify the 545 endometrial cancer patients in the TCGA cohort into a high-risk and low-risk-cohorts. Additionally, four genes within the 45 upregulated chemoresistance-associated genes are ADAMTS5, MICAL2, STAT5A, and PTP4A3 codes for proteins for which small-molecule inhibitors already exist. We identified these proteins as molecular targets for chemoresistant endometrial cancer and showed that treatment with their correspondent inhibitors effectively killed otherwise chemoresistant cells. Collectively, these findings underline the utility of matched pair of chemosensitive and chemoresistant cancer cells to identify markers for endometrial cancer risk stratification and to serve as a pharmacogenomics model for identification of alternative chemotherapy approaches for treatment of patients with recurrent disease.
Electronic supplementary material
The online version of this article (10.1007/s12672-018-0337-6) contains supplementary material, which is available to authorized users.
Keywords: Endometrial cancer, Recurrence, Gene expression, Chemoresistance
Introduction
Endometrial cancer (EC) is the most common gynecologic malignancy diagnosed in the Western world. The majority of patients are diagnosed at an early stage with an overall favorable prognosis. Unfortunately, approximately 20% of patients, despite early stage and seemingly excellent prognosis, will recur [1–3]. Recurrent endometrial cancer is generally treated with a combination of carboplatin and paclitaxel, and while the initial response rate is around 45%, the vast majority of patients relapses and develops a chemoresistant disease, leading to a low survival rate [4]. A number of signaling pathways have been associated with endometrial cancer chemoresistance, including repair mechanisms and pro-survival pathways as well as the upregulation of efflux pumps which have been specifically associated with chemoresistance to paclitaxel. Tumor biomarkers, including USP14 and L1CAM, have also been associated with poor patient outcome and have predictive value of endometrial cancer recurrence [5–8]. Despite this knowledge, a deep understanding of the cellular mechanisms leading to endometrial cancer chemoresistance is still lacking, and the development of tools for stratifying patients based on their likelihood to respond to chemotherapy and for identification of novel treatments for patients with recurrent disease is still urgently needed. We believe that the paucity of studies focusing on the comprehension of endometrial cancer chemoresistance at the molecular level is in part due to the difficulties of establishing chemoresistant endometrial cancer cells in vitro and has greatly limited our understanding of this disease.
In this study, we established what to our knowledge is the first matched pair of carboplatin-sensitive/carboplatin-resistant and paclitaxel-sensitive/paclitaxel-resistant endometrial cancer cell lines. The obtained matched pairs of cells were subjected to bulk RNA sequencing and their expression profile was compared to the one of their chemosensitive counterpart. We found that 45 genes were commonly upregulated in both paclitaxel- and carboplatin-resistant cells that are compared to the corresponding sensitive cells. Of these, we found that LIF, PTP4A3, and TGFB1 are strong predictors of poor overall survival in the endometrial cancer cohort from The Cancer Genome Atlas (TCGA) and that PTP4A3, ADAMTS5, STAT5A, and MICAL2 are potential molecular targets for chemoresistant endometrial cancer. We believe that our characterized chemoresistant endometrial cancer cells could be a useful platform for further investigating the mechanisms governing endometrial cancer chemoresistance and for preclinical models to test and develop novel chemotherapy agents for chemoresistant endometrial cancer.
Results
Establishment and Characterization of Matching Pairs of Carboplatin- and Paclitaxel-Resistant Human Endometrial Cancer Cells
To gain a better understanding of the pathways that are responsible for chemoresistance, we generated matched pairs of carboplatin-sensitive/carboplatin-resistant and paclitaxel-sensitive/paclitaxel-resistant endometrial cancer cells. To do this, ECC-1 endometrial cancer cells were either mock treated (vehicle) or treated with increasing concentrations of carboplatin (carbo) or paclitaxel (pacli) over a period of several weeks as shown in Scheme 1. At the end of each 6-week period, clones were isolated, allowed to grow, and then re-treated with increasing concentrations of the drugs for a total of four cycles (representative pictures are shown in Fig. 1a, b). This choice of intermittent scheduling of the drugs was made so to mimic the intermittent chemotherapy treatment endometrial cancer patients undergo. Two weeks after the end of the last treatment, carboplatin-resistant clones were approximatively eightfold less sensitive to the drug as compared to vehicle-treated cells (Fig. 2a) and paclitaxel-resistant cells were approximatively tenfold less sensitive to the drug as compared to vehicle cells (Fig. 2b). No cross-resistance was observed. To exclude that the difference in drug sensitivity was due to a difference in proliferation rate, we measured the total cell number of chemosensitive and chemoresistant cells over a period of 4 days. As shown in Fig. 2c, sensitive and resistant cells grew at a comparable rate, suggesting that the difference in drug sensitivity is not due to differences in their proliferation rate.
Scheme 1.

Exposure of endometrial cancer cells to increasing concentrations of drugs. Cells were initially exposed to their IC20–30 concentration of carboplatin (carbo) or paclitaxel (pacli) over a period of 6 weeks with the drug being added twice a week. At the end of each cycle, clones were isolated, allowed to grown in absence of drug over a 2-week recovery period prior being re-exposed to increasing concentrations of drugs
Fig. 1.

Multi-staged process during the development of chemoresistant cells. Representative pictures are shown depicting the different stages during the process leading to a carboplatin- or b paclitaxel-resistant cells. First windows (a, b) are vehicle-treated parental cells at the beginning of the treatment. Second windows (a, b) are treated cells after two rounds of chemotherapy which killed about 99% of cells (inserts are the magnification of the surviving cells). Third windows (a, b) are chemoresistant cells growing in presence of the relative drugs (insert are magnifications of the growing chemoresistant clones). Fourth windows (a, b) are expansion of chemoresistant cells
Fig. 2.
