

Abstract
Thyroid hormones (TH) play a fundamental role in diverse processes, including cellular movement. Cell migration requires the integration of events that induce changes in cell structure towards the direction of migration. These actions are driven by actin remodeling and stabilized by the development of adhesion sites to extracellular matrix via transmembrane receptors linked to the actin cytoskeleton. Focal adhesion kinase (FAK) is a non-receptor tyrosine kinase that promotes cell migration and invasion through the control of focal adhesion turnover. In this work, we demonstrate that the thyroid hormone triiodothyronine (T3) regulates actin remodeling and cell movement in breast cancer T-47D cells through the recruitment of FAK. T3 controls FAK phosphorylation and translocation at sites where focal adhesion complexes are assembled. This process is triggered via rapid signaling to integrin αV/β3, Src, phosphatidylinositol 3-OH kinase (PI3K), and FAK. In addition, we established a cellular model with different concentration of T3 levels: normal, absence, and excess in T-47D breast cancer cells. We found that the expression of Src, FAK, and PI3K remained at normal levels in the excess of T3 model, while it was significantly reduced in the absence model. In conclusion, these results suggest a novel role for T3 as an important modulator of cell migration, providing a starting point for the development of new therapeutic strategies for breast cancer treatment.
Electronic supplementary material
The online version of this article (doi:10.1007/s12672-016-0280-3) contains supplementary material, which is available to authorized users.
Keywords: Triiodothyronine, Src/FAK/PI3K, Focal adhesion complex, Breast cancer cell migration
Introduction
Breast cancer is the most common malignancy among women worldwide and an important public health concern. According to the American Cancer Society, one out of eight women will develop breast cancer at some stage in her life [1]. Metastasis of tumor cells is the major cause of mortality among women with breast cancer. Given this reality, strategies to prevent this process are needed in breast cancer therapies [2]. Cell migration is fundamental for cancer spread, invasion, and metastasis. It is achieved by means of dynamic actin cytoskeleton remodeling and development of focal adhesion sites [3]. These structural modifications are required for the protrusion of the leading edge of the cell, formation of adhesion complexes, myosin/actin-mediated cell contraction, and the release of adhesions at the rear of the cell [4], all of which are necessary for cell movement [5]. These actions are driven by the recruitment of several regulatory proteins that are part of rapid intracellular signaling pathways and whose activation promotes actin cytoskeleton reorganization, driving cell migration and invasion [6].
The focal adhesion kinase (FAK) protein is an essential regulator of the actin cytoskeleton [7, 8]. FAK is a non-receptor tyrosine kinase that acts as a key mediator in intracellular signaling, which in turn leads to activation of several cellular processes, including cell motility [9]. FAK plays a major role in focal adhesion complex formation [10]. In this position, it not only serves as a structural anchor but also regulates this dynamic process, thereby modulating the steps involved in cell migration and invasion [11, 12].
The Src protein, a member of the Src family, is another non-receptor tyrosine kinase. This protein interacts physically and functionally with FAK to promote a variety of cellular responses [13, 14]. Formation and activation of a signaling complex between Src and FAK modulates the actin cytoskeleton [15]. The Src/FAK complex induces the recruitment of several proteins involved in cell survival and motility, resulting in a metastatic phenotype in invasive cancer cells [16]. Numerous studies have found an increased activity of the Src/FAK complex in tumor cells involved in the signals that lead to tumor growth and metastasis [17]. They have shown its strict association with tumor metastasis and that there is a strong correlation between increased expression levels and phosphorylation of Src/FAK in human tumors [18–20].
Once this complex is activated by extracellular signals, recruitment of phosphoinositide 3-kinase (PI3K) occurs. PI3K belongs to a prominent family of enzymes whose activation is a key event in signaling pathways involved in the control of diverse cellular responses, such as proliferation, survival, metabolism, and cellular mobility [21]. The control mechanism of this enzyme is, however, often dysregulated in pathological processes such as cancer, where PI3K contributes to different aspects of tumorigenesis, including tumor growth, progression, invasiveness, and metastasis processes [22, 23]. Breast cancer is a hormone-dependent disease; studying the influence of different hormones on this pathology is therefore a key step in cancer research.
Thyroid hormones (TH) have known effects on mitogenesis and apoptosis [24, 25]. Several research groups have addressed the question of whether TH may be associated with an increased risk of cancer development [26]. Cristofanilli et al. observed that hypothyroidism was associated with a decreased risk of breast cancer and a more indolent invasive disease [27]. Similarly, Tosovic et al. carried out a prospective study of T3 levels in correlation to breast cancer risk in postmenopausal women. They found that T3 levels were positively correlated with breast cancer risk in a dose-dependent manner [28]. Different studies have subsequently provided solid evidence about the role of TH in carcinogenesis. In particular, T3 may have a considerable impact on the metastatic stage of the disease [25, 29]. Thyroid hormones exert most of their actions through classical genomic pathways. Nevertheless, a large number of non-genomic effects have been described for T3 in recent years [30]. They suggest that T3 is involved in the regulation of ion channels as well as in the metabolism of phospholipids [31] through activation of different intracellular signaling pathways [32]. In light of this evidence, in this study, we aimed at identifying whether T3 exerts an effect on T-47D breast cancer cell motility through non-genomic mechanisms by remodeling the actin cytoskeleton. In addition, we characterized the intracellular signaling cascade recruited during this process.
Materials and Methods
Cell Culture and Treatments
MCF-7, T-47D, and SKBR-3 human breast cancer cells were obtained from the American Type Culture Collection. MCF-7 cells were cultured in minimal essential medium (MEM) with l-glutamine (2 mM) and 10% fetal bovine serum (FBS) and 10% penicillin/streptomycin. T-47D and SKBR-3 cells were grown in RPMI 1640 supplemented with l-glutamine (2 mM), 10% fetal bovine serum, and 10% penicillin/streptomycin. Before long treatments, breast cancer cells were kept 24 h in medium containing steroid-deprived FBS. Before experiments investigating non-transcriptional effects, cancer cells were kept in medium containing no FBS for 8 h. T3, PD98059 (PD), wortmannin (WM), and LY294002 (LY) were from Sigma-Aldrich (Saint Louis, MO); 4-amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo (3,4-d) pyrimidine (PP2) was from Calbiochem (La Jolla, CA) and Tetraidothyroacetic acid (Tetrac) and FAK inhibitor (FAKi) from Santa Cruz Biotechnology (Santa Cruz, CA). Whenever an inhibitor was used, the compound was added 30–45 min before starting the active treatments. PP2, FAKi, WM, LY, and PD were dissolved in DMSO, Tetrac was dissolved in acetone, and triiodothyronine (T3) was dissolved in RPMI 1640 Medium. In control cultures, the DMSO, acetone, or RPMI medium vehicle was added at the same final dilution. The final concentration of the solvents was 1 μL of solvent per 1 mL of medium.
