

Virtually since its inception, hypertrophic cardiomyopathy (HCM) has been regarded as a genetically transmitted disease, often demonstrating an autosomal dominant pattern of inheritance, and supported by the monogenic sarcomere mutation hypothesis. 1 , 2 , 3 This construct has been promoted universally for HCM with respect to both disease cause and transmission of clinically overt phenotype within families. The principle that HCM can occur in families and be responsible for heart failure disability or arrhythmic sudden death often serves as a source of both anxiety and guilt on the part of families. 1
However, it has been our experience that many families with HCM may not demonstrate an autosomal dominant pattern of transmission. Notably, the concept of a nonfamilial form of HCM first emerged in the 1960s, 3 and was more recently revisited by Ingles et al in 2017,4 when the authors reported 40% of families as having nonfamilial HCM, based on absent HCM family history and without a sarcomeric mutation on genetic testing in the proband. 4 Nonfamilial HCM was associated with benign clinical course, although this study did not evaluate HCM phenotypes in all relatives.
With these considerations in mind, we embarked on a systematic analysis in 2022 at the Lahey HCM Center interrogating 304 consecutive families to determine whether there was evidence of inherited HCM. Each family surveyed included a referred patient (representing the proband) in whom an HCM diagnosis was confirmed at our center. Data that support the findings of this study are available from the authors on reasonable request. The study was reviewed and approved by Institutional Review Board at Lahey Hospital and Medical Center, allowing retrospective review of medical records and granted a waiver of informed consent in accord with 45 CFR 164.512(i)2(ii).
This analysis of 304 families with HCM identified 130 (43%) with some evidence of a genetic disease (ie, either a relative or a proband with a pathogenic/likely pathogenic HCM sarcomere mutation [n=55], ≥1 relative of any degree with known or probable HCM phenotype [n=53], or both [n=22]).
In addition, 163 other families could not be judged reliably as having either familial or nonfamilial HCM because of a variety of indicators: insufficient compliance or impractical logistics for acquiring comprehensive family information; offspring in last generation aged <18 years or a noninformative small pedigree; or sudden death in young relatives with or without HCM confirmation.
Ultimately, we were able to identify 11 individual families (4% of 304), all composed of 3 generations, who met our predefined criterion of nonfamilial disease (ie, absence of the HCM phenotype in first‐degree relatives by echocardiography and/or cardiac magnetic resonance, clinical events, or other evidence of HCM) (Figure). Also, in the extended family beyond first‐degree relatives, no patient had a personal history consistent with HCM. Because of the self‐imposed restrictions in our conservative study design, we have likely underestimated nonfamilial HCM in our study population.
Figure 1. Four study pedigrees demonstrating the nonfamilial form of hypertrophic cardiomyopathy (HCM).
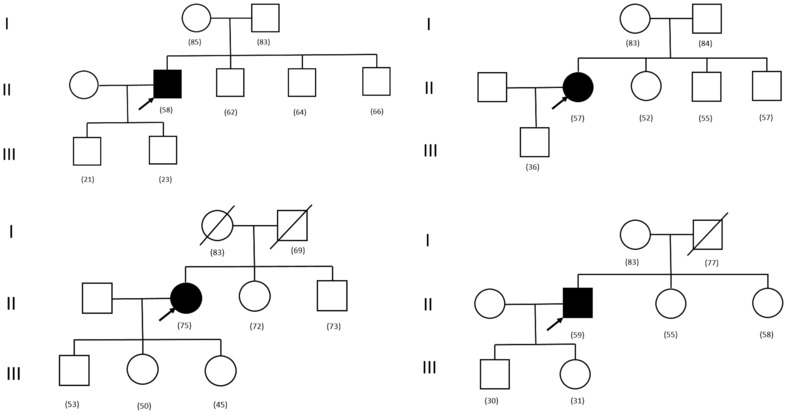
Clear symbols indicate absence of evidence of HCM; solid symbols, clinically confirmed HCM; and symbols with slash, died of non‐HCM disease. Subject ages are in parentheses. Arrows indicate probands.