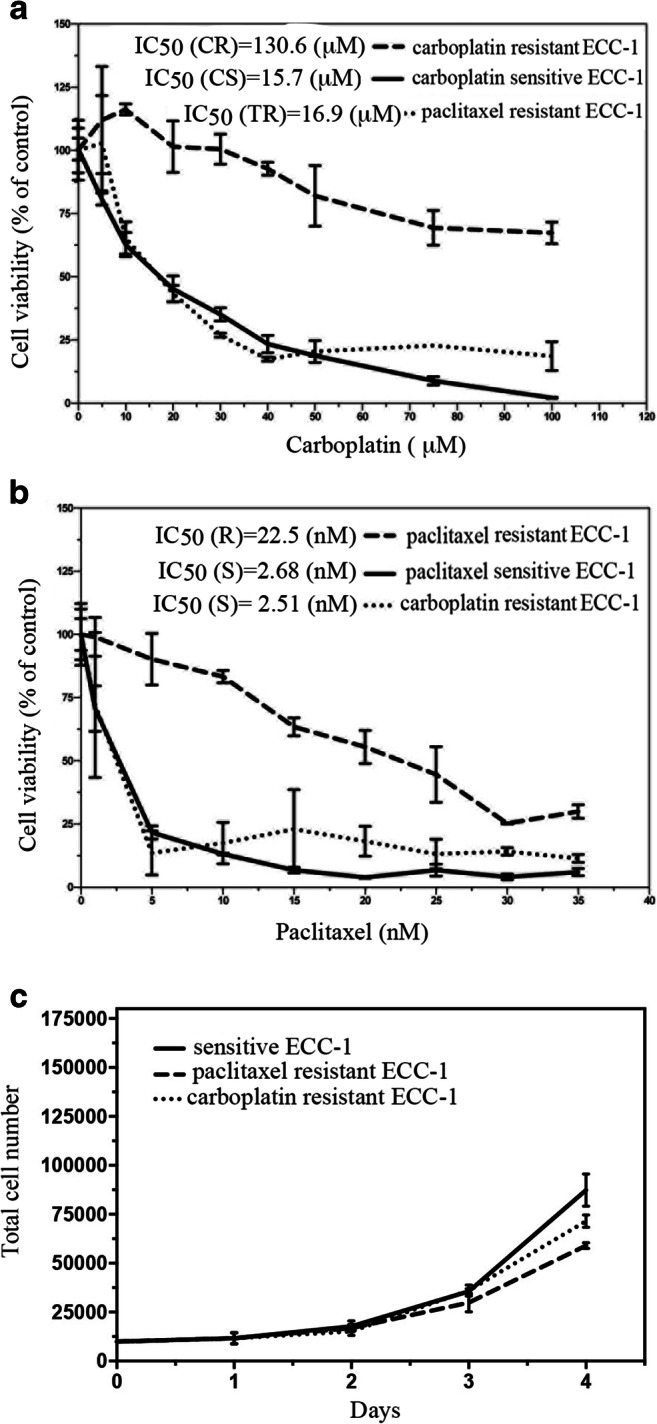
Residual cell viability in sensitive and resistant cells exposed to carboplatin and paclitaxel. Dose-dependent inhibition of cell viability of ECC-1-sensitive and -resistant cells exposed to the indicated concentrations of carboplatin (a) or paclitaxel (b) over a period of 48 and 96 h, respectively. Percentage of viable cells is relative to mock-treated controls. c Proliferation rate of sensitive and resistant endometrial cancer cells as measured by manually counting cells over a period of 4 days using an inverted microscope. Results are expressed as total cell number
RNA-seq Analysis in Chemosensitive Versus Chemoresistant Cancer Cells
In order to identify genes that are potentially relevant to chemoresistance in endometrial cancer, we performed gene expression analysis on ECC-1-sensitive (vehicle treated), carboplatin-resistant (ECC-1C), and paclitaxel-resistant (ECC-1P) cell lines via RNA sequencing. Per each cell line, three independent clones were submitted to RNA sequencing. As shown in Fig. 3a, comparison of the gene expression profile between chemosensitive and chemoresistant cells revealed that 691 genes were differentially expressed in paclitaxel-resistant cells versus vehicle-treated cells, and 731 genes were differentially expressed in carboplatin-resistant cells versus vehicle-treated cells (Supplementary Table 1). Of these genes, 175 were commonly altered in both carboplatin- and paclitaxel-resistant cells as compared to vehicle cells. Specifically, 45 of the 175 genes were upregulated in both cases, 89 were downregulated, and 41 genes showed a combination of up-or downregulation. Importantly, comparison of the gene expression of all submitted samples via principal component analysis (PCA) showed reproducible gene expression profile between biological triplicates and very different profiles between the three different conditions (Fig. 3b). Differentially expressed genes whose absolute fold change between conditions was less than twofold and did not show any significant difference after Bonferroni correction (p = 0.05) were not considered to be significant and therefore excluded from further analysis. A complete list of all genes is given in Supplementary Table 1.
Fig. 3.

Gene expression profiling of carboplatin- and paclitaxel-resistant endometrial cancer cells. Venn diagram representing the number of genes significantly altered (upregulated or downregulated) in carboplatin- or paclitaxel-resistant cells as compared to vehicle-treated cells. A total of 45 genes were found to be upregulated for both drugs. b Principal component analysis (PCA) of RNA-seq data based on normalized read counts. Sample to sample distances after treatment are illustrated for each cell line
Upregulation of Genes in Resistant Cells Correlates with Poor Outcome in Endometrial Cancer Patients
Next, we evaluated the prognostic power of the 45 commonly upregulated genes in ECC-1C and ECC-1P cell lines compared with the parental ECC-1 cell line in the endometrial cancer cohort from The Cancer Genome Atlas (TCGA). Specifically, the database was interrogated for correlation between mRNA upregulation and/or DNA amplification and patients’ overall survival. Shown in Table 1 is a list of the genes that, out of the 45 upregulated ones, had the highest correlation between gene expression and overall survival (OS). Of those, the leukemia inhibitory factor (LIF), the protein tyrosine phosphatase type IVA, member 3 (PTP4A3), and the transforming growth factor beta 1 (TGFB1) genes showed a highly significant correlation between expression level and OS. Importantly, each of these three genes was able to stratify the 545 endometrial cancer patients into a high-risk and low-risk-cohorts (Fig. 4). Patients with high LIF expression had a statistically significant lower median survival (63.86 months) as compared with patients low LIF expression (> 225.33-month median survival) and a hazard ratio (HR) for death of 3.43 (95% CI, 0.9739 to 12.13; p = 0.0004). Patients with high PTP4A3 expression had a statistically significant lower median survival (102.23 months) as compared with patients low PTP4A3 expression (> 225.33-month median survival) and a hazard ratio (HR) for death of 2.59 (95% CI, 0.9739 to 7.09; p = 0.0009). Patients with high TGFB1 expression had a statistically significant lower median survival (31.11 months) as compared to patients with low TGFB1 expression (> 225.33-month median survival) and a HR for death of 4.2 (95% CI, 0.9739 to 17.23; p = 0.0001).
Table 1.
Correlation between commonly upregulated genes in carboplatin- and paclitaxel-resistant cells and poor outcome in endometrial cancer patients. Upregulation of genes for which a correlation between gene expression and endometrial cancer overall survival (OS) was found based on the TCGA database. Those in italic are the genes for which the correlation was highly significant
| Gene | Prediction of OS |
|---|---|
| ABLIM1 | p = 2.1375 |
| ANK 1 | p = 0.5085 |
| APOE | p = 1.224 |
| FABP6 | p = 1.2825 |
| FSTL3 | p = 1.1025 |
| LBH | p = 0.6043 |
| LGALS3BP | p = 0.2101 |
| LIF | p = 0.0182 |
| NEAT1 | p = 0.666 |
| OSBPL7 | p = 0.6795 |
| PODN | p = 0.3676 |
| PTP4A3 | p = 0.035 |
| SLC9A4 | p = 1.5255 |
| TGFB1 | p = 0.0054 |
| TNFRSF12A | p = 0.0882 |
| TNFSF10 | p = 1.7415 |
| ZBED6CL | p = 1.071 |
| ZNF853 | p = 0.5895 |
Fig. 4.