The “in vitro” model consisted in treating T-47D cells for 48 h with different T3 concentrations in order to simulate three states: normal or physiological concentration (T3 10−9 M in RPMI with 0% FBS), absence of T3 (only RPMI with 0% FBS), and excess of T3 (T3 10−7 M in RPMI with 0% FBS). Expression levels of FAK, Src, p85, and Akt were analyzed by Western blot with specific antibodies for each protein.
Immunoblottings
Cell lysates were separated by SDS-PAGE. Antibodies used were as follows: p-FAKY397 (611807), FAK (610088) (BD Transduction Laboratories, Lexington, KY); p-FAK (Tyr397) (sc-11765-R), Akt1/2/3 (sc-81434), p-Akt1/2/3 (Thr 308)-R (sc-16646-R), Src (sc-5266), p-SrcY419 (sc-101802), integrin αV/β3 (sc-7312), Akt1 (sc-1618), TRα1/β1 (sc-740), and ERα (sc-7207) (Santa Cruz Biotechnology, Santa Cruz, CA); p-AktS473, p44/42 MAPK (Erk1/2) (Cell Signaling Technology, Danvers, MA); α-tubulin (T6074) (Sigma-Aldrich, Saint Louis, MO); integrin αV/β3 (NB600–1342) (Novus Biologicals, USA); p85α (Pharmingen, BD Biosciences); and Anti-MAP Kinase ERK1/ERK2 Phospho-Specific (Thr202/Tyr204) (Calbiochem). Primary and secondary antibodies were incubated with the membranes using standard techniques. Immunodetection was accomplished using enhanced chemiluminescence.
Cell Immunofluorescence
T-47D cells were grown on coverslips. Cells were fixed with 4% paraformaldehyde for 30 min and permeabilized with 0.1% Triton for 5 min. Blocking was performed with PBS containing 3% bovine serum albumin for 30 min. Cells were incubated with antibodies against FAK and p-FAKY397 (BD Transduction Laboratories, Lexington, KY) overnight at 4 °C, followed by incubation with a fluorescein-conjugated secondary antibody (1:150; Vector Laboratories, Burlingame, CA). Cells were then incubated with Texas Red-phalloidin (Sigma-Aldrich, Saint Louis, MO) for 30 min. After washing, the nuclei were counterstained with or 4′-6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, Saint Louis, MO) and mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA). Immunofluorescence was visualized using a Nikon Eclipse E200 microscope and recorded with a high-resolution DP70 Olympus digital camera.
Co-immunoprecipitation Assay
Breast cancer cells were washed with ice-cold PBS and lysed with the following: 20 mM Tris-HCl, pH 7.4, 10 mM EDTA, 100 mM NaCl, 1% Igepal, 1 mM Na3VO4, 50 mM NaF, 0.1 mg/L PMSF, 0.3 mg/L aprotinin, and 0.01% protease inhibitor mixture (Sigma-Aldrich, Saint Louis, MO). Immunoprecipitating antibodies vs. integrin αV/β3, FAK, and p85α in 500 μL of lysis buffer were then added and incubated for 1 h at 4 °C with gentle rocking. Forty microliters of 1:1 protein-A-agarose was then added and gently rocked for 2 h at 4 °C. The mixture was centrifuged at 12,000×g for 5 min at 4 °C. The supernatant was removed and the immunoprecipitates washed with 500 mL of the following: 20 mM Tris-HCl, pH 7.4, 10 mM EDTA, 150 mM NaCl, 1% IGEPAL, 1 mM Na3VO4, 50 mM NaF, 0.1 mg/L PMSF, 0.3 mg/L aprotinin, and 0.01% protease inhibitor mixture (Sigma-Aldrich, Saint Louis, MO). Immunoprecipitated proteins were separated under reducing and denaturing conditions by 10% SDS-PAGE and transferred to a polyvinylidene difluoride membrane. Non-specific binding was blocked with 3% BSA (Bovine serum albumin) in PBS–Tween 20. Membranes were incubated with anti-integrin αV/β3, Src, p85α, and FAK antibodies.
Cell Adhesion Assays
Five hundred thousand cells per well were seeded into six-well plates on coverslips previously coated with 1% sterile gelatin and exposed to different treatments. The cells were incubated at 37 °C for 2 h. Non adherent T-47D cells were then removed by gently washing with PBS. The attached cells were fixed with 4% formaldehyde and stained with Giemsa. Cells attached images were captured and counted in ten randomly chosen fields per well using a Nikon Eclipse E200 microscope coupled to a high-resolution CCD digital camera, as previously described [33].
Cell Migration Assays
Cell migration was assayed with razor scrape assays as previously described [15]. Briefly, a razor blade was pressed through the confluent T-47D breast cancer cell monolayer into the plastic plate to mark the starting line. T-47D cells were swept away on one side of that line. Cells were washed and 2.0 mL of RPMI containing steroid-deprived FBS and gelatin (1 mg/mL) added. Cytosine β-d-arabinofuranoside hydrochloride (Sigma) (10 μM), a selective inhibitor of DNA synthesis that does not inhibit RNA synthesis, was added 1 h before the test to prevent cell proliferation. Absence of cell proliferation and viability of the cells were checked in preliminary experiments with MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) tests. Migration was monitored for 48 h. Fresh medium and treatment were replaced after 24 h. Digital images from cells were taken and the distance of migration was analyzed by phase-contrast microscopy.
Real-Time PCR
Cell cultures were homogenized in 0.5 mL of TRIzol (GIBCO-BRL); total RNA was isolated according to the manufacturer’s instructions. Total RNA concentration was determined spectrophotometrically and RNA integrity examined by 1% agarose gel electrophoresis. First-strand cDNA synthesis from 2.5 μg RNA per sample was performed using Moloney murine leukemia virus retrotranscriptase and random hexamer primers (Invitrogen/Life Technologies, Argentina) in 20 μL reaction mixture. Real-time quantification was monitored by measuring the increase in fluorescence caused by the binding of EvaGreen dye to double-stranded DNA at the end of each amplification cycle. Amplification of the genes of interest was performed using human-specific primers in the conditions described in Table 1 (Supporting Information S1). Samples were run in duplicate. Each PCR run included a no-template control and a sample without reverse transcriptase. Real-time PCR was performed with a Corbett-Rotor Gene 6000. Cycle threshold (CT) was plotted against input concentration and the efficiency of amplification for each primer pair calculated using the equation E = 10 − 1/s − 1, where s is the slope. Melt curve analysis (60–95 °C in 0.2 °C increments) was performed at the end of the amplification. Some samples were subjected to 1.5% agarose gel electrophoresis to examine product purity and to verify the correct size of the PCR product. We analyzed the experiment with two housekeeping “s18 mRNA” and “GPDH”( the glycerol phosphate dehydrogenase) that is generally accepted to normalize real-time PCR in eukaryotic cells, at any rate the two housekeeping have not changed with the treatment.
