

Abstract
Background
Prolonged activation of angiotensin II is the main mediator that contributes to the development of heart diseases, so converting angiotensin II into angiotensin 1‐7 has emerged as a new strategy to attenuate detrimental effects of angiotensin II. Prolylcarboxypeptidase is a lysosomal pro‐X carboxypeptidase that is able to cleave angiotensin II at a preferential acidic pH optimum. However, insufficient attention has been given to the cardioprotective functions of prolylcarboxylpeptidase.
Methods and Results
We established a CRISPR/CRISPR‐associated protein 9–mediated global prolylcarboxylpeptidase‐knockout and adeno‐associated virus serotype 9–mediated cardiac prolylcarboxylpeptidase overexpression mouse models, which were challenged with the angiotensin II infusion (2 mg/kg per day) for 4 weeks, aiming to investigate the cardioprotective effect of prolylcarboxylpeptidase against hypertensive cardiac hypertrophy. Prolylcarboxylpeptidase expression was upregulated after 2 weeks of angiotensin II infusion and then became downregulated afterward in wild‐type mouse myocardium, suggesting its compensatory function against angiotensin II stress. Moreover, angiotensin II–treated prolylcarboxylpeptidase‐knockout mice showed aggravated cardiac remodeling and dampened cardiac contractility independent of hypertension. We also found that prolylcarboxylpeptidase localizes in cardiomyocyte lysosomes, and loss of prolylcarboxylpeptidase led to excessive angiotensin II levels in myocardial tissue. Further screening demonstrated that hypertrophic prolylcarboxylpeptidase‐knockout hearts showed upregulated extracellular signal‐regulated kinases 1/2 and downregulated protein kinase B activities. Importantly, adeno‐associated virus serotype 9–mediated restoration of prolylcarboxylpeptidase expression in prolylcarboxylpeptidase‐knockout hearts alleviated angiotensin II–induced hypertrophy, fibrosis, and cell death. Interestingly, the combination of adeno‐associated virus serotype 9–mediated prolylcarboxylpeptidase overexpression and an antihypertensive drug, losartan, likely conferred more effective protection than a single treatment protocol to mitigate angiotensin II–induced cardiac dysfunction.
Conclusions
Our data demonstrate that prolylcarboxylpeptidase protects the heart from angiotensin II–induced hypertrophic remodeling by controlling myocardial angiotensin II levels.
Keywords: hypertension, hypertrophy, lysosome, prolylcarboxylpeptidase
Subject Categories: Hypertension, Hypertrophy, Remodeling
Nonstandard Abbreviations and Acronyms
- AAV9
adeno‐associated virus serotype 9
- AT1R
angiotensin type 1 receptor
- AT2R
angiotensin type 2 receptor
- ERK1/2
extracellular signal‐regulated kinases 1/2
- GFP
green fluorescent protein
- MAPK
mitogen‐activated protein kinase
- PKB
protein kinase B
- RAS
renin‐angiotensin system
Research Perspective.
What Is New?
The causation of prolylcarboxylpeptidase loss‐induced hypertensive remodeling and cardiac dysfunction is independent of angiotensin II–induced hypertension, indicating that prolylcarboxylpeptidase might not be responsible for the regulation of angiotensin II at the systemic level like angiotensin‐converting enzyme 2.
Prolylcarboxylpeptidase localizes in lysosomes, and loss of prolylcarboxylpeptidase leads to excessive production of myocardial angiotensin II, indicating prolylcarboxylpeptidase's roles in the regulation of the renin‐angiotensin system system at the intracellular level.
What Question Should Be Addressed Next?
In this study, we showed that the combined therapy of prolylcarboxylpeptidase and losartan demonstrated a better efficiency compared with losartan only to slow down the progression of hypertrophic remodeling toward heart failure.
In the future, we will investigate whether prolylcarboxylpeptidase and a broader choice of renin‐angiotensin system–based drugs could be used to treat ischemic cardiac disease and diabetic cardiomyopathy.
The renin‐angiotensin system (RAS) is a coordinated hormonal signaling pathway regulating cardiovascular, renal, and adrenal function. Angiotensin II is the key effector peptide with pleiotropic actions via the angiotensin type I receptor (AT1R), including stimulation of oxidative stress, hypertrophic growth, fibrogenesis, inflammation, and cell death. 1 Pharmacological angiotensin II antagonists, including angiotensin‐converting enzyme inhibitors (ACEis) and angiotensin type 1 receptor blockers, are some of the most successful clinical interventions in the treatment of hypertension and heart failure. 2 Over the past decades, the existence of a counterregulatory arm of the RAS, including angiotensin 1‐7, angiotensin 1‐9, and angiotensin III, has been discovered. Of those, evidence over the past decade has shown that angiotensin 1‐7 via the Mas1 receptor cascade is able to counteract and prevent the detrimental effects caused by angiotensin II overactivity. 3
There are 3 enzymes that negatively regulate angiotensin II, including angiotensin‐converting enzyme 2 (ACE2), prolylcarboxypeptidase, and prolyloligopeptidase. 4 There is increasing evidence in rodent models that human recombinant ACE2 could prevent many cardiac diseases such as angiotensin II–induced hypertension, myocardial hypertrophy, diastolic and systolic dysfunction, and myocardial fibrosis by lowering angiotensin II and increasing angiotensin 1‐7 levels. 3 Moreover, a pilot study involving 5 patients with pulmonary arterial hypertension demonstrated beneficial effects of angiotensin 1‐7 on cardiac performance and pulmonary vascular resistance in company with increased superoxide dismutase 2 and reduced inflammatory markers levels in plasma. 5 In another study, 44 patients with acute respiratory distress syndrome receiving intravenous administration of human recombinant ACE2 were found to be well tolerated, but the improvement in oxygenation in these patients was not detected. 6 Thus, further clinical trials of ACE2 need a larger sample size to powerfully evaluate clinical outcomes. In addition, ACE2 has recently been identified as the SARS‐CoV‐2 receptor to allow viral cell entry. This raises the awareness of the urgent need to examine the regulatory functions of the other 2 enzymes in heart diseases in detail for safer treatment options. 7 Interestingly, a recent study showed that angiotensin II conversion to angiotensin 1‐7 in plasma in the circulation is prolyloligopeptidase dependent and ACE2 independent. 8 Thus, these discrepancies suggest a lack of knowledge of the physiological and pathophysiological mechanisms involved in this noncanonical cascade.
Prolylcarboxylpeptidase is a lysosomal Pro‐X carboxypeptidase that is capable of cleaving angiotensin II to produce angiotensin 1‐7 at a preferential acidic pH optimum. Prolylcarboxylpeptidasegt/gt mice experienced significantly elevated blood pressure and increased reactive oxygen species. 9 Furthermore, spontaneous hypertensive rats showed decreased prolylcarboxylpeptidase mRNA and protein levels in left ventricles, along with the development of ventricular hypertrophy. 10 Similarly, alcohol‐mediated left ventricular systolic dysfunction in spontaneous hypertensive rats also was associated with the downregulation of prolylcarboxylpeptidase. 11 However, a study by Maier et al 12 indicated that cardiac dysfunction in global prolylcarboxylpeptidase deficiency mice was mainly due to the lack of prolylcarboxylpeptidase activity in the kidney collecting tubules, where the prevailing pH is low. These conflicting findings have inspired us to explore the mechanistic action of prolylcarboxylpeptidase in metabolizing angiotensin II in the heart and its therapeutic potential for the treatment of heart diseases.
Increasing evidence supports the notion that some organs, such as hearts and kidneys, are able to express or uptake different components of the RAS, leading to the local angiotensin II synthesis. 13 In addition, increasing evidence showing the intracellular expression of several RAS components such as renin and intracellular angiotensin II and its receptors indicates the presence of functional intracrine RAS. 14 However, the regulatory mechanism of the intracrine angiotensin II cascade and its roles in the progression of heart failure are not fully understood. In contrast with ACE2 and prolyloligopeptidase, prolylcarboxylpeptidase is expressed predominantly in lysosomes, where its activity is essentially optimal for angiotensin II degradation under the acidic range. 12 , 15 , 16 Therefore, we proposed that prolylcarboxylpeptidase is important for protecting hearts from angiotensin II–mediated hypertrophy through the lysosomal control of intracellular angiotensin II.
In the present study, we aimed to investigate the role and the underlying mechanism of prolylcarboxylpeptidase in pathological cardiac hypertrophy subjected to 4 weeks of angiotensin II infusion. Moreover, we also characterize the therapeutic values of prolylcarboxylpeptidase in alleviating the progression of pathological hypertrophy.