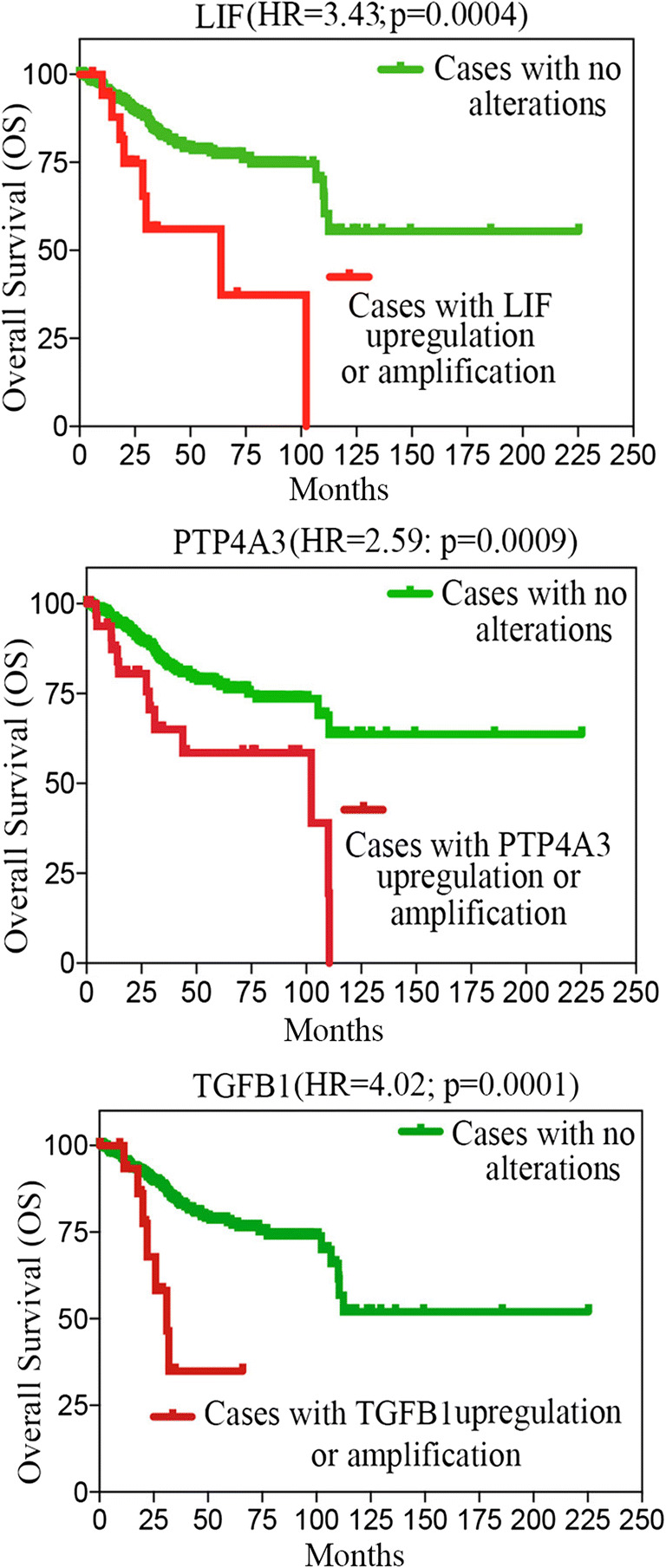
LIF, PTP4A3, and TGFB1 expression stratified endometrial cancer patients in low- and high-risk groups. Kaplan–Meier showing stratification of the TCGA endometrial cancer cohort into high- and low-risk groups based on the indicated gene expression
We further classified the 175 commonly altered genes by using ingenuity pathway analysis (IPA) and integrated them into canonical pathways (CP). As shown in Supplementary Table 2, ECC-1C cells showed engagement of the tight junction, osteoarthritis, gamma-glutamyl cycle, epithelial-mesenchymal transition, and folate glutamylation pathways. The ECC-1P cells on the other hand showed activation of CP such as the hepatic fibrosis/hepatic stellate cell activation, osteoarthritis, axonal guidance, atherosclerosis, and netrin signaling pathways. When looking at CP that are significantly modified among the 175 commonly altered genes in both the ECC-1C and ECC-1P cells, the gamma-glutamyl cycle, atherosclerosis, histidine degradation IV, pregnenolone biosynthesis, and ubiquinol-10 biosynthesis signaling pathways appeared to have primarily changed. Importantly, the abovementioned signaling pathways, with the exception of the osteoarthritis pathway, have been previously shown to be associated with human cancers’ chemoresistance, progression, and/or poor outcome [9–13]. Taken together, this suggests that our model of chemoresistance can recapitulate some of the clinical features of endometrial cancer patients with respect to transcript levels of the commonly upregulated genes and patients’ overall survival. This also suggests that chemoresistant endometrial cancer cells share some of the same activated pathways as other human cancer cells.
Validation of RNA-seq Results via qRT-PCR Analysis and of Protein Expression via Western Blot
Next, we validated our RNA-seq data via quantitative real-time PCR (qRT-PCR). In doing this, we have chosen to focus on validation of genes for which a small-molecule inhibitor is available. This restricted the field to four genes: ADAMTS5, PTP4A3, STAT5A, and MICAL2. As shown in Fig. 5, and consistent with our findings in the gene expression analysis, qRT-PCR showed an upregulation for ADAMTS5, MICAL2, STAT5A, and PTP4A3 in the chemoresistant versus the chemosensitive endometrial cancer cells. Next, we determined whether gene upregulation corresponds to protein upregulation. As shown in Fig. 6, Western blot analysis confirmed that the upregulation of the mRNA corresponded well with an overexpression of each associated protein in the chemoresistant cell lines as compared to the chemosensitive. Taken together, this suggests that ADAMTS5, MICAL2, STAT5A, and PTP4A3 are potential molecular targets for treatment of chemoresistant endometrial cancer using small-molecule inhibitor approach.
Fig. 5.
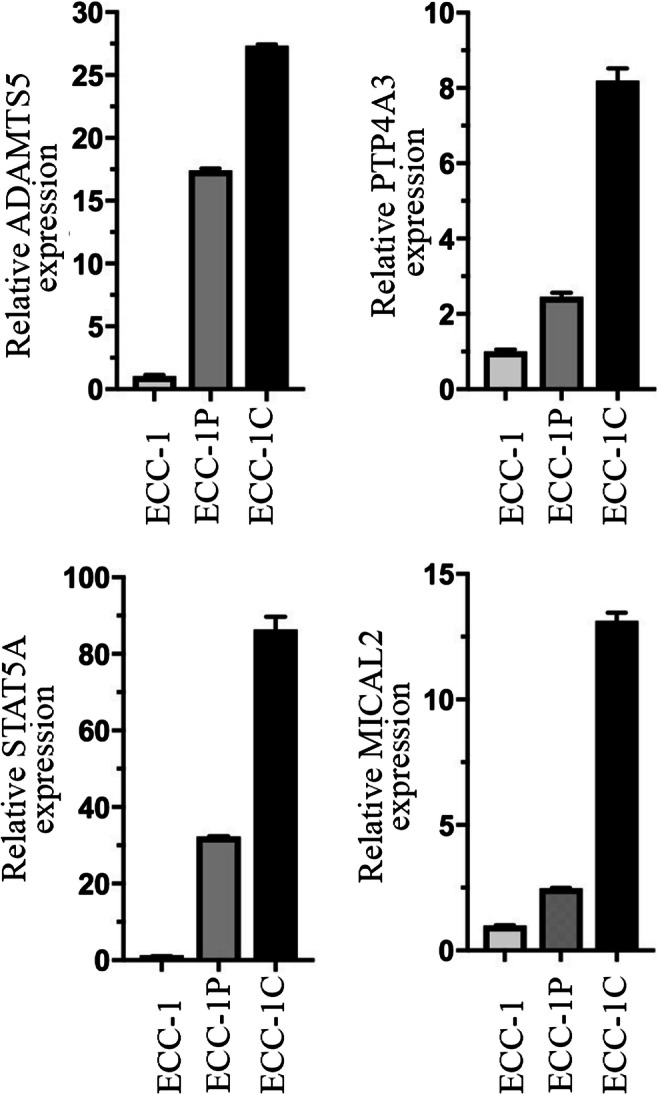
Validation of gene expression. qRT-PCR analysis of ADAMTS5, PTP4A3, STAT5A, and MICAL2 in vehicle-treated (ECC-1) versus chemoresistant (ECC-1P and ECC-1C) endometrial cancer cells. Three independent experiments with means ± standard deviations are presented for each protein
Fig. 6.