Radioligand Binding Assay
Membranes from T-47D breast cancer cells were prepared by method as described previously [34, 35]. The harvested cells were washed with PBS, harvested, and resuspended in buffer ice-cold. The samples were centrifuged, and Na2CO3 (100 mM) was added to the resulting supernatant and shaken at 4 °C, and then centrifuged at 100,000g for 15 min. The pellet obtained was resuspended in 500 μL of a buffer and separated into aliquots, as described [35]. The protein content was quantitated using the Micro BCA™ Protein Assay Kit (Thermo Scientific, USA). Thyroid hormone binding displacement studies were performed as follows: stock solutions of T3 were diluted to their final concentrations in 0.04 N KOH with 0.4% polyethylene glycol to ensure that the effect was independent of the solvent used. Purified cell membrane protein preparation (60 μg) was mixed with 10 μL of [125I]T3 (3 μCi) for 10 min, and the indicated concentrations of test compounds were then added; the mixture was allowed to incubate an additional 30 min at room temperature. The mixtures were precipitated with TCA (5%) and the precipitate was washed with cold ethanol (2 times), and the resulting TCA-precipitable radioactivity was quantitated in a liquid scintillation counter.
Gene Silencing with RNA Interference
Synthetic small interfering RNAs targeting estrogen receptor α (small interfering RNA (siRNA) SMARTpool ESR1) and control siRNAs (D-001810-01-05) were purchased from Dharmacon (Thermo Fisher Scientific Inc., USA). Thyroid hormone receptor alpha (TRα) and beta 1 (TRβ1) siRNAs were from Santa Cruz Biotechnology (Santa Cruz, CA). The siRNAs were used at the final concentration of 50 nM. Breast cancer cells were treated 48 h after siRNAs transfection. Efficacy of gene silencing was checked with western analysis and found to be optimal at 48 h.
Statistical Analysis
All values are expressed as mean ± SD. Statistical analyses and graphs were done using InStat from GraphPad Prism Software. Statistical differences between mean values were determined by ANOVA, followed by Fisher’s protected least significance difference (PLSD).
Results
Effects of Different T3 Doses on Src, FAK, and PI3K Proteins in Breast Cancer Cells
To determine whether different doses of thyroid hormone could alter the expression patterns of the kinases involved in cell movement, we designed an in vitro model using T-47D breast cancer cells. In this model, the cells were exposed during 48 h to different doses of T3 to recreate a normal, excess, and absence state of T3 (described in “Materials and Methods”), which allowed measuring the expression of Src, FAK, p85α, and Akt1/2/3 (PI3K substrate) proteins and mRNA were then measured by Western blot and quantitative RT-PCR, respectively (Fig. 1a–h). We found no significant differences in the expression of Src, FAK, p85α, and Akt proteins between normal and excess T3 levels. We did, however, observe a marked decrease in the expression of the proteins mentioned above in cells without T3 compared to the other T3 states (Fig. 1a–e). We also found a similar behavior of mRNA and protein expression for total Akt, but the changes were not significant. We did not observe correlation between mRNA and protein expression for Src and FAK, suggesting that post-transcriptional control mechanisms may be operating.
Fig. 1.
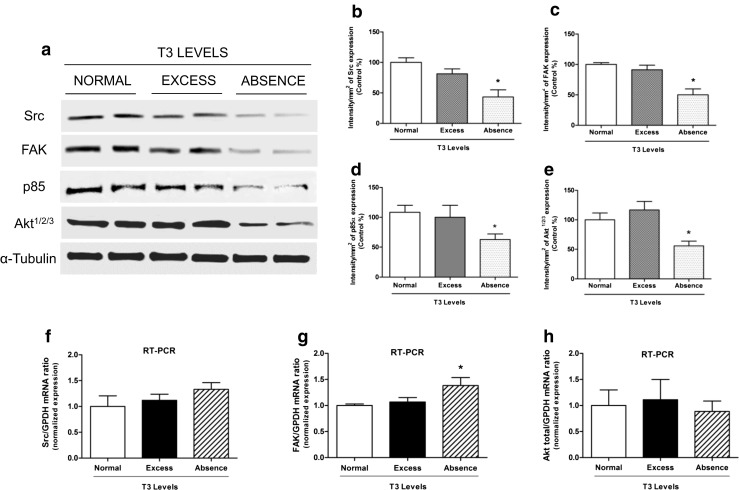
T3 controls the expression levels of Src, FAK, p85α, and Akt1/2/3 in breast cancer cells. a T-47D cells were pre-treated during 48 h with T3 in different concentrations: T3 1 nM (normal state), T3 100 nM (excess state of T3) and only RPMI medium (absence state of T3). b–e The blots were quantified using Quantity One software (BIO-RAD). f–h mRNA abundance was determined by RT-PCR on samples of total of breast cancer cells T-47D. Results are expressed as mean ± SD of the measurements. * = P < 0.05 vs. control. All experiments were performed in triplicate; representative images are shown
T3 Induces Phosphorylation of FAK and AKT in T-47D Cells
In order to determine if T3 could activate this protein kinases, we performed a rapid treatment of T-47D breast cancer cells with thyroid hormone (T3, 1 nM) resulting in an increased Tyr397-FAK phosphorylation, which corresponds to activation [7–9]. The same effect was observed in Thr308-Akt1/2/3 protein phosphorylation as a result of PI3K activation. This phenomenon was time-dependent and transient; it was maximal after 20 min and returned to baseline after 60 min (Fig. 2a–c).
Fig. 2.
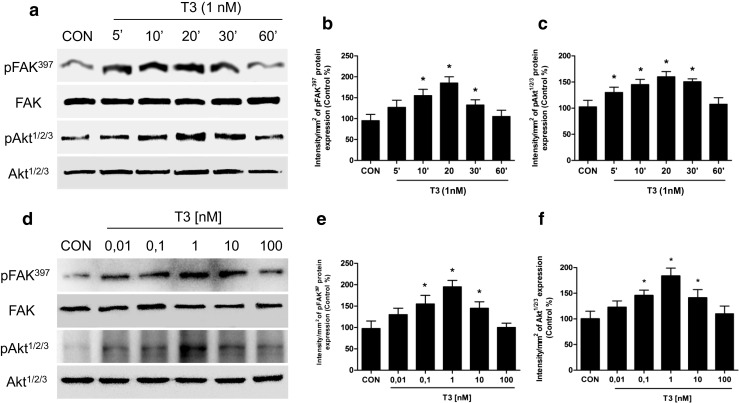
T3 induces FAK and Akt phosphorylation/activation in T-47D breast cancer cells. Time (a) and dose-dependent (d) FAK and Akt activation of T-47D cells after T3 treatment. Total cell amount of wild-type FAK and Akt or Tyr397-phosphorylated FAK (p-FAK) and phosphorylated-Akt (p-Akt) are shown by Western blot analysis. Phospho-FAK397 and p-Akt densitometry values were adjusted to FAK and Akt intensity, respectively, and then normalized to the control sample. * = P < 0.05 vs. corresponding control. All experiments were performed in triplicate with consistent results; representative images are shown (b, c, e, f)
To establish the T3 dose capable of stimulating FAK and Akt activation, T-47D cells were treated for 20 min with increasing concentrations of T3 from 0.01 to 100 nM (Fig. 2d–f). Maximum Tyr397-FAK and Akt1/2/3 phosphorylation was observed with 1 nM T3 (Fig. 2d–f).