Methods
All laboratory mice and rats used in this study were maintained in a pathogen‐free facility at the University of Manchester. Animal studies were performed in accordance with the United Kingdom Animals (Scientific Procedures) Act 1986 and were approved by the University of Manchester Ethics Committee. Data, methods, and study materials are available from the corresponding author upon reasonable request. Additional details of experimental procedures are included in Data S1 and Table S1.
Statistical Analysis
The power calculation was performed on the basis of pilot data of systolic blood pressure from wild‐type mice after 2 weeks of angiotensin II infusion, where that in the vehicle group was 120.25±4.92 mm Hg versus that in angiotensin II–infused mice was 131±5.47 mm Hg (pooled SD=5.19). Based on the calculation at the power of 0.9 and α=0.05, the minimum number of 5 mice was required per group for functional study. For all in vivo and in vitro analyses, normal distribution (Gaussian distribution) was first determined by the Shapiro–Wilk test. Data were then analyzed using Student's t‐test for comparisons between 2 experimental groups, and 1‐way or 2‐way ANOVA with Bonferroni post hoc tests for comparisons among multiple experimental groups. For nonnormal distribution data, the nonparametric Mann–Whitney U test and the Kruskal–Wallis test were used as alternatives for the t‐test and ANOVA, respectively. Data were analyzed using GraphPad software (La Jolla, CA) and expressed as mean±SEM, and a P<0.05 was considered statistically significant.
Results
Alteration of Prolylcarboxylpeptidase Expression During Angiotensin II–Mediated Hypertrophic Development
To determine how prolylcarboxylpeptidase expression responds during the development of hypertrophic growth of the heart, adult wild‐type mice were subjected to 4 weeks of angiotensin II infusion (2 mg/kg per day). As a result, wild‐type hearts under angiotensin II–induced hypertrophic stress displayed an increase (≈1.5‐fold) in prolylcarboxylpeptidase protein expression level after a 2‐week angiotensin II infusion, which was then dramatically reduced in the fourth week of angiotensin II infusion. In contrast, ACE2 expression progressively declined during the time course of angiotensin II treatment (Figure 1A). Moreover, we also tested whether these changes in prolylcarboxylpeptidase expression could be mirrored in human induced pluripotent stem cell–derived cardiomyocytes under angiotensin II treatment (100 nmol/L). As expected, prolylcarboxylpeptidase expression was significantly increased after 24 hours of angiotensin II treatment and then decreased at 48 hours as compared with control groups (Figure 1B). Next, we examined the change of prolylcarboxylpeptidase expression in the hearts of patients with end‐stage dilated cardiomyopathy. Similarly, prolylcarboxylpeptidase expression was decreased by 50% in dilated cardiomyopathy hearts (Figure 1C). These results suggest that the upregulation of prolylcarboxylpeptidase expression might be essential to protect the heart from the angiotensin II stress, and loss of prolylcarboxylpeptidase might contribute to the failure stage of hypertrophic myocardium.
Figure 1. Alteration of cardiac prolylcarboxylpeptidase expression in response to angiotensin II stress.

A, While cardiac ACE2 expression was continuously downregulated, cardiac prolylcarboxylpeptidase expression was upregulated after 2 weeks of angiotensin II infusion and then became downregulated (n=4 per group). One‐way ANOVA with Bonferroni correction for post hoc comparisons was used for analysis. Data are expressed as means±SEM. B, ACE2 expression was decreased after angiotensin II (100 nmol/L) treatments, whereas prolylcarboxylpeptidase was increased after 24 hours of angiotensin II treatment and then decreased in human induced pluripotent stem cell–derived cardiomyocytes. One‐way ANOVA with Bonferroni correction for post hoc comparisons was used for analysis. Data are expressed as means±SEM (n=4 per group). C, Prolylcarboxylpeptidase expression was downregulated in heart tissue samples of patients with CDM, compared with controls. Unpaired 2‐tailed Student's t‐test was used for analysis. Data are expressed as means±SEM (n=4 for control, n=8 for CDM). ACE2 indicates angiotensin‐converting enzyme 2; AngII, angiotensin II; A.U, Arbitrary unit; CDM, cardiomyopathy; and PRCP, prolylcarboxylpeptidase.
Characterization of Blood Pressure and Cardiac Function of Prolylcarboxylpeptidase‐Knockout Mice at 2 and 6 Months Old
First, we recruited CRISPR/CRISPR‐associated protein 9 technology to produce the global prolylcarboxylpeptidase‐knockout mice. The deletion of prolylcarboxylpeptidase expression in different organs was confirmed by Western blot, which showed a >90% removal of prolylcarboxylpeptidase in different organs (Figure S1A through S1E). A trace of prolylcarboxylpeptidase protein residual was observed in prolylcarboxylpeptidase‐knockout mice, which was likely due to a small degree of mosaicism that occurred during pronuclear coinjection of guided RNA and CRISPR‐associated protein 9 transcript rather than CRISPR‐associated protein 9.
Because angiotensin II plays an important role in the regulation of blood pressure and cardiac function, we first examined whether knockout of prolylcarboxylpeptidase alters blood pressure and cardiac functions in the 2‐ and 6‐month‐old prolylcarboxylpeptidase‐knockout mice at a basal level. Blood pressure measured by tail‐cuff method revealed that the 2‐month prolylcarboxylpeptidase‐knockout mice exhibited no alteration in diastolic and systolic blood pressures compared with the wild‐type littermates (Figure S2A). To assess any mild alterations of blood pressure caused by prolylcarboxylpeptidase loss, arterial blood pressure was quantified by invasive hemodynamic assessment at the 6‐month‐old time point. However, we still could not detect any hypertensive phenotypes developed in 6‐month‐old prolylcarboxylpeptidase‐knockout mice (Figure S2D). Consistently, no significant changes were detected in echocardiographic assessments (Figure S2B, S2C, S2E, and S2F).
Prolylcarboxylpeptidase Deficiency Accelerates Hypertrophic Remodeling in Response to Long‐Term Angiotensin II Infusion
Prolylcarboxylpeptidase was previously proposed to possess a negative regulating function of angiotensin II. 16 Thus, to determine the functional significance of prolylcarboxylpeptidase to the angiotensin II signaling cascades, wild‐type and prolylcarboxylpeptidase‐knockout littermates were subjected to 4 weeks of angiotensin II infusion (2 mg/kg per day−1). Both the tail‐cuff method and invasive hemodynamic assessment showed that although angiotensin II infusion for 28 days resulted in dramatic increases in diastolic and systolic blood pressures in both wild‐type and prolylcarboxylpeptidase‐knockout mice compared with vehicle groups, angiotensin II–treated groups did not elicit significant differences in their blood pressure (Figure 2A and Figure S3A). Despite this, after 4 weeks of angiotensin II infusion, we found significantly enhanced left ventricular hypertrophy in angiotensin II–treated prolylcarboxylpeptidase‐knockout mice, indicated by a significant increase in heart weight over tibial length ratio and the cardiac hypertrophic index (Figure S3B). In addition, the quantification of the cross‐sectional area of cardiomyocytes by wheat germ agglutinin staining showed that greater cell size was present in angiotensin II–treated prolylcarboxylpeptidase‐knockout mice compared with angiotensin II–treated wild‐type (Figure 2B). Moreover, the expressions of the hypertrophic gene markers atrial natriuretic peptide and brain natriuretic peptide were significantly upregulated in prolylcarboxylpeptidase‐knockout mice in comparison with the wild‐type group (Figure S3C and S3D). Additionally, echocardiographic M‐mode assessments revealed that the left ventricular end‐diastolic posterior wall was also significantly increased in angiotensin II–treated prolylcarboxylpeptidase‐knockout mice compared with angiotensin II–treated wild‐type mice, indicating the greater concentric remodeling in the prolylcarboxylpeptidase‐knockout mice (Figure 2C). Notably, within 4 weeks of angiotensin II infusion, prolylcarboxylpeptidase‐knockout mice showed greater diastolic dysfunction, assessed by transmitral Doppler filling, than wild‐type mice since the second week, indicated by significant increases in the E/A ratio and left ventricle isovolumetric relaxation time (Figure 2D, Figure S3E). However, contractile function, determined by fractional shortening, remained unchanged among groups within the first 2 weeks, and that started to progressively decrease from the third week. As a result, the prolylcarboxylpeptidase‐knockout mice had dramatically lower fractional shortening compared with wild‐type littermates after 4 weeks of angiotensin II infusion (Figure 2E). In accordance with the exacerbation of angiotensin II–mediated pathological effects in prolylcarboxylpeptidase‐knockout mice, Masson's trichrome staining demonstrated that interstitial fibrosis area in the angiotensin II–treated prolylcarboxylpeptidase‐knockout mice was almost doubled compared with the angiotensin II–treated wild‐type littermates (Figure 2F). Consistently, upregulation of the transcripts of the fibrosis marker genes procollagen type 1α, procollagen type 3α, and transforming growth factor‐beta 1 were significantly upregulated in prolylcarboxylpeptidase‐knockout ventricles (Figure S3F and S3H).