Validation of protein expression. Western blot analysis for ADAMTS5 (panel a), PTP4A3 (panel b), STAT5A (panel c), and MICAL2 (panel d) in vehicle-treated (ECC-1) versus chemoresistant (ECC-1P and ECC-1C) endometrial cancer cells. Equal protein loading in each line was verified using amido blots. Quantification of protein expression is given at the bottom of each panel. Three independent experiments with means ± standard deviations are presented for each protein
Small-Molecule Inhibitors of PTP4A3, ADAMTS5, STAT5A, or MICAL2 Kill Chemoresistant Endometrial Cancer Cells
Next, we tested the feasibility of targeting PTP4A3, ADAMTS5, STAT5A, or MICAL2 for treatment of chemoresistant endometrial cancer cells. To this end, carboplatin-resistant ECC-1 cells were treated with either carboplatin or the FDA-approved STAT5A-inhibitor pimozide [14–19], the FDA-approved PTP4A3 inhibitor pentamidine [20–22], the ADAMTS5A-inhibitor 5-((4-chlorobenzylthio-3-trifluoromethyl-N-methyl-1H-pyrazol-4-yl)methylene)-2 thioxothiazolidin-4-one [23, 24], or the MICAL2-inhibitor N-(4-chlorophenyl)-1-(3-(furan-2-yl)benzoyl)piperidine-3-carboxamide [25, 26].
Specifically, cells were treated with increasing concentration of the drugs over a period of 72 h and the residual cell viability was measured by WST1 assay as previously described [27]. As shown in Fig. 7, treatment with pentamidine, pimozide, ADAMTS5, or MICAL2 inhibitors resulted in a dose-dependent inhibition of cell viability of otherwise carboplatin-resistant cells. Specifically, the IC50 were 3.35, 12.7, 14.9, and 43.70 μM for pentamidine, pimozide, ADAMTS5, or MICAL2 inhibitors, respectively. Similar results were obtained when we tested the paclitaxel-resistant cancer cells (Supplementary Table 3). IC50 of the parental (sensitive) ECC-1 cells lines to pentamidine, pimozide, ADAMTS5, or MICAL2 inhibitors are shown in Supplementary Table 4. This suggests that inhibition of PTP4A3, ADAMTS5, STAT5A, or MICAL2 could represent a novel chemotherapy approach for the treatment of chemoresistant endometrial cancer.
Fig. 7.

PTP4A3, ADAMTS5, STAT5A, and MICAL2 are molecular targets for carboplatin-resistant endometrial cancer cells. Dose-dependent inhibition of cell viability of carboplatin-resistant ECC-1 cells exposed to increasing concentrations of pentamidine (a), pimozide (b), ADAMTS-5 inhibitor (c), or MICAL-2-inhibitor (d) over a period of 72 h. Percentage of viable cells is relative to mock-treated controls. Three independent experiments performed in triplicate; means ± standard deviations are presented
Discussion
The use of matched pairs of chemosensitive and chemoresistant cancer cells has greatly improved our understanding of the molecular mechanisms regulating human cancers’ chemoresistance in ovarian, lung, prostate, and brain cancers among others [28–32]. Unfortunately, the paucity of matched pairs of chemosensitive and chemoresistant endometrial cancer cells has greatly limited our understanding of this disease whose poor prognosis has been rising over the years mainly due to recurrence and chemoresistance.
Here, we developed and characterized matched pairs of carboplatin-sensitive/carboplatin-resistant and paclitaxel-sensitive/paclitaxel-resistant endometrial cancer cells. This was achieved via cyclic and incremental drug exposure so as to simulate the in vivo development of resistance through multiple cycles of chemotherapy. The resulting chemoresistant cells are approximatively tenfold less sensitive to the drug as compared to their sensitive counterparts. While others have reported the generation of chemoresistant cancer cells that are hundred folds less sensitive to chemotherapy than their sensitive counterpart [29], we think our model is still relevant to the disease. This is because in patients increases in drug dosage are not common practice due to limiting side effects. Importantly, we did not observe cross-resistance, suggesting that different molecular pathways are involved in the development of chemoresistance to carboplatin and paclitaxel.
For the past 20 years, microarray analysis of gene expression has been a widely used technique and precious source of information for understanding the complexities regulating cancer progression, including onset of chemoresistance in both cell lines and primary tumor specimens [33–38]. Despite its many advantages, including the possibility to measure the expression levels of thousands of genes simultaneously, microarray analysis suffers a major drawback in that it can only provide information about the genes that are included in a specific array. The recent development of the high-throughput RNA sequencing (RNA-seq) has provided a novel and powerful tool for understanding cancer cells’ transcriptome [39, 40]. While still relatively expensive, this technique allows for detection of both known and yet to be discovered transcripts even when their expression is relatively low.
Here, the matched pairs were subjected to bulk RNA-seq and screened for differentially expressed genes (DEGs) commonly found in both carboplatin- and paclitaxel-resistant endometrial cancer cells. These commonly upregulated genes were of particular interest given that endometrial cancer is typically resistant to both drugs. Our analysis of The Cancer Genome Atlas (TCGA) database revealed a strong correlation between the leukemia inhibitory factor (LIF), the protein tyrosine phosphatase type IVA, member 3 (PTP4A3), and the transforming growth factor beta 1 (TGFB1) gene expression and OS in endometrial cancer patients with each of these genes being able to stratify the 545 endometrial cancer patients in the TGCA into high-risk and low-risk cohorts. This is consistent with previous studies showing a correlation between LIF expression and poor outcome in additional human cancers including melanoma and oral squamous cell carcinoma [41, 42]. Furthermore, PTP4A3 has been shown to be overexpressed in a number of human cancers and its levels to be associated with the severity of the disease and patients’ poor survival. This is the case for ovarian cancer [43], lymphoblastic leukemia [44], and breast [45, 46] and gastric [47] cancers. Lastly, aberrant regulation of the TGFB1 pathway has been associated with stemness and metastasis in human cancer, including endometrial cancer [48], via promoting epithelial to mesenchymal transition (EMT) [49].
With the goal of identifying novel therapeutic targets for chemoresistant endometrial cancer, we interrogated our list of 45 commonly upregulated genes to identify the ones for whose products are small-molecule inhibitors available either FDA approved or used in vitro and preclinically. This restricted the field to the previously identified protein tyrosine phosphatase type IVA, member 3 (PTP4A3) and its FDA-approved inhibitor pentamidine, the disintegrin and metalloproteinase with thrombospondin motifs 5 (ADAMTS5) and its FDA-approved inhibitor pimozide, the signal transducer and activator of transcription 5A (STAT5A) and its inhibitor 5-((4-chlorobenzylthio-3-trifluoromethyl-N-methyl-1H-pyrazol-4-yl)methylene)-2 thioxothiazolidin-4-one, and the microtubule associated monooxygenase, calponin and LIM domain containing 2 (MICAL2) and its inhibitor N-(4-chlorophenyl)-1-(3-(furan-2-yl)benzoyl)piperidine-3-carboxamide.