T3 Induces FAK Phosphorylation and Translocation via Src Kinase and Promotes Formation of Focal Adhesion Complexes
Treatment of T-47D breast cancer cells with T3 (1 nM, 20 min) led to a rapid increase of Tyr419- Src and Tyr397-FAK phosphorylation, which corresponds to activation of these proteins compared to control [6] (Fig. 3a–c). Using the specific Src kinase inhibitor PP2 (10 μM), we determined that Src kinase was involved in FAK phosphorylation (Fig. 3d, e) and translocation (Fig. 3f) during T3 exposure.
Fig. 3.
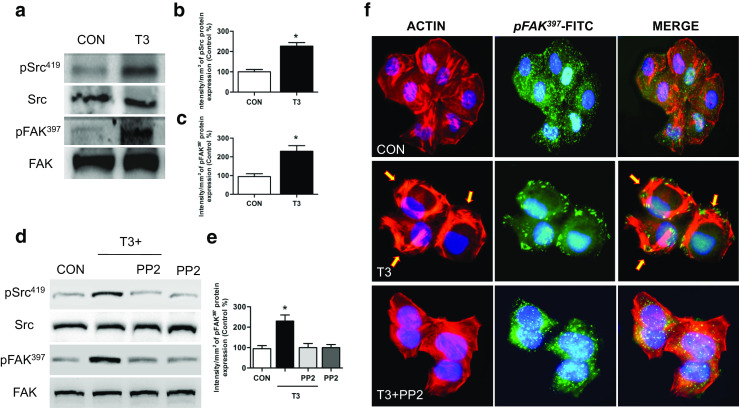
Src is involved in T3-induced FAK phosphorylation and translocation towards the plasma membrane. T-47D cells were treated with T3 (1 nM) for 20 min. a Total cell amount of wild-type Src and FAK, or Tyr397-FAK and phosphorylated-Src (Tyr419), are shown with Western blot. b, c Phospho-Src and phospho-FAK densitometry values were adjusted to Src and FAK intensity, respectively, and then normalized to the control sample. d–f T-47D breast cancer cells were exposed to T3 (1 nM) for 20 min, in the presence or absence of PP2 (10 μM), and Src and FAK phosphorylation were analyzed through Western blot assay. Phospho-FAK densitometry values were adjusted to FAK intensity and normalized to the control. Cells were stained with anti-phospho-Tyr397 FAK linked to FITC, filamentous actin was stained with phalloidin linked to Texas Red, and nuclei were counterstained with DAPI. CON control. Yellow arrows indicate membrane-localized Tyr397-FAK. * = P < 0.05 vs. corresponding control. All experiments were performed in triplicate with consistent results; representative images are shown
Treatment with T3 (1 nM) for 20 min led to a rapid change of the spatial organization of actin fibers and subcellular FAK localization. After T3 treatment, we observed a reorganization of the actin cytoskeleton towards the periphery of the membrane, resulting in membrane thickening (Fig. 3f, yellow arrows). Actin translocated from the cytoplasm towards the edge of the cell membrane, where it co-localized with phosphorylated FAK forming typical focal adhesion complexes (Fig. 3f).
T3 Controls a Multi-protein Complex with Src, PI3K, and FAK
Integrins typically initiate signaling via cell adhesion to the extracellular matrix, where they interact with matrix proteins and cluster on the surface of the membrane. This action promotes the assembly of a focal contact containing integrins together with tyrosine kinases, such as Src or FAK, and adaptor proteins that mediate downstream signaling, leading to a wide array of cellular activities [36]. In fact, previous studies suggest that T3 could bind integrin αV/β3 and transmit the signal to Src, FAK, and PI3K [35]. To determine whether T3 initiates its signaling pathway via integrin αV/β3, we treated the cells with T3 in the presence or absence of the integrin αV/β3 receptor antagonist tetraiodothyroacetic acid (Tetrac—10 μM). As mentioned before, treatment with T3 (1 nM) for 20 min induces FAK activation. However, T3 is not able to activate it in the presence of Tetrac, which suggests that αV/β3 integrin receptor signal to FAK after T3 treatment (Fig. 4a, b). Consistent with this hypothesis, we test whether thyroid hormone receptor alpha (TRα) or thyroid hormone receptor beta 1 (TRβ1) affects this post-translational modification in T-47D breast cancer cells. We silenced TRα and TRβ1 with siRNAs. In TRα and TRβ1-silenced cells, T3 induced FAK phosphorylation (Supporting Information S4), suggesting that TRα/β1 is not involved in this rapid signaling cascade.
Fig. 4.