Figure 2. Prolylcarboxylpeptidase deficiency led to reduced cardiac function with augmented hypertrophic remodeling upon 4 weeks of angiotensin II infusion.

A, Diastolic and systolic blood pressure after 4 weeks of angiotensin II infusion. B, WGA staining of heart cross‐sections (scale bar: 50 μm). C, Left ventricle end‐diastolic posterior wall. D, Mitral Doppler showed the ratio of peak velocity blood flow from left ventricular relaxation in early diastole (the E wave) to peak velocity flow in late diastole caused by atrial contraction (the A wave) indicated worsen diastolic function in prolylcarboxylpeptidase‐knockout mice. E, Fractional shortening (%) was significantly decreased in prolylcarboxylpeptidase‐knockout mice. F, Masson's trichrome‐staining of cross‐sections showed significantly increased interstitial fibrosis in prolylcarboxylpeptidase‐knockout mice compared with the same cohort of control mice (scale bar: 30 μm), followed by quantification of fibrosis area against total tissue area (n=8 mice per group). Data are expressed as means±SEM. Two‐way ANOVA with Bonferroni correction for post hoc comparisons was used for analysis. AngII indicates angiotensin II; PRCP‐KO, prolylcarboxylpeptidase knockout; and WGA, wheat germ agglutinin.
To delineate whether prolylcarboxylpeptidase is required for angiotensin II–induced cardiomyocyte hypertrophy, prolylcarboxylpeptidase expression in adult rat cardiomyocytes was knocked down by small interfering RNA for 48 hours (Figure S4A). Then, the cells were treated with angiotensin II (100 nmol/L) for 48 hours. The result demonstrated that the knockdown of prolylcarboxylpeptidase led to an increase in the cell size in adult rat cardiomyocytes in response to angiotensin II, showing a significantly increased cell surface area concomitant with an ≈1.5‐fold upregulation of atrial natriuretic peptide expression relative to controls (Figure S4B through S4D). These findings together suggest that prolylcarboxylpeptidase is involved in cardioprotection against angiotensin II–induced hypertrophy.
Subcellular Localization of Prolylcarboxylpeptidase and an Excess of Myocardial Angiotensin II Level in Prolylcarboxylpeptidase‐Knockout Hearts
Prolylcarboxylpeptidase was previously reported to localize mainly in lysosomes, where it has the capability of degrading angiotensin II in an acidic condition. 12 , 15 , 16 Therefore, to discern whether loss of prolylcarboxylpeptidase will lead to alterations in angiotensin II and angiotensin 1‐7 levels in response to angiotensin II stress, we first confirmed the localization of prolylcarboxylpeptidase in cardiac lysosomes where lysosomal‐associated membrane protein 1 protein was used as a marker. As expected, coimmunofluorescent staining showed prolylcarboxylpeptidase is colocalized with lysosomal‐associated membrane protein 1 in lysosomes (Figure 3A). This was further confirmed by examining prolylcarboxylpeptidase expression in the isolated cardiac lysosomes. Interestingly, prolylcarboxylpeptidase does not solely reside in lysosomes but also in the cytosol (Figure 3B). Next, we employed ELISA assays to measure alterations of angiotensin II and angiotensin 1‐7 in plasma and myocardial tissues. After 4 weeks of exogenous angiotensin II infusion, angiotensin II and angiotensin 1‐7 significantly increased in both plasma and cardiac tissues in both wild‐type and prolylcarboxylpeptidase‐knockout mice (Figure 3C through 3F). However, prolylcarboxylpeptidase deficiency did not cause any significant change in angiotensin II and angiotensin 1‐7 concentrations in plasma in prolylcarboxylpeptidase‐knockout mice compared with wild‐type mice (Figure 3C and 3D). In contrast, prolylcarboxylpeptidase‐knockout myocardial tissues displayed a significant increase in angiotensin II level compared with wild‐type hearts (Figure 3E). To explore the prolylcarboxylpeptidase‐mediated angiotensin II conversion in vitro, small interfering RNA–mediated knockdown of prolylcarboxylpeptidase was performed in human induced pluripotent stem cells–derived cardiomyocytes, followed by 48 hours of angiotensin II treatment (100 nmol/L). Angiotensin II and angiotensin 1‐7 levels from cell culture media and cell lysis were quantified by ELISA. Consistent with the above results, only the angiotensin II level from the prolylcarboxylpeptidase knockdown group was significantly higher in the control group (Figure S5A through S5D). Moreover, we assessed lysosomal acidic condition–dependent prolylcarboxylpeptidase function by treating induced pluripotent stem cell–derived cardiomyocytes with chloroquine (10 μmol/L), which is a lysosome inhibitor, by altering the acidic environment of lysosomes, for 24 hours, followed by 48 hours of angiotensin II treatment. Quantification of angiotensin II and angiotensin 1‐7 levels from human induced pluripotent stem cell–derived cardiomyocytes showed that angiotensin II but not angiotensin 1‐7 level was significantly increased in the group of chloroquine and angiotensin II treatment (Figure S5E through S5F). These observations suggest that lysosomal prolylcarboxylpeptidase might play an important role in the prevention of excessive accumulation of angiotensin II in the cardiomyocytes.
Figure 3. Prolylcarboxylpeptidase localizes in lysosomes and mediates intracardiac angiotensin II degradation through the lysosomal pathway.
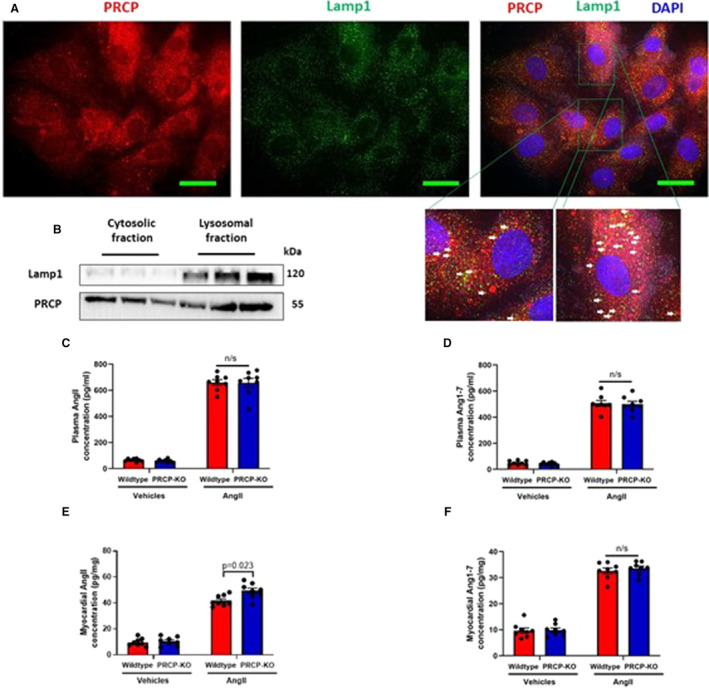
A, Immunostaining of prolylcarboxylpeptidase (red) and Lamp1 (lysosome marker) in H9C2 cells shows prolylcarboxylpeptidase partially resides in lysosomes. White arrows indicate colocalization of prolylcarboxylpeptidase and Lamp1 (yellow dots) (scale bar: 15 μm). B, Cytosolic and lysosomal fractions of wild‐type hearts show the localization of prolylcarboxylpeptidase in both cytosol and lysosome. Lamp1 is used as a marker for lysosomes. C, Angiotensin II levels in plasma. D, Angiotensin 1‐7 levels in plasma. E, Angiotensin II levels in myocardial tissues. F, Angiotensin 1‐7 levels in myocardial tissues (n=8 mice per group). Data are expressed as means±SEM. Two‐way ANOVA with Bonferroni correction for post hoc comparisons was used for analysis. AngII indicates angiotensin II; DAPI, 4′, 6‐diamidino‐2‐phenylindole; Lamp1, lysosomal‐associated membrane protein 1; PRCP, prolylcarboxylpeptidase; and PRCP‐KO, prolylcarboxylpeptidase knockout.