The PTP4A3 inhibitor pentamidine is an FDA-approved antifungal and antiprotozoal agent. Importantly, pentamidine and its derivatives, as well as a novel class of PTP4A3 allosteric inhibitors, have been shown to have anticancer activity in human cancer cell lines and in preclinical models of human cancers [50, 51]. This includes chemoresistant ovarian cancer [43, 52], renal cancer [20], melanoma [22], and leukemia [21]. The mechanism/s through which small-molecule anti-PTP4A3 exert anticancer activity is/are still poorly understood, but several studies suggest they might do so via inhibiting angiogenesis and the Rho signaling pathway [53, 54]. The STAT5 inhibitor pimozide is an FDA-approved antipsychotic agent which has been recently shown to have antineoplastic effects in human cancers including prostate cancer [14, 15], lung cancer [55], and leukemia [19] via inhibition of STAT5 and wnt/β-catenin signaling pathways. ADAMTS5 has been shown to promote cancer cell migration and invasion and to be a promising molecular target for colorectal cancer [56] and melanoma [5] via a mechanism consistent with inhibition of pro-angiogenic factors [6]. Lastly, MICAL2 has been recently shown to play a role in controlling EMT transition via regulating cytoskeletal dynamics [7, 8]. Interestingly, MICAL2 overexpression has been associated with a subpopulation of cells within a tumor with highly migratory behavior, suggesting that its selective targeting could prevent cancer metastasis [57].
In conclusion, we found that the matched pair of chemosensitive and chemoresistant endometrial cancer cells recapitulates the features of human cancers, including endometrial cancer, and can be used to discover biomarkers for risk stratification and as a pharmacogenomics model for identification of alternative cancer treatments. This is particularly true for PTP4A3, which we propose as a novel biomarker that alone or in combination with other markers could be used to stratify endometrial cancer patients based on the likelihood to recur as well as a molecular target for the treatment of chemoresistant disease.
Methods and Material
ECC-1 Cell Line
The endometrial cancer cell line ECC-1 was provided by Dr. Gottfried Konecny (UCLA Medical Center). Cells were cultured in DMEM supplemented with 10% fetal bovine serum at 5% CO2.
Establishment of Carboplatin- and Paclitaxel-Chemoresistant ECC-1 Cells
Chemoresistant ECC-1 sub-lines were generated by treatment with increasing doses of carboplatin or paclitaxel over a 6-week period followed by a 2-week recovery for a total of four cycles. Chemosensitive, original ECC-1 cells were treated with vehicle (saline).
Crystal Violet Staining and Phase Contrast Microscopy
Cells were stained with crystal violet and images were taken with a Nikon Eclipse T200 microscope and acquired using a NIS-Element F 3.3 camera and software. Magnification was set at ×20 and the scale bar is set at 50 μm.
Cell Viability Assay
Cell viability was determined by (4-[3-(4-iodophenyl)-2-(4-nitro-phenyl)-2H-5-tetrazolio] 1,3-benzene disulfonate) (WST-1) as previously described [58–60]. Briefly, cells were seeded at the concentration of 2000 cells per well in 100 μl medium in a 96-well plate and treated with the indicated concentrations of drugs. At the indicated time points (48, 72, 96 h), cells were incubated according to the manufacturer’s protocol with the WST-1 labeling mixture for 4 h. Formazan dye was quantified using a spectrophotometric plate reader to measure the absorbance at 480 nm (ELISA reader 190; Molecular Devices). Each experiment was performed in triplicate.
Proliferation Assay
For the cell proliferation assay, 10,000 cells/well were seeded in 24-well plates in triplicate and allowed to grow over a period up to 4 days. At each time point, cells were trypsinized, stained with trypan blue, and counted using a hemocytometer.
Chemicals
Carboplatin and paclitaxel were purchased from Sagent Pharmaceuticals (Schaumburg, IL) and Pharmachemie B.V (Haarlem, Netherlands) respectively. The MICAL2 inhibitor CCG-203971 and the ADAMTS5 inhibitor CAS# 929634-33-3 were purchased by Calbiochem and resuspended in DMSO. The PTP4A3 inhibitor pentamidine and the STAT5A inhibitor pimozide were purchased from Sigma-Aldrich and resuspended in DMSO. The (4-[3-(4-iodophenyl)-2-(4-nitro- phenyl)-2H-5-tetrazolio] 1,3-benzene disulfonate) (WST-1) was purchased from Cayman Chemicals.
Western Blot Analysis
Total cellular protein (10–20 μg) from each sample was separated by SDS-PAGE, transferred to PVDF membranes, and subjected to Western blot analysis. Antibodies used for Western blot analysis were obtained from the following commercial sources: rabbit polyclonal anti-ADAMTS5 antibody (abcam), mouse monoclonal anti STAT5A antibody (Thermo Fisher Scientific), rabbit polyclonal anti-MICAL2 antibody (proteintech), and mouse monoclonal PTP4A3 (PRL-3) antibody (Santa Cruz Biotechnology). Peroxidase-linked anti-mouse immunoglobulin G and peroxidase-linked anti-rabbit immunoglobulin G were from Amersham. Each experiment was performed in triplicates.
Statistical Analysis
Per gene, data including mRNA upregulation and/or DNA amplification and patients’ overall survival were obtained from the TGCA project via the data portal on 02/13/2018. Kaplan–Meier survival curves were created using the log-rank test for TCGA data to compare high-risk and low-risk groups. Hazard ratio was calculated using GraphPad and statistical significance was set at p = 0.05.
RNA-seq Data Analysis
50-bp FastQ paired-end reads (n = 12.3 million per sample) were trimmed using Trimmomatic (v 0.33) enabled with the optional “-q” option; 3-bp sliding-window trimming from 3′ end requiring minimum Q30. Quality control on raw sequence data for each sample was performed with FastQC. Read mapping was performed via Hisat2 (v2.0.2) using the UCSC human genome (hg38) as reference. Gene quantification was done via Cuffquant for FPKM values and Feature Counts for raw read counts. Differentially expressed genes were identified using the edgeR (negative binomial) feature in CLCGWB (Qiagen, Valencia, CA) using raw read counts. We filtered the generated list based on a minimum 2× absolute fold change and Bonferroni corrected p < 0.05. Principal component analysis (PCA) was performed in CLCGWB to identify and quantify variability in the data. Volcano plots and Venn diagrams were used to visualize the expression data and the results of significance testing. Pair-wise comparisons between treatment groups were made in the Bioconductor (3.2) R package following the standard workflow.
Quantitative Real-Time PCR
Total RNA was extracted using the RNeasy plus kit (Qiagen) according to the manufacturer’s instructions. Total RNA concentration and quality were measured using the Epoch microplate spectrophotometer (Biotek). One microgram of total RNA from each cell line was used to generate cDNA using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). SYBR green-based RT-qPCR was performed in triplicate using diluted cDNA. Samples were run in the LightCycler 96 (Roche). The comparative Ct method was used to determine the relative expression in each sample using TBP as normalization control. Primers were as follows: STAT5A, forward: 5′- ACATGTACCCACAGAACCCTGACC-3′, reverse: 5′- CACAACACGACCGCTTCACATTGC-3′; PTP4A3, forward: 5′-GGGACTTCTCAGGTCGTGTC-3′, reverse: 5′-AGCCCCGTACTTCTTCAGGT-3′; ADAMTS5, forward: 5′CAC TGT GGC TCA CGA AAT CG 3′, reverse: 5′CGC TTA TCT TCT GTG GAA CCA AA 3′; and MICAL2, forward: 5′-CTCACACGACACCTGGACCTA-3′, reverse: 5′-CCACGCTTATCCAATTTGTACCA-3′.