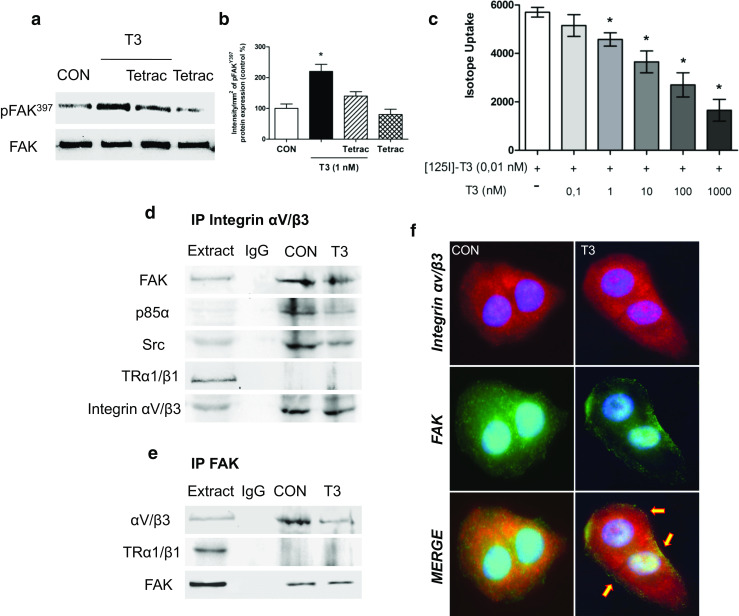
Integrin αV/β3 signals to Focal Adhesion Kinase via interaction with Src and PI3K. a, b T-47D breast cancer cells were exposed to T3 (1 nM) for 20 min in the presence or absence of the specific antagonist Tetrac (10 μM); Tyr397-FAK was analyzed through Western blot assay. Phospho-FAK densitometry values were adjusted to FAK intensity and normalized to the control. c Unlabeled T3 displace [125I]T3 from T-47D plasma membranes. [125I]T3 was added to purified plasma membrane proteins (60 μg/sample) before the addition of unlabeled T3 (0.1 to 1000 nM). Unlabeled T3 was effective in the displacement of radiolabeled T3 conformed best to the bind with the integrin αvβ3 in breast cancer plasma membranes. d, e Breast cancer cells were exposed to 1 nM T3 for 20 min and the cell protein extracts immunoprecipitated with an antibody vs. integrin αV/β3 (d) and FAK (e). IPs were assayed for co-immunoprecipitation of Src, p85α, FAK, and integrin αV/β3. Membranes were re-blotted for the immunoprecipitated protein to show equal loading. Protein extracts without immunoprecipitation (input) were used to identify the correct band. f Cells were incubated in the presence of T3 for 20 min in baseline conditions and co-localization of integrin αV/β3 vs. FAK was measured with immunofluorescence after staining of FAK with FITC and integrin αV/β3 with Texas Red. Nuclei were counterstained with DAPI. Experiments were performed in triplicate; representative images are shown
In order to prove whether T3 binds to integrin αV/β3 at the cell membrane, we studied this effect through the radiolabel displacement assays using T-47D breast cancer cell membranes. To evaluate this possibility, 60 μg of plasma membrane protein/sample was incubated with [125I] T3 (0.01 nM) for 10 min, and varying concentrations of unlabeled T3 (0.1–1000 nM) were then added. Membrane-bound radiolabeled T3 was displaced by unlabeled T3 in a concentration-dependent manner in T-47D plasmatic membranes, as previously described Lin et al. [35] (Fig. 4c).
On the other hand, previous studies have linked thyroid hormone signaling with integrin αV/β3 and estrogen receptor alpha (ERα) suggesting that T3 activates crosstalk between both receptors in human lung carcinoma cells [37]. To test whether ERα is involved in this rapid signaling system in T-47D (ER+) breast cancer cells, we silenced ERα with siRNAs. In ERα-silenced cells, T3 induced FAK phosphorylation, suggesting that ERα is not involved in this non-genomic signaling cascade (Supporting Information S6).
We performed co-immunoprecipitation assays (IP) in breast cancer cells to further explore the signaling pathways through which integrin αV/β3 transmits the signal to Src, FAK, and PI3K. With this approach we found that integrin αV/β3, Src, p85α, and FAK interacted in T-47D breast cancer control cells (Fig. 4d, e). This interaction was partially disrupted after treatment with T3 for 20 min (Fig. 4d, e) and membrane translocation of FAK was observed (Figs. 3f and 4f) in the presence of T3. Furthermore, the dissociation of integrin αV/β3 with this complex (Src/FAK/p85α) was prevented by the use of Tetrac (Supporting Information S8). These findings support the concept that T3 signals to FAK through an integrin αV/β3/Src/PI3K cascade.
Integrin αV/β3 Signals to FAK through a Src and PI3K-Dependent Signaling Pathway
We used specific pharmacological inhibitors in cells exposed or not to T3 to identify the signaling intermediates involved in FAK activation by T3 hormone in T-47D breast cancer cells. The Src kinase inhibitor PP2 (10 μM), the focal adhesion kinase inhibitor (FAKi 1 μM), and wortmannin (WM 30 nM)—an inhibitor of phosphatidylinositol 3-OH kinase (PI3K)—all significantly inhibited Tyr397-FAK phosphorylation induced by T3 (Fig. 5a, b and Supporting Information S1), suggesting that in the presence of T3, integrin αV/β3 signals to FAK via Src and PI3K.
Fig. 5.

T3 regulates Src/FAK/PI3K complex formation. T-47D breast cancer cells were exposed for 20 min to T3 (1 nM) in the presence or absence of the Src inhibitor (PP2 10 μM), the PI3K inhibitor wortmannin (WM 30 nM), FAK inhibitor (FAKi 1 μM), or the MAPK inhibitor PD98059 (PD 5 mM). a, b Tyr397- FAK phosphorylation was evaluated through Western blot analysis. c Breast cancer cells treated with T3 for 20 min in the presence or absence of FAK inhibitor and the inhibitor of the extracellular regulated kinase 1/2 (ERK1/2), PD98059. Cell contents of total or phosphorylated ERK1/2 are shown. d, e T-47D breast cancer cells were exposed to T3 (1 nM) for 20 min in the presence or absence of PP2, WM, and FAKi. Cell protein extracts were immunoprecipitated with an antibody vs. FAK (d) and p85alpha (e). IPs were assayed for co-immunoprecipitation with Src, FAK, and p85α. Membranes were re-blotted for the immunoprecipitated protein to show equal input. The experiments were performed in triplicate; representative images are shown. CON control
To examine the effect of T3 on PI3K kinase activation (via AKT phosphorylation, AKT1S473) in more detail, we used three different breast cancer cell lines with or without mutations in the catalytic (p110α) subunit of PI3K (PI3KCA), the lines SKBR-3 (wild type for PI3KCA), MCF-7 (with helical PI3KCA mutations), and T-47D (with kinase-domain PI3KCA mutation), in order to determine the AKT pathway activation. By means of Western blot analysis, we performed phosphoprotein profiles associated with distinct alterations affecting the PI3K pathway. Additionally, we examined the effects of T3 using another specific PI3K inhibitor (LY294002) that is wild type for PIK3CA (Supporting Information S7). Our results have shown that the PI3K kinase activation was promoted by the effects of T3 in three different breast cancer cells, and this induction was disrupted by the use of the specific PI3K inhibitor LY independent of the PIK3CA mutations.
The thyroid hormone analog (Tetrac 10 μM) and MAPK inhibitor (PD 5 mM) markedly decreased ERK1/2 phosphorylation (Supporting Information S5 and Fig. 5c), but did not affect FAK activation during T3 exposure (Fig. 5a, b), indicating that the MAPK pathway is not involved in Src/FAK/PI3K signaling induced by T3 (Fig. 5a–c).
In order to evaluate the participation of direct associations between Src, p85α (regulatory subunit of PI3K), and FAK after T3 treatment, we performed IP using or not the specific inhibitors PP2, WM, and FAKi. With immunoprecipitation assays, we found that, in the presence of T3, Src increased its interaction with FAK and this interaction was disrupted by the use of PP2, WM and FAKi (Fig. 5d). On the other hand, in the presence of T3, Src kinase increased its interaction with p85α and this interaction was disrupted by the use of PP2, WM, and FAKi (Fig. 5f), suggesting that T3 induces the formation of a multi-protein complex between Src with p85α and FAK (Fig. 5d, e).