Increased Extracellular Signal‐Regulated Kinases 1/2 Activity and Reduced Protein Kinase B/Endothelial Nitric Oxide Synthase Signaling Lead to Profound Cell Death in Prolylcarboxylpeptidase‐Knockout Hearts
The angiotensin II pathway plays important role in the regulation of oxidative stress and cell death. 3 After 4 weeks of angiotensin II infusion, dihydroethidium staining showed that angiotensin II–treated prolylcarboxylpeptidase‐knockout mice had a greater level of superoxide production than angiotensin II–treated wild‐type mice (Figure 4A). Moreover, the accumulation of reactive oxygen species is known as a critical trigger for cardiomyocyte loss. As expected, terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling staining demonstrated that prolylcarboxylpeptidase‐knockout hearts displayed a pronounced increase in cell death, nearly twice more than that of wild‐type hearts (Figure 4B and 4C). To confirm a higher level of cardiomyocyte loss, molecular pathways responsible for apoptosis were examined. Dramatic upregulation of proapoptotic proteins including p53 and Bax was observed, whereas the expression of antiapoptotic protein, B‐cell lymphoma 2, was downregulated in the prolylcarboxylpeptidase‐knockout hearts. In line with that, the enhanced activity of caspase 3 was detected in association with prolylcarboxylpeptidase deletion (Figure S6A).
Figure 4. Prolylcarboxylpeptidase‐knockout hearts revealed more reactive oxygen species production and apoptosis than wild‐type hearts in response to 2 weeks of angiotensin II infusion.

A, Dihydroethidium staining was used to detect levels of reactive oxygen species production (scale bar: 30 μm), followed by quantification (n=8 per group). B, TUNEL staining assay was applied to detect levels of apoptotic cells in heart cross sections (scale bar: 30 μm). TUNEL: green; nuclei: blue/DAPI; a‐actinin: red. Arrows indicate TUNEL‐positive nuclei. C, The quantification of TUNEL positive nuclei is shown in bar graphs (n=7–8 per group). D, Immunoblot analysis shows expression levels of several mitogen‐activated protein kinase proteins, including eNOS, pPKB, tPKB, pERK1/2, tERK1/2, pJNK, tJNK, pMKK7, tMKK7, pMKK4, tMKK4. Student's t‐test or 2‐way ANOVA with Bonferroni correction for post hoc comparisons was used for analysis. AngII indicates angiotensin II; DAPI, 4′, 6‐diamidino‐2‐phenylindole; eNOS, endothelial nitric oxide synthase; ERK1/2, extracellular signal‐regulated kinases1/2; JNK, c‐Jun N‐terminal kinase; MKK4, mitogen‐activated protein kinase kinase 4; MKK7, mitogen‐activated protein kinase kinase 7; p, phhosphorylation; PKB, protein kinase B; PRCP‐KO, Prolylcarboxylpeptidase knockout; t, total protein; and TUNEL, terminal deoxynucleotidyl transferase‐mediated deoxyuridine triphosphate nick end labeling.
To further dissect the mechanism by which loss of prolylcarboxylpeptidase enhances the hypertrophic response, we examined the expression and phosphorylation of several prohypertrophic MAPKs (mitogen‐activated protein kinases) that are also downstream of the angiotensin II signaling pathway, including ERK1/2, MAPK kinase 7, MAPK kinase 4, and c‐Jun N‐terminal kinase 1/2. While we found the other MAPK proteins expression and phosphorylation levels, including MAPK kinase 4, MAPK kinase 7, c‐Jun N‐terminal kinase 1/2, remained unchanged between wild‐type littermates and prolylcarboxylpeptidase‐knockout mice, prolylcarboxylpeptidase‐knockout hearts demonstrated a significant increase in phosphorylation of ERK1/2 at p44/p42 sites (Figure 4D and Figure S6B). In addition, a previous study has shown that prolylcarboxylpeptidase is critical for PI3K/PKB (phosphoinositide‐3‐kinase/protein kinase B) signaling pathway by stabilizing insulin receptor substrate 1 through which it can maintain survival in breast cancer cells. 17 Using Western blotting, we also discerned that phosphorylation levels of PKB at Ser437 and its downstream target, endothelial nitric oxide synthase, dramatically downregulated in prolylcarboxylpeptidase‐knockout hearts compared with wild‐type hearts (Figure 4D and Figure S6C and S6D). Taken together, our screening results suggest that while the high level of cell death detected in prolylcarboxylpeptidase‐knockout hearts might be attributable to reduced PKB activities, the greater hypertrophic phenotypes might be due to increased ERK1/2 activities.
Adeno‐Associated Virus Serotype 9–Mediated Overexpression of Prolylcarboxylpeptidase Attenuated Hypertrophic Remodeling in Prolylcarboxylpeptidase‐Knockout Hearts
To investigate whether restoration of cardiac prolylcarboxylpeptidase expression would protect the heart from angiotensin II–induced hypertrophic stress, prolylcarboxylpeptidase‐knockout mice were first injected with adeno‐associated virus serotype 9 (AAV9)‐prolylcarboxylpeptidase with a dose of 1×1011 viral genome per mouse via tail‐vein route 3 days in advance, which was followed by 4 weeks of angiotensin II infusion (2 mg/kg per day), and AAV9‐GFP (green fluorescent protein) was used as a control. The cardiac overexpression of exogenous prolylcarboxylpeptidase was regulated under the control of cardiac troponin T promoter to ensure cardiac specificity. The amount of AAV9‐mediated prolylcarboxylpeptidase overexpression in the heart was validated by Western blot (Figure S7A through S7C). After 4 weeks of angiotensin II infusion, the blood pressures were not notably different between angiotensin II–treated groups (Figure 5A). Hence, overexpression of prolylcarboxylpeptidase did not detain raised blood pressures in response to angiotensin II.
Figure 5. AAV9‐mediated restoration of cardiac prolylcarboxylpeptidase expression alleviated the progression of angiotensin II–induced hypertrophic remodeling in the prolylcarboxylpeptidase‐knockout heart.

A, Diastolic and systolic blood pressure. B, Myocardial angiotensin II concentration. C, Wheat germ agglutinin staining of heart cross‐sections (scale bar: 50 μm). D, Echocardiographic assessment of fractional shortening %. E, Masson's trichome staining of representative myocardial sections (scale bar: 45 μm) and quantification of interstitial fibrosis. Data are presented as means±SEM. Student's t‐test or 2‐way ANOVA with Bonferroni correction for post hoc comparisons was used for analysis. AngII indicates angiotensin II; AAV9, adeno‐associated virus serotype 9; GFP, green fluorescent protein; PRCP, prolylcarboxylpeptidase; and PRCP‐KO, prolylcarboxylpeptidase‐knockout.
ELISA assays on myocardial tissues showed that angiotensin II concentration was dramatically decreased by ≈30% in prolylcarboxylpeptidase‐knockout/AAV9‐prolylcarboxylpeptidase compared with that in prolylcarboxylpeptidase‐knockout/AAV9‐GFP mice (Figure 5B). However, overexpression of prolylcarboxylpeptidase had no effects on myocardial angiotensin 1‐7, plasma angiotensin II, and angiotensin 1‐7 concentrations in response to chronic angiotensin II infusion (Figure S8A through S8C).
Regarding cardiac morphology and function, prolylcarboxylpeptidase‐knockout/AAV9‐GFP mice showed a significant increase in heart weight/tibia length compared with prolylcarboxylpeptidase‐knockout/AAV9‐prolylcarboxylpeptidase mice (Figure S8D). Furthermore, a smaller cross‐sectional area of cardiomyocytes was observed in prolylcarboxylpeptidase‐knockout/AAV9‐prolylcarboxylpeptidase mice, compared with prolylcarboxylpeptidase‐knockout/AAV9‐GFP mice (Figure 5C). Moreover, echocardiography demonstrated that prolylcarboxylpeptidase‐knockout/AAV9‐prolylcarboxylpeptidase mice exhibited significant decreases in posterior wall thickness compared with the angiotensin II–treated prolylcarboxylpeptidase‐knockout/AAV9‐GFP mice (Figure S8E). Consistent with these results, the mRNA levels of the hypertrophic gene markers including atrial natriuretic peptide and brain natriuretic peptide were significantly decreased in prolylcarboxylpeptidase‐knockout/AAV9‐prolylcarboxylpeptidase mice in comparison with the controls (Figure S8F and S8G). Regarding cardiac function, cardiac overexpression of prolylcarboxylpeptidase alleviated the progression of diastolic dysfunction, which was indicated by the lower E/A ratio and isovolumetric relaxation time compared with prolylcarboxylpeptidase‐knockout/AAV9‐GFP mice after 4 weeks of angiotensin II infusion (Figure S8H). Also, prolylcarboxylpeptidase‐knockout/AAV9‐prolylcarboxylpeptidase displayed improved contractility compared with prolylcarboxylpeptidase‐knockout/AAV9‐GFP mice (Figure 5D). Masson's trichrome staining illustrated less fibrosis in prolylcarboxylpeptidase‐knockout/AAV9‐prolylcarboxylpeptidase hearts (Figure 5E). Besides, prolylcarboxylpeptidase‐knockout/AAV9‐prolylcarboxylpeptidase hearts showed the downregulation of the fibrosis marker genes including procollagen type 1α and transforming growth factor‐beta (Figure S8I).