Electronic supplementary material
Complete list of the commonly DEGs in chemosenstive versus chemoresistant endometrial cancer cells. (DOCX 103 kb)
{kind=link}
Top 5 pathways enriched in IPA (Ingenuity Pathway Analysis) for each of the chemoresistant cell lines and for the commonly altered genes. (PNG 891 kb)
{kind=link}
Sensitivity of paclitaxel resistant ECC-1 (ECC-1P) endometrial cancer cells to ADAMTS5, PTP4A3, STAT5A, and MICAL2 inhibitors. (PNG 129 kb)
{kind=link}
Sensitivity of parental (sensitive) ECC-1 endometrial cancer cells to ADAMTS5, PTP4A3, STAT5A, and MICAL2 inhibitors. (PNG 120 kb)
Acknowledgements
We would like to thank Mrs. Dinesha Walek (Genomic Center, University of Minnesota) for the help with preparation of the samples for RNA-seq analysis. We are grateful to Mr. Juri Habicht (Brandenburg Medical School, Neuruppin, Germany) for the critical reading of the manuscript and to Dr. Boris Winterhoof (University of Minnesota) for the helpful discussion.
Funding
This work was supported by the Department of Defense Ovarian Cancer Research Program (OCRP) OC093424, by the Minnesota Ovarian Cancer Alliance, and by the Randy Shaver Cancer Foundation to MB. Ashley Mooneyham was supported by Cancer Biology Training Grant NIH T32 CA009138. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Compliance with Ethical Standards
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.DeSantis CE, et al. Cancer statistics for African Americans, 2016: progress and opportunities in reducing racial disparities. CA Cancer J Clin. 2016;66(4):290–308. doi: 10.3322/caac.21340. [DOI] [PubMed] [Google Scholar]
- 2.Lheureux S, Wilson M, Mackay HJ. Recent and current Phase II clinical trials in endometrial cancer: review of the state of art. Expert Opin Investig Drugs. 2014;23(6):773–792. doi: 10.1517/13543784.2014.907272. [DOI] [PubMed] [Google Scholar]
- 3.Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, Stein KD, Alteri R, Jemal A. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66(4):271–289. doi: 10.3322/caac.21349. [DOI] [PubMed] [Google Scholar]
- 4.Burstein HJ, Krilov L, Aragon-Ching JB, Baxter NN, Chiorean EG, Chow WA, de Groot JF, Devine SM, DuBois SG, el-Deiry WS, Epstein AS, Heymach J, Jones JA, Mayer DK, Miksad RA, Pennell NA, Sabel MS, Schilsky RL, Schuchter LM, Tung N, Winkfield KM, Wirth LJ, Dizon DS. Clinical cancer advances 2017: annual report on progress against cancer from the American Society of Clinical Oncology. J Clin Oncol. 2017;35(12):1341–1367. doi: 10.1200/JCO.2016.71.5292. [DOI] [PubMed] [Google Scholar]
- 5.Kumar S, Sharghi-Namini S, Rao N, Ge R. ADAMTS5 functions as an anti-angiogenic and anti-tumorigenic protein independent of its proteoglycanase activity. Am J Pathol. 2012;181(3):1056–1068. doi: 10.1016/j.ajpath.2012.05.022. [DOI] [PubMed] [Google Scholar]
- 6.Nissinen L, Kahari VM. ADAMTS5: a new player in the vascular field. Am J Pathol. 2012;181(3):743–745. doi: 10.1016/j.ajpath.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 7.Cai Y, Lu J, Tang F. Overexpression of MICAL2, a novel tumor-promoting factor, accelerates tumor progression through regulating cell proliferation and EMT. J Cancer. 2018;9(3):521–527. doi: 10.7150/jca.22355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lundquist MR, Storaska AJ, Liu TC, Larsen SD, Evans T, Neubig RR, Jaffrey SR. Redox modification of nuclear actin by MICAL-2 regulates SRF signaling. Cell. 2014;156(3):563–576. doi: 10.1016/j.cell.2013.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song Y, Kim SH, Kim KM, Choi EK, Kim J, Seo HR. Activated hepatic stellate cells play pivotal roles in hepatocellular carcinoma cell chemoresistance and migration in multicellular tumor spheroids. Sci Rep. 2016;6:36750. doi: 10.1038/srep36750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harburg GC, Hinck L. Navigating breast cancer: axon guidance molecules as breast cancer tumor suppressors and oncogenes. J Mammary Gland Biol Neoplasia. 2011;16(3):257–270. doi: 10.1007/s10911-011-9225-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ziel JW, Hagedorn EJ, Audhya A, Sherwood DR. UNC-6 (netrin) orients the invasive membrane of the anchor cell in C. elegans. Nat Cell Biol. 2009;11(2):183–189. doi: 10.1038/ncb1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tapia-Vieyra JV, Delgado-Coello B, Mas-Oliva J. Atherosclerosis and cancer; a resemblance with far-reaching implications. Arch Med Res. 2017;48(1):12–26. doi: 10.1016/j.arcmed.2017.03.005. [DOI] [PubMed] [Google Scholar]
- 13.Salvador E, Burek M, Forster CY. Tight junctions and the tumor microenvironment. Curr Pathobiol Rep. 2016;4:135–145. doi: 10.1007/s40139-016-0106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mohanty SK, Yagiz K, Pradhan D, Luthringer DJ, Amin MB, Alkan S, Cinar B. STAT3 and STAT5A are potential therapeutic targets in castration-resistant prostate cancer. Oncotarget. 2017;8(49):85997–86010. doi: 10.18632/oncotarget.20844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou W, et al. The antipsychotic drug pimozide inhibits cell growth in prostate cancer through suppression of STAT3 activation. Int J Oncol. 2016;48(1):322–328. doi: 10.3892/ijo.2015.3229. [DOI] [PubMed] [Google Scholar]
- 16.Cai N, Zhou W, Ye LL, Chen J, Liang QN, Chang G, Chen JJ. The STAT3 inhibitor pimozide impedes cell proliferation and induces ROS generation in human osteosarcoma by suppressing catalase expression. Am J Transl Res. 2017;9(8):3853–3866. [PMC free article] [PubMed] [Google Scholar]
- 17.Chen JJ, Cai N, Chen GZ, Jia CC, Qiu DB, du C, Liu W, Yang Y, Long ZJ, Zhang Q. The neuroleptic drug pimozide inhibits stem-like cell maintenance and tumorigenicity in hepatocellular carcinoma. Oncotarget. 2017;8(11):17593–17609. doi: 10.18632/oncotarget.4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santo L, Hideshima T, Cirstea D, Bandi M, Nelson EA, Gorgun G, Rodig S, Vallet S, Pozzi S, Patel K, Unitt C, Squires M, Hu Y, Chauhan D, Mahindra A, Munshi NC, Anderson KC, Raje N. Antimyeloma activity of a multitargeted kinase inhibitor, AT9283, via potent Aurora kinase and STAT3 inhibition either alone or in combination with lenalidomide. Clin Cancer Res. 2011;17(10):3259–3271. doi: 10.1158/1078-0432.CCR-10-3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nelson EA, Walker SR, Weisberg E, Bar-Natan M, Barrett R, Gashin LB, Terrell S, Klitgaard JL, Santo L, Addorio MR, Ebert BL, Griffin JD, Frank DA. The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood. 2011;117(12):3421–3429. doi: 10.1182/blood-2009-11-255232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zerbini LF, Bhasin MK, de Vasconcellos JF, Paccez JD, Gu X, Kung AL, Libermann TA. Computational repositioning and preclinical validation of pentamidine for renal cell cancer. Mol Cancer Ther. 2014;13(7):1929–1941. doi: 10.1158/1535-7163.MCT-13-0750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qiu G, Jiang J, Liu XS. Pentamidine sensitizes chronic myelogenous leukemia K562 cells to TRAIL-induced apoptosis. Leuk Res. 2012;36(11):1417–1421. doi: 10.1016/j.leukres.2012.07.017. [DOI] [PubMed] [Google Scholar]