T3 Controls T-47D Breast Cancer Cell Adhesion and Migration via Src/FAK/PI3K
We performed horizontal migration assays with T-47D cells to relate the T3-induced actin cytoskeleton remodeling and Src/FAK/PI3K activation with increased cell motility. A selective inhibitor of DNA strand separation that does not block RNA synthesis, cytosine β-darabinofuranoside hydrochloride (10 μM), was used to arrest cell proliferation, to distinguish cell migration from cell proliferation. Treatment with T3 (1 nM) for 48 h significantly increased the number of T-47D breast cancer cells that migrated through the starting line as well as the mean length of migration compared to control. This effect was blocked by PP2, WM and FAKi but not by PD (Fig. 6a, b), suggesting that the Src/FAK/PI3K cascade is implicated in the signaling of T3 to T-47D cell migration.
Fig. 6.

T3 modulates breast cancer cell adhesion and migration through Src/FAK/PI3K pathway. Cells were treated with T3 (1 nM) for 48 h in the presence or absence of the Src inhibitor (PP2; 0.2 μM), the PI3kinase inhibitor wortmannin (WM; 30 nM), the FAK inhibitor (FAKi 1 μM), and MAP Kinase inhibitor PD98059 (PD 5 mM). a, b The arrows indicate the direction of migration. The upper black lines indicate the starting line and the lower black lines the mean migration distance. c, d Representative images of T-47D cell adhesion to gelatin after T3 treatment (1 nM/2 h) are shown. Experiments were performed in triplicate; representative images are shown
In addition to promoting T-47D cell migration, treatment with T3 (1 nM/48 h) led to significantly higher cellular adhesion to gelatin (Fig. 6c, d). As control, we tested the effect of the different inhibitors individually, demonstrating the specific action of T3 on cellular migration (Supporting Information S2) and adhesion (Fig. 6c, d).
Discussion
The most common malignancy in women is breast cancer. It affects approximately 1.38 million new patients and causes 459,000 deaths/year worldwide (http://globocan.iarc.fr/). Despite improvements in early diagnosis as well as in surgical and systemic adjuvant therapies, the mortality rate for breast cancer still remains high. Breast cancer is an easily curable disease if detected early, but prognosis is severe when metastasis occurs [33]. The metastasis is the final stage of neoplastic progression. It is the primary cause of death from solid tumors [33, 38]. It is therefore fundamental for the development of novel therapeutic strategies in oncology to study the molecular actions linked to the metastasis process.
Breast cancer is a hormone-dependent pathology, and the effect of thyroid hormones on this disease has been studied for years [25, 39]. Hypothyroidism has been repeatedly associated with a reduced breast cancer risk, a more indolent invasive disease [17, 27, 28], and lower occurrence of metastasis and cell invasion [25]. Goodman et al. [40] showed that it was very difficult to induce breast cancer with 7,12-dimethylbenz(a)anthracene (DMBA) in hypothyroid rats. On the other hand, other groups have supported the idea that hypothyroidism enhances tumor metastasis and invasion [41]. The association between thyroid hormones and carcinogenesis therefore remains unclear.
Thyroid hormones exert their actions through thyroid hormone receptors that mediate their genomic and non-genomic mechanisms [30, 42]. In addition to this scenario, recent studies have indicated that T3 is able to bind to integrin αV/β3, a plasma membrane protein. αV/β3 contains two TH binding sites, denoted as S1 and S2, which translate the TH signal differently [43, 44]. S1 exclusively binds T3 at physiological concentrations and leads to PI3K activation, while S2 activates the ERK1/2 pathway [35]. Lin et al. showed in human glioblastoma U-87 MG cells that T3, but not T4, activates Src and PI3K/Akt via integrin αV/β3 and stimulates hypoxia-inducible factor (HIF-1α) gene expression [35]. HIF-1α, a regulator of the hypoxic response, has been extensively studied during a hypoxic microenvironment, playing a key role in regulating breast cancer progression and metastasis [45]. In this way, they demonstrated that the plasma membrane integrin receptor for thyroid hormone may be involved in the hormonal effect, being an example of the genomic consequences of a non-genomic action of the hormone.
Because integrin αV/β3 binds TH and transmits their signal into the cell, it clearly qualifies as a cell surface TR. In fact, our results show that signaling is initiated with integrin αV/β3, as T3 binds integrin αV/β3 and induces FAK activation while the specific integrin αV/β3 receptor antagonist Tetrac inhibits this action.
Our study reveals that in basal conditions, a multi-protein complex exists where integrin αV/β3/Src/PI3K/FAK interact. Our pull-down experiments indicate that T3 treatment leads to the abolishment of the interaction between integrin αV/β3 and Src/FAK/p85α complex, and this complex remains associated by a short period of time to finally dissociate completely. As a consequence, Src activation by T3 probably induces FAK phosphorylation and creates some steric hindrance in FAK. The rapid disruption of this interaction of this complex could lead to a time-consistent membrane translocation of FAK (Fig. 3c, d). This is consistent with findings of previous studies [36, 46] and suggests that thyroid hormones control the interaction via a direct association/disruption of integrin αV/β3 with Src, PI3K, and FAK. (Fig. 7).
Fig. 7.

Squematic signaling pathway proposed in T-47D breast cancer cells motility. Binding of T3 to the specific membrane integrin αv/β3 induces phosphorylation of the Src, FAK, and PI3K cascade, promoting the breast cancer cell motility
In parallel, we identified the recruitment of the Src/PI3K/FAK complex by T3, which is a step required for FAK activation. This is consistent with previous reports [47, 48] that have shown that the Src/PI3K pathway is involved in Tyr397 FAK phosphorylation. The autophosphorylation on Tyr397 creates a docking site for Src kinase and other SH2-containing proteins [6]. Src would thus trigger phosphorylation of other Tyr residues and modulate FAK activity [7]. Our pull-down experiments establish that Src kinase interacts with p85α in the presence of T3. After T3 treatment, Src is activated, interacts with FAK and produces FAK phosphorylation at the Tyr397 site, thereby activating this protein as well. FAK phosphorylation in Tyr397 is therefore fundamental for a conformational change that allows FAK protein to expose the Tyr397/407/576/577/861/925 residues for autophosphorylation [6, 7].
We also show that T3 modulates breast cancer cell morphology through actin cytoskeleton remodeling. FAK activation in breast cancer cells is involved in the rapid and dynamic remodeling of the actin cytoskeleton induced by T3 as well as in the formation of specialized cell membrane structures that mediate the interaction with the extracellular matrix and cell movement. Remodeling of the actin cytoskeleton by TH has previously been demonstrated by Leonard and Farwell who concluded that T4 and reverse T3 altered actin [49, 50]. This effect has been prominently shown in neurons and glial cells and is important for brain development.