In line with this, overexpression of prolylcarboxylpeptidase also attenuated the increases in oxidative stress and cardiomyocyte loss in response to chronic angiotensin II stress. After 4 weeks of angiotensin II infusion, the number of terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling–positive nuclei in the heart of prolylcarboxylpeptidase‐knockout/AAV9‐prolylcarboxylpeptidase mice was almost half that in controls, indicating less cardiomyocyte apoptosis (Figure S8J). Our study further showed that the restoration of prolylcarboxylpeptidase in prolylcarboxylpeptidase‐knockout hearts also blocked downstream effects of angiotensin II overactivation in which increased phosphorylated ERK1/2 and decreased phosphorylated PKB levels were reversed (Figure S8K). Together, prolylcarboxylpeptidase restoration was able to slow down the progress of cardiac hypertrophy and remodeling in response to angiotensin II stress by the control of myocardial angiotensin II level.
Combination Therapy of AAV9‐Mediated Prolylcarboxylpeptidase Overexpression and Losartan Conferred More Cardioprotection Against Angiotensin II–Induced Cardiac Remodeling
Although the restoration of prolylcarboxylpeptidase expression could alleviate the progression of cardiac hypertrophic remodeling and improve cell survival in prolylcarboxylpeptidase‐knockout hearts, it could not correct a high level of plasma angiotensin II and resultant hypertension in response to angiotensin II stress. In addition, angiotensin II can be produced and works at both intracellular and systemic levels, and the notion of the intracardiac angiotensin II regulation by prolylcarboxylpeptidase suggests that lone prolylcarboxylpeptidase use as the monotherapy might not provide a complete treatment regimen by blocking excess angiotensin II activities at different levels. Thus, we further hypothesized that the combination of the gene therapy‐mediated prolylcarboxylpeptidase overexpression with other RAS‐based antihypertensive drugs might reduce the susceptibility to angiotensin II–induced cardiac hypertrophy more effectively. To test this hypothesis, wild‐type C57BL/6J mice were subjected to 4 weeks of angiotensin II (2 mg/kg per day) and of the treatment course, followed by either AAV9‐GFP or AAV9‐prolylcarboxylpeptidase plus losartan (50 mg/kg per day) at the fourth week. The AAV9 was injected 3 days in advance to allow sufficient overexpression of prolylcarboxylpeptidase, and losartan was delivered daily by intraperitoneal injections for 7 days (Figure S9A).
As expected, the wild‐type mice that received either the losartan or the combination treatment groups exhibited a significant reduction in systolic blood pressure compared with other treatment groups (Figure 6A). This indicated that the losartan treatment attenuated the progressive development of hypertension. In addition, echocardiographic assessments showed that AAV9‐mediated prolylcarboxylpeptidase overexpression or losartan treatment could reduce the hypertrophic growth of the posterior wall and mitigate the angiotensin II–induced diastolic and systolic dysfunction compared with angiotensin II–treated wild‐type mice, and these beneficial effects tended to be further enhanced in the combination treatment despite no statistical significance indicated (Figure 6B and 6C, Figure S9B and S9C). In line with this, wheat germ agglutinin staining showed that the combination therapy tends to have a smaller cross‐sectional area compared with the angiotensin II–treated monotherapy groups (Figure 6D). Masson's trichrome and terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling staining showed that angiotensin II–treated wild‐type hearts, which received the combination therapy, tended to display less fibrosis and apoptotic cells compared with those receiving a single AAV9‐prolylcarboxylpeptidase gene therapy (Figure 6E).
Figure 6. The combination therapy of AAV9‐mediated prolylcarboxylpeptidase overexpression and LSRT gave better efficacy than the monotherapies to treat angiotensin II–induced cardiac malfunction.

A, Diastolic and systolic blood pressure. B, Left ventricle end‐diastolic posterior wall. C, Echocardiographic assessment of fractional shortening percentage. D, Wheat germ agglutinin staining of heart cross sections. E, Masson's trichome staining for the quantification of interstitial fibrosis and terminal deoxynucleotidyl transferase‐mediated deoxyuridine triphosphate nick end labeling assay to detect levels of apoptotic cells. F, Angiotensin II levels in plasma and myocardial tissues. n=5 for angiotensin II + LSRT group and n=8 for other groups. Data are presented as means±SEM. One‐ or 2‐way ANOVA with Bonferroni correction for post hoc comparisons was used for analysis. *P value: angiotensin II + AAV9‐prolylcarboxylpeptidase vs angiotensin II + AAV9‐prolylcarboxylpeptidase + LSRT; **P value: angiotensin II + LSRT vs angiotensin II + AAV9‐prolylcarboxylpeptidase + LSRT. AAV9 indicates adeno‐associated virus serotype 9; angiotensin II, angiotensin II; GFP, green fluorescent protein; LSRT, losartan; and PRCP, prolylcarboxylpeptidase.
Interestingly, while the AAV9‐prolylcarboxylpeptidase‐treated group showed comparable levels of plasma angiotensin II to the AAV9‐GFP‐treated group, the combination therapy could reduce the angiotensin II level in plasma. Moreover, prolylcarboxylpeptidase overexpression could alleviate the high level of angiotensin II in myocardial tissues, which was further relieved as combined with losartan (Figure 6F, Figure S9D). Collectively, the combination treatment of AAV9‐mediated prolylcarboxylpeptidase overexpression and losartan might reduce the susceptibility to cardiac hypertrophy induced by angiotensin II more effectively than a single therapy via blood pressure control and myocardial angiotensin II level control. However, longer treatment is required to further validate the efficiency of the combination for the treatment of heart diseases and any potential side effects.
Discussion
Prolylcarboxylpeptidase is a serine protease that can metabolize angiotensin II to generate angiotensin 1‐7 in an acidic condition. With the support of novel technologies, we were able to create various in vivo models including the prolylcarboxylpeptidase‐knockout mice and AAV9‐mediated cardiac prolylcarboxylpeptidase overexpression mice to investigate the cardioprotective roles of prolylcarboxylpeptidase in response to angiotensin II–induced cardiac hypertrophy.
Although our prolylcarboxylpeptidase‐knockout model is not a complete knockout likely due to mosaicism caused by a pronuclear injection of transcript form of CRISPR‐associated protein 9, the pathological phenotypes observed in the heart are evident compared with wild‐type littermates. This highlights the physiological importance of prolylcarboxylpeptidase in the heart. Similar to the global ACE2 knockout model, 18 2‐month‐old prolylcarboxylpeptidase‐knockout mice did not exhibit hypertension and ventricular remodeling at a basal condition, so either the presence of prolylcarboxylpeptidase or ACE2 expression is sufficient to maintain the physiology of the heart. In this study, noninvasive tail‐cuff and invasive hemodynamic assessments showed that prolylcarboxylpeptidase‐knockout mice did not display a significant difference in blood pressure compared with the wild‐type littermates. This phenotype seems to be contradictory to a previous finding by Adams et al, 9 where blood pressure was detected by telemetry and showed notably higher blood pressure in the 6‐month‐old prolylcarboxylpeptidasegt/gt mice compared with the wild‐type mice. However, they did not describe the phenotypes of younger mice. Interestingly, we observed that 4 weeks of angiotensin II infusion did not induce a significant difference in blood pressure between prolylcarboxylpeptidase‐knockout and wild‐type mice despite the hypertension phenotypes observed, which is consistent with a previous result by Maier et al. 12 The difference in basal blood pressure between our results and previous findings could be explained by the genetic background of the mouse strain used to produce the lines. While prolylcarboxylpeptidasegt/gt mice were generated with 129svj background, our prolylcarboxylpeptidase‐knockout mice were produced from the DBA/2J strain. It has been previously reported that 129svj mice develop higher blood pressure compared with C57 mice, whereas blood pressure and cardiac function are within the normal range in the DBA/2J mice. 19 , 20
In addition, our data showed that prolylcarboxylpeptidase expression was significantly increased after 2 weeks of angiotensin II induction despite a decrease in ACE2 expression, which was consistent with previous results. 18 Some studies have reported that either overexpression of angiotensin type 2 receptor (AT2R) or stimulating AT2R with the agonist CGP42112A increased prolylcarboxylpeptidase expression, so AT2R is required for prolylcarboxylpeptidase upregulation, which might be necessarily important during a compensatory stage of cardiac hypertrophy. 21 , 22 Moreover, the downregulation of both ACE2 and prolylcarboxylpeptidase expression after 4 weeks of angiotensin II infusion led to deleterious function in both diastole and systole. Thus, the presence of both ACE2 and prolylcarboxylpeptidase is essential for maintaining cardiac functions under stress conditions.