The 11 pedigrees consisted of 7 relatives on average (range, 5–8), aged 57±19 years. The 11 probands were aged 45 to 77 years (mean±SD, 62±10 years); 73% were men; left ventricular thickness was 17±2 mm; and left ventricular outflow tract obstruction (gradient ≥30 mm Hg) occurred in 64%, including 5 with surgical septal myectomy.
For molecular genetic testing, a commercial panel of 30 genes associated with HCM and its most common phenocopies was used: ACTC1, ACTN2, ALPK3, ANKRD1, CSRP3, FHL1, FLNC, GLA, JPH2, LAMP2, MYBPC3, MYH6, MYH7, MYL2, MYL3, MYPN, NEXN, PLN, PRKAG2, PTPN11, RAF1, RIT1, SOS1, TCAP, TNNC1, TNNI3, TNNT2, TPM1, TTR, and VCL. No pathogenic/likely pathogenic mutations or variants of uncertain significance were identified in any probands or relatives.
Our novel data from Lahey HCM Center suggest that not all families with HCM demonstrate autosomal dominance, and by inference HCM may not be a familial or inherited disease in all cases. 5 This demonstration of sarcomere‐negative and nonfamilial HCM pedigrees is an important observation from a scientific perspective, as it may relate directly to the fundamental basis of HCM. 2 , 5 In addition, there are important clinical inferences from our findings for families. Recognizing that evidence for an autosomal dominant disease may not be apparent in all families is highly relevant given that this patient population deserves a realistic understanding of the risk (or lack thereof) for transmitting their clinical disease to offspring or others, a concern that is often unavoidably associated with substantial anxiety. We also acknowledge that pedigree size and other competing factors could be relevant to our conclusions about penetrance and lack of familial inheritance.
Therefore, in conclusion, our identification of novel documentation of individual pedigrees with nonfamilial HCM is highly relevant to the overriding principle that patients with HCM deserve access to the most relevant clinical information about their complex heterogeneous disease, including the possibility that this disease may not be transmitted to other family members.
Sources of Funding
This research was funded by the Conceptual Development of Research Organization, Motol University Hospital, Prague, Czech Republic (grant 00064203).
Disclosures
Dr M.S. Maron is a consultant for Cytokinetics, iRhythm (with grant), Imbria Pharmaceuticals, and TAKEDA Pharmaceuticals (with grant). Dr Rowin reports research funding from Pfizer and iRhythm. The remaining authors have no disclosures to report.
This article was sent to Sakima A. Smith, MD, MPH, Associate Editor, for review by expert referees, editorial decision, and final disposition.
For Sources of Funding and Disclosures, see page 3.
REFERENCES
- 1. Maron BJ. Clinical course and management of hypertrophic cardiomyopathy. N Engl J Med. 2018;379:655–666. doi: 10.1056/NEJMra1710575 [DOI] [PubMed] [Google Scholar]
- 2. Chou C, Chin MT. Pathogenic mechanisms of hypertrophic cardiomyopathy beyond sarcomere dysfunction. Int J Mol Sci. 2021;22:8933. doi: 10.3390/ijms22168933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Braunwald E, Lambrew CT, Rockoff SD, Ross J Jr, Morrow AG. Idiopathic hypertrophic subaortic stenosis. I. A description of the disease based upon an analysis of 64 patients. Circulation. 1964;30:3–119. doi: 10.1161/01.cir.29.5s4.iv-3 [DOI] [PubMed] [Google Scholar]
- 4. Ingles J, Burns C, Bagnall RD, Lam L, Yeates L, Sarina T, Puranik R, Briffa T, Atherton JJ, Driscoll T, et al. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ Cardiovasc Genet. 2017;10:e001620. doi: 10.1161/CIRCGENETICS.116.001620 [DOI] [PubMed] [Google Scholar]
- 5. Maron BA, Wang RS, Carnethon MR, Rowin EJ, Loscalzo J, Maron BJ, Maron MS. What causes hypertrophic cardiomyopathy? Am J Cardiol. 2022;179:74–82. doi: 10.1016/j.amjcard.2022.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]