Additionally, we investigated whether different T3 doses could alter Src, FAK, p85, and Akt protein expression. We developed an “in vitro” model to simulate normal, excess and absence states of T3. We did not find significant differences in protein expression levels between excess and normal states of T3. A marked decrease in Src, FAK, p85α, and total Akt1/2/3 protein expression is, however, obtained in absence of T3. Our results suggest a key regulation of these proteins by T3 and could explain, at least in part, clinical observations of a reduced incidence and progression of breast cancer in hypothyroid women. For instance, if T3 was indeed a contributor to breast cancer cell adhesion and motility, then the nonthyroidal illness syndrome of low circulating T3 levels would be advantageous for cancer patients. This signal pathway may thus offer potential for specific interference with suitable pharmacological or biological tools, which could be of interest for new strategies to alter the metastatic potential of hormone-sensitive cancers. On the other hand, Heublein et al. [51] proposed that TR might be interesting biomarkers, especially in the absence of classical hormone receptors. These authors identified TRβ to be upregulated in BRCA1-associated breast cancer and revealed TR to be associated with patients’ prognosis. They demonstrated that TR are active in BRCA1-associated breast cancer; that contrary to our findings, TRβ expression in BRCA1 mutant tumor samples is associated with a prolonged overall survival; and that both TR may arise as interesting alternative targets for endocrine treatment of BRCA1-associated triple negative breast cancer [51].
In conclusion, our findings provide new insights about non-genomic mechanisms of thyroid hormone in breast cancer cell migration. T3 leads to rapid changes in the cell membrane, leading to a rearrangement of the actin cytoskeleton and consequent formation of focal adhesion complexes at sites where structures related with cell motility are formed. These findings could be useful to develop new tools that interfere with the ability of breast tumors to diffuse locally or at distant sites. Our results provide new evidence for T3 effects in breast cancer. The development of hormonal therapies is crucial for patients with thyroid disorders who also suffer breast cancer. The identification of T3 actions may thus enhance the development of therapies that counteract breast cancer cell progression through the reduction of circulating T3 levels in patients. Also, the use of specific inhibitors of the kinases involved in cell migration may be helpful to prevent breast cancer migration and invasion. Finally, we intend to pursue additional research on the role of thyroid hormones in breast cancer in order to achieve a better understanding of the disease.
Electronic Supplementary Material
(PPT 1259 kb).
Acknowledgements
We thank Ph.D. Mariella Superina for correcting this manuscript.
Compliance with Ethical Standards
Funding Information
This work has been supported by the National Cancer Institute, Ministry of Health of Argentina (Ministerial Resolution n° 489 - Expte. N° 2002-20551/11-0) and by the Bristol Myers Squibb grant # 64749 to AMS.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Disclosure Statement
The authors have nothing to disclose.
References
- 1.Siegel R, et al. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61(4):212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 2.Stevanovic A, Lee P, Wilcken N. Metastatic breast cancer. Aust Fam Physician. 2006;35(5):309–312. [PubMed] [Google Scholar]
- 3.Acconcia F, Barnes CJ, Kumar R. Estrogen and tamoxifen induce cytoskeletal remodeling and migration in endometrial cancer cells. Endocrinology. 2006;147(3):1203–1212. doi: 10.1210/en.2005-1293. [DOI] [PubMed] [Google Scholar]
- 4.Wu X, et al. Focal adhesion kinase regulation of N-WASP subcellular localization and function. J Biol Chem. 2004;279(10):9565–9576. doi: 10.1074/jbc.M310739200. [DOI] [PubMed] [Google Scholar]
- 5.Sanchez AM, et al. Estrogen receptor-{alpha} promotes endothelial cell motility through focal adhesion kinase. Mol Hum Reprod. 2011;17(4):219–226. doi: 10.1093/molehr/gaq097. [DOI] [PubMed] [Google Scholar]
- 6.McLean GW, et al. The role of focal-adhesion kinase in cancer—a new therapeutic opportunity. Nat Rev Cancer. 2005;5(7):505–515. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- 7.Sanchez AM, et al. Estrogen receptor-alpha promotes breast cancer cell motility and invasion via focal adhesion kinase and N-WASP. Mol Endocrinol. 2010;24(11):2114–2125. doi: 10.1210/me.2010-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanchez AM, et al. Effects of progesterone and medroxyprogesterone on actin remodeling and neuronal spine formation. Mol Endocrinol. 2013;27(4):693–702. doi: 10.1210/me.2012-1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fu XD, et al. Progesterone receptor enhances breast cancer cell motility and invasion via extranuclear activation of focal adhesion kinase. Endocr Relat Cancer. 2010;17(2):431–443. doi: 10.1677/ERC-09-0258. [DOI] [PubMed] [Google Scholar]
- 10.van Nimwegen MJ, van de Water B. Focal adhesion kinase: a potential target in cancer therapy. Biochem Pharmacol. 2007;73(5):597–609. doi: 10.1016/j.bcp.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 11.Watermann DO, et al. Specific induction of pp125 focal adhesion kinase in human breast cancer. Br J Cancer. 2005;93(6):694–698. doi: 10.1038/sj.bjc.6602744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28(1–2):35–49. doi: 10.1007/s10555-008-9165-4. [DOI] [PubMed] [Google Scholar]
- 13.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 14.Kim LC, Song L, Haura EB. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol. 2009;6(10):587–595. doi: 10.1038/nrclinonc.2009.129. [DOI] [PubMed] [Google Scholar]
- 15.Flamini MI, et al. Estrogen regulates endometrial cell cytoskeletal remodeling and motility via focal adhesion kinase. Fertil Steril. 2011;95(2):722–726. doi: 10.1016/j.fertnstert.2010.08.039. [DOI] [PubMed] [Google Scholar]