Prolylcarboxylpeptidase Degrades Angiotensin II at the Cellular Level
The synergetic actions of both ACE2 and prolylcarboxylpeptidase for the metabolism of angiotensin II are essential for preserving the heart function upon angiotensin II stress. However, the exact mechanism remains elusive. ACE2 is classified as an ectoenzyme that is found to be mainly membrane bound and works optimally at a neutral pH. 12 Membranous ACE2 has a catalytic site exposed outward to the extracellular surface, where angiotensin II conversion happens. 23 Loss of ACE2 leads to increased angiotensin II concentration in both plasma and myocardial tissues. 18 In contrast, prolylcarboxylpeptidase is not only found in the membrane and plasma but is also found to locate predominantly in lysosomes, where an acidic pH condition is optimum for its angiotensin II–cleaving activity. 15 , 24 , 25 This might suggest an alternate mechanism of metabolizing angiotensin II in the heart, in which prolylcarboxylpeptidase is likely to degrade angiotensin II intracellularly through a lysosomal pathway rather than extracellularly like ACE2. Consistently, our biochemical analysis demonstrated that loss of prolylcarboxylpeptidase results in increased angiotensin II levels but not angiotensin 1‐7 in myocardial tissues. No change in angiotensin 1‐7 levels suggests that lysosomal angiotensin II degradation may also lead to angiotensin 1‐7 degradation. However, the mechanism of how lysosomal prolylcarboxylpeptidase regulates myocardial angiotensin II level has not been fully elucidated. Several studies showed that the angiotensin II could be degraded through the β‐arrestin1/2–dependent desensitization and endocytosis of the binding complex of angiotensin II and AT1R. 26 , 27 Furthermore, other studies showed that the AT1R–angiotensin II complex was dissociated after endocytosis, in which AT1R is recycled and angiotensin II is transferred to lysosomes via microautophagy. 28 , 29 Thus, lysosomal prolylcarboxylpeptidase might be important in the regulation of angiotensin II circulating in the cardiac microenvironment through AT1R internalization. Additionally, lysosomal prolylcarboxylpeptidase might be critical in regulating intracardiac angiotensin II. Increasing evidence supports the notion that some organs, such as hearts and kidneys, express different components of the RAS, leading to the local angiotensin II synthesis. 13 , 30 Moreover, angiotensin II acts not only as an endocrine/paracrine factor but also as an intracrine peptide, with the ability to trigger signaling cascades in the cell, without stimulating plasma membrane receptors. 31 , 32 Interestingly, Abadir et al 14 described a functional mitochondrial angiotensin system in a variety of cells of human and mouse origin, such as skeletal muscle cells, mouse cardiac myocytes, renal tubular cells, neuronal cells, and hepatocytes, where the colocalization of endogenous angiotensin II and AT2R on the inner mitochondrial membrane was reported to regulate mitochondrial NO production. Moreover, another study confirmed the presence of both AT1R and AT2R in mitochondria from dopaminergic neurons by immunofluorescence and confocal microscopy. 33 Interestingly, a recent study has shown that prolylcarboxylpeptidase plays an important role in mitophagy. 32 These indicate that prolylcarboxylpeptidase might negatively regulate the angiotensin II level inside cardiomyocytes through the mitophagy pathway, where mitochondrial membrane‐bound angiotensin II could be delivered to the lysosome and degraded there.
Our findings demonstrated that loss of prolylcarboxylpeptidase leads to increased cell death, which is an important indicator of cardiac remodeling. An increase in apoptosis is directly derived from elevated reactive oxygen species production. Interestingly, prolylcarboxylpeptidase‐knockout hearts showed less PKB activity upon the angiotensin II stress. Because the phosphoinositide‐3‐kinase/PKB signaling pathway is responsible for cardiomyocyte growth and survival at physiological conditions, impairment of PKB activity can reduce cell survival. 34 , 35 Moreover, another study demonstrated that prolylcarboxylpeptidase inhibition caused destabilization of insulin receptor substrate 1 in response to rapamycin‐induced cytotoxicity, resulting in a disruption in the PKB pathway in pancreatic cancer cells. 17 In addition, several studies indicated that PKB is responsible for physiological cardiomyocyte growth and survival, so the reduced PKB activity might result in maladaptive responses. 36 , 37 In addition, our screening in prolylcarboxylpeptidase‐knockout mice revealed greater levels of phosphorylated ERK1/2. The enhanced ERK1/2 phosphorylation in cardiomyocytes can promote the growth pathway in hearts, resulting in hypertrophic development. 38 , 39 It is worth mentioning that increased ERK1/2 could associate with increased p53 transcriptional activity, enhanced proapoptotic pathways, and inhibited antiapoptotic proteins. 40 , 41 Our data so far suggest that prolylcarboxylpeptidase might protect the heart against angiotensin II–mediated cardiac hypertrophy via its interaction with the RAS at the intracellular level. However, prolylcarboxylpeptidase is a multifunctional peptidase that targets other substrates, which might relate to the involvement of prolylcarboxylpeptidase in the regulation of Des‐Arg 9 ‐bradykinin, (Pyr)‐apelin‐13, and α‐melanocyte‐stimulating hormone 1–13. 42 Thus, it could not be excluded that our prolylcarboxylpeptidase‐knockout mice might have alterations of these peptides, which could contribute to the development of observed phenotypes.
Therapeutic Potential of Prolylcarboxylpeptidase
To investigate the therapeutic potential of cardiac prolylcarboxylpeptidase, AAV9‐mediated restoration of cardiac prolylcarboxylpeptidase expression in prolylcarboxylpeptidase‐knockout hearts by AVV9 was first employed. The study revealed that restoration of cardiac prolylcarboxylpeptidase leads to mitigation of the development of hypertrophic remodeling of prolylcarboxylpeptidase‐knockout hearts in response to angiotensin II infusion. This evidence reinforces our concept that maintenance of cardiac prolylcarboxylpeptidase expression holds a promising therapy for cardiovascular diseases.
Although the single use of gene therapy–mediated prolylcarboxylpeptidase overexpression could ameliorate cardiac performance and hypertrophic remodeling to a certain extent, it could not halt stressed hearts from progressing to failing hearts due to sustained systemic high blood pressure. To reinforce the therapeutic purpose of prolylcarboxylpeptidase in the treatment of cardiovascular diseases, we further designed the experiment as a proof‐of‐concept strategy that blocks angiotensin II action at both systemic and intracellular levels to improve the efficacy of the single use of classic anti‐RAS drugs. We combined the cardiac‐specific overexpression of prolylcarboxylpeptidase and losartan, a classic angiotensin type 1 receptor blocker, and investigated their efficacy compared with the lone treatment. In our study, losartan was chosen because it is more advantageous than an ACEi by not affecting the bradykinin pathway and having a better tolerability profile. 43 , 44 , 45 In contrast, ACEi has been shown to block the degradation of bradykinin, leading to an increase in bradykinin levels and troublesome side effects in patients such as dry cough and angioedema. Therefore, the combined use of prolylcarboxylpeptidase and ACEi might cause an overload of bradykinin levels and induce undesirable side effects. Additionally, angiotensin II breakthrough and aldosterone breakthrough are described as factors that potentially happen during long‐term use of ACEis. 46 In addition, the generation of plasma and tissue angiotensin II by non–ACE‐related enzymes such as chymase suggests that alternative options of complement therapy may be required in the future of the RAS‐based new drug discovery. Our observations in the combinatory use of prolylcarboxylpeptidase overexpression and losartan against angiotensin II–induced cardiac hypertrophy provide the first line of evidence supporting a novel notion for the treatment of heart disease via limiting the pathological effects of an activated systemic and local RAS. However, our combination treatment was conducted for only 7 days, so this outcome might not reflect the complete picture of the long‐term treatment. For example, a long‐term losartan treatment might lead to an unprecedented surge of plasma angiotensin II, which was not detected in our experimental setting. 47 Hence, this work, hopefully, will inspire further studies to investigate the synergistic functions of each component of the RAS signaling pathway that can provide a refined therapeutic guideline.
Conclusions
The present study demonstrates that prolylcarboxylpeptidase exerts a cardioprotective effect against angiotensin II stress‐induced hypertrophic remodeling through the negative regulation of angiotensin II at the tissue level. This finding thus adds another level of complexity to the RAS signaling pathway in the heart. With chronic heart diseases, excessive angiotensin II levels can occur in both tissues and blood, and we believe that the treatment of a single agent, either an ACEi or angiotensin type 1 receptor blocker, might only delay the development of heart failure through the management of hypertension caused by systemic angiotensin II. Thus, to provide a more comprehensive therapy, we combined the gene therapy–mediated cardiac‐specific prolylcarboxylpeptidase overexpression with losartan, an angiotensin type 1 receptor blocker, to evaluate whether this combination therapy could protect angiotensin II–induced stressed hearts more effectively. Hence, the synergetic actions of exogenous cardiac prolylcarboxylpeptidase and losartan have limited adverse myocardial remodeling through the controls of tissue angiotensin II level and hypertensive effects of plasma angiotensin II (Figure 7). We conclude that prolylcarboxylpeptidase plays an important role in the alleviation of myocardial hypertrophy, and the use of prolylcarboxylpeptidase represents a novel therapeutic strategy for cardiovascular diseases.