- 22.Hartman KG, McKnight LE, Liriano MA, Weber DJ. The evolution of S100B inhibitors for the treatment of malignant melanoma. Future Med Chem. 2013;5(1):97–109. doi: 10.4155/fmc.12.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bursavich MG, Gilbert AM, Lombardi S, Georgiadis KE, Reifenberg E, Flannery CR, Morris EA. 5′-Phenyl-3′H-spiro[indoline-3,2′-[1,3,4]thiadiazol]-2-one inhibitors of ADAMTS-5 (aggrecanase-2) Bioorg Med Chem Lett. 2007;17(20):5630–5633. doi: 10.1016/j.bmcl.2007.07.048. [DOI] [PubMed] [Google Scholar]
- 24.Gilbert AM, Bursavich MG, Lombardi S, Georgiadis KE, Reifenberg E, Flannery CR, Morris EA. 5-((1H-Pyrazol-4-yl)methylene)-2-thioxothiazolidin-4-one inhibitors of ADAMTS-5. Bioorg Med Chem Lett. 2007;17(5):1189–1192. doi: 10.1016/j.bmcl.2006.12.020. [DOI] [PubMed] [Google Scholar]
- 25.Haak AJ, Tsou PS, Amin MA, Ruth JH, Campbell P, Fox DA, Khanna D, Larsen SD, Neubig RR. Targeting the myofibroblast genetic switch: inhibitors of myocardin-related transcription factor/serum response factor-regulated gene transcription prevent fibrosis in a murine model of skin injury. J Pharmacol Exp Ther. 2014;349(3):480–486. doi: 10.1124/jpet.114.213520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson LA, Rodansky ES, Haak AJ, Larsen SD, Neubig RR, Higgins PDR. Novel Rho/MRTF/SRF inhibitors block matrix-stiffness and TGF-beta-induced fibrogenesis in human colonic myofibroblasts. Inflamm Bowel Dis. 2014;20(1):154–165. doi: 10.1097/01.MIB.0000437615.98881.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vogel RI, Pulver T, Heilmann W, Mooneyham A, Mullany S, Zhao X, Shahi M, Richter J, Klein M, Chen L, Ding R, Konecny G, Kommoss S, Winterhoff B, Ghebre R, Bazzaro M. USP14 is a predictor of recurrence in endometrial cancer and a molecular target for endometrial cancer treatment. Oncotarget. 2016;7(21):30962–30976. doi: 10.18632/oncotarget.8821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ai Z, Lu Y, Qiu S, Fan Z. Overcoming cisplatin resistance of ovarian cancer cells by targeting HIF-1-regulated cancer metabolism. Cancer Lett. 2016;373(1):36–44. doi: 10.1016/j.canlet.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sherman-Baust CA, Becker KG, Wood III WH, Zhang Y, Morin PJ. Gene expression and pathway analysis of ovarian cancer cells selected for resistance to cisplatin, paclitaxel, or doxorubicin. J Ovarian Res. 2011;4(1):21. doi: 10.1186/1757-2215-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Servidei T, Riccardi A, Mozzetti S, Ferlini C, Riccardi R. Chemoresistant tumor cell lines display altered epidermal growth factor receptor and HER3 signaling and enhanced sensitivity to gefitinib. Int J Cancer. 2008;123(12):2939–2949. doi: 10.1002/ijc.23902. [DOI] [PubMed] [Google Scholar]
- 31.Jia P, Jin H, Meador CB, Xia J, Ohashi K, Liu L, Pirazzoli V, Dahlman KB, Politi K, Michor F, Zhao Z, Pao W. Next-generation sequencing of paired tyrosine kinase inhibitor-sensitive and -resistant EGFR mutant lung cancer cell lines identifies spectrum of DNA changes associated with drug resistance. Genome Res. 2013;23(9):1434–1445. doi: 10.1101/gr.152322.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brookman-Amissah N, Duchesnes C, Williamson MP, Wang Q, Ahmed A, Feneley MR, Mackay A, Freeman A, Fenwick K, Iravani M, Weber B, Ashworth A, Masters JR. Genome-wide screening for genetic changes in a matched pair of benign and prostate cancer cell lines using array CGH. Prostate Cancer Prostatic Dis. 2005;8(4):335–343. doi: 10.1038/sj.pcan.4500826. [DOI] [PubMed] [Google Scholar]
- 33.Ong PS, Chan SY, Ho PC. Microarray analysis revealed dysregulation of multiple genes associated with chemoresistance to As(2)O(3) and increased tumor aggressiveness in a newly established arsenic-resistant ovarian cancer cell line, OVCAR-3/AsR. Eur J Pharm Sci. 2012;45(3):367–378. doi: 10.1016/j.ejps.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 34.Lu HP, Chao CC. Cancer cells acquire resistance to anticancer drugs: an update. Biom J. 2012;35(6):464–472. doi: 10.4103/2319-4170.104411. [DOI] [PubMed] [Google Scholar]
- 35.Chon HS, Lancaster JM. Microarray-based gene expression studies in ovarian cancer. Cancer Control. 2011;18(1):8–15. doi: 10.1177/107327481101800102. [DOI] [PubMed] [Google Scholar]
- 36.Chanrion M, Negre V, Fontaine H, Salvetat N, Bibeau F, Grogan GM, Mauriac L, Katsaros D, Molina F, Theillet C, Darbon JM. A gene expression signature that can predict the recurrence of tamoxifen-treated primary breast cancer. Clin Cancer Res. 2008;14(6):1744–1752. doi: 10.1158/1078-0432.CCR-07-1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chao A, Wang TH, Lai CH. Overview of microarray analysis of gene expression and its applications to cervical cancer investigation. Taiwan J Obstet Gynecol. 2007;46(4):363–373. doi: 10.1016/S1028-4559(08)60005-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Russo G, Zegar C, Giordano A. Advantages and limitations of microarray technology in human cancer. Oncogene. 2003;22(42):6497–6507. doi: 10.1038/sj.onc.1206865. [DOI] [PubMed] [Google Scholar]
- 39.Zhu S, Qing T, Zheng Y, Jin L, Shi L. Advances in single-cell RNA sequencing and its applications in cancer research. Oncotarget. 2017;8(32):53763–53779. doi: 10.18632/oncotarget.17893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cieslik M, Chinnaiyan AM. Cancer transcriptome profiling at the juncture of clinical translation. Nat Rev Genet. 2018;19(2):93–109. doi: 10.1038/nrg.2017.96. [DOI] [PubMed] [Google Scholar]