- 16.Luo M, Guan JL. Focal adhesion kinase: a prominent determinant in breast cancer initiation, progression and metastasis. Cancer Lett. 2010;289(2):127–139. doi: 10.1016/j.canlet.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arold ST. How focal adhesion kinase achieves regulation by linking ligand binding, localization and action. Curr Opin Struct Biol. 2011;21(6):808–813. doi: 10.1016/j.sbi.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bolos V, et al. The dual kinase complex FAK-Src as a promising therapeutic target in cancer. Onco Targets Ther. 2010;3:83–97. doi: 10.2147/OTT.S6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18(5):516–523. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 20.Vultur A, et al. SKI-606 (bosutinib), a novel Src kinase inhibitor, suppresses migration and invasion of human breast cancer cells. Mol Cancer Ther. 2008;7(5):1185–1194. doi: 10.1158/1535-7163.MCT-08-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McLean BA, et al. PI3K inhibitors as novel cancer therapies: implications for cardiovascular medicine. J Card Fail. 2013;19(4):268–282. doi: 10.1016/j.cardfail.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 22.Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4(4):257–262. doi: 10.1016/S1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- 23.Stephens L, Williams R, Hawkins P. Phosphoinositide 3-kinases as drug targets in cancer. Curr Opin Pharmacol. 2005;5(4):357–365. doi: 10.1016/j.coph.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 24.Guigon CJ, et al. Mutation of thyroid hormone receptor-beta in mice predisposes to the development of mammary tumors. Oncogene. 2011;30(30):3381–3390. doi: 10.1038/onc.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moeller LC, Fuhrer D. Thyroid hormone, thyroid hormone receptors, and cancer: a clinical perspective. Endocr Relat Cancer. 2013;20(2):R19–R29. doi: 10.1530/ERC-12-0219. [DOI] [PubMed] [Google Scholar]
- 26.Ditsch N, et al. Thyroid hormone receptor (TR)alpha and TRbeta expression in breast cancer. Histol Histopathol. 2013;28(2):227–237. doi: 10.14670/HH-28.227. [DOI] [PubMed] [Google Scholar]
- 27.Cristofanilli M, et al. Thyroid hormone and breast carcinoma. Primary hypothyroidism is associated with a reduced incidence of primary breast carcinoma. Cancer. 2005;103(6):1122–1128. doi: 10.1002/cncr.20881. [DOI] [PubMed] [Google Scholar]
- 28.Tosovic A, et al. Prospectively measured triiodothyronine levels are positively associated with breast cancer risk in postmenopausal women. Breast Cancer Res. 2010;12(3):R33. doi: 10.1186/bcr2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hercbergs AH, Ashur-Fabian O, Garfield D. Thyroid hormones and cancer: clinical studies of hypothyroidism in oncology. Curr Opin Endocrinol Diabetes Obes. 2010;17(5):432–436. doi: 10.1097/MED.0b013e32833d9710. [DOI] [PubMed] [Google Scholar]
- 30.Furuya F, et al. Nongenomic activation of phosphatidylinositol 3-kinase signaling by thyroid hormone receptors. Steroids. 2009;74(7):628–634. doi: 10.1016/j.steroids.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kavok NS, Krasilnikova OA, Babenko NA. Thyroxine signal transduction in liver cells involves phospholipase C and phospholipase D activation. Genomic independent action of thyroid hormone. BMC Cell Biol. 2001;2:5. doi: 10.1186/1471-2121-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hiroi Y, et al. Rapid nongenomic actions of thyroid hormone. Proc Natl Acad Sci U S A. 2006;103(38):14104–14109. doi: 10.1073/pnas.0601600103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flamini, M.I., et al. 2014 Retinoic acid reduces migration of human breast cancer cells: role of retinoic acid receptor beta. J Cell Mol Med [DOI] [PMC free article] [PubMed]
- 34.Lin HY, et al. Thyroid hormone induces activation of mitogen-activated protein kinase in cultured cells. Am J Phys. 1999;276(5 Pt 1):C1014–C1024. doi: 10.1152/ajpcell.1999.276.5.C1014. [DOI] [PubMed] [Google Scholar]
- 35.Lin HY, et al. L-thyroxine vs. 3,5,3′-triiodo-L-thyronine and cell proliferation: activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Am J Physiol Cell Physiol. 2009;296(5):C980–C991. doi: 10.1152/ajpcell.00305.2008. [DOI] [PubMed] [Google Scholar]
- 36.Desgrosellier JS, et al. An integrin alpha(v)beta(3)-c-Src oncogenic unit promotes anchorage-independence and tumor progression. Nat Med. 2009;15(10):1163–1169. doi: 10.1038/nm.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meng R, et al. Crosstalk between integrin alphavbeta3 and estrogen receptor-alpha is involved in thyroid hormone-induced proliferation in human lung carcinoma cells. PLoS One. 2011;6(11):e27547. doi: 10.1371/journal.pone.0027547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sporn MB. The war on cancer. Lancet. 1996;347(9012):1377–1381. doi: 10.1016/S0140-6736(96)91015-6. [DOI] [PubMed] [Google Scholar]
- 39.Aranda A, et al. Thyroid receptor: roles in cancer. Trends Endocrinol Metab. 2009;20(7):318–324. doi: 10.1016/j.tem.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 40.Goodman AD, Hoekstra SJ, Marsh PS. Effects of hypothyroidism on the induction and growth of mammary cancer induced by 7,12-dimethylbenz(a)anthracene in the rat. Cancer Res. 1980;40(7):2336–2342. [PubMed] [Google Scholar]
- 41.Martinez-Iglesias O, et al. Hypothyroidism enhances tumor invasiveness and metastasis development. PLoS One. 2009;4(7):e6428. doi: 10.1371/journal.pone.0006428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J, Lazar MA. The mechanism of action of thyroid hormones. Annu Rev Physiol. 2000;62:439–466. doi: 10.1146/annurev.physiol.62.1.439. [DOI] [PubMed] [Google Scholar]
- 43.Freindorf M, et al. Combined QM/MM study of thyroid and steroid hormone analogue interactions with alphavbeta3 integrin. J Biomed Biotechnol. 2012;2012:959057. doi: 10.1155/2012/959057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davis PJ, et al. Membrane receptor for thyroid hormone: physiologic and pharmacologic implications. Annu Rev Pharmacol Toxicol. 2011;51:99–115. doi: 10.1146/annurev-pharmtox-010510-100512. [DOI] [PubMed] [Google Scholar]
- 45.Liu ZJ, Semenza GL, Zhang HF. Hypoxia-inducible factor 1 and breast cancer metastasis. J Zhejiang Univ Sci B. 2015;16(1):32–43. doi: 10.1631/jzus.B1400221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cohen, K., et al. 2014 Thyroid hormone regulates adhesion, migration and matrix metalloproteinase 9 activity via alphavbeta3 integrin in myeloma cells. Oncotarget [DOI] [PMC free article] [PubMed]
- 47.Reiske HR, et al. Requirement of phosphatidylinositol 3-kinase in focal adhesion kinase-promoted cell migration. J Biol Chem. 1999;274(18):12361–12366. doi: 10.1074/jbc.274.18.12361. [DOI] [PubMed] [Google Scholar]
- 48.Thamilselvan V, Craig DH, Basson MD. FAK association with multiple signal proteins mediates pressure-induced colon cancer cell adhesion via a Src-dependent PI3K/Akt pathway. FASEB J. 2007;21(8):1730–1741. doi: 10.1096/fj.06-6545com. [DOI] [PubMed] [Google Scholar]
- 49.Leonard JL, Farwell AP. Thyroid hormone-regulated actin polymerization in brain. Thyroid. 1997;7(1):147–151. doi: 10.1089/thy.1997.7.147. [DOI] [PubMed] [Google Scholar]
- 50.Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010;31(2):139–170. doi: 10.1210/er.2009-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heublein S, et al. Thyroid hormone receptors predict prognosis in BRCA1 associated breast cancer in opposing ways. PLoS One. 2015;10(6):e0127072. doi: 10.1371/journal.pone.0127072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PPT 1259 kb).