Figure 7. Schematic figure proposing prolylcarboxylpeptidase cardioprotection against hypertensive cardiac remodeling.

A, At a normal state, the cardiac function is regulated by angiotensin II, which is produced in the circulatory system and in cardiomyocytes. ACE2 and prolylcarboxylpeptidase work synergically to protect the hearts from the overaction of angiotensin II at both systemic and intracellular levels. B, In hypertensive diseases, the angiotensin II levels, however, are significantly upregulated due to loss of cardiac ACE2 and prolylcarboxylpeptidase expressions, leading to the development of hypertrophic remodeling and heart failure. C, In this study, we proposed that a combination treatment of AAV9‐mediated overexpression of prolylcarboxylpeptidase and losartan could ameliorate cardiac functions by improving the regulation of angiotensin II at both systemic and intracellular levels. AAV9 indicates adeno‐associated virus serotype 9; ACE, angiotensin‐converting enzyme; ACE2, angiotensin‐converting enzyme 2; AGT, angiotensinogen; AngII, angiotensin II; and ROS, reactive oxygen species.
Sources of Funding
This study was supported by the British Heart Foundation (PG/17/31/32988 and PG/19/53/34499 to X. Wang).
Disclosures
None.
Supporting information
This manuscript was sent to Daniel T. Eitzman, MD, Senior Guest Editor, for review by expert referees, editorial decision, and final disposition.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.122.028298
For Sources of Funding and Disclosures, see page 16.
References
- 1. Santos RAS, Oudit GY, Verano‐Braga T, Canta G, Steckelings UM, Bader M. The renin‐angiotensin system: going beyond the classical paradigms. Am J Physiol Heart Circ Physiol. 2019;316:H958–H970. doi: 10.1152/ajpheart.00723.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zaman MA, Oparil S, Calhoun DA. Drugs targeting the renin‐angiotensin‐aldosterone system. Nat Rev Drug Discov. 2002;1:621–636. doi: 10.1038/nrd873 [DOI] [PubMed] [Google Scholar]
- 3. Paz Ocaranza M, Riquelme JA, Garcia L, Jalil JE, Chiong M, Santos RAS, Lavandero S. Counter‐regulatory renin‐angiotensin system in cardiovascular disease. Nat Rev Cardiol. 2020;17:116–129. doi: 10.1038/s41569-019-0244-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Silva‐Aguiar RP, Peruchetti DB, Rocco PRM, Schmaier AH, PMR ES, Martins MA, Carvalho VF, Pinheiro AAS, Caruso‐Neves C. Role of the renin‐angiotensin system in the development of severe covid‐19 in hypertensive patients. Am J Physiol Lung Cell Mol Physiol. 2020;319:L596–L602. doi: 10.1152/ajplung.00286.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hemnes AR, Rathinasabapathy A, Austin EA, Brittain EL, Carrier EJ, Chen X, Fessel JP, Fike CD, Fong P, Fortune N, et al. A potential therapeutic role for angiotensin‐converting enzyme 2 in human pulmonary arterial hypertension. Eur Respir J. 2018;51:1702638. doi: 10.1183/13993003.02638-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Khan A, Benthin C, Zeno B, Albertson TE, Boyd J, Christie JD, Hall R, Poirier G, Ronco JJ, Tidswell M, et al. A pilot clinical trial of recombinant human angiotensin‐converting enzyme 2 in acute respiratory distress syndrome. Crit Care. 2017;21:234. doi: 10.1186/s13054-017-1823-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heyman SN, Kinaneh S, Abassi Z. The duplicitous nature of ACE2 in covid‐19 disease. EBioMedicine. 2021;67:103356. doi: 10.1016/j.ebiom.2021.103356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Serfozo P, Wysocki J, Gulua G, Schulze A, Ye M, Liu P, Jin J, Bader M, Myohanen T, Garcia‐Horsman JA, et al. Ang II (angiotensin II) conversion to angiotensin‐(1‐7) in the circulation is POP (prolyloligopeptidase)‐dependent and ACE2 (angiotensin‐converting enzyme 2)‐independent. Hypertension. 2020;75:173–182. doi: 10.1161/HYPERTENSIONAHA.119.14071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Adams GN, LaRusch GA, Stavrou E, Zhou Y, Nieman MT, Jacobs GH, Cui Y, Lu Y, Jain MK, Mahdi F, et al. Murine prolylcarboxypeptidase depletion induces vascular dysfunction with hypertension and faster arterial thrombosis. Blood. 2011;117:3929–3937. doi: 10.1182/blood-2010-11-318527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marangoni RA, Santos RA, Piccolo C. Deficient prolylcarboxypeptidase gene and protein expression in left ventricles of spontaneously hypertensive rats (SHR). Peptides. 2014;61:69–74. doi: 10.1016/j.peptides.2014.08.016 [DOI] [PubMed] [Google Scholar]
- 11. Liu J, Hakucho A, Fujimiya T. Angiotensinase C mRNA and protein downregulations are involved in ethanol‐deteriorated left ventricular systolic dysfunction in spontaneously hypertensive rats. Biomed Res Int. 2015;2015:409350. doi: 10.1155/2015/409350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maier C, Schadock I, Haber PK, Wysocki J, Ye M, Kanwar Y, Flask CA, Yu X, Hoit BD, Adams GN, et al. Prolylcarboxypeptidase deficiency is associated with increased blood pressure, glomerular lesions, and cardiac dysfunction independent of altered circulating and cardiac angiotensin II. J Mol Med (Berl). 2017;95:473–486. doi: 10.1007/s00109-017-1513-9 [DOI] [PubMed] [Google Scholar]
- 13. De Mello WC, Frohlich ED. On the local cardiac renin angiotensin system. Basic and clinical implications. Peptides. 2011;32:1774–1779. [DOI] [PubMed] [Google Scholar]
- 14. Abadir PM, Foster DB, Crow M, Cooke CA, Rucker JJ, Jain A, Smith BJ, Burks TN, Cohn RD, Fedarko NS, et al. Identification and characterization of a functional mitochondrial angiotensin system. Proc Natl Acad Sci USA. 2011;108:14849–14854. doi: 10.1073/pnas.1101507108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jackman HL, Tan F, Schraufnagel D, Dragovic T, Dezso B, Becker RP, Erdos EG. Plasma membrane‐bound and lysosomal peptidases in human alveolar macrophages. Am J Respir Cell Mol Biol. 1995;13:196–204. doi: 10.1165/ajrcmb.13.2.7626287 [DOI] [PubMed] [Google Scholar]
- 16. De Hert E, Bracke A, Lambeir AM, Van der Veken P, De Meester I. The c‐terminal cleavage of angiotensin II and III is mediated by prolyl carboxypeptidase in human umbilical vein and aortic endothelial cells. Biochem Pharmacol. 2021;192:114738. doi: 10.1016/j.bcp.2021.114738 [DOI] [PubMed] [Google Scholar]
- 17. Duan L, Motchoulski N, Danzer B, Davidovich I, Shariat‐Madar Z, Levenson VV. Prolylcarboxypeptidase regulates proliferation, autophagy, and resistance to 4‐hydroxytamoxifen‐induced cytotoxicity in estrogen receptor‐positive breast cancer cells. J Biol Chem. 2011;286:2864–2876. doi: 10.1074/jbc.M110.143271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhong J, Basu R, Guo D, Chow FL, Byrns S, Schuster M, Loibner H, Wang XH, Penninger JM, Kassiri Z, et al. Angiotensin‐converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation. 2010;122:717–728, 718 p following 728. doi: 10.1161/CIRCULATIONAHA.110.955369 [DOI] [PubMed] [Google Scholar]
- 19. Lum C, Shesely EG, Potter DL, Beierwaltes WH. Cardiovascular and renal phenotype in mice with one or two renin genes. Hypertension. 2004;43:79–86. doi: 10.1161/01.HYP.0000107401.72456.50 [DOI] [PubMed] [Google Scholar]