- 41.Guo H, Cheng Y, Martinka M, McElwee K. High LIFr expression stimulates melanoma cell migration and is associated with unfavorable prognosis in melanoma. Oncotarget. 2015;6(28):25484–25498. doi: 10.18632/oncotarget.4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohata Y, Tsuchiya M, Hirai H, Yamaguchi S, Akashi T, Sakamoto K, Yamaguchi A, Ikeda T, Kayamori K. Leukemia inhibitory factor produced by fibroblasts within tumor stroma participates in invasion of oral squamous cell carcinoma. PLoS One. 2018;13(2):e0191865. doi: 10.1371/journal.pone.0191865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McQueeney KE, Salamoun JM, Burnett JC, Barabutis N, Pekic P, Lewandowski SL, Llaneza DC, Cornelison R, Bai Y, Zhang ZY, Catravas JD, Landen CN, Wipf P, Lazo JS, Sharlow ER. Targeting ovarian cancer and endothelium with an allosteric PTP4A3 phosphatase inhibitor. Oncotarget. 2018;9(9):8223–8240. doi: 10.18632/oncotarget.23787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hjort MA, Abdollahi P, Vandsemb EN, Fenstad MH, Lund B, Slørdahl TS, Børset M, Rø TB. Phosphatase of regenerating liver-3 is expressed in acute lymphoblastic leukemia and mediates leukemic cell adhesion, migration and drug resistance. Oncotarget. 2018;9(3):3549–3561. doi: 10.18632/oncotarget.23186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Radke I, Götte M, Smollich M, Scharle N, Kiesel L, Wülfing P. Expression of PRL-3 regulates proliferation and invasion of breast cancer cells in vitro. Arch Gynecol Obstet. 2017;296(6):1153–1160. doi: 10.1007/s00404-017-4542-2. [DOI] [PubMed] [Google Scholar]
- 46.den Hollander P, Rawls K, Tsimelzon A, Shepherd J, Mazumdar A, Hill J, Fuqua SAW, Chang JC, Osborne CK, Hilsenbeck SG, Mills GB, Brown PH. Phosphatase PTP4A3 promotes triple-negative breast cancer growth and predicts poor patient survival. Cancer Res. 2016;76(7):1942–1953. doi: 10.1158/0008-5472.CAN-14-0673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiong J, Li Z, Zhang Y, Li D, Zhang G, Luo X, Jie Z, Liu Y, Cao Y, le Z, Tan S, Zou W, Gong P, Qiu L, Li Y, Wang H, Chen H. PRL-3 promotes the peritoneal metastasis of gastric cancer through the PI3K/Akt signaling pathway by regulating PTEN. Oncol Rep. 2016;36(4):1819–1828. doi: 10.3892/or.2016.5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiong S, et al. TGF-beta1 stimulates migration of type II endometrial cancer cells by down-regulating PTEN via activation of SMAD and ERK1/2 signaling pathways. Oncotarget. 2016;7(38):61262–61272. doi: 10.18632/oncotarget.11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bellomo C, Caja L, Moustakas A. Transforming growth factor beta as regulator of cancer stemness and metastasis. Br J Cancer. 2016;115(7):761–769. doi: 10.1038/bjc.2016.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bardelli A, Saha S, Sager JA, Romans KE, Xin B, Markowitz SD, Lengauer C, Velculescu VE, Kinzler KW, Vogelstein B. PRL-3 expression in metastatic cancers. Clin Cancer Res. 2003;9(15):5607–5615. [PubMed] [Google Scholar]
- 51.Yu ZH, Zhang ZY. Regulatory mechanisms and novel therapeutic targeting strategies for protein tyrosine phosphatases. Chem Rev. 2018;118(3):1069–1091. doi: 10.1021/acs.chemrev.7b00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Polato F, et al. PRL-3 phosphatase is implicated in ovarian cancer growth. Clin Cancer Res. 2005;11(19 Pt 1):6835–6839. doi: 10.1158/1078-0432.CCR-04-2357. [DOI] [PubMed] [Google Scholar]
- 53.Zimmerman MW, McQueeney KE, Isenberg JS, Pitt BR, Wasserloos KA, Homanics GE, Lazo JS. Protein-tyrosine phosphatase 4A3 (PTP4A3) promotes vascular endothelial growth factor signaling and enables endothelial cell motility. J Biol Chem. 2014;289(9):5904–5913. doi: 10.1074/jbc.M113.480038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bai Y, Luo Y, Liu S, Zhang L, Shen K, Dong Y, Walls CD, Quilliam LA, Wells CD, Cao Y, Zhang ZY. PRL-1 protein promotes ERK1/2 and RhoA protein activation through a non-canonical interaction with the Src homology 3 domain of p115 Rho GTPase-activating protein. J Biol Chem. 2011;286(49):42316–42324. doi: 10.1074/jbc.M111.286302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fortney K, Griesman J, Kotlyar M, Pastrello C, Angeli M, Sound-Tsao M, Jurisica I. Prioritizing therapeutics for lung cancer: an integrative meta-analysis of cancer gene signatures and chemogenomic data. PLoS Comput Biol. 2015;11(3):e1004068. doi: 10.1371/journal.pcbi.1004068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen Q, Tian S, Zhu J, Li KT, Yu TH, Yu LH, Bai DQ. Exploring a novel target treatment on breast cancer: aloe-emodin mediated photodynamic therapy induced cell apoptosis and inhibited cell metastasis. Anti Cancer Agents Med Chem. 2016;16(6):763–770. doi: 10.2174/1871520615666150821093323. [DOI] [PubMed] [Google Scholar]
- 57.Mariotti S, Barravecchia I, Vindigni C, Pucci A, Balsamo M, Libro R, Senchenko V, Dmitriev A, Jacchetti E, Cecchini M, Roviello F, Lai M, Broccoli V, Andreazzoli M, Mazzanti CM, Angeloni D. MICAL2 is a novel human cancer gene controlling mesenchymal to epithelial transition involved in cancer growth and invasion. Oncotarget. 2016;7(2):1808–1825. doi: 10.18632/oncotarget.6577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anchoori RK, Khan SR, Sueblinvong T, Felthauser A, Iizuka Y, Gavioli R, Destro F, Isaksson Vogel R, Peng S, Roden RBS, Bazzaro M. Stressing the ubiquitin-proteasome system without 20S proteolytic inhibition selectively kills cervical cancer cells. PLoS One. 2011;6(8):e23888. doi: 10.1371/journal.pone.0023888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bazzaro M, et al. α,β-Unsaturated carbonyl system of chalcone-based derivatives is responsible for broad inhibition of proteasomal activity and preferential killing of human papilloma virus (HPV) positive cervical cancer cells. J Med Chem. 2011;54(2):449–456. doi: 10.1021/jm100589p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bazzaro M, Lee MK, Zoso A, Stirling WLH, Santillan A, Shih IM, Roden RBS. Ubiquitin-proteasome system stress sensitizes ovarian cancer to proteasome inhibitor-induced apoptosis. Cancer Res. 2006;66(7):3754–3763. doi: 10.1158/0008-5472.CAN-05-2321. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete list of the commonly DEGs in chemosenstive versus chemoresistant endometrial cancer cells. (DOCX 103 kb)
Top 5 pathways enriched in IPA (Ingenuity Pathway Analysis) for each of the chemoresistant cell lines and for the commonly altered genes. (PNG 891 kb)
Sensitivity of paclitaxel resistant ECC-1 (ECC-1P) endometrial cancer cells to ADAMTS5, PTP4A3, STAT5A, and MICAL2 inhibitors. (PNG 129 kb)
Sensitivity of parental (sensitive) ECC-1 endometrial cancer cells to ADAMTS5, PTP4A3, STAT5A, and MICAL2 inhibitors. (PNG 120 kb)