- 20. Zhao W, Zhao T, Chen Y, Zhao F, Gu Q, Williams RW, Bhattacharya SK, Lu L, Sun Y. A murine hypertrophic cardiomyopathy model: the DBA/2J strain. PLoS One. 2015;10:e0133132. doi: 10.1371/journal.pone.0133132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhu L, Carretero OA, Liao TD, Harding P, Li H, Sumners C, Yang XP. Role of prolylcarboxypeptidase in angiotensin II type 2 receptor‐mediated bradykinin release in mouse coronary artery endothelial cells. Hypertension. 2010;56:384–390. doi: 10.1161/HYPERTENSIONAHA.110.155051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhu L, Carretero OA, Xu J, Wang L, Harding P, Rhaleb NE, Yang JJ, Sumners C, Yang XP. Angiotensin II type 2 receptor‐stimulated activation of plasma prekallikrein and bradykinin release: role of shp‐1. Am J Physiol Heart Circ Physiol. 2012;302:H2553–H2559. doi: 10.1152/ajpheart.01157.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Patel VB, Zhong JC, Grant MB, Oudit GY. Role of the ACE2/angiotensin 1‐7 axis of the renin‐angiotensin system in heart failure. Circ Res. 2016;118:1313–1326. doi: 10.1161/CIRCRESAHA.116.307708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shariat‐Madar Z, Mahdi F, Schmaier AH. Recombinant prolylcarboxypeptidase activates plasma prekallikrein. Blood. 2004;103:4554–4561. doi: 10.1182/blood-2003-07-2510 [DOI] [PubMed] [Google Scholar]
- 25. de Voer G, Peters D, Taschner PE. Caenorhabditis elegans as a model for lysosomal storage disorders. Biochim Biophys Acta. 2008;1782:433–446. doi: 10.1016/j.bbadis.2008.04.003 [DOI] [PubMed] [Google Scholar]
- 26. Hunyady L, Catt KJ, Clark AJ, Gaborik Z. Mechanisms and functions of AT(1) angiotensin receptor internalization. Regul Pept. 2000;91:29–44. doi: 10.1016/S0167-0115(00)00137-3 [DOI] [PubMed] [Google Scholar]
- 27. Bkaily G, Sleiman S, Stephan J, Asselin C, Choufani S, Kamal M, Jacques D, Gobeil F Jr, DOrleans‐Juste P. Angiotensin II AT1 receptor internalization, translocation and de novo synthesis modulate cytosolic and nuclear calcium in human vascular smooth muscle cells. Can J Physiol Pharmacol. 2003;81:274–287. doi: 10.1139/y03-007 [DOI] [PubMed] [Google Scholar]
- 28. Hein L, Barsh GS, Pratt RE, Dzau VJ, Kobilka BK. Behavioural and cardiovascular effects of disrupting the angiotensin II type‐2 receptor in mice. Nature. 1995;377:744–747. doi: 10.1038/377744a0 [DOI] [PubMed] [Google Scholar]
- 29. Deliu E, Tica AA, Motoc D, Brailoiu GC, Brailoiu E. Intracellular angiotensin II activates rat myometrium. Am J Physiol Cell Physiol. 2011;301:C559–C565. doi: 10.1152/ajpcell.00123.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ferrario CM. Cardiac remodelling and ras inhibition. Ther Adv Cardiovasc Dis. 2016;10:162–171. doi: 10.1177/1753944716642677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kumar R, Singh VP, Baker KM. The intracellular renin‐angiotensin system: a new paradigm. Trends Endocrinol Metab. 2007;18:208–214. doi: 10.1016/j.tem.2007.05.001 [DOI] [PubMed] [Google Scholar]
- 32. Re RN, Cook JL. Mechanisms of disease: intracrine physiology in the cardiovascular system. Nat Clin Pract Cardiovasc Med. 2007;4:549–557. doi: 10.1038/ncpcardio0985 [DOI] [PubMed] [Google Scholar]
- 33. Valenzuela R, Costa‐Besada MA, Iglesias‐Gonzalez J, Perez‐Costas E, Villar‐Cheda B, Garrido‐Gil P, Melendez‐Ferro M, Soto‐Otero R, Lanciego JL, Henrion D, et al. Mitochondrial angiotensin receptors in dopaminergic neurons. Role in cell protection and aging‐related vulnerability to neurodegeneration. Cell Death Dis. 2016;7:e2427. doi: 10.1038/cddis.2016.327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kang PM, Izumo S. Apoptosis in heart: basic mechanisms and implications in cardiovascular diseases. Trends Mol Med. 2003;9:177–182. doi: 10.1016/S1471-4914(03)00025-X [DOI] [PubMed] [Google Scholar]
- 35. Olivares‐Reyes JA, Arellano‐Plancarte A, Castillo‐Hernandez JR. Angiotensin II and the development of insulin resistance: implications for diabetes. Mol Cell Endocrinol. 2009;302:128–139. doi: 10.1016/j.mce.2008.12.011 [DOI] [PubMed] [Google Scholar]
- 36. Walsh K. Akt signaling and growth of the heart. Circulation. 2006;113:2032–2034. doi: 10.1161/CIRCULATIONAHA.106.615138 [DOI] [PubMed] [Google Scholar]
- 37. Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol. 2013;14:38–48. doi: 10.1038/nrm3495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barry SP, Davidson SM, Townsend PA. Molecular regulation of cardiac hypertrophy. Int J Biochem Cell Biol. 2008;40:2023–2039. doi: 10.1016/j.biocel.2008.02.020 [DOI] [PubMed] [Google Scholar]
- 39. Mutlak M, Kehat I. Extracellular signal‐regulated kinases 1/2 as regulators of cardiac hypertrophy. Front Pharmacol. 2015;6:149. doi: 10.3389/fphar.2015.00149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lu Z, Xu S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life. 2006;58:621–631. doi: 10.1080/15216540600957438 [DOI] [PubMed] [Google Scholar]
- 41. Liu J, Mao W, Ding B, Liang CS. ERKS/p53 signal transduction pathway is involved in doxorubicin‐induced apoptosis in H9c2 cells and cardiomyocytes. Am J Physiol Heart Circ Physiol. 2008;295:H1956–H1965. doi: 10.1152/ajpheart.00407.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kehoe K, Van Elzen R, Verkerk R, Sim Y, Van der Veken P, Lambeir AM, De Meester I. Prolyl carboxypeptidase purified from human placenta: its characterization and identification as an apelin‐cleaving enzyme. Biochim Biophys Acta. 2016;1864:1481–1488. doi: 10.1016/j.bbapap.2016.07.004 [DOI] [PubMed] [Google Scholar]
- 43. McDowell SE, Coleman JJ, Ferner RE. Systematic review and meta‐analysis of ethnic differences in risks of adverse reactions to drugs used in cardiovascular medicine. BMJ. 2006;332:1177–1181. doi: 10.1136/bmj.38803.528113.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bangalore S, Kumar S, Wetterslev J, Messerli FH. Angiotensin receptor blockers and risk of myocardial infarction: meta‐analyses and trial sequential analyses of 147 020 patients from randomised trials. BMJ. 2011;342:d2234. doi: 10.1136/bmj.d2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Saglimbene V, Palmer SC, Ruospo M, Natale P, Maione A, Nicolucci A, Vecchio M, Tognoni G, Craig JC, Pellegrini F, et al. The long‐term impact of renin‐angiotensin system (RAS) inhibition on cardiorenal outcomes (LIRICO): a randomized, controlled trial. J Am Soc Nephrol. 2018;29:2890–2899. doi: 10.1681/ASN.2018040443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bomback AS, Klemmer PJ. The incidence and implications of aldosterone breakthrough. Nat Clin Pract Nephrol. 2007;3:486–492. doi: 10.1038/ncpneph0575 [DOI] [PubMed] [Google Scholar]
- 47. Minas JN, Thorwald MA, Conte D, Vazquez‐Medina JP, Nishiyama A, Ortiz RM. Angiotensin and mineralocorticoid receptor antagonism attenuates cardiac oxidative stress in angiotensin II‐infused rats. Clin Exp Pharmacol Physiol. 2015;42:1178–1188. doi: 10.1111/1440-1681.12473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu W, Zi M, Jin J, Prehar S, Oceandy D, Kimura TE, Lei M, Neyses L, Weston AH, Cartwright EJ, et al. Cardiac‐specific deletion of mkk4 reveals its role in pathological hypertrophic remodeling but not in physiological cardiac growth. Circ Res. 2009;104:905–914. doi: 10.1161/CIRCRESAHA.108.188292 [DOI] [PubMed] [Google Scholar]
- 49. Jungmann A, Leuchs B, Rommelaere J, Katus HA, Muller OJ. Protocol for efficient generation and characterization of adeno‐associated viral vectors. Hum Gene Ther Methods. 2017;28:235–246. doi: 10.1089/hgtb.2017.192 [DOI] [PubMed] [Google Scholar]
- 50. Binder P, Wang S, Radu M, Zin M, Collins L, Khan S, Li Y, Sekeres K, Humphreys N, Swanton E, et al. Pak2 as a novel therapeutic target for cardioprotective endoplasmic reticulum stress response. Circ Res. 2019;124:696–711. doi: 10.1161/CIRCRESAHA.118.312829